The present state of coupling of dispersive liquid–liquid microextraction with atomic absorption spectrometry
Vasil
Andruch
a,
Ioseph S.
Balogh
b,
Lívia
Kocúrová
*a and
Jana
Šandrejová
a
aDepartment of Analytical Chemistry, University of P.J. Šafárik, SK-04154 Košice, Slovak Republic. E-mail: liviamonika.kocurova@gmail.com
bDepartment of Chemistry, College of Nyíregyháza, HU-4400 Nyíregyháza, Hungary
First published on 1st October 2012
Abstract
As a new sample pre-treatment technique, dispersive liquid–liquid microextraction (DLLME) is attracting the attention of a growing number of researchers. This review is devoted to the coupling of DLLME with atomic spectroscopy. The basic principles of the DLLME technique are briefly explained. Experimental factors affecting the extraction efficiency, such as type and volume of the extraction and dispersive solvent, ionic strength, extraction time, centrifugation time and rate, as well as applications of surfactants and chelating reagents, are discussed. Recent developments in the automation of DLLME are also presented. The applications of dispersive liquid–liquid microextraction coupled with atomic spectrometry are given in a Table.
 Vasil Andruch | Vasil Andruch works at the Department of Analytical Chemistry at Pavol Jozef Šafárik University in Košice. His research is devoted to green analytical chemistry, to the miniaturization and automation of liquid–liquid extraction and to the application of alternative energy sources for improving analytical procedures. |
 Ioseph S. Balogh | Ioseph S. Balogh has been working as a university professor of chemistry at the College of Nyíregyháza. His research has been devoted to a wide range of fields in analytical chemistry, including spectrophotometry, separation techniques, ionometry, environmental chemistry and the analytical chemistry of semiconductors. He is the author of numerous articles, patents and monographs. |
 Lívia Kocúrová | Lívia Kocúrová is a fourth-year PhD student at the Department of Analytical Chemistry, Faculty of Science, Pavol Jozef Šafárik University in Košice, working under the supervision of Vasil Andruch. Her research is devoted to miniaturized extraction systems, with special attention given to dispersive liquid–liquid microextraction and its modifications. |
 Jana Šandrejová | Jana Šandrejová (born name Škrlíková) works at the Department of Analytical Chemistry, Faculty of Science, Pavol Jozef Šafárik University in Košice. Her research is focused on the development of novel, environmentally friendly extraction procedures, primarily on new trends in liquid–liquid extraction procedures as well as their automation in flow systems. |
1 Introduction
An analytical procedure in its entirety consists of a number of steps, such as sampling, sample pre-treatment, the measurement itself and evaluation of the results, and each of these steps can affect the quality of the analytical results. Among the challenges analytical chemists often face in the determination of trace elements are the low concentration of the analyte in the sample and the significant impact of matrix. An appropriate tool for addressing these issues may be the inclusion of a separation and preconcentration step in the entire analytical procedure. And while the wide use of modern instrumental techniques for performing determinations does not exclude the need for separation and/or preconcentration methods, as one could easily believe, it can offer new advantages enabling the possible use of these methods in the future.1 It has been shown that preconcentration improves detection limits. Moreover, employing a preconcentration step could also increase the accuracy and precision of a determination due to the elimination of interferences. Then again, it could also decrease them due to the loss of the analyte to be determined or sample contamination.1 Therefore, preconcentration/separation methods should be carefully considered in each case and employed only when really necessary. Ideally, a sample pretreatment method should be simple, fast, low-cost, free from hazardous chemicals, with minimum waste generation, and be of potential consistent with the instrumental technique used for determination.One of the oldest sample pretreatment methods in analytical chemistry is liquid–liquid extraction (LLE) based on the transfer of analytes from an aqueous sample to a water-immiscible organic solvent phase. Despite its simplicity, however, LLE does not always act in accordance with the requirements of modern analytical chemistry. For instance, it requires high consumption of often hazardous organic solvents, which suggests the high production of organic waste. As a result, this technique began being used less and less and was replaced by other techniques, such as solid-phase extraction (SPE). However, SPE is not always a suitable substitute for LLE. Thus, the search began for new ways of sample preparation, leading to the miniaturisation of LLE.
The miniaturisation of liquid–liquid extraction began in the mid-to-late 1990s. In 1995 Liu and Dasgupta2 suggested a renewable gas sampling interface – a droplet collector – based on the diffusion and dissolution of soluble constituents contained in a gas, when a gas stream flowed across a liquid droplet. In 1996 the same authors3 described a drop-in-drop system consisting of a microdrop of chloroform suspended in an aqueous drop of sample. In the same year, Jeannot and Cantwell4 described a system in which a water-immiscible organic solvent drop was located at the end of a Teflon rod holder immersed in a stirred aqueous sample. In 1999 Pedersen-Bjergaard and Rasmussen5 suggested a microextraction method in which the analyte was extracted from an alkaline donor solution through an organic solvent in the pores of a hollow fiber into an acidic acceptor solution. These works led to the intensive development of microextraction techniques, and since then, various modes of miniaturised extraction procedures have been reported.6–9
In 2006 Rezaee et al.10 introduced a novel microextraction technique called “dispersive liquid–liquid microextraction” (DLLME). The first articles about DLLME were devoted to the coupling of this technique with gas chromatography. However, already in early 2007 the first article describing the connection of DLLME with graphite furnace atomic absorption spectrometry appeared,11,12 and the combination of dispersive liquid–liquid microextraction with flame atomic absorption spectrometry appeared only slightly later, in early 2008.13 To date, the DLLME procedure coupled with atomic spectrometry has been suggested for a variety of elements (Fig. 1); examples of its application are given in Table 1.
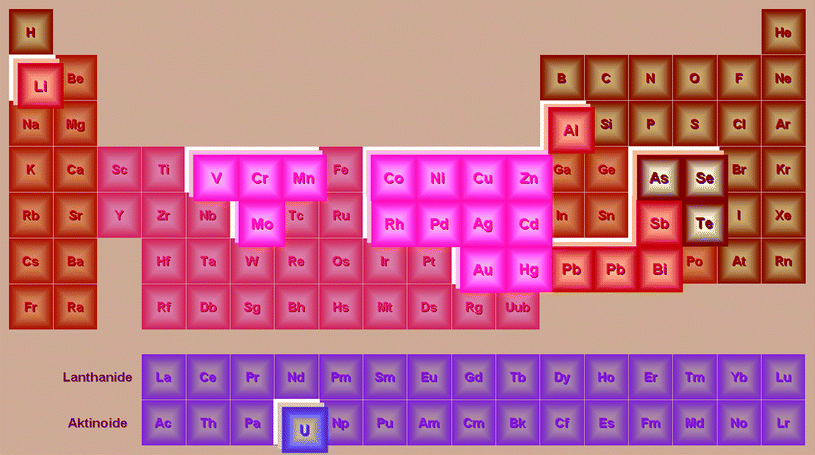 | ||
| Fig. 1 Applications of dispersive liquid–liquid microextraction coupled with atomic spectroscopy based on the data in Table 1. | ||
| Analyte | Sample | Procedure | Detection | Reagent | Extraction solvent | Dispersive solvent | Linear range | LOD | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Abbr. [C4Mim][PF6], 1-butyl-3-methylimidazolium hexafluorophosphate; [C6Mim][PF6], 1-hexyl-3-methylimidazolium hexafluorophosphate; [C6Mim][Tf2N], 1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide; [C6Py][PF6], 1-hexylpyridinium hexafluorophosphate; [C8Mim][Tf2N], 1-octyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide; [C8Mim]Cl, 1-octyl-3-methylimidazolium chloride; Li[NTf2], lithium bis(trifluoromethanesulfonyl)imide; 1N2N, 1-nitroso-2-naphthol; 5-Br-PADAP, 2-(5-bromo-2-pyridylazo)-5-(diethylamino)phenol; 8-HQ, 8-hydroxyquinoline; ACDA, 2-amino-1-cyclohexene-1-dithiocarboxylic acid; APDC, ammonium pyrrolidine dithiocarbamate; B15C5, benzo-15-crown-5; BPHA, N-benzoyl-N-phenylhydroxylamine; BTAC, 2-(2′-benzothiazolylazo)-p-cresol; DDPA, ammonium diethyldithiophosphate; DDTC, diethyldithiocarbamate; DDTP, diethyldithiophosphoric acid; DPTH, 2,2′-bis(di-2-pyridinyl-methylene)-thiocarbohydrazone; Dtz, dithizone; PAN, 1-(2-pyridylazo)-2-naphthol; PAR, 4-(2-pyridylazo)-resorcinol; PMBP, 1-phenyl-3-methyl-4-benzoyl-5-pyrazolone; TBAB, tetrabutylammonium bromide; TDMBA, tetradecyldimethylbenzylammonium chloride; TMK, 4,4′-bis(dimethylamino)thiobenzophenone; TRH, thioridazine HCl; TTA, 1-(2-thenoyl)-3,3,3-trifluoroacetone. | |||||||||
| Ag | Water samples | DLLME | FAAS | Dtz | 65 μL carbon tetrachloride | 4 mL ethanol | 0.1–7 μg L−1 | 0.018 μg L−1 | 14 |
| Ag | Water samples | DLLME | FAAS | Ligandless | 15 μL carbon tetrachloride | 0.5 mL ethanol | 5.0–2000 ng mL−1 | 1.2 ng mL−1 | 15 |
| Ag | Water samples | DLLME | GFAAS | DDTC | 35 μL carbon tetrachloride | 0.5 mL methanol | 0.1–10 ng mL−1 | 12 ng L−1 | 16 |
| Ag | Environmental and geological samples | DLLME | GFAAS | Cu–DDTC | 48 μL carbon tetrachloride | 0.5 mL methanol | 0.1–5.0 ng mL−1 | 20 ng L−1 | 17 |
| Ag | Water samples | UA-DLLME-SFO | GFAAS | 5-(4-dimethylaminobenzyliden)-rhodamine | 30 μL 1-undecanol | — | 0.10–10.0 ng mL−1 | 0.056 ng mL−1 | 18 |
| Ag | Water samples | SI-DLLME | FAAS | DDTC | 1-Dodecanol | Methanol | 0.40–20.0 μg L−1 | 0.15 μg L−1 | 19 |
| Ag | Drinking water and hair samples | TC-IL-DLPME | GFAAS | Disulfiram | [C6Mim][PF6] | — | 6.0–100.0 ng L−1 | 5.2 ng L−1 | 20 |
| Al | Water samples | DLLME-SFO | ICP-OES | Morin | 132 μL 1-undecanol | 2 mL acetone | 1.0–250.0 μg L−1 | 0.8 μg L−1 | 21 |
| As | Urine and whole blood | IL-DLLME | FI-HGAAS | APDC | 0.2 g [BMim][PF6] | — | 0.02–10 μg L−1 | 5 ng L−1 | 22 |
| As | Water samples | DLLME | GFAAS | APDC | 35 μL carbon tetrachloride | 0.5 mL methanol | 0.1–10 ng mL−1 | 36 ng L−1 | 23 |
| As | Water and urine samples | IL-DLLME | ETAAS | APDC | 0.06 g [C6Mim][PF6] | — | 50–400 ng L−1 | 10 ng L−1 | 24 |
| As, Sb | Water samples | DLLME | ETAAS | APDC | 50 μL carbon tetrachloride | 0.4 mL methanol | 0.06–2 (As), 0.05–5 (Sb) μg L−1 | 0.01 (As), 0.05 (Sb) μg L−1 | 25 |
| Au | Silica ore and water samples | DLLME | GFAAS | Victoria blue R | 40 μL chlorobenzene | 1 mL acetone | 0.03–0.5 ng mL−1 | 0.005 ng mL−1 | 26 |
| Au | Platinum metal and its alloy and effluent | DLLME | ETAAS | Dicyclohexylamine | 100 μL chloroform | 1 mL acetone | 0.02–1.0 μg L−1 | 0.002 μg L−1 | 27 |
| Au | Pharmaceutical sample | AS-DLLME | GFAAS | Astra Phloxine | Toluene–tetrachloromethane | Methanol | 0.5–39.4 μg L−1 | — | 28 |
| Au | Water samples, soils and river sediments | DLLME | ETAAS | TBAB | 35 μL chlorobenzene | 0.5 mL acetone | 0.075–0.75 μg L−1 | 42 ng L−1 | 29 |
| Bi | Water samples | DLLME | FAAS | 5-Br-PADAP | 75 μL dichlorobenzene | 700 μL acetone | 30–1700 ng mL−1 | 3.0 ng mL−1 | 30 |
| Cd | Water samples | DLLME | GFAAS | APDC | 34 μL carbon tetrachloride | 500 μL methanol | 2–20 ng L−1 | 0.6 ng L−1 | 11 |
| Cd | Water samples | DLLME | GFAAS | Salen | 34 μL carbon tetrachloride | 500 μL methanol | 2–21 ng L−1 | 0.5 ng L−1 | 31 |
| Cd | Water samples | UA-IL-DLLME | ETAAS | DDTC | 73 μL [C6Mim][PF6] | — | 20–150 ng L−1 | 7.4 ng L−1 | 32 |
| Cd | Water, beverage and cereal samples | DLLME-SFO | FAAS | 8-HQ | 100 μL 1-dodecanol | 1 mL methanol | 1–50 ng mL−1 | 0.3 ng mL−1 | 33 |
| Cd | Water samples | DLLME | AFS | 8-HQ | Carbon tetrachloride | Acetonitrile | 1–40 ng mL−1 | 19.4 ng L−1 | 34 |
| Cd | Water samples | DLLME | GFAAS | BPHA | 34 μL carbon tetrachloride | 500 μL methanol | 2–21 ng L−1 | 0.6 ng L−1 | 35 |
| Cd | Water samples | DLLME | ICP-OES | APDC | 200 μL chloroform | 2 mL ethanol | — | 0.8 ng L−1 | 36 |
| Cd | Water and food samples | DLLME | FAAS | DPTH | 0.5 mL chloroform | 1 mL methanol | 5–100 μg L−1 | 0.4 μg L−1 | 37 |
| Cd | Water samples | DLLME | FAAS | Dtz | 30 μL tetrachloroethylene | 0.5 mL tetrahydrofuran | 5–150 μg L−1 | 1.2 μg L−1 | 38 |
| Cd, Pb | Human teeth samples | DLLME | GFAAS | DDTP | 35 μL carbon tetrachloride | 0.5 mL methanol | 0.02–1.0 (Cd), 0.1–10 (Pb) ng mL−1 | 5.6 (Cd), 45 (Pb) ng L−1 | 39 |
| Cd, Pb, Bi | Water samples | DLLME | ICP-MS | DDTC | 17 μL carbon tetrachloride | 0.5 mL methanol | 10–1000 ng L−1 | 0.5 (Cd), 1.6 (Pb), 4.7 (Bi) ng L−1 | 40 |
| Co | Water samples | DLLME | FAAS | Br–TAO | 50 μL carbon tetrachloride | 2 mL methanol | 3–100 μg L−1 | 0.9 μg L−1 | 41 |
| Co | Waters, urine and saliva samples | IL-DLLME | ETAAS | 1N2N | 60 mg [C6Mim][PF6] | 500 μL methanol | 0.038–3.5 μg L−1 | 3.8 ng L−1 | 42 |
| Co | Water samples | IL-DLLME | FAAS | PMBP | 0.3 g [C6Py][PF6] | 2 mL acetone | 2–166 μg L−1 | 0.70 μg L−1 | 43 |
| Co | Water and saline samples | IL-DLLME | FAAS | PAN | 75 mg [C6Mim][PF6] | 500 μL ethanol | 0.4–120 μg L−1 | 0.1 μg L−1 | 44 |
| Co | Water and pharmaceutical samples | FI-IL-DLLME | ETAAS | PAR | 35 mg tetradecyl(trihexyl)phosphonium chloride | 100 μL acetone | 0.080–7 μg L−1 | 8 ng L−1 | 45 |
| Co | Water samples | DLLME-SFO | FAAS | APDC | 80 μL 1-dodecanol | 1 mL ethanol | 1.15–110 μg L−1 | 0.35 μg L−1 | 46 |
| Co | Water samples | UA-DLLME | FAAS | APDC | 80 μL carbon tetrachloride | — | 2.5–500 μg L−1 | 0.8 μg L−1 | 47 |
| Co | Nutritional supplements | UA-TC-IL-DLPME | ETAAS | 1N2N | 80 mg [C6Mim][PF6] | — | 0.050–3 μg L−1 | 5.4 ng L−1 | 48 |
| Co, Cr, Cu, Mn | Water samples | DLLME-SFO | ICP-OES | TTA | 140 μL 1-undecanol | 2 mL acetone | 1.25–250 (Co, Cr, Cu), 0.5–250 (Mn) μg L−1 | 0.1–0.3 μg L−1 | 49 |
| Co, Cu, Mn, Ni, Zn | Water samples | IL-DLLME | ICP-OES | TTA | 210 μL [C6Mim][Tf2N] | 4 mL ethanol | From 0.25 (Co), 0.50 (Ni), 0.75 (Mn), 5.00 (Cu and Zn) to 200 ng mL−1 | 0.10–0.20 ng mL−1 | 50 |
| Co, Ni | Environmental water and rice samples | DLPME | GFAAS | PAN | 15 μL carbon tetrachloride | 1 mL acetone | — | 21 (Co), 33 (Ni) pg mL−1 | 51 |
| Co, Ni | Water samples and vitamin B12 | DLLME | FAAS | 1N2N | 45 or 67 μL 1,2-dichlorobenzene | 2 mL ethanol | 10–150 (Co), 10–250 (Ni) μg L−1 | 1.59 (Ni), 2.42 (Co) μg L−1 | 52 |
| Cr | Water | UA-IL-DLLME | ETAAS | APDC | 50 μL [C6Mim][PF6] | — | 0.5–8.0 ng mL−1 | 0.07 ng mL−1 | 53 |
| Cr | Water and hair samples | DLLME | ETAAS | APDC | 35 μL carbon tetrachloride | 500 μL methanol | 0.1–15.0 μg L−1 | 5.96 ng L−1 | 54 |
| Cr | Water samples | IL-DLLME | ETAAS | APDC | 100 μL [C8Mim]Cl and LiNTf2 | — | 0.005–0.1 μg L−1 | 0.002 μg L−1 | 55 |
| Cr | Water samples | DLLME | FAAS | APDC | 60 μL carbon tetrachloride | 2 mL ethanol | 0.3–20 μg L−1 | 0.07 μg L−1 | 56 |
| Cr | Water samples | DLLME | ETAAS | BPHA | 45 μL carbon tetrachloride | 1 mL ethanol | 0.1–5 ng mL−1 | 0.01 ng mL−1 | 57 |
| Cr, Co, Ni, Pb | Wastewaters | DLLME-SFO | GFAAS | 5-Br-PADAP | 80 μL 1-undecanol | 1.5 mL ethanol | 5.0–55 (Co), 5.0–40 (Ni), 5.0–50 (Pb), 1.0–25 (Cr) ng L−1 | 0.2 (Cr), 1.3 (Co, Ni, Pb) ng L−1 | 58 |
| Cr, Cu, Ni, Zn | Water samples | DLLME | ICP-OES | DDTC | 113 μL carbon tetrachloride | 1 mL methanol | 1–1000 (Cu, Ni, Zn), 1–750 (Cr) μg L−1 | 0.23–0.55 μg L−1 | 59 |
| Cu | Water samples | DLLME | FAAS | 8-HQ | 250 μL chloroform | 1.5 mL methanol | 50–2000 μg L−1 | 3 μg L−1 | 60 |
| Cu | Water samples | DLLME | FAAS | Ligandless | 15 μL 1,2-dichlorobenzene | 1.5 mL ethanol | 1.0 ng mL−1 to 0.6 μg mL−1 | 0.5 ng mL−1 | 61 |
| Cu | Water samples | IL-DLLME | FAAS | TMK | 65 mg [C6Mim][Tf2N] | 550 μL acetone | 2–50 μg L−1 | 0.45 μg L−1 | 62 |
| Cu | Water and beverage samples | DLLME-SFO | FAAS | 8-HQ | 150 μL 1-dodecanol | 1.25 mL methanol | 0.5–300 ng mL−1 | 0.1 ng mL−1 | 63 |
| Cu | Cereals | DLLME–SFO | FAAS | 8-HQ | 150 μL 1-dodecanol | 1.25 mL methanol | 0.5–500 ng mL−1 | 0.1 ng mL−1 | 64 |
| Cu, Pb | Water samples | SI-DLLME | FAAS | DDPA | Xylene | Methanol | 0.16–12.0 (Cu), 2.3–160.0 (Pb) μg L−1 | 0.04 (Cu), 0.54 (Pb) μg L−1 | 65 |
| Hg | Commercial food supplements of marine origin | DLLME | ICP-OES | Diphenyl-3-formazathiol | 130 μL carbon tetrachloride | 8 mL acetone | 1–500 μg L−1 | 0.24 μg L−1 | 66 |
| Li | Water samples | DLLME | FAAS | B15C5 | 90 μL tetrachloroethene | 8 mL acetone | 0.047–5.547 mg L−1 | 0.014 mg L−1 | 67 |
| Mo | Water samples | DLLME | GFAAS | DDTC | 30 μL carbon tetrachloride | 1 mL acetone | 0.04–0.8 ng mL−1 | 0.007 ng mL−1 | 68 |
| Ni | Water and biological samples | IL-DLLME | ETAAS | DDTC | 0.1 g [C4Mim][PF6] | — | 20–700 ng L−1 | 5 ng L−1 | 69 |
| Ni | Water samples | DLLME-SFO | FAAS | APDC | 80 μL 1-dodecanol | 0.75 mL ethanol | 4.23–250 μg L−1 | 1.27 μg L−1 | 70 |
| Ni | Water samples | DLLME | FAAS | BTAC | 50 μL carbon tetrachloride | 2 mL ethanol | 4.7–100.00 μg L−1 | 1.4 μg L−1 | 71 |
| Ni, Mn | Soil, vegetable, and water samples | IL-DLLME | FAAS | 8-HQ | 65 mg [C6Mim][Tf2N] | 550 μL ethanol | 2–100 (Mn), 4–200 (Ni) μg L−1 | 0.52 (Mn), 0.93 (Ni) μg L−1 | 72 |
| Pb | Water samples | DLLME | FAAS | DDTP | 52 μL carbon tetrachloride | 2.5 mL methanol | 1–70 μg L−1 | 0.5 μg L−1 | 13 |
| Pb | Water samples | DLLME | ETAAS | DDTP | 35 μL carbon tetrachloride | 0.5 mL acetone | 0.05–1 μg L−1 | 0.02 μg L−1 | 73 |
| Pb | Human urine and water samples | DLLME | GFAAS | PMBP | 40 μL carbon tetrachloride | 0.5 mL ethanol | 0.1–20 ng mL−1 | 39 ng L−1 | 74 |
| Pb | Water samples | DLLME | AFS | Dtz | 250 μL carbon tetrachloride | 3 mL ethanol | 0.01–100 ng mL−1 | 0.95 ng L−1 | 75 |
| Pb | Water samples | IL-DLLME | FAAS | APDC | 500 μL [C6Mim][PF6] | — | — | 1.5 μg L−1 | 76 |
| Pb | Water samples | DLLME-SFO | FAAS | APDC | 90 μL 1-dodecanol | 1 mL ethanol | 8.43–400 μg L−1 | 2.53 μg L−1 | 77 |
| Pb | Blood samples | TC-IL-DLPME | FAAS | APDC | 45 μL [C4Mim][PF6] | — | 5–20 μg L−1 | 0.13 μg L−1 | 78 |
| Pb, Cd | Water samples | DLLME | ETAAS | APDC | 50 μL carbon tetrachloride | 0.4 mL methanol | 0.03–1 (Pb), 0.01–0.3 (Cd) μg L−1 | 10 (Pb), 4 (Cd) ng L−1 | 79 |
| Pb, Cd | Water samples | SI-DLLME | ETAAS | APDC | Xylene | Methanol | 0.04–1.50 (Pb) 0.006–0.150 (Cd) μg L−1 | 10 (Pb), 2 (Cd) ng L−1 | 80 |
| Pb, Cd | Saline samples | IL-DLLME | FAAS | DDTC | 75 mg [C6Mim][PF6] | 500 μL ethanol | 2–100 (Pb), 0.1–15 (Cd) μg L−1 | 0.6 (Pb), 0.03 (Cd) μg L−1 | 81 |
| Pd | Water samples | DLLME | GFAAS | DDTC | 40 μL carbon tetrachloride | 0.5 mL ethanol | 0.1–5 ng mL−1 | 2.4 ng L−1 | 82 |
| Pd | Water and soil samples | DLLME | GFAAS | ACDA | 40 μL carbon tetrachloride | 800 μL acetone | 0.02–0.6 μg L−1 | 0.007 μg L−1 | 83 |
| Pd | Water samples | DLLME | FAAS | TRH | 0.15 mL chloroform | 1.5 mL ethanol | 100–2000 μg L−1 | 90 μg L−1 | 84 |
| Pd | Water and standard samples | DLLME | FAAS | Ligandless | 15 μL carbon tetrachloride | 2 mL ethanol | 0.015–7.0 μg mL−1 | 1.4 ng mL−1 | 85 |
| Pd | Environmental certified reference materials | DLLME | GFAAS | Cu–DDTC | 38 μL carbon tetrachloride | 0.2 mL methanol | 0.05–8.0 ng mL−1 | 7.6 ng L−1 | 86 |
| Pd | Water and road dust sample | SD-DLLME | ETAAS | TMK | 75 μL 1-octanol | 750 μL acetonitrile | 0.025–0.500 μg L−1 | 0.007 μg L−1 | 87 |
| Rh | Water and alloys | UA-IL-DLLME | FAAS | 5-Br-PADAP | 30 mg [C8MIm][Tf2N] | — | 4.0–500.0 ng mL−1 | 0.37 ng mL−1 | 88 |
| Rh | Water and road dust samples | DLLME | FAAS | TDMBA | 50 μL chlorobenzene | 1.5 mL ethanol | 0.6–500 ng mL−1 | 0.10 ng mL−1 | 89 |
| Sb | Water samples | DLLME | ETAAS | BPHA | 65 μL chloroform | 0.5 mL ethanol | 0.02–0.8 μg L−1 | 0.005 μg L−1 | 90 |
| Se | Water samples | DLLME | GFAAS | APDC | 35 μL carbon tetrachloride | 0.5 mL ethanol | 0.1–3 μg L−1 | 0.05 μg L−1 | 12 |
| Se | Water and garlic samples | FI-IL-DLLME | ETAAS | APDC | Tetradecyl(trihexyl)phosphonium chloride | Methanol | Up to 12.5 μg L−1 | 15 ng L−1 | 91 |
| Te | Water samples | DLLME | ETAAS | APDC | 30 μL carbon tetrachloride | 550 μL ethanol | 0.015–1 ng mL−1 | 0.004 ng mL−1 | 92 |
| Tl | Water and biological samples | SI-IL-DLLME | FAAS | — | [C6Mim][PF6] | Methanol | 2.8–120.0 μg L−1 | 0.86 μg L−1 | 93 |
| Tl | Water samples | IL-DLLME | ETAAS | Tetradecyl(trihexyl)phosphonium chloride | 60 mg [C6Mim][PF6] | 500 μL ethanol | 0.033–4 μg L−1 | 3.3 ng L−1 | 94 |
| Tl | Hair samples | DLLME-SFO | FAAS | PAN | 100 μL 1-dodecanol | 2.5 mL ethanol | 6.0–900.0 ng mL−1 | 2.1 ng mL−1 | 95 |
| U | Water samples | DLLME | ICP-OES | APDC | 200 μL chloroform | 500 μL methanol | 5–200 μg L−1 | 2.0 μg L−1 | 96 |
| FI-ICP-MS | 50–5000 ng L−1 | 30 ng L−1 | |||||||
| V | Water and parsley samples | DLLME-SFO | ETAAS | BPHA | 100 μL 1-undecanol | 200 μL acetone | 0.02–1 μg L−1 | 0.007 μg L−1 | 97 |
| V | Environmental and biological samples | TC-IL-DLME | ETAAS | 5-Br-PADAP | 40 μL [C4Mim][PF6] | 250 μL ethanol | Up to 15 μg L−1 | 4.8 ng L−1 | 98 |
| V | Water samples | TC-IL-DLME | ETAAS | 5-Br-PADAP | 45 μL [C4Mim][PF6] | 100 μL ethanol | Up to at least 5000 ng L−1 | 4.9 ng L−1 | 99 |
| Zn | Water and milk samples | IL-DLLME | FAAS | 8-HQ | 0.3 g [C6Py][PF6] | Acetonitrile | 0.8–33 μg L−1 | 0.22 μg L−1 | 100 |
| Zn | Water and food samples | IL-CIA-DLLME | FAAS | 8-HQ | 95 mg [C6Mim][PF6] | 650 μL ethanol | 0.7–26 μg L−1 | 0.18 μg L−1 | 101 |
| Zn, Cd | Water samples | DLLME | FAAS | Ligandless | 30 μL carbon tetrachloride | 2 mL ethanol | 1.0 ng mL−1 to 1.5 μg mL−1 (Zn), 1.0 ng mL−1 to 0.4 μg mL−1 (Cd) | 0.3 (Zn), 0.4 (Cd) ng mL−1 | 102 |
2 Principle of DLLME
The basic principle of the method lies in the rapid injection of a mixture of an extraction and a dispersive solvent into a sample solution. At the moment of injection, a cloudy solution is formed consisting of very fine droplets of extraction solvent dispersed throughout the whole aqueous phase with the aid of a dispersive solvent. Since the interfacial area between the many tiny droplets of extraction solvent and the bulk aqueous phase is infinitely large, the transfer of analyte from the aqueous-donor phase to the organic-acceptor phase occurs very rapidly, leading very quickly to a state of equilibrium and thus to a very short extraction time, which can be considered as the prime advantage of DLLME. As the volume of the extraction organic phase involved in DLLME is on the microlitre level, the high donor-to-acceptor phase volume ratio results in high analyte enrichment, a second important goal of DLLME. In addition, the merits of DLLME are its simplicity of operation, rapidity and low cost. While in a cloudy state the solution must be exposed to centrifugation in order to completely separate the phases and to get the extraction solvent with the target analyte to the bottom of the test tube. After centrifugation, the organic drop containing the analyte of interest is withdrawn and is exposed to instrumental analysis.3 Factors affecting the DLLME procedure
There are certain factors which can highly affect the extraction efficiency and which therefore should be investigated: type and volume of extraction solvent, type and volume of dispersive solvent, effect of adding a salt, effect of a surfactant, extraction time, time and rate of centrifugation and effect of chelating agents.3.1 Type and volume of extraction solvent
Selecting an extraction solvent is the major task in DLLME, since it can affect the extraction efficiency to such a great extent. In general, the extraction solvent in DLLME has to fulfil several requirements: its extraction capability for the target analyte should be as high as possible; its miscibility with the aqueous phase should be as low as possible; it has to be of density higher than water for easy phases separation by centrifugation. Aside from the mentioned general requirements for extraction solvents, when DLLME is coupled with atomic spectrometry, special requirements resulting from the electrothermal atomic absorption spectrometry (ETAAS) and flame atomic absorption spectrometry (FAAS) equipment must be taken into consideration. The most commonly employed extraction solvents in DLLME procedures coupled with atomic spectrometry include halogenated hydrocarbons such as carbon tetrachloride, chloroform, chlorobenzene, dichlorobenzene, tetrachloroethylene and tetrachloroethane as well as various ionic liquids, and only in a few cases solvents less dense than water, such as dodecanol, undecanol, octanol, xylene and toluene (Fig. 2).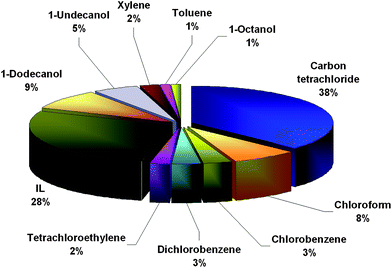 | ||
| Fig. 2 The frequency of use of various extraction solvents in DLLME coupled with atomic spectrometric detection based on the data in Table 1. | ||
Variations of enrichment factors using different extraction solvents do not necessarily make a significant difference. Nevertheless, researchers frequently prefer carbon tetrachloride because this solvent forms a stable cloudy solution, requires smaller volume consumption to obtain the necessary volume of sedimented phase, and its sedimented phase can be easily removed by a sampler (or a micropipette) to be introduced into a graphite furnace. In contrast, chloroform forms an unstable cloudy solution and carbon disulfide is difficult to remove with a sampler.11,31,73
It is to be anticipated that increasing the volume of extraction solvent will lead to an increase in the sedimented phase, with a consequent decrease in the enrichment factor. Therefore, at a low volume of extraction solvent, a higher enrichment factor can be expected.73 On the other hand, too small volume of extraction solvent leads to a decrease in the sedimented phase volume and consequently makes removal for injection into GFAAS difficult, meaning the procedure is potentially accompanied by a systematic error.11,83 Usually a microlitre volume of the extraction solvent is employed.
In FAAS, the extraction solvent should also fulfil additional requirements; namely it should exhibit good nebulisation and burning characteristics.13 Moreover, contrary to ETAAS, when the detection equipment requires a small and defined sample volume, the sample volume required for the FAAS equipment is usually greater than that obtained in the DLLME procedure.41 These difficulties can be overcome by dilution of the sedimented phase with a proper diluting agent (which is often acidified in order to induce dissociation of analyte complexes and further release the analyte into solution),62,72 evaporation of the organic solvent and replacement by another solvent,84 or the use of various microsample introduction systems.13,41
In conventional DLLME, the extraction solvent is limited to those solvents of density higher than water, which is moreover hazardous. To overcome this drawback, a dispersive liquid–liquid microextraction based on the solidification of a floating organic drop (DLLME-SFO) was developed.103 In this technique, the extraction solvent must have a melting point near room temperature in a range from 10–30 °C, and an additional solidification step is required, which can be considered as a limitation. Despite this fact, some articles devoted to coupling DLLME-SFO with atomic spectrometry can be found in the scientific literature (see Table 1).
Another solution which allows the application of extraction solvents lighter than water in DLLME coupled with ETAAS has been reported by our group;28 it is based on an auxiliary solvent (AS) assisted DLLME methodology. The purpose of the auxiliary solvent is to alter the density of the extraction phase such that it becomes heavier than water and is thus easily separable from the aqueous sample by centrifugation, as is usual in conventional DLLME procedures. The efficiency of the suggested approach was demonstrated through the determination of gold based on the formation of the ion pair [Au(CN)2]− with Astra Phloxine reagent and its extraction using the DLLME procedure with subsequent graphite furnace atomic absorption spectrometric detection.
The application of ionic liquids (ILs) offers many benefits in comparison with organic solvents.104,105 Nevertheless, using ILs in DLLME coupled with atomic spectrometry can also be a source of problems. Direct automatic injection of the ionic liquid phase into a graphite furnace is difficult due to the high viscosity of the sedimented phase. Therefore, dilution with an appropriate solvent is often required.32,42,94,106 Moreover, in the presence of IL, due to its organic nature, the background signal can be increased; therefore, several parameters, such as thermal treatment in a graphite furnace42,94 and the effect of a modifier, should be investigated.32,42,106
3.2 Type and volume of dispersive solvent
Selection of the dispersive solvent is another important task in DLLME, because this solvent promotes the formation of fine droplets of extraction solvent in the aqueous phase, thus increasing the interfacial surface. Like the extraction solvent, the dispersive solvent also has to satisfy certain conditions, the most important of which is its miscibility with both the extraction solvent and the aqueous sample. The dispersive solvents most frequently used in DLLME procedures coupled with atomic spectrometry thus include ethanol, methanol, acetone and acetonitrile (Fig. 3).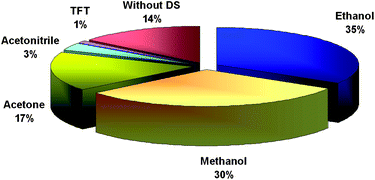 | ||
| Fig. 3 The frequency of use of various dispersive solvents in DLLME coupled with atomic spectrometric detection; DS, dispersive solvent based on the data in Table 1. | ||
Dispersive solvent volume has a direct impact on the dispersion of extraction solvent in the aqueous phase and thus influences the extraction efficiency. At a low volume the dispersive solvent is unable to disperse the extraction solvent throughout the entire aqueous sample properly, and the cloudy solution does not entirely form.25,26,74,85 With an increase in the volume of dispersive solvent, two competing processes may occur.15 The solubility of the analyte complex in aqueous phase can increase, leading to the diminished partition of polar compounds into the extraction solvent droplets. On the other hand, the increased volume of the partitioned dispersive solvent into the extraction solvent phase can lead to an increase in the extraction efficiency. Typically, 0.5–1.5 mL of dispersive solvent is applied, usually in great excess of the extraction solvent.
Besides conventional DLLME with an organic solvent as dispersive agent, various alternatives have been reported to produce a cloudy state, such as the application of vortex agitation or ultrasound energy32,53,88 or the formation of fine droplets of a water-immiscible IL generated in situ.55
3.3 Salt addition
The addition of electrolytes to the sample solution causes a decrease in the solubility of analytes in the aqueous phase and thus enhances their partitioning into the organic phase.107 This process is known as the salting-out effect and is commonly used in conventional LLE. At the same time, a reduction in the mutual miscibility of the two liquids can be observed,30 causing an increase in the volume of the sedimented phase. The combination of these opposing phenomena has no significant impact on the extraction efficiency of the DLLME characterised by the recovery and enrichment factor,83 as reported by a majority of authors, and extraction experiments are therefore usually performed without salt addition. However, in some papers authors have reported a significant effect of salt addition on the enrichment factor or recovery.40,423.4 Role of surfactant
Surfactants can improve formation of the cloudy state and the extraction efficiency96 and can also play the role of anti-sticking agent – minimising the interaction between the ionic liquid and the wall of the centrifuge tube42 or the inner walls of tubes in on-line flow based systems, as can be confirmed by some examples given below.Chandrasekaran et al.96 reported a DLLME method for the determination of uranium(VI) in groundwater/seawater by ICP-OES and FI-ICPMS based on the complexation of uranium(VI) with APDC in the presence of cetyltrimethyl ammonium bromide, which enhanced the hydrophobicity of the ion-associate complex, resulting in its improved extraction into chloroform. The addition of CTAB improved the extraction of the uranium(VI)–APDC complex from 56% (without CTAB) to 95–98%.
In on-line extraction systems the high viscosity of some ILs can lead to partial adherence of the IL droplets on the inner walls of the tubes. Since surfactant molecules surround the fine droplets of the IL, the adherence of the IL to the inner walls of the tubes is thus avoided, which reduces analyte dispersion in the whole FI system.45,91,98 Nevertheless, higher surfactant concentrations could negatively affect the retention of the IL phase into the microcolumn, which could, on the other hand, cause problems with reproducibility of the results.
3.5 Extraction time
In DLLME the extraction time can be defined as the interval between the injection of the mixture of solvents and the start of centrifugation. Because the interfacial surface between the extraction solvent and aqueous phase in the cloudy state is so large, equilibrium is achieved very quickly. The majority of researchers are of the opinion that the extraction time is not really important in DLLME; thus, the technique can be considered as time-independent. Nevertheless, some authors do use either manual shaking, vortex agitation, ultrasound or a combination of these techniques in order to ensure better mixing. In these cases, the extraction time is defined as the time spent shaking the mixture after the organic phase was added to the sample solution. However, herein we have to note that the frequency and amplitude of shaking when performed manually may be different for every extraction, and thus manual performance can affect the extraction efficiency as well as the amount of extraction solvent dissolved in water.108,1093.6 Centrifugation time and rate
DLLME is based on the formation of a cloudy state which is stable for a more or less long period of time. Therefore, centrifugation is necessary for the thorough separation of the extraction phase prior to the detection step. Authors tend to merely state the centrifugation conditions and only in exceptional cases mention their detailed examination. It must be emphasised that centrifugation is the most time-consuming step in DLLME. Too short a centrifugation time may not provide satisfactory separation, while a longer centrifugation time can cause problems due to local heat generation,20 which can lead to the dissolution of the complex or the ionic liquid in aqueous phase or to the particular evaporation of the extraction solvent. Therefore, centrifugation time that is either too short or too long can affect the quality of the determination. Moreover, too long a centrifugation time needlessly prolongs the analysis and thus reduces the sample throughput.20,75,88,94,99 The higher the centrifugation rate, the shorter the centrifugation time, and consequently the shorter the time needed for the whole analytical procedure.110 However, the centrifugation rate is also limited by the material of the centrifugal tube.3.7 Chelating reagents
Chelation is a well-known and widely used methodology for trace metal preconcentration. The basic principles of the application of chelating agents in DLLME are similar as in conventional LLE and have been described in a variety of monographs and reviews.111,112 Generally, the chelating reagent should form with target analyte a sufficiently stable complex that is easily extractable into the organic phase. The most frequently used reagents for chelation of metals in DLLME coupled with atomic spectroscopic methods are ammonium pyrrolidinedithiocarbamate, sodium diethyldithiocarbamate, 8-hydroxyquinoline, 1-(2-pyridylazo)-2-naphthol, and dithizone (Fig. 4). Nevertheless, some approaches have also been reported that do not employ ligands.15,61,85,102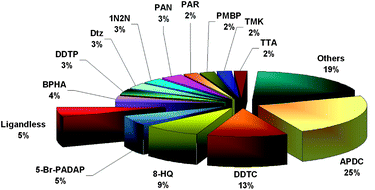 | ||
| Fig. 4 The frequency of use of various chelating agents in DLLME coupled with atomic spectrometric detection based on the data in Table 1. | ||
The reagents used are usually poorly selective and form complexes with many metals, which can cause interferences as a result of other metal ions competing for the chelating agent and their subsequent co-extraction.113 Liang et al. suggested a displacement-dispersive liquid–liquid microextraction coupled with graphite furnace atomic absorption spectrometry for the determination of silver17 and palladium86 in complicated matrices. The technique is based on the different stability of metal complexes and thus improves the selectivity of the determination. The method includes two steps. First, Cu(II) is complexed by DDTC and extracted using the DLLME procedure. Afterwards, the sedimented phase is dispersed into the sample solution with the aid of a dispersive solvent and a second DLLME procedure is performed. Since the target analyte forms a complex with DDTC that is more stable than the Cu–DDTC complex, the analyte can displace Cu(II) from the pre-extracted Cu–DDTC complex. Only Hg(II), Pd(II) and Ag(I) are able to displace Cu(II) from the Cu–DDTC complex. Consequently, in comparison with the conventional DLLME method, the interference caused by the competition for DDTC by other co-existing metals can be largely eliminated.17
4 Speciation analysis
According to Florence, “Speciation of an element is the determination of the individual physico-chemical forms of that element which together make up its total concentration in a sample”.114 Speciation analysis is important for studying the bioavailability, bioaccumulation and toxicological properties of a particular element. Atomic absorption spectroscopic methods can be applicable for speciation studies only if during preparatory stages measures are taken to prevent any change occurring in the form of the species, or if more separation stages are added to separate the different analyte species.115Liang et al.23 suggested a method for arsenic speciation based on the complexation of As(III) with ammonium pyrrolidinedithiocarbamate followed by extraction into carbon tetrachloride droplets formed during the DLLME procedure and subsequent graphite furnace atomic absorption spectrometric determination. Total inorganic arsenic was then determined after reduction of As(V) to As(III); As(V) was calculated by the difference. The method was applied to the speciation of arsenic in river, lake and tap water samples. Shirkhanloo et al.24 applied 1-hexyl-3-methylimidazolium hexafluorophosphate as an ionic liquid for the same purpose.
López-García55 reported a method for the determination of very low amounts of Cr(III) and Cr(VI) using APDC as a chelating agent followed by dispersive liquid–liquid microextraction by in situ formation of an ionic liquid and subsequent electrothermal atomic absorption spectrometry. Other papers can be found devoted to the coupling of DLLME with atomic absorption spectrometric determination for speciation of chromium,57 antimony,90 selenium,91 tellurium,92 thallium94,95 and vanadium.99
5 Automation of DLLME
In addition to miniaturisation, we can at present also observe another trend in analytical chemistry – toward automation. While the pursuit of miniaturisation of conventional LLE led to the development of DLLME, which enabled the consumption of sample and organic solvent to be reduced, thus reducing the volume of waste generated as well as increasing the sensitivity of the methods, automation allows for the elimination of tedious, laborious and time-consuming steps, thus helping to increase the precision of a determination.Generally speaking, the automation of LLE is difficult, mainly due to the complexity of the manifolds which consist of three key parts, namely a segmentor, an extraction coil and a phase separator. The phase separator is the most crucial of these components,116,117 which could explain why the applications of on-line LLE systems are rather limited in comparison with other on-line preconcentration systems such as solid-phase extraction.
In regard to DLLME we can mention two shortcomings: firstly, the procedure is performed manually and secondly, the procedure requires centrifugation for separation of the phases, which can be considered as the most time-consuming step. To overcome these drawbacks, automation based on sequential injection systems seems to be very suitable. Herein we would like to underline the works of Anthemidis and Ioannou, in which DLLME was fully automated and was successfully coupled with flame atomic absorption spectrometry (Fig. 5)65 and electrothermal atomic absorption spectrometry (Fig. 6).80
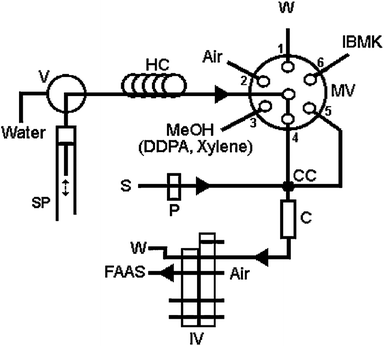 | ||
| Fig. 5 Schematic manifold for SI-DLLME metal determination by FAAS. S, sample; MeOH, solution containing 2.0% (v/v) xylene and 0.3% (m/v) DDPA; W, waste; P, peristaltic pump; SP, syringe pump; MV, multiposition valve; IV, injection valve in “load” position; V, valve in “out” position; HC, holding coil; C, microcolumn; CC, confluence connector.65 Reprinted from ref. 65 with permission from Elsevier. | ||
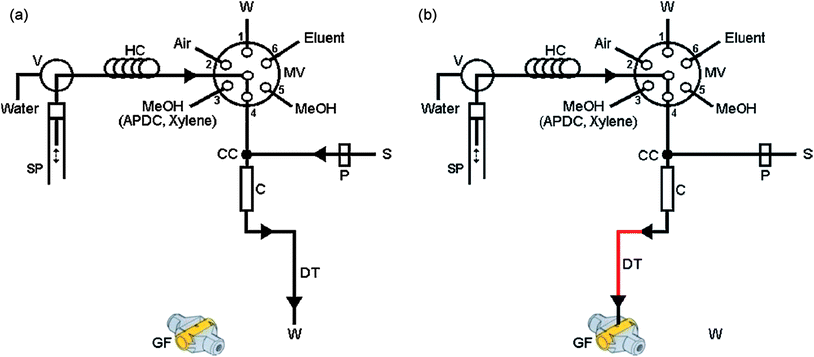 | ||
| Fig. 6 Schematic manifold for SI-DLLME metal determination by ETAAS. (a) Extraction step and (b) elution step. S, sample; MeOH, solution containing 2.0% (v/v) xylene and 0.2% (m/v) APDC; W, waste; P, peristaltic pump; SP, syringe pump; MV, multi-position valve; HC, holding coil; C, microcolumn; CC, confluence connector; GF, graphite furnace of ETAAS.80 Reprinted from ref. 80 with permission from Elsevier. | ||
In their system a 5 μL segment of air and several hundred microlitres of disperser solution – methanol containing extraction solvent and a chelating agent – are aspirated into a holding coil. Afterwards, the sample is transported toward a microcolumn through a confluence connector using a peristaltic pump. Simultaneously, a syringe pump dispenses the disperser solution toward the microcolumn, also through the confluence connector. Thus, a cloudy solution is formed, and the metal complex is extracted continuously from the aqueous to organic phase. The droplets of extraction solvent which contain the metal complex are retained on PTFE-turnings in the microcolumn. Next, a few hundred microliters of IBMK are delivered through the microcolumn in order to elute the analyte. The eluent is then forwarded to an FAAS nebuliser or into the graphite tube of the ETAAS for automation and measurement. The potential of the system was demonstrated for copper, lead, cadmium and silver determination.19,65,80
Very recently, an SI-DLLME manifold with some modifications was employed for automation of ionic liquid dispersive liquid–liquid microextraction.93 In this approach, the ionic liquid is fully dispersed into the sample solution on-line, and the resulting cloudy solution flows through a microcolumn, where the fine droplets containing extracted analyte are retained on the surface of polyurethane foam packed into the microcolumn. Then, a micro-volume of 300 μL MIBK were delivered through the microcolumn for elution and transportation of analyte to the FAAS nebuliser. As an advantage we can point to the fact that no heating/cooling step is required. A method for trace thallium determination based on SI-IL-DLLME and flame atomic absorption spectrometry was developed and applied for the analysis of water and biological samples.
6 Concluding remarks
Despite the technical limitations of some detection systems (due mainly to requirements regarding the extraction solvent and the volume of sedimented phase needed to perform a measurement), the DLLME method has now been coupled with the majority of analytical instrumentation. Electrothermal and flame atomic absorption spectrometry are no exception. DLLME procedures coupled with atomic absorption spectrometry have been reported for a number of elements. On the other hand, the number of papers dedicated to the coupling of DLLME with plasma methods is limited, as is clear from Fig. 7.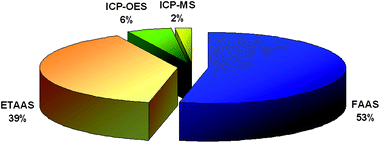 | ||
| Fig. 7 Coupling of DLLME with atomic detection techniques based on the data in Table 1. | ||
Notwithstanding the generally acclaimed high sensitivity of atomic absorption spectrometric methods, they can only offer information on the total concentration of elements in a sample; they are not satisfactorily selective and fail to provide us with sufficient data about speciation.118 In the past decades, we can see a clearly increasing trend in the number of articles devoted to speciation analysis.119 However, very few articles have focused on the application of DLLME for speciation analysis, and we did not find any articles devoted to its exploitation for indirect determination of non-metals or organic compounds employing ETAAS and FAAS techniques.
The application of ionic liquids is “a hot research topic in analytical chemistry”,120 and one can find numerous reviews121–123 and research articles124–126 devoted to this subject. Ionic liquids are already well represented in the DLLME technique, as can be seen in Fig. 2, and we can expect to see a further increase in publications in this field.
In addition, further developments in the automation of DLLME can be expected in the future.
Abbreviations
| AS-DLLME | Auxiliary solvent assisted dispersive liquid–liquid microextraction; |
| DLLME | Dispersive liquid–liquid microextraction; |
| DLLME-SFO | Dispersive liquid–liquid microextraction based on the solidification of a floating organic drop; |
| DLPME | Dispersive liquid phase microextraction; |
| ETAAS | Electrothermal atomic absorption spectrometry; |
| FAAS | Flame atomic absorption spectrometry; |
| FI | Flow injection; |
| GFAAS | Graphite furnace atomic absorption spectrometry; |
| HGAAS | Hydride generation atomic absorption spectrometry; |
| ICP-MS | Inductively coupled plasma mass spectrometry; |
| ICP-OES | Inductively coupled plasma-optical emission spectrometry; |
| IL-CIA-DLLME | Ionic liquid cold-induced aggregation dispersive liquid–liquid microextraction; |
| IL-DLLME | Ionic liquid dispersive liquid–liquid microextraction; |
| SD-DLLME | Solvent-based de-emulsification dispersive liquid–liquid microextraction; |
| SI-DLLME | Sequential injection dispersive liquid–liquid microextraction; |
| TC | Temperature-controlled; |
| UA | Ultrasound-assisted. |
Acknowledgements
Partial financial support from the Scientific Grant Agency of the Ministry of Education of the Slovak Republic and the Slovak Academy of Sciences (grant no. 1/0226/11) is gratefully acknowledged.Notes and references
- E. Jackwerth, A. Mizuike, Y. A. Zolotov, H. Berndt, R. Hohn and N. M. Kuzmin, Pure Appl. Chem., 1979, 51, 1195 Search PubMed.
- S. Liu and P. K. Dasgupta, Anal. Chem., 1995, 67, 2042 CrossRef CAS.
- H. Liu and P. K. Dasgupta, Anal. Chem., 1996, 68, 1817 CrossRef CAS.
- M. A. Jeannot and F. F. Cantwell, Anal. Chem., 1996, 68, 2236 CrossRef CAS.
- S. Pedersen-Bjergaard and K. E. Rasmussen, Anal. Chem., 1999, 71, 2650 CrossRef CAS.
- M. A. Jeannot and F. F. Cantwell, Anal. Chem., 1997, 69, 235 CrossRef CAS.
- A. L. Theis, A. J. Waldack, S. M. Hansen and M. A. Jeannot, Anal. Chem., 2001, 73, 5651 CrossRef CAS.
- M. Ma and F. F. Cantwell, Anal. Chem., 1999, 71, 388 CrossRef CAS.
- W. Liu and H. K. Lee, Anal. Chem., 2000, 72, 4462 CrossRef CAS.
- M. Rezaee, Y. Assadi, M.-R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr., A, 2006, 1116, 1 CrossRef CAS.
- E. Zeini Jahromi, A. Bidari, Y. Assadi, M. R. Milani Hosseini and M. R. Jamali, Anal. Chim. Acta, 2007, 585, 305 CrossRef.
- A. Bidari, E. Zeini Jahromi, Y. Assadi and M. R. Milani Hosseini, Microchem. J., 2007, 87, 6 CrossRef CAS.
- M. T. Naseri, P. Hemmatkhah, M. R. M. Hosseini and Y. Assadi, Anal. Chim. Acta, 2008, 610, 135 CrossRef.
- S. Jafarvand, A. Bidari, P. Hemmatkhah, M. R. M. Hosseini and Y. Assadi, Anal. Lett., 2009, 42, 2214 CrossRef CAS.
- S. Z. Mohammadi, D. Afzali, M. A. Taher and Y. M. Baghelani, Talanta, 2009, 80, 875 CrossRef.
- P. Liang and L. Peng, Microchim. Acta, 2010, 168, 45 CrossRef CAS.
- P. Liang, L. Zhang and E. Zhao, Talanta, 2010, 82, 993 CrossRef CAS.
- D. Afzali, A. R. Mohadesi, B. B. Jahromi and M. Falahnejad, Anal. Chim. Acta, 2011, 684, 45 Search PubMed.
- A. N. Anthemidis and K.-I. G. Ioannou, Talanta, 2011, 84, 1215 CrossRef CAS.
- G. Absalan, M. Akhond, L. Sheikhian and D. M. Goltz, Anal. Methods, 2011, 3, 2354 RSC.
- M. Rezaee, Y. Yamini, A. Khanchi, M. Faraji and A. Saleh, J. Hazard. Mater., 2010, 178, 766 CrossRef CAS.
- H. Shirkhanloo, A. Rouhollahi and H. Z Mousavi, Bull. Korean Chem. Soc., 2011, 32, 3923 Search PubMed.
- P. Liang, L. Peng and P. Yan, Microchim. Acta, 2009, 166, 47 CrossRef CAS.
- H. Shirkhanloo, H. Z. Mousavi and A. Rouhollahi, J. Chin. Chem. Soc., 2011, 58, 623 Search PubMed.
- R. E. Rivas, I. López-García and M. Hernández-Córdoba, Spectrochim. Acta, Part B, 2009, 64, 329 CrossRef.
- M. Shamsipur and M. Ramezani, Talanta, 2008, 75, 294 CrossRef CAS.
- S. Kagaya, D. Takata, T. Yoshimori, T. Kanbara and K. Tohda, Talanta, 2010, 80, 1364 CrossRef CAS.
- L. Kocúrová, I. S. Balogh, J. Škrlíková, J. Posta and V. Andruch, Talanta, 2010, 82, 1958 CrossRef CAS.
- I. De La Calle, F. Pena-Pereira, N. Cabaleiro, I. Lavilla and C. Bendicho, Talanta, 2011, 84, 109 CrossRef CAS.
- M. Fayazi, D. Afzali and A. Mostafavi, J. Anal. At. Spectrom., 2011, 26, 2064 RSC.
- A. Moghimi, J. Chin. Chem. Soc., 2008, 55, 369 CAS.
- S. Li, S. Cai, W. Hu, H. Chen and H. Liu, Spectrochim. Acta, Part B, 2009, 64, 666 CrossRef.
- Q. Wu, C. Wu, C. Wang, X. Lu, X. Li and Z. Wang, Anal. Methods, 2011, 3, 210 RSC.
- Q. Zhou, N. Zhao and J. Xiao, At. Spectrosc., 2011, 32, 62 Search PubMed.
- A. Moghimi and M. J. Poursharifi, Asian J. Chem., 2011, 23, 1429 Search PubMed.
- M. Salahinejad and F. Aflaki, Environ. Monit. Assess., 2011, 177, 115 CrossRef CAS.
- F. S. Rojas, C. B. Ojeda and J. M. C. Pavón, Anal. Methods, 2011, 3, 1652 RSC.
- G. Karim-Nezhad, M. Ahmadi and B. Zare-Dizajdizi, J. Braz. Chem. Soc., 2011, 22, 1816 Search PubMed.
- C. Zhang, Y. Wang, X. Cheng, H. Xia and P. Liang, J. Chin. Chem. Soc., 2011, 58, 919 Search PubMed.
- X. Jia, Y. Han, X. Liu, T. Duan and H. Chen, Microchim. Acta, 2010, 171, 49 CrossRef CAS.
- P. X. Baliza, L. S. G. Teixeira and V. A. Lemos, Microchem. J., 2009, 93, 220 CrossRef CAS.
- P. Berton and R. G. Wuilloud, Anal. Chim. Acta, 2010, 662, 155 CrossRef CAS.
- H. Abdolmohammad-Zadeh and E. Ebrahimzadeh, Cent. Eur. J. Chem., 2010, 8, 617 Search PubMed.
- S. R. Yousefi and S. J. Ahmadi, Microchim. Acta, 2011, 172, 75 Search PubMed.
- P. Berton and R. G. Wuilloud, Anal. Methods, 2011, 3, 664 RSC.
- J.-W. Zhang, X.-J. Ke, Y.-K. Wang, X. Du, J.-J. Ma and J.-C. Li, J. Chin. Chem. Soc., 2011, 58, 911 Search PubMed.
- Y. Wang, X. Ke, J. Zhang, X. Du, J. Ma and J. Li, Bull. Chem. Soc. Ethiop., 2012, 26, 9 Search PubMed.
- P. Berton, E. M. Martinis, L. D. Martinez and R. G. Wuilloud, Anal. Chim. Acta, 2012, 713, 56 CrossRef CAS.
- Y. Yamini, M. Rezaee, A. Khanchi, M. Faraji and A. Saleh, J. Chromatogr., A, 2010, 1217, 2358 CrossRef CAS.
- L. Ranjbar, Y. Yamini, A. Saleh, S. Seidi and M. Faraji, Microchim. Acta, 2012, 177, 119 CrossRef CAS.
- H. Jiang, Y. Qin and B. Hu, Talanta, 2008, 74, 1160 CrossRef CAS.
- M.-H. Sorouraddin and L. Khoshmaram, J. Chin. Chem. Soc., 2010, 57, 1346 Search PubMed.
- H. Chen, P. Du, J. Chen, S. Hu, S. Li and H. Liu, Talanta, 2010, 81, 176 CrossRef.
- M. S. Tehrani, P. A. Azar, S. W. Husain and F. Shafaei, Asian J. Chem., 2010, 22, 6302.
- I. López-García, Y. Vicente-Martínez and M. Hernández-Córdoba, J. Anal. At. Spectrom., 2012, 27, 874 RSC.
- P. Hemmatkhah, A. Bidari, S. Jafarvand, M. R. M. Hosseini and Y. Assadi, Microchim. Acta, 2009, 166, 69 CrossRef CAS.
- Y. Li, G. Hou, Z. Zhanga and Y. Zhaoc, At. Spectrosc., 2011, 32, 160 CAS.
- M. Mirzaei, M. Behzadi, N. M. Abadi and A. Beizaei, J. Hazard. Mater., 2011, 186, 1739 CrossRef CAS.
- H. Sereshti, V. Khojeh and S. Samadi, Talanta, 2011, 83, 885 CrossRef CAS.
- M. A. Farajzadeh, M. Bahram, B. G. Mehr and J. A. Jönsson, Talanta, 2008, 75, 832 CrossRef CAS.
- S. Z. Mohammadi, D. Afzali and Y. M. Baghelani, Anal. Chim. Acta, 2009, 653, 173 CrossRef CAS.
- R. Khani, F. Shemirani and B. Majidi, Desalination, 2011, 266, 238 CrossRef CAS.
- C. Wu, B. Zhao, Y. Li, Q. Wu, C. Wang and Z. Wang, Bull. Korean Chem. Soc., 2011, 32, 829 Search PubMed.
- C. X. Wu, Q. H. Wu, C. Wang and Z. Wang, Chin. Chem. Lett., 2011, 22, 473 CrossRef CAS.
- A. N. Anthemidis and K.-I. G. Ioannou, Talanta, 2009, 79, 86 CrossRef CAS.
- A. Bidari, M. R. Ganjali, Y. Assadi, A. Kiani and P. Norouzi, Food Anal. Methods, 2012, 5, 695 Search PubMed.
- M. H. Mallah and M. Davoudi, J. Radioanal. Nucl. Chem., 2012, 293, 247–254 Search PubMed.
- M. Shamsipur and S. Habibollahi, Microchim. Acta, 2010, 171, 267 CrossRef CAS.
- H. Shirkhanloo, A. Rouhollahi and H. Z. Mousavi, J. Chin. Chem. Soc., 2010, 57, 1035 Search PubMed.
- Y. Wang, J. Zhang, B. Zhao, X. Du, J. Ma and J. Li, Biol. Trace Elem. Res., 2011, 144, 1381 Search PubMed.
- V. A. Lemos, E. V. Dos Santos Vieira, E. Dos Santos Silva and L. O. Dos Santos, Clean: Soil, Air, Water, 2012, 40, 268 Search PubMed.
- R. Khani and F. Shemirani, Clean: Soil, Air, Water, 2010, 38, 1177 Search PubMed.
- M. T. Naseri, M. R. Milani Hosseini, Y. Assadi and A. Kiani, Talanta, 2008, 75, 56 CrossRef CAS.
- P. Liang and H. Sang, Anal. Biochem., 2008, 380, 21 CrossRef CAS.
- Q. Zhou, N. Zhao and G. Xie, J. Hazard. Mater., 2011, 189, 48 CrossRef CAS.
- M. Soylak and E. Yilmaz, Desalination, 2011, 275, 297 CrossRef CAS.
- J.-W. Zhang, Y.-K. Wang, H.-Y. Du, X. Du, J.-J. Ma and J.-C. Li, Clean: Soil, Air, Water, 2011, 39, 1095 Search PubMed.
- F. Shah, T. G. Kazi, H. I. Naeemullah, Afridi and M. Soylak, Microchem. J., 2012, 101, 5 Search PubMed.
- R. E. Rivas, I. López-García and M. Hernández-Córdoba, Microchim. Acta, 2009, 166, 355 CrossRef CAS.
- A. N. Anthemidis and K. I. G. Ioannou, Anal. Chim. Acta, 2010, 668, 35 CrossRef CAS.
- S. R. Yousefi and F. Shemirani, Anal. Chim. Acta, 2010, 669, 25 CrossRef CAS.
- P. Liang, E. Zhao and F. Li, Talanta, 2009, 77, 1854 CrossRef CAS.
- M. Shamsipur, M. Ramezani and M. Sadeghi, Microchim. Acta, 2009, 166, 235 CrossRef CAS.
- T. A. Kokya and K. Farhadi, J. Hazard. Mater., 2009, 169, 726 CrossRef CAS.
- S. Z. Mohammadi, D. Afzali, M. A. Taher and Y. M. Baghelani, Microchim. Acta, 2010, 168, 123 CrossRef CAS.
- P. Liang and E. Zhao, Microchim. Acta, 2011, 174, 153 CrossRef CAS.
- B. Majidi and F. Shemirani, Talanta, 2012, 93, 245 CrossRef CAS.
- E. Molaakbari, A. Mostafavi and D. Afzali, J. Hazard. Mater., 2011, 185, 647 CrossRef CAS.
- D. Afzali, A. Ghazizadeh and H. Salari, Quim. Nova, 2011, 34, 1124 Search PubMed.
- S. R. Yousefi, F. Shemirani and M. R. Jamali, Anal. Lett., 2010, 43, 2563 CrossRef CAS.
- E. M. Martinis, L. B. Escudero, P. Berton, R. P. Monasterio, M. F. Filippini and R. G. Wuilloud, Talanta, 2011, 85, 2182 CrossRef CAS.
- N. M. Najafi, H. Tavakoli, R. Alizadeh and S. Seidi, Anal. Chim. Acta, 2010, 670, 18 CrossRef CAS.
- A. N. Anthemidis and K.-I. G. Ioannou, Anal. Bioanal. Chem., 2012, 404, 685 CrossRef CAS.
- L. B. Escudero, P. Berton, E. M. Martinis, R. A. Olsina and R. G. Wuilloud, Talanta, 2012, 88, 277 CrossRef CAS.
- S. Z. Mohammadi, A. Sheibani, F. Abdollahi and E. Shahsavani, Arabian J. Chem., 2012 DOI:10.1016/j.arabjc.2012.03.008 , in press.
- K. Chandrasekaran, D. Karunasagar and J. Arunachalam, Anal. Methods, 2011, 3, 2140 RSC.
- T. Asadollahi, S. Dadfarnia and A. M. H. Shabani, Talanta, 2010, 82, 208 CrossRef CAS.
- P. Berton, E. M. Martinis and R. G. Wuilloud, J. Hazard. Mater., 2010, 176, 721 CrossRef CAS.
- P. Berton, E. M. Martinis, L. D. Martinez and R. G Wuilloud, Anal. Chim. Acta, 2009, 640, 40 CrossRef CAS.
- H. Abdolmohammad-Zadeh and G. H. Sadeghi, Anal. Chim. Acta, 2009, 649, 211 CrossRef CAS.
- M. Zeeb and M. Sadeghi, Microchim. Acta, 2011, 175, 159 CrossRef CAS.
- S. Z. Mohammadi, Y. M. Baghelani, F. Mansori, T. Shamspur and D. Afzali, Quim. Nova, 2012, 35, 198 Search PubMed.
- M.-I. Leong and S.-D. Huang, J. Chromatogr., A, 2008, 1211, 8 CrossRef CAS.
- P. Sun and D. W. Armstrong, Anal. Chim. Acta, 2010, 661, 1 CrossRef CAS.
- A. Berthod, M. J. Ruiz-Angel and S. Carda-Broch, J. Chromatogr., A, 2008, 1184, 6 CrossRef CAS.
- H. Ashkenani and M. A.Taher, Microchem. J., 2012, 103, 185 Search PubMed.
- M. Mohamadi and A. Mostafavi, J. AOAC Int., 2011, 94, 959 Search PubMed.
- W.-C. Tsai and S.-D. Huang, J. Chromatogr., A, 2009, 1216, 7846 CrossRef CAS.
- W.-C. Tsai and S.-D. Huang, J. Chromatogr., A, 2009, 1216, 5171 CrossRef CAS.
- B. Majidi and F. Shemirani, Anal. Methods, 2012, 4, 1072 RSC.
- Z. Marczenko and M. Balcerzak, Separation, Preconcentration and Spectrophotometry in Inorganic Analysis, Elsevier, 1st edn. 2000 Search PubMed.
- T. Prasada Rao, M. L. P. Reddy and A. Ramalingom Pillai, Talanta, 1998, 46, 765 CrossRef.
- Y. Han, X. Jia, T. Duan and H. Chen, Anal. Methods, 2011, 3, 842 RSC.
- T. M. Florence, Talanta, 1982, 29, 345 CrossRef.
- A. M. Ure, L. R. P. Butler, R. O. Scoti and R. Junkins, Spectrochim. Acta, Part B, 1997, 52, 409 Search PubMed.
- A. N. Anthemidis and M. Miró, Appl. Spectrosc. Rev., 2009, 44, 140 CrossRef CAS.
- C. I. C. Silvestre, J. L. M. Santos, J. L. F. C. Lima and E. A. G. Zagatto, Anal. Chim. Acta, 2009, 652, 54 CrossRef CAS.
- A. Gonzalvez, S. Armenta, M. L. Cervera and M. de la Guardia, TrAC, Trends Anal. Chem., 2010, 29, 260 CrossRef CAS.
- L. Ruzik, Talanta, 2012, 93, 18 CrossRef CAS.
- A. Martın-Calero, V. Pino and A. M. Afonso, TrAC, Trends Anal. Chem., 2011, 30, 1598 CrossRef CAS.
- J. Liu, J. Jonsson and G. Jiang, TrAC, Trends Anal. Chem., 2005, 24, 20 CrossRef CAS.
- E. M. Martinis, P. Berton, R. P. Monasterio and R. G. Wuilloud, TrAC, Trends Anal. Chem., 2010, 29, 1184 CrossRef CAS.
- Zh.-q. Tan, J.-f. Liu and L. Pang, TrAC, Trends Anal. Chem., 2012, 39, 218 CrossRef CAS.
- M. H. Mallah, F. Shemirani, M. G. Maragheh and M. R. Jamali, J. Mol. Liq., 2010, 151, 122 CrossRef CAS.
- M. H. Mallah, F. Shemirani and M. G. Maragheh, Environ. Sci. Technol., 2009, 43, 1947 CrossRef CAS.
- M. H. Mallah, F. Shemirani and M. G. Maragheh, J. Radioanal. Nucl. Chem., 2008, 278, 97 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |