Ru–Zn supported on hydroxyapatite as an effective catalyst for partial hydrogenation of benzene†
Peng
Zhang
,
Tianbin
Wu
,
Tao
Jiang
,
Weitao
Wang
,
Huizhen
Liu
,
Honglei
Fan
,
Zhaofu
Zhang
and
Buxing
Han
*
Beijing National Laboratory for Molecular Sciences (BNLMS), CAS Key Laboratory of Colloid and Interface and Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: Hanbx@iccas.ac.cn; Tel: +86 10 62562821
First published on 25th October 2012
Abstract
Design and preparation of efficient and greener catalytic systems for partial hydrogenation of benzene to cyclohexene is an interesting topic in green chemistry. In this work, Ru and Ru–Zn catalysts supported on hydroxyapatite (HAP), which is nontoxic and abundant in nature, were prepared via the simple ion-exchange method. The catalysts were characterized by powder X-ray diffraction (XRD), transmission electron spectroscopy (TEM), X-ray photoelectron spectroscopy (XPS) and nitrogen adsorption–desorption methods. The influences of Ru/Zn molar ratio, reaction temperature, pressure, reaction time, and amount of modifier NaOH on the partial hydrogenation of benzene were studied in detail. It was demonstrated that metallic nanoparticles of less than 2 nm were dispersed uniformly on the surface of the HAP, and the bimetallic Ru–Zn/HAP catalysts showed high activity and selectivity. The yield of cyclohexene could reach 33% over Ru–Zn/HAP at the optimized conditions, and the catalyst could be reused at least four times without obvious loss of the activity and selectivity.
Introduction
Cyclohexene is the raw material for producing adipic acid, Nylon 6, Nylon 66, caprolactam, and other fine chemicals. The traditional routes for producing cyclohexene, such as dehydrogenation of cyclohexane, dehydration of cyclohexanol and dehalogenation of cyclohexane halide, suffer from complicated procedures, high energy consumption, and low efficiency.1–3 Compared to other routes, preparation of cyclohexene by benzene partial hydrogenation is superior in that the feedstock is abundant and inexpensive, and the procedures are simple. However, hydrogenation of benzene to cyclohexane is much easier than that of benzene to cyclohexene (Gibbs free energy: benzene → cyclohexane −98 kJ mol−1, benzene → cyclohexene −23 kJ mol−1). Therefore, achieving a high yield of cyclohexene is very difficult.Much research has been devoted to this reaction in the past few decades, focusing on heterogeneous catalysis using Ru based catalysts in a four-phase reaction system.4–8 This reaction system consists of vapor (hydrogen), oil (benzene and products), aqueous (water and modifiers) and solid (catalyst) phases. Water in the system is important for enhancing the selectivity of cyclohexene because the solubility of cyclohexene in water is rather low, and therefore cyclohexene is withdrawn from the surface of the hydrophilic catalyst and further hydrogenation is inhibited. It has been proved that the modifiers, which are dissolved in water, can improve the cyclohexene selectivity effectively.9 Various substances have been used as the modifiers, including inorganic salts,10 organic compounds,11,12 ionic liquids,13etc. Struijk et al.10 studied the effect of a variety of inorganic salts on the reaction. Liu et al.14 employed RuLa/SBA-15 as the catalyst incorporating ZnSO4 and CdSO4 as co-modifiers. Fan et al.11 and Sun et al.12 investigated the effect of organic modifiers on the reaction. Schwab et al.13 utilized small amounts (ppm range) of ionic liquid with [N(CN)2]− as the modifier to block the consecutive hydrogenation to cyclohexane. It has also been reported that adding some alkaline agents into the reaction system, especially sodium hydroxide (NaOH), enhances the selectivity to cyclohexene.5,6,15 NaOH is capable of improving the hydrophilicity of the catalyst, and reducing the rate of cyclohexene hydrogenation. Ronchin and Toniolo5 have found that the benzene hydrogenation rate, selectivity and yield of cyclohexene can be enhanced by treating the unreduced catalyst with NaOH.
Preparing effective catalysts taking both activity and selectivity into consideration is critical to this reaction. A series of unsupported catalysts has been prepared by the co-precipitation method and the direct reduction method.16 It is known that the supported Ru catalysts are advantageous in that less Ru is required. Niwa et al.17 prepared Ru–Cu catalysts supported on SiO2 by the chemical mixing procedure. Qiao and co-workers18,19 synthesized a series of catalysts supported on SBA-15. It has been demonstrated by different researchers that addition of zinc,20 boron,21 copper,6 cerium,19 lanthanum,14 barium,18 cobalt,22 iron,23 manganese24 can improve the selectivity towards cyclohexene effectively. In addition, the hydrophilicity of the support is also an important factor because the catalysts are required to disperse in aqueous phase in the reaction. Up to now, a variety of hydrophilic materials have been employed as the supports, such as SiO2,25 Al2O3,26 ZrO2,20 ZnO,6 clays,8etc. Cobo and co-workers27 reported that Ru/C catalysts containing a larger quantity of hydrophilic functional groups give a higher yield of cyclohexene.
Hydroxyapatite (Ca10(PO4)6(OH)2, HAP) is the main component of bones and teeth; it is nontoxic and abundantly available in nature. It is receiving more and more attention for potential use as biomaterials, adsorbents, and ion-exchangers. In recent years, utilization of HAP as the support of catalysts has also attracted much attention. It has been reported that Co,28 Ru,29 Pd,30 Au,31 Cu32 and Zn33 supported on HAP exhibit excellent catalytic performances in the oxidation and dihydroxylation of alkenes, synthesis of imine, cross-coupling reactions, and CO2 cycloaddition reactions. Zahmakıran et al.34,35 prepared Ru(0) and Rh(0) nanoclusters supported on HAP for hydrogenation of aromatics to their corresponding cyclohexane derivatives under mild conditions.
Design and preparation of highly efficient, greener and economical catalytic systems for partial hydrogenation of benzene to cyclohexene is an interesting topic in green chemistry. HAP has high ion-exchange and adsorption abilities, and therefore the active species can be immobilized on its surface. Its nonporous structure makes the mass transfer easier in the reactions. The low surface acid–base property prevents the side reactions induced by the support itself.36 Moreover, HAP contains abundant hydroxyl groups on its surface, which makes it well dispersed in aqueous phase. All of these properties indicate that HAP is a promising catalyst support for partial hydrogenation of benzene to cyclohexene. However, to the best of our knowledge, HAP as a catalyst support for this reaction has not been reported. In this work, we prepared bimetallic Ru–Zn catalysts supported on HAP by a simple ion-exchange method. It was demonstrated that the catalysts were very active, selective, and stable for the reaction.
Experimental
Materials
Ruthenium(III) chloride trihydrate (RuCl3·3H2O), zinc sulfate heptahydrate (ZnSO4·7H2O), sodium hydroxide (NaOH), dichloromethane, benzene, cyclohexene, cyclohexane were all A. R. grade and purchased from Sinopharm Chemical Reagent Co. Ltd. Hydroxyapatite ([Ca5(PO4)3OH], A. R. grade) was obtained from Aldrich and used as received. H2 (99.99%) was provided by Beijing Analytical Instrument Company.Catalyst preparation
The bimetallic Ru–Zn catalysts supported on HAP with different Ru/Zn molar ratios were prepared by the ion-exchange method. In a typical procedure, 0.5 g HAP was dispersed in 150 mL double deionized water under vigorous stirring for 2 h. Then a certain amount of RuCl3 aqueous solution (Ru: 2.5 wt%) with different amounts of ZnSO4 was added into the HAP suspension. After being stirred for 2 h at room temperature, the slurry was heated and refluxed at 100 °C for 12 h. The obtained solid was collected by filtration and washed with deionized water until the Cl− and SO42− were not detectable. The as-prepared catalyst was dried under vacuum at 60 °C overnight and was then reduced in pure H2 (99.99%, 60 cm3 min−1) at 300 °C for 1 h. The catalysts are denoted as Ru–Zn/HAP-x, where x indicates the nominal Ru/Zn molar ratio. The catalysts without Zn or Ru are denoted as Ru/HAP and Zn/HAP, respectively. When the nominal Ru/Zn molar ratio in the catalyst was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the actual loadings of Ru and Zn analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) were 2.54 wt% and 1.66 wt%, respectively, which corresponded to a molar ratio of 1:1, indicating that all the metal ions were exchanged onto the HAP at the experimental conditions.
:1, the actual loadings of Ru and Zn analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) were 2.54 wt% and 1.66 wt%, respectively, which corresponded to a molar ratio of 1:1, indicating that all the metal ions were exchanged onto the HAP at the experimental conditions.
For comparison, Ru–Zn/MgO, Ru–Zn/CeO2, and Ru–Zn/ZrO2 catalysts were prepared by the incipient wetness impregnation method. The Ru content was 2.5 wt% and the nominal Ru/Zn molar ratios for these catalysts were 1:1.
Catalyst characterization
The contents of the Ru and Zn in the catalysts were analyzed by ICP-AES (PROFILE. SPEC, Leeman). Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku D/max-2500 X-ray diffractometer using Cu Kα radiation (λ = 0.15406 nm). The tube voltage was 40 kV and the current was 200 mA. The specific surface area of the samples was determined by the N2 adsorption technique (Quantachrome Autosorb-1). The samples were degassed at 573 K for 12 h and adsorption–desorption isotherms were measured at 77 K. The structural properties were characterized by transmission electron microscopy (TEM, JEOL JEM-2100F). The X-ray photoelectron spectroscopy (XPS) data were obtained with an ESCALab 220i-XL electron spectrometer from VG Scientific using 300 W Al Kα radiation. The base pressure was about 3 × 10−9 mbar. The binding energies were referenced to the C 1s line at 284.8 eV from adventitious carbon.Reaction
The partial hydrogenation of benzene was performed in a 6 mL Teflon-lined stainless steel autoclave equipped with a magnetic stirrer, which was similar to that used previously.37 In a typical experiment, 0.5 mL benzene, 20 mg catalyst, and 1.5 mL aqueous sodium hydroxide solution were introduced into the reactor. After being purged by low pressure hydrogen several times to remove the air, the autoclave was heated to the reaction temperature. Then hydrogen was charged into the reactor to the desired pressure and the reaction mixture was vigorously stirred. During the reaction hydrogen was continuously supplied to maintain the pressure. After a suitable reaction time, the reactor was placed in ice water to quench the reaction and n-hexane was added as an internal standard. The organic phase was extracted from the mixture by dichloromethane, and analyzed by an Agilent 6890 gas chromatograph equipped with an HP-INNOWax capillary column and an FID.Reuse of the catalysts
To test the reusability, the catalyst was separated from the liquid by centrifugation, washed with water until pH = 7 and dried under vacuum at 60 °C, and was then directly reused for the next run without further reduction.Results and discussion
Catalyst characterization
Fig. 1 shows the XRD patterns of the HAP and Ru–Zn/HAP catalysts with different Ru/Zn molar ratios. It can be seen that all the catalysts exhibited Ca10(PO4)6(OH)2 phase, indicating that there were no transformations of the catalyst support framework during the catalyst preparation. In addition, the XRD patterns of Ru–Zn/HAP catalysts were in good agreement with the parent HAP, suggesting that the ion-exchange had no obvious influence on the crystallinity of the HAP support. The diffraction peak at 2θ = 44° for Ru was not observed, mainly due to the small size and high dispersion of the Ru particles on the HAP.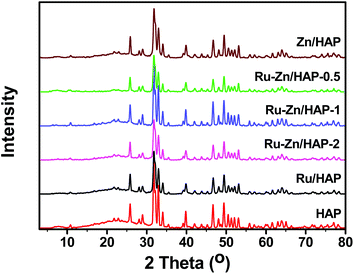 | ||
| Fig. 1 XRD patterns of HAP and Ru–Zn/HAP catalysts with different Ru/Zn ratios. | ||
Surface areas of HAP and Ru–Zn/HAP-1 were measured by N2 physisorption. The adsorption–desorption isotherms are given in Fig. S1.† The specific surface area of the Ru–Zn/HAP-1 was 50.3 m2 g−1, and that of the parent HAP was 48.0 m2 g−1. Both of the samples showed type H3 sorption hysteresis loops, and BJH pore size distribution curves both give the maximum at 150 Å (pore radius). This is consistent with the non-porous nature of the HAP.
The TEM images of Ru/HAP, Zn/HAP, and Ru–Zn/HAP-1 are shown in Fig. 2a–d. It can be seen from Fig. 2a and 2c that the Ru and Ru–Zn nanoparticles were uniformly dispersed on the surface of the HAP, indicating that the HAP had excellent ability to absorb the precursors and prevent the aggregation of metallic particles during the reduction process at 300 °C. No metallic Zn particles were observed in the TEM image of the Zn/HAP (Fig. 2b). Fig. 2d shows the high resolution TEM (HRTEM) image of the Ru–Zn/HAP-1 and the histogram of Ru–Zn particle size distribution. The average diameter of the Ru–Zn particles on the Ru–Zn/HAP-1 was around 1.6 nm.
 | ||
| Fig. 2 TEM images of Ru/HAP (a), Zn/HAP (b), Ru–Zn/HAP-1 (c), HRTEM and Ru particle size distribution of Ru–Zn/HAP-1 (d). | ||
The XPS spectra of the Ru–Zn/HAP-1 catalyst are shown in Fig. 3. No peak for Cl 2p or S 2p was observed, showing that the Cl− and SO42− were completely removed during washing. The narrow scan spectra of Ru 3d and Zn 2p are shown in Fig. 3b and 3c. In Fig. 3b, the peak centered at 281.0 eV was assigned to Ru(0) 3d5/2, demonstrating that the metallic Ru was formed during the reduction process. No peak for Ru2+, Ru3+, or Ru4+ was observed in the spectrum. The peak of Ru 3d3/2 overlapped with the C 1s peak. For comparison, the Ru 3d spectrum of the Ru/HAP is given in Fig. S2.† The binding energies (BE) of Ru(0) 3d5/2 in Ru–Zn/HAP-1 (281.0 eV) and Ru/HAP (281.2 eV) were higher than that of metallic Ru (280.0 eV).38 The higher energy value may be attributed to the small particle size of the Ru (∼1.6 nm), the electron properties of the HAP framework,36 and the oxidation of Ru particles during the XPS sampling procedure. Furthermore, there existed interactions between Ru and the oxygen in the HAP framework,35 the electronic interaction makes the Ru surface electron-deficient. This would increase the BE of Ru(0) particles.
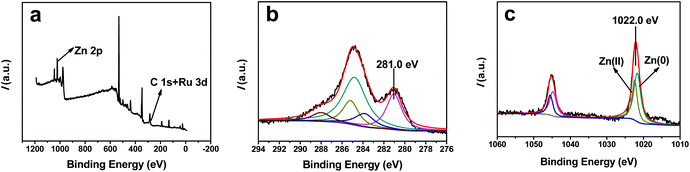 | ||
| Fig. 3 XPS spectra of the Ru–Zn/HAP-1 catalyst (a) full spectrum; (b) Ru 3d spectrum; (c) Zn 2p spectrum. | ||
Fig. 3c shows the Zn 2p spectrum of the Ru–Zn/HAP-1. The BE of Zn 2p3/2 (1022.0 eV) was lower than that of Zn(II) species (1022.5 eV) on the HAP support reported by Li et al.,39 but higher than that of metallic zinc (1021.7 eV). The Zn 2p spectrum of Zn/HAP is given in Fig. S3.† The BE of Zn 2p3/2 (1022.4 eV) in the Zn/HAP was close to the value reported in the literature,39 indicating that Zn existed in the form of Zn2+ in the Zn/HAP, which is consistent with the result of TEM observation in Fig. 2b.
As shown in the TEM image (Fig. 2b) and the XPS spectrum (Fig. S2†) of the Zn/HAP, Zn2+ exchanged onto HAP could not be reduced by hydrogen at 300 °C. However, when Ru3+ and Zn2+ are introduced to the surface of HAP simultaneously, it can be seen that the BE of Zn 2p3/2 shifted to lower BE area (Fig. 3c). By deconvolution of the Zn 2p spectrum in Fig. 3c, it can be deduced that both Zn(0) and Zn(II) species existed in the catalyst, which indicates that part of the Zn2+ ions on the catalyst surface were reduced to metallic Zn. This results partially from hydrogen spillover during the reduction process. Ru3+ ions were reduced to Ru nanoparticles by H2 and hydrogen molecules were dissociatively adsorbed on metallic Ru.40 Those active hydrogen atoms will spill over from metallic Ru to reduce Zn2+. The electronegativity of Ru is higher than that of Zn and the Ru particles have a tendency to attract the electrons from adjacent Zn. In addition, the interaction of Ru and Zn favors the reduction of Zn2+ because the Zn in the Ru–Zn particles may be more stable thermodynamically than monometallic Zn particles. The interaction between Ru and Zn causes a slight difference of the Ru 3d BE between Ru/HAP (281.2 eV) and Ru–Zn/HAP (281.0 eV). Similar electron transfer effects were also observed on the Pt–Zn/TiO2 catalyst41 and the Au(0)/ZnO catalyst.42
The effect of Ru/Zn molar ratios in the catalysts
Table 1 shows the catalytic performances of Ru–Zn/HAP catalysts with different Ru/Zn molar ratios for the reaction. In this work, the yield is the product of the conversion of benzene and the selectivity to cyclohexene. As can be seen, the Ru/Zn molar ratio influenced the catalytic activity and selectivity significantly. Ru/HAP showed the highest activity. The conversion of benzene reached 46.0% within 2.5 minutes. However, the selectivity to cyclohexene was poor on the Ru/HAP, which was as low as 13.3%. Zn/HAP had no activity towards the reaction, indicating Ru nanoparticles were responsible for the hydrogenation of benzene. The presence of Zn could enhance the selectivity to cyclohexene. Although the catalytic activity decreased with the increase of Zn content in the catalyst, the selectivity towards cyclohexene increased obviously. When the Ru/Zn molar ratio was 1:1, the HAP supported catalyst exhibited better performance than those with other Ru/Zn molar ratios. 58.2% selectivity and 30.5% yield of cyclohexene were reached. Extending the reaction time, the highest cyclohexene yield 33.0% was achieved. The catalytic performance declined dramatically at higher Zn load. The conversion of benzene was only 10.4% in 120 minutes when the Ru/Zn molar ratio was 1:2, while the selectivity to cyclohexene did not increase further.
| Entry | Catalyst | Ru/Zn ratio | Time (min) | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|---|---|---|
| Reaction conditions: benzene, 0.5 mL; reaction temperature, 150 °C; H2 pressure, 5.0 MPa; catalyst Ru–Zn/HAP, 20 mg; NaOH concentration, 0.5 M; water, 1.5 mL.a The results given in Tables 1–4 are mainly provided at close to isoconversion conditions (45–55%) to allow a suitable comparison of the selectivity data. The yield of cyclohexene is expressed as the product of the conversion of benzene by the selectivity of cyclohexene. The reaction time is included to give an idea of the catalysts activity. | ||||||
| 1 | Ru/HAP | — | 2.5 | 46.0 | 13.3 | 6.1 |
| 2 | Ru–Zn/HAP-2 | 2:1 |
12.5 | 54.1 | 31.9 | 17.3 |
| 3 | Ru–Zn/HAP-1 | 1:1 |
26.5 | 52.4 | 58.2 | 30.5 |
| 4 | Ru–Zn/HAP-1 | 1:1 |
50.0 | 69.8 | 47.3 | 33.0 |
| 5 | Ru–Zn/HAP-0.5 | 1:2 |
120.0 | 10.4 | 52.2 | 5.4 |
| 6 | Zn/HAP | — | 120.0 | 0 | 0 | 0 |
| 7 | Ru–Zn/MgO-1 | 1:1 |
18.0 | 52.7 | 36.1 | 19.0 |
| 8 | Ru–Zn/CeO2-1 | 1:1 |
65.0 | 49.5 | 26.5 | 13.1 |
| 9 | Ru–Zn/ZrO2-1 | 1:1 |
80.0 | 50.1 | 30.6 | 15.3 |
As aforementioned, both Zn and Zn2+ existed in the Ru–Zn/HAP catalysts. The contact between Zn and Ru influenced the catalytic performance by adjusting the property of active sites. The adsorption behaviors of benzene and cyclohexene on Ru were affected due to metallic Zn. Yuan et al.43 studied the catalytic behavior of Ru (0001) when Zn existed on its surface by a DFT method. The number of sites for the chemisorption of cyclohexene decreased sharply. The modified catalyst was partly passivated for the adsorption of cyclohexene, while the presence of Zn did not influence the adsorption of benzene significantly. Therefore, metallic Zn in the catalyst enhanced the yield of cyclohexene.
Zn2+ in the HAP framework is also positive to cyclohexene production. Liu et al.14 combined experimental and theoretical approaches to study the effect of Zn2+. In their reaction system ZnSO4 dissolved in water was used as the modifier to study this effect. Herein, we propose that Zn2+ exchanged onto the HAP has a similar function. On the one hand, the Zn2+ ion is effective in improving the hydrophilicity of the catalyst. On the other hand, it is able to form a complex with cyclohexene in the presence of water. The Zn2+ ions existed on the support surface which was not occupied by the Ru–Zn particles. Therefore, formation of the complex facilitates desorption of cyclohexene from the Ru surface and retards the re-adsorption of cyclohexene onto the active sites. Thus, the selectivity to cyclohexene was improved.
Ru–Zn catalysts supported on MgO, CeO2, and ZrO2 were also prepared. The Ru contents were 2.5 wt% and Ru/Zn molar ratios were 1:1, as were confirmed by ICP analysis. As shown in Table 1 (entries 7–9), Ru–Zn catalysts on the other supports were less active and selective than that on the HAP support. The better performance of the Ru–Zn/HAP catalyst may be due to the excellent hydrophilicity and adsorption ability of the support, the high dispersion of Ru–Zn nanoparticles, and the synergistic effect of metallic Zn and Zn2+ ions.
The effect of reaction time
Fig. 4 shows the plots of the amounts of benzene, cyclohexene, and cyclohexane during the reaction process as a function of reaction time. Benzene converted to cyclohexene and cyclohexane continuously with the extension of reaction time. At the initial stage the content of cyclohexene is higher than that of cyclohexane. Selectivity towards cyclohexene decreased gradually with the increase of benzene conversion. The maximum yield of cyclohexene was 33.0% at a benzene conversion of 69.8% and a cyclohexene selectivity of 47.3%. These results exhibited the characteristic behavior of a consecutive reaction.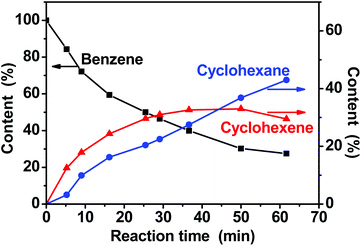 | ||
| Fig. 4 Time course of the benzene hydrogenation. | ||
The effect of NaOH concentrations
The presence of NaOH greatly enhanced the selectivity and yield of cyclohexene.5,6Table 2 illustrates the catalytic performances of Ru–Zn/HAP-1 in NaOH solutions with different concentrations. The catalytic activity gradually decreased with the increase of NaOH concentration, while the selectivity of cyclohexene improved dramatically. The selectivity to cyclohexene reached 35.8% at a benzene conversion of 50.5% when the NaOH concentration was 0.1 M (entry 2), while the selectivity was only 5.1% at a similar conversion without NaOH (entry 1). The highest selectivity reached 58.2% at a benzene conversion of 52.4% at the NaOH concentration of 0.5 M (entry 5). However, adding too much NaOH led to lower cyclohexene selectivity. In order to elucidate the positive effect of NaOH, the control experiments of cyclohexene hydrogenation were conducted with and without NaOH (Table 2, entries 10 to 12). It could be observed that NaOH retarded the cyclohexene hydrogenation to cyclohexane. Therefore the selectivity to cyclohexene could be improved when conducting the partial hydrogenation of benzene in NaOH solution.| Entry | NaOH (M) | Time (min) | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|---|---|
| Reaction conditions: benzene, 0.5 mL; reaction temperature, 150 °C; H2 pressure, 5.0 MPa; catalyst Ru–Zn/HAP-1, 20 mg; water, 1.5 mL.a Cyclohexene is the reactant at the same reaction conditions. Cyclohexane is the only product. | |||||
| 1 | 0 | 3.2 | 50.9 | 5.1 | 2.6 |
| 2 | 0.1 | 4.5 | 50.5 | 35.8 | 18.1 |
| 3 | 0.2 | 12.3 | 48.1 | 49.2 | 23.7 |
| 4 | 0.3 | 16.5 | 49.7 | 50.2 | 24.9 |
| 5 | 0.4 | 21.6 | 50.4 | 51.1 | 25.8 |
| 6 | 0.5 | 26.5 | 52.4 | 58.2 | 30.5 |
| 7 | 0.6 | 33.6 | 57.6 | 52.5 | 30.2 |
| 8 | 0.7 | 40.0 | 47.9 | 30.1 | 14.4 |
| 9 | 1.0 | 50.0 | 47.2 | 27.0 | 12.7 |
| 10a | 0 | 1.3 | 45.0 | — | — |
| 11a | 0 | 5.0 | 83.2 | — | — |
| 12a | 0.5 | 5.0 | 43.6 | — | — |
The promoting effect of NaOH towards the reaction is mainly ascribed to the following factors. First, the presence of NaOH enhanced the hydrophilicity of the catalyst. Ronchin and Toniolo5 reported that treating the unreduced Ru catalyst with NaOH could enhance the surface hydrophilicity. As benzene has larger solubility in water or NaOH solution than cyclohexene,44 further hydrogenation of cyclohexene to cyclohexane is depressed and cyclohexene is withdrawn from the water domain as soon as it is formed. In addition, with the enhancement of the hydrophilicity, water molecules compete with cyclohexene to adsorb on the Ru surface and occupy some active sites for cyclohexene hydrogenation. So the rate of cyclohexene hydrogenation is decreased.
The effect of reaction temperature
Table 3 demonstrates cyclohexene yield at different temperatures ranging from 120 °C to 170 °C over the Ru–Zn/HAP-1 catalyst. The yield of cyclohexene increased from 23.9% at 120 °C to 30.5% at 150 °C. However, the yield reduced to 26.8% as the temperature rose to 170 °C, i.e., the highest cyclohexene yield occurred at 150 °C.| Entry | Temperature (°C) | Time (min) | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|---|---|
| Reaction conditions: benzene, 0.5 mL; H2 pressure, 5.0 MPa; catalyst Ru–Zn/HAP-1, 20 mg; NaOH concentration, 0.5 M; water, 1.5 mL. | |||||
| 1 | 120 | 63.3 | 51.5 | 50.5 | 26.0 |
| 2 | 130 | 53.0 | 48.5 | 50.6 | 24.5 |
| 3 | 140 | 44.0 | 48.0 | 49.7 | 23.9 |
| 4 | 145 | 31.7 | 54.3 | 51.4 | 27.9 |
| 5 | 150 | 26.5 | 52.4 | 58.2 | 30.5 |
| 6 | 155 | 24.6 | 52.7 | 53.7 | 28.3 |
| 7 | 160 | 22.6 | 50.6 | 54.1 | 27.4 |
| 8 | 170 | 22.0 | 54.0 | 49.7 | 26.8 |
The apparent activation energy for benzene hydrogenation over Ru catalysts is very low, which is reported as 15 ± 3 kJ mol−1 from the literature, which was calculated from the dependence of consumption of H2 in the hydrogenation of benzene on temperature.45 This indicates that the diffusivity can affect the reactions significantly. As the temperature increased, desorption of cyclohexene from the catalyst surface is facilitated and surface coverage of hydrogen is lowered. These are favorable to the increase of cyclohexene yield.45 However, the elevated temperature results in an increase of cyclohexene solubility in the stagnant water layer around the catalyst. The surface coverage of cyclohexene and the rate of cyclohexene hydrogenation increased, so that the yield of cyclohexene is lower.20 The competition of the opposite factors resulted in the highest value of the yield at 150 °C.
The effect of H2 pressure
H2 pressure is another crucial factor that should be taken into account. The reactions were conducted at 150 °C at H2 pressures ranging from 2 to 7 MPa. As can be seen from Table 4, both the reaction rate and cyclohexene yield reached the highest values at 5 MPa. It is known that hydrogenation of benzene occurs in a consecutive manner. Hydrogen pressure affects the different steps of benzene hydrogenation in different ways. Odenbrand and Lundin46 reported that cyclohexene hydrogenation to cyclohexane is less dependent on H2 pressure than benzene hydrogenation to cyclohexene. With the increase of hydrogen pressure, the rate for benzene hydrogenation to cyclohexene grows faster than that of cyclohexene hydrogenation to cyclohexane, resulting in the higher cyclohexene yield. Hu and Chen15 investigated the influence of H2 pressure on the catalytic performances of Ru/La2O3–ZnO and Ru/Ga2O3–ZnO catalysts. They supposed that hydrogen, benzene, and cyclohexene competitively adsorb on the same active sites, which depends on the pressure of hydrogen. So there exists the optimum hydrogen pressure. Moreover, recent study showed that there is a relationship between H2 coverage and the hydrophilicity of the Ru surface. At high or low hydrogen coverage on the Ru surface, the hydrophilicity was depressed and resulted in lower cyclohexene yield.47| Entry | H2 pressure (MPa) | Time (min) | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|---|---|
| Reaction conditions: benzene, 0.5 mL; reaction temperature, 150 °C; catalyst Ru–Zn/HAP-1, 20 mg; NaOH concentration, 0.5 M; water, 1.5 mL. | |||||
| 1 | 2 | 105.0 | 53.2 | 42.7 | 22.7 |
| 2 | 3 | 65.0 | 51.9 | 48.5 | 25.2 |
| 3 | 4 | 44.0 | 52.2 | 55.1 | 28.8 |
| 4 | 5 | 26.5 | 52.4 | 58.2 | 30.5 |
| 5 | 6 | 40.0 | 46.9 | 55.2 | 25.9 |
| 6 | 7 | 75.0 | 44.8 | 54.6 | 24.5 |
Reuse of the catalyst
The reusability of the Ru–Zn/HAP-1 catalyst for the hydrogenation of benzene was investigated under the optimized reaction conditions, and the results are given in Fig. 5. The catalyst could be reused at least four times without notable changes in activity and selectivity to cyclohexene. Fig. 6a and 6b give the TEM images of the fresh catalyst and that of the catalyst after four cycles, and it can be shown from the images that the change of the morphology of the catalyst was not considerable. Fig. S4† compared the XRD patterns of the fresh and used catalyst. No obvious peaks of Ru were observed for the catalyst after using four times, indicating that ruthenium was still highly dispersed on HAP, which is the same as the fresh catalyst. The excellent stability of the catalyst may result from the strong interaction between the support and active species.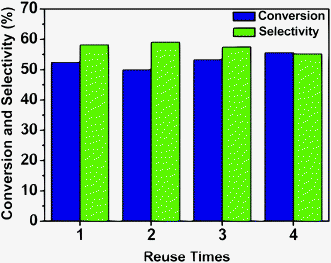 | ||
| Fig. 5 The performance of the catalyst in the recycle experiment. Reaction conditions: benzene, 0.5 mL; reaction temperature, 150 °C; H2 pressure, 5.0 MPa; catalyst Ru–Zn/HAP-1, 20 mg; NaOH concentration, 0.5 M; water, 1.5 mL. | ||
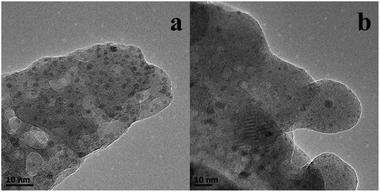 | ||
| Fig. 6 TEM images of fresh Ru–Zn/HAP-1 (a) and Ru–Zn/HAP-1 used four times (b). | ||
Conclusions
The Ru–Zn/HAP catalysts have been prepared and utilized to catalyze the partial hydrogenation of benzene to cyclohexene. Compared to the Ru/HAP catalyst, the addition of Zn in the catalyst can greatly increase the selectivity to cyclohexene. NaOH in the solution can retard the hydrogenation of cyclohexene and improve the yield of cyclohexene. The yield of cyclohexene can reach 33.0% over Ru–Zn/HAP with a Ru/Zn molar ratio of 1:1 at 150 °C and 5 MPa of hydrogen. The catalyst can be used for at least four times without decreasing the yield of cyclohexene notably. We believe that the efficient, stable, and greener catalytic system has great potential of application with obvious advantages.
Acknowledgements
The authors thank the National Natural Science Foundation of China (Nos 20973177, 20903103, 20932002, 21021003), the Chinese Academy of Sciences (KJCX2.YW.H30), and the Ministry of Science and Technology of China (2011CB808603).Notes and references
- H. Feng, J. W. Elam, J. A. Libera, M. J. Pellin and P. C. Stair, J. Catal., 2010, 269, 421 CrossRef CAS.
- N. Akiya and P. E. Savage, Ind. Eng. Chem. Res., 2001, 40, 1822 CrossRef CAS.
- G. Tavoularis and M. A. Keane, Appl. Catal., A, 1999, 182, 309 CrossRef CAS.
- H. Nagahara, M. Ono, M. Konishi and Y. Fukuoka, Appl. Surf. Sci., 1997, 121, 448 CrossRef.
- L. Ronchin and L. Toniolo, Appl. Catal., A, 2001, 208, 77 CrossRef CAS.
- H. Liu, S. Liang, W. Wang, T. Jiang and B. Han, J. Mol. Catal. A: Chem., 2011, 341, 35 CrossRef CAS.
- H. Z. Liu, T. Jiang, B. X. Han, S. G. Liang, W. T. Wang, T. B. Wu and G. Y. Yang, Green Chem., 2011, 13, 1106 RSC.
- W. Wang, H. Liu, T. Wu, P. Zhang, G. Ding, S. Liang, T. Jiang and B. Han, J. Mol. Catal. A: Chem., 2012, 355, 174 CrossRef CAS.
- J. Struijk and J. J. F. Scholten, Appl. Catal., A, 1992, 82, 277 CrossRef CAS.
- J. Struijk, R. Moene, T. Vanderkamp and J. J. F. Scholten, Appl. Catal., A, 1992, 89, 77 CrossRef CAS.
- G. Y. Fan, R. X. Li, X. J. Li and H. Chen, Catal. Commun., 2008, 9, 1394 CrossRef CAS.
- H. Sun, Z. Chen, W. Guo, X. Zhou, Z. Liu and S. Liu, Chin. J. Chem., 2011, 29, 369 CrossRef CAS.
- F. Schwab, M. Lucas and P. Claus, Angew. Chem., Int. Ed., 2011, 50, 10453 CrossRef CAS.
- J. L. Liu, Y. Zhu, J. Liu, Y. Pei, Z. H. Li, H. Li, H. X. Li, M. H. Qiao and K. N. Fan, J. Catal., 2009, 268, 100 CrossRef CAS.
- S. C. Hu and Y. W. Chen, Ind. Eng. Chem. Res., 1997, 36, 5153 CrossRef CAS.
- S. C. Hu and Y. W. Chen, Ind. Eng. Chem. Res., 2001, 40, 3127 CrossRef CAS.
- S. Niwa, F. Mizukami, S. Isoyama, T. Tsuchiya, K. Shimizu, S. Imai and J. Imamura, J. Chem. Technol. Biotechnol., 1986, 36, 236 CrossRef CAS.
- J. Bu, J. L. Liu, X. Y. Chen, J. H. Zhuang, S. R. Yan, M. H. Qiao, H. Y. He and K. N. Fan, Catal. Commun., 2008, 9, 2612 CrossRef CAS.
- J. L. Liu, L. J. Zhu, Y. Pei, J. H. Zhuang, H. Li, H. X. Li, M. H. Qiao and K. N. Fan, Appl. Catal., A, 2009, 353, 282 CrossRef CAS.
- J. Q. Wang, Y. Z. Wang, S. H. Zhe, M. H. Qiao, H. X. Li and K. N. Fan, Appl. Catal., A, 2004, 272, 29 CrossRef CAS.
- S. H. Xie, M. H. Qiao, H. X. Li, W. J. Wang and J. F. Deng, Appl. Catal., A, 1999, 176, 129 CrossRef CAS.
- G. Y. Fan, W. D. Jiang, J. B. Wang, R. X. Li, H. Chen and X. J. Li, Catal. Commun., 2008, 10, 98 CrossRef CAS.
- J. W. da-Silva and A. J. G. Cobo, Appl. Catal., A, 2003, 252, 9 CrossRef CAS.
- X. Zhou, H. Sun, W. Quo, Z. Liu and S. Liu, J. Nat. Gas Chem., 2011, 20, 53 CrossRef CAS.
- J. B. Ning, J. Xu, J. Liu and F. Lu, Catal. Lett., 2006, 109, 175 CrossRef CAS.
- V. Mazzieri, N. Figoli, F. C. Pascual and P. L'Argentiere, Catal. Lett., 2005, 102, 79 CrossRef CAS.
- C. Zanutelo, R. Landers, W. A. Carvalho and A. J. Gomez Cobo, Appl. Catal., A, 2011, 409, 174 CrossRef.
- Z. Opre, T. Mallat and A. Baiker, J. Catal., 2007, 245, 482 CrossRef CAS.
- K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144 CrossRef CAS.
- K. Mori, K. Yamaguchi, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2002, 124, 11572 CrossRef CAS.
- H. Sun, F. Z. Su, J. Ni, Y. Cao, H. Y. He and K. N. Fan, Angew. Chem., Int. Ed., 2009, 48, 4390 CrossRef CAS.
- B. M. Choudary, C. Sridhar, M. L. Kantam, G. T. Venkanna and B. Sreedhar, J. Am. Chem. Soc., 2005, 127, 9948 CrossRef CAS.
- K. Mori, Y. Mitani, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Commun., 2005, 3331 RSC.
- M. Zahmakıran, Y. Tonbul and S. Ozkar, Chem. Commun., 2010, 46, 4788 RSC.
- M. Zahmakıran, Y. Roman-Leshkov and Y. Zhang, Langmuir, 2012, 28, 60 CrossRef.
- K. Kaneda and T. Mizugaki, Energy Environ. Sci., 2009, 2, 655 CAS.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250 CrossRef CAS.
- V. Mazzieri, F. Coloma-Pascual, A. Arcoya, P. L'Argentiere and N. S. Figoli, Appl. Surf. Sci., 2003, 210, 222 CrossRef CAS.
- J. Li, Y. Li, L. Zhang and Y. Zuo, Appl. Surf. Sci., 2008, 254, 2844 CrossRef CAS.
- H. Shimizu, K. Christmann and G. Ertl, J. Catal., 1980, 61, 412 CrossRef CAS.
- J. Silvestre-Albero, A. Sepulveda-Escribano, F. Rodriguez-Reinoso and J. A. Anderson, J. Catal., 2004, 223, 179 CrossRef CAS.
- X. Liu, M. H. Liu, Y. C. Luo, C. Y. Mou, S. D. Lin, H. Cheng, J. M. Chen, J. F. Lee and T. S. Lin, J. Am. Chem. Soc., 2012, 134, 10251 CrossRef CAS.
- P. Q. Yuan, B. Q. Wang, Y. M. Ma, H. M. He, Z. M. Cheng and W. K. Yuan, J. Mol. Catal. A: Chem., 2009, 309, 124 CrossRef CAS.
- F. Mizukami, S. Niwa, S. Ohkawa and A. Katayama, Stud. Surf. Sci. Catal., 1993, 78, 337 CrossRef CAS.
- J. Struijk, M. Dangremond, W. J. M. Lucasderegt and J. J. F. Scholten, Appl. Catal., A, 1992, 83, 263 CrossRef CAS.
- C. U. I. Odenbrand and S. T. Lundin, J. Chem. Technol. Biotechnol., 1980, 30, 677 CrossRef CAS.
- P. Q. Yuan, Y. M. Ma, X. K. Li, B. Q. Wang, Z. M. Cheng and W. K. Yuan, J. Mol. Struct. (THEOCHEM), 2010, 942, 77 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2gc36596k |
| This journal is © The Royal Society of Chemistry 2013 |