Cyclization of o-phenylenediamines by CO2 in the presence of H2 for the synthesis of benzimidazoles†
Bo
Yu
,
Hongye
Zhang
,
Yanfei
Zhao
,
Sha
Chen
,
Jilei
Xu
,
Changliang
Huang
and
Zhimin
Liu
*
Beijing National Laboratory for Molecular Sciences, Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liuzm@iccas.ac.cn; Fax: +8610-62562821; Tel: +8610-62562852
First published on 15th November 2012
Abstract
The cyclization of o-phenylenediamines by CO2 in the presence of H2 was presented to directly synthesize benzimidazoles, and a series of benzimidazoles were obtained in excellent yields using RuCl2(dppe)2 as the catalyst.
Introduction
The chemical conversion of carbon dioxide (CO2) into value-added chemicals such as formic acid, methanol, carbonates, amides and so on has attracted much attention in recent years since CO2 is an economical, abundant and nontoxic renewable C1 resource and its utilization is of significance for sustainable development.1,2 However, CO2 is chemically inert, and a large energy input or special catalysts are usually required for its chemical transformation. For example, in the hydrogenation of CO2 to formic acid or its derivatives, a base and catalysts (e.g., RuCl2(dppe)2, RuCl2(PPh3)3, Ru3(CO)12) are necessary.3 Therefore, to activate a CO2 multi-component reaction system in which each component has a synergistic effect on CO2 conversion may be promising candidates for efficient CO2 chemical transformation and designing such systems is highly desirable.Catalytic C–N bond formation reactions between the N-containing compounds with CO2 are important in both industry and academia because they offer economical and environmental advantages.4 Benzimidazoles are important intermediates in the synthesis of pharmaceutical compounds, and heterocycles with a benzimidazole structure are omnipresent in biologically active compounds.5,6 The conventional methods to synthesize benzimidazoles are based on the condensation reactions of 1,2-diaminobenzenes with formic acid or its derivatives (imidates, esters, orthoesters, or nitriles) under strong acidic conditions at high temperature7 or under microwave irradiation.8 Recently, benzimidazoles have been reported to be synthesized from aldehydes instead of carboxylic acids in the presence of different oxidants.9 Although much progress has been made in the synthesis of benzimidazoles, it is still highly desirable to develop simple and green routes using clean and renewable materials instead of harmful compounds, which is of paramount importance from a standpoint of green chemistry and sustainable development.
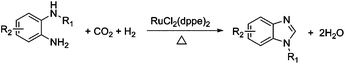 | (1) |
Herein, we proposed a new route to directly synthesize benzimidazoles via the cyclization of o-phenylenediamines by CO2 in the presence of H2 and a catalyst under the solvent-free condition, as illustrated in eqn (1). A series of benzimidazoles were obtained in excellent yields by cyclizing the corresponding o-phenylenediamines with CO2 under reductive conditions using RuCl2(dppe)2 as the catalyst. Though benzimidazoles have been synthesized using various starting materials via different routes,10 this is the first example to synthesize benzimidazoles directly from renewable carbon resource CO2 to the best of our knowledge. This strategy is very appealing due to the following obvious advantages: (1) using CO2 instead of harmful compounds as the starting material; (2) water is the only byproduct. More importantly, it opens a new route for CO2 fixation.
Results and discussion
The reaction of o-phenylenediamine with CO2 and H2 was first investigated in detail, and the results are listed in Table 1. All reactions were performed at 120 °C since o-phenylenediamine is in liquid state and can dissolve CO2 and H2. A blank test was performed in the absence of catalyst, and no benzimidazole was obtained (Table 1, entry 1), while in the presence of the RuCl2(dppe)2 catalyst benzimidazole was successfully achieved. This suggests that RuCl2(dppe)2 catalyzed the formation of benzimidazole. We explored the influence of system pressures on this reaction. It was found that the conversion of o-phenylenediamine increased with pressure remarkably, approaching 100% at 20 MPa under the other identical conditions (Table 1, entries 2–5). The isolated yields of benzimidazole increased with pressure, and a high product yield of 87% was obtained at a pressure of 20 MPa and a catalyst amount of 0.06 mol% (Table 1, entry 5). The catalyst amount considerably affected the conversion of the substrate. For example, increasing the catalyst amount from 0.06 to 0.20 mol%, o-phenylenediamine was completely converted under the same conditions, affording a 92% isolated yield of benzimidazole (2a) (Table 1, entries 4 and 6). It is noteworthy that a byproduct, 2-hydroxybenzimidazole (3), identified by 1H and 13C NMR (see ESI, Fig. S3 and S4†), was detectable with a trace amount accompanied with benzimidazole. The formation of this byproduct was proved to be the sole product of o-phenylenediamine reacting with CO2 (see ESI†). The maximum isolated yield of this byproduct was 7% at 20 MPa and a catalyst amount of 0.06%. The above experiments indicate that controlling the system pressure and the catalyst amount can tune the competing reactions among o-phenylenediamine, CO2 and H2, and benzimidazole could be obtained in excellent yield.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | mol% | P/MPa | Conv./%b | Isolated yield/%c |
| a Reaction conditions: o-phenylenediamine, 5.0 mmol; 120 °C; 40 h. b Conversion is defined as the percentage of the converted substrate, which was calculated based on the results analyzed by LC. c The isolated yield of the product is the percentage of the obtained amount divided by the theoretical value of the product. | |||||
| 1 | — | — | 15 | 0 | 0 |
| 2 | RuCl2(dppe)2 | 0.06 | 5 | 7 | 4 |
| 3 | RuCl2(dppe)2 | 0.06 | 10 | 29 | 24 |
| 4 | RuCl2(dppe)2 | 0.06 | 15 | 81 | 74 |
| 5 | RuCl2(dppe)2 | 0.06 | 20 | >99 | 87 |
| 6 | RuCl2(dppe)2 | 0.20 | 15 | >99 | 92 |
| 7 | RuCl2(PPh3)3 | 0.06 | 15 | 67 | 63 |
| 8 | PdCl2(dppe)2 | 0.06 | 15 | 19 | 15 |
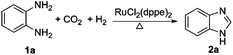
Two other homogeneous catalysts, RuCl2(PPh3)3 and PdCl2(dppe)2, which show high reactivity in the CO2 hydrogenation were selected to catalyze the cyclization of o-phenylenediamine with CO2 in the presence of H2. It was demonstrated that these catalysts could also catalyze this reaction, and the product yields were 63% and 15% using RuCl2(PPh3)3 and PdCl2(dppe)2, respectively (Table 1, entries 7 and 8). From these results, it may be deduced that the catalysts effective in the CO2 hydrogenation can catalyze the production of benzimidazoles from o-phenylenediamines reacting with CO2 and H2.
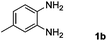
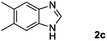
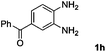
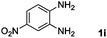
Encouraged by the initial success in production of benzimidazole, to investigate the general scope and versatility of this strategy in the synthesis of substituted benzimidazoles, we examined the cyclization of a variety of structurally different phenylenediamines possessing functional groups containing both electron-donating and electron-withdrawing groups (1a–k) by CO2 in the presence of H2 catalyzed by RuCl2(dppe)2 (0.2 mol%). Excitingly, the corresponding substituted benzimidazole derivatives were successfully obtained (see ESI,†1H and 13C NMR data), and the results are listed in Table 2. From this table, it can be observed that most of the substituted benzimidazoles were obtained in excellent yields no matter if the substituents were electron-donating (Table 2, entries 2 and 3) or electron-withdrawing groups (Table 2, entries 3–9), similar to benzimidazole (Table 2, entry 1). These findings indicate that most of the substituted groups in the phenyl ring of diamines had little influence on the cyclization of the substrates by CO2 in the presence of H2. However, the CF3 group was an exception, which had a considerable effect on the yield of the corresponding benzimidazole (Table 2, entry 10). It was demonstrated that the competing reaction of 5-trifluoromethylphenylenediamine with CO2 resulted in the reduction in 5-trifluoromethylbenzimidazole yield. In addition, the steric hindrance of N-phenyl-o-phenylenediamine (1k) seemed not to hamper the cyclization by CO2, confirmed by the fact that N-phenyl-benzimidazole (2k) was produced in 92% yield (Table 2, entry 11). It is worth noticing that the dehalogenation (Table 2, entries 4, 5) and the ketone reduction (Table 2, entry 8) of the substituted diamines did not occur under the experimental conditions, which is important for the production of these kinds of benzimidazoles. It should be pointed out that in all the above reactions the corresponding byproducts from the reactions of diamine substrates with CO2 were detectable, most with a trace amount.
| Entry | Substrate | Product | Isolated yield/% |
|---|---|---|---|
| a Reaction conditions: substrate, 5.0 mmol; RuCl2(dppe)2, 0.2 mol%; 120 °C; pressure of mixed gas of CO2 and H2, 15 MPa; 40 h. The conversions of the substrates were >99%. | |||
| 1 |
|
|
92 |
| 2 |
|
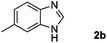
|
95 |
| 3 |
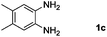
|
|
93 |
| 4 |
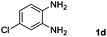
|
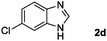
|
91 |
| 5 |
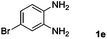
|
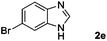
|
90 |
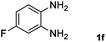
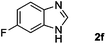
| 6 |
|
|
91 |
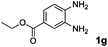
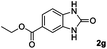
| 7 |
|
|
93 |
| 8 |
|
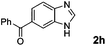
|
92 |
| 9 |
|
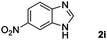
|
87 |
| 10 |
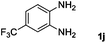
|
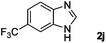
|
73 |
| 11 |
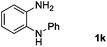
|
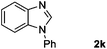
|
92 |
It is noteworthy that when 2′-aminoacetanilide (1l) was used as the diamine substrate under similar conditions, neither the desired product (i.e., N-acetyl benzimidazole) nor byproduct (i.e., 2-hydroxybenzimidazole) was obtained. Instead, 2-methyl benzimidazole (2l) was obtained in a yield of 98% (Scheme 1), which may be attributed to the good tendency of 2′-aminoacetanilide to undergo cyclization under the experimental conditions (Scheme 1).
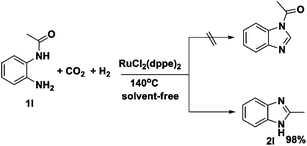 | ||
| Scheme 1 Reaction of 2′-aminoacetanilide with H2 and CO2. | ||
To explore the reaction mechanism of the benzimidazole production, a control experiment, the hydrogenation of 2-hydroxybenzimidazole, was performed in the presence of RuCl2(dppe)2 under similar conditions, and no benzimidazole was obtained. This indicates that 2-hydroxybenzimidazole was not the intermediate of the final product, benzimidazole. We checked the reaction solution at the o-phenylenediamine conversion around 30%. Unfortunately, no intermediates, such as formic acid and formamide, were detectable, implying the formation of benzimidazole was very fast. We performed the reaction of o-phenylenediamine with formic acid without RuCl2(dppe)2. It was found that o-phenylenediamine could react with formic acid to form benzimidazole within 1 min at 120 °C, and even at room temperature the reaction could occur. From these findings, it can be deduced that the ruthenium catalyst was not involved in the formation of the intermediate product in this work. To get information about the intermediate from the reaction of o-phenylenediamine with formic acid, in situ13C NMR investigations were performed on the reaction solution as the reaction proceeded, and the results are shown in Fig. 1. In the 13C NMR spectra of Fig. 1b and 1c, the signals at δ = 161.8, 132.9, 131.0, 127.5 126.4, 121.3, and 117.3 ppm are attributed to the intermediate formamide (4), while the signals at δ = 141.9, 135.9, 124.1, 115.7 ppm to the final product benzimidazole. Moreover, as the reaction proceeded, the 13C NMR signal intensity of benzimidazole gradually increased, and the intensity of the intermediate declined until disappearing. This indicates that benzimidazole was produced via the formation of the intermediate formamide (4) from o-phenylenediamine reacting with formic acid. Considering that CO2 can be hydrogenated to formic acid catalyzed by RuCl2(dppe)2 with the assistance of a base3 and benzimidazole can be produced via the reaction of o-phenylenediamine with formic acid7,8, we proposed the reaction pathway of benzimidazole production from o-phenylenediamine cyclization by CO2 in the presence of H2, as depicted in Scheme 2. The reaction may in principle proceed in two steps. In step 1, the aromatic diamine serves as a base to promote the CO2 hydrogenation to formic acid catalyzed by RuCl2(dppe)2, and subsequently formamide (4) may form fast via dehydration from the diamine with formic acid in step 2, followed by quick intramolecular cyclization to the final product benzimidazole (2a). The process in which CO2 hydrogenation is combined with further formation of the imidazole ring takes place in several steps wherein each product becomes the substrate for the next reaction. According to this possible reaction pathway, o-phenylenediamines should be completely converted into corresponding benzimidazoles. However, due to the presence of the competing reaction of o-phenylenediamines with CO2, byproducts are inevitably formed in small amounts.
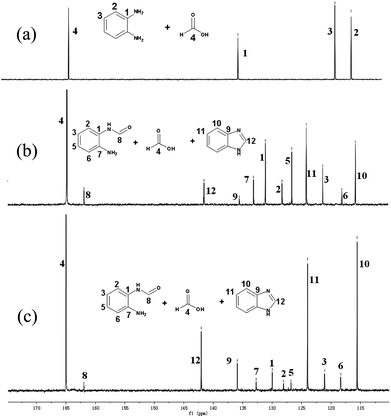 | ||
| Fig. 1 13C NMR spectra (D6-DMSO) of the reaction solution of o-phenylenediamine with HCOOH at different reaction times, (a) 0 min, (b) 40 min, (c) 60 min. | ||
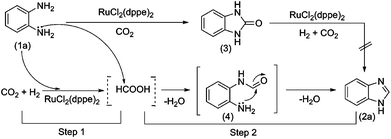 | ||
| Scheme 2 Proposed reaction pathway for benzimidazole production. | ||
Conclusions
The cyclization of o-phenylenediamines by CO2 was achieved in the presence of H2, producing benzimidazoles. The catalyst RuCl2(dppe)2 is highly active and selective for the production of a series of benzimidazoles in excellent yields. In addition, the reactions can be performed under solvent-free conditions, rendering the strategy to synthesize benzimidazoles from CO2 highly valuable from both environmental and economical points of view. The present synthetic route of benzimidazoles starting from CO2 represents a novel alternative to the traditional routes, and opens a new way for the CO2 utilization as well.Experimental section
Hydrogen (99.99%) and CO2 (99.99%) were provided by Beijing Analytical Instrument Company. o-Phenylenediamine (1a: 98%), 3,4-diaminotoluene(1b: 97%), 4,5-dimethyl-o-phenylenediamine (1c: 98%), 4-chloro-o-phenylenediamine (1d: 97%), 4-bromo-o-phenylenediamine (1e: 97%), 4-fluoro-o-phenylenediamine (1f: 97%), ethyl-3,4-diaminobenzoate (1g: 97%), 3,4-diaminobenzophenone (1h: 97%), 4-nitro-o-phenylenediamine (1i: 98%), 4-trifluoromethyl-o-phenylenediamine (1j: 98%), N-phenyl-o-phenylenediamine (1k: 98%), 2′-aminoacetanilide (1l: 98%) were purchased from Alfa Aesar and used without further purification. RuCl2(dppe)2 (dppe = 1,2-bis(diphenylphosphino)-ethane) was prepared according to a reported procedure.31H (400 MHz) and 13C (100 MHz) NMR spectra were collected in CDCl3 or (CD3)2SO on a Bruker Avance NMR (400 MHz) at ambient temperature, and chemical shifts were recorded relative to tetramethylsilane (TMS). 1H and 13C NMR chemical shifts are reported in ppm downfield from tetramethylsilane. Abbreviations used in the NMR follow-up experiments: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Melting points of new products were recorded using an XT4 microscopy melting point determinator. The quantity analysis of the products was conducted on an HPLC (Shimadzu, LC-15C) with a UV detector set at a wavelength of 254 nm.
Procedure for the synthesis of benzimidazoles from o-phenylenediamines and CO2
The cyclization of o-phenylenediamines by CO2 in the presence of H2 was carried out in a Teflon-lined stainless steel reactor of 22 mL coupled with a magnetic stirrer. In a typical experiment to synthesize benzimidazole, o-phenylenediamine (5.0 mmol) and RuCl2(dppe)2 were loaded into the reactor, and moved subsequently to an oil bath of 120 °C, which was controlled by a Haake-D3 temperature controller. Then the mixed gas of CO2 and H2 (the molar ratio of CO2 to H2 was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was charged into the reactor up to the desired pressure (e.g., 5, 10, 15, 20 MPa), and the stirrer started. After the reaction, the reactor was cooled in ice water and the gas inside was slowly vented. The reaction mixture was dissolved in methanol and transferred into a volumetric flask. The quantitative analysis was conducted by HPLC using a Shimadzu LC-20AT pump, a Hypersil ODS2 5 μm column, and a Soma UV-Vis LC-830 detector at 282 nm. A methanol–water (50:50 V/V) solution was used as the mobile phase with a flow rate of 0.8 mL min−1. The pure products were obtained via column chromatography separation. The pure products were identified by 1H and 13C NMR. Similarly, substituted benzimidazoles were obtained using the corresponding o-phenylenediamines as the substrates.
:2) was charged into the reactor up to the desired pressure (e.g., 5, 10, 15, 20 MPa), and the stirrer started. After the reaction, the reactor was cooled in ice water and the gas inside was slowly vented. The reaction mixture was dissolved in methanol and transferred into a volumetric flask. The quantitative analysis was conducted by HPLC using a Shimadzu LC-20AT pump, a Hypersil ODS2 5 μm column, and a Soma UV-Vis LC-830 detector at 282 nm. A methanol–water (50:50 V/V) solution was used as the mobile phase with a flow rate of 0.8 mL min−1. The pure products were obtained via column chromatography separation. The pure products were identified by 1H and 13C NMR. Similarly, substituted benzimidazoles were obtained using the corresponding o-phenylenediamines as the substrates.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (Nos. 21125314, 21021003, 21073202) and the Chinese Academy of Sciences (KJXC2-YW-H30) for financial support.Notes and references
- (a) T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS; (b) P. Munshi, A. D. Main, J. C. Linehan, C. C. Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963 CrossRef CAS; (c) O. Krocher, R. A. Koppel and A. Baiker, Chem. Commun., 1997, 453 RSC; (d) C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254 CrossRef CAS; (e) O. Krocher, R. A. Koppel and A. Baiker, J. Mol. Catal. A: Chem., 1999, 140, 185 CrossRef CAS; (f) P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231 CrossRef CAS.
- I. Omae, Catal. Today, 2006, 115, 33 CrossRef CAS; J. S. Chang, V. P. Vislovskiy, M. S. Park, D. Y. Hong, J. S. Yoo and S. E. Park, Green Chem., 2003, 5, 587 RSC; C. Y. Hao, S. P. Wang, M. S. Li, L. Q. Kang and X. B. Ma, Catal. Today, 2011, 160, 184 CrossRef.
- O. Krocher, R. A. Koppel, M. Froba and A. Baiker, J. Catal., 1998, 178, 284 CrossRef CAS; P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259 CrossRef; O. Krocher, R. A. Koppel and A. Baiker, Chem. Commun., 1996, 1497 RSC; M. A. Fox, J. E. Harris, S. Heider, V. Perez-Gregorio, M. E. Zakrzewska, J. D. Farmer, D. S. Yufit, J. A. K. Howard and P. J. Low, J. Organomet. Chem., 2009, 694, 2350 CrossRef.
- T. Kimura, K. Kamata and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 6700 CrossRef CAS.
- C. G. Mortimer, G. Wells, J. P. Crochard, E. L. Stone, T. D. Bradshaw, M. F. G. Stevens and A. D. Westwell, J. Med. Chem., 2006, 49, 179 CrossRef CAS.
- C. S. Lin, R. Q. Zhang, C. S. Lee, T. A. Niehaus and T. Frauenheim, J. Phys. Chem. B, 2006, 110, 20847 CrossRef CAS; J. Bhaumik, Z. Yao, K. E. Borbas, M. Taniguchi and J. S. Lindsey, J. Org. Chem., 2006, 71, 8807 CrossRef.
- W. J. Ebenezer, M. G. Hutchings, K. Jones, D. A. Lambert and I. Watt, Tetrahedron Lett., 2007, 48, 1641 CrossRef CAS; H. Thakuria and G. Das, ARKIVOC, 2008, 321 Search PubMed; K. F. Ansari and C. Lal, Eur. J. Med. Chem., 2009, 44, 4028 CrossRef.
- R. Dubey and N. S. H. N. Moorthy, Chem. Pharm. Bull., 2007, 55, 115 CrossRef CAS; C. Mukhopadhyay, S. Ghosh, S. Sengupta and S. De, RSC Adv., 2011, 1, 1033 RSC.
- M. A. Chari, D. Shobha and T. Sasaki, Tetrahedron Lett., 2011, 52, 5575 CrossRef CAS; A. J. Blacker, M. M. Farah, M. I. Hall, S. P. Marsden, O. Saidi and J. M. J. Williams, Org. Lett., 2009, 11, 2039 CrossRef; G. M. Raghavendra, A. B. Ramesha, C. N. Revanna, K. N. Nandeesh, K. Mantelingu and K. S. Rangappa, Tetrahedron Lett., 2011, 52, 5571 CrossRef.
- K. R. Hornberger, G. M. Adjabeng, H. D. Dickson and R. G. Davis-Ward, Tetrahedron Lett., 2006, 47, 5359 CrossRef CAS; C. Mukhopadhyay and P. K. Tapaswi, Tetrahedron Lett., 2008, 49, 6237 CrossRef; C. Lin, P. T. Lai, S. K. S. Liao, W. T. Hung, W. B. Yang and J. M. Fang, J. Org. Chem., 2008, 73, 3848 CrossRef; X. J. Wang, M. Xiao, S. J. Wang, Y. X. Lu and Y. Z. Meng, J. Mol. Catal. A: Chem., 2007, 278, 92 CrossRef; D. L. Yang, D. Fokas, J. Z. Li, L. B. Yu and C. M. Baldino, Synthesis, 2005, 47 CrossRef; M. P. Surpur, P. R. Singh, S. B. Patil and S. D. Samant, Synth. Commun., 2007, 37, 1375 CrossRef; B. L. Zou, Q. L. Yuan and D. W. Ma, Angew. Chem., Int. Ed., 2007, 46, 2598 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and full compound characterization. See DOI: 10.1039/c2gc36517k |
| This journal is © The Royal Society of Chemistry 2013 |