The anomalous sodium doublet D2/D1 spectral line intensity ratio – a manifestation of CCD's presaturation effect
Bećko
Kasalica
a,
Stevan
Stojadinović
a,
Ivan
Belča
a,
Mirjana
Sarvan
a,
Ljubiša
Zeković
a and
Jelena
Radić-Perić
*b
aUniversity of Belgrade, Faculty of Physics, Studentski trg 12–16, 11000 Belgrade, Serbia
bUniversity of Belgrade, Faculty of Physical Chemistry, Studentski trg 12–16, 11000 Belgrade, Serbia. E-mail: len@ffh.bg.ac.rs; Fax: +381-11-2187133; Tel: +381-11-3336632
First published on 24th October 2012
Abstract
In this paper we present the results of intensity measurements of D1 (589.5224 nm) and D2 (588.9950 nm) sodium doublet spectral lines emitted from a short-lifetime plasma randomly appearing across the aluminum anode surface during its electrolytic oxidation from the water solution of boric acid with sodium tetraborate. We found that the D2 to D1 intensity ratios were not constant, varying from 2 to 1.2. Assuming that the plasma is in local thermal equilibrium it is expected that the intensity ratio of sodium doublet components equals the ratio of the statistical weights of the atomic sublevels corresponding to the electronic transitions (D1: 32P1/2–32S1/2, D2: 32P3/2–32S1/2) i.e. 2. After detailed analyses of a series of spectra obtained by applying different detection parameters, like exposure time, slit width, detection mode, and number of accumulations, we attributed the anomalous D2/D1 intensity ratio to the effect of approaching a saturation condition of the charge coupled device detector and thus to an early indication of saturation occurrence.
Introduction
Plasma electrolytic oxidation (PEO) of valve metals like Al, Ti, Zr, Ta, etc. is a processing technique in which surfaces of metals are transformed into oxide coatings.1 The thickness of the coating film varies from tens to hundreds of micrometers, which is influenced by the processing parameters like power supply, electric field strength and the type of the metals and electrolytes used in the PEO process. In the conventional electrolytic oxidation process, the oxidation of the metal is a consequence of electrochemical reactions in the electrolyte (one phase) and on the electrode (the other phase), including the phase-boundary processes. In the PEO processing, due to dielectric breakdown of an anode oxide film under high electric field, a plasma state is generated in the form of visible microdischarges or (plasma) sparks. They are of short duration, typically of a few microseconds, appearing stochastically in time and in space (on the surface of the coating). The oxidation mechanism becomes now more complex, including also plasma chemical reactions.2 Due to these plasma processes, the roughness of the coating as well as its morphology, the coating microstructure and composition are modified to some extent, which can be controlled by process parameters.3,4The investigation and characterization of PEO microdischarges (plasmas) are important steps for understanding of the PEO process. In a number of papers the results of optical emission spectroscopic studies of plasma generated during the PEO process have been presented.5–8 In these studies the recorded optical emission spectra have been used to determine the plasma composition. Based on the identification of a number of atomic spectral lines (e.g. of Al, Mg, H, O, Na, K, Si, etc.), ionic lines (e.g. of Al, Si, etc.), and molecular bands (OH, AlO, MgO, etc.), the presence of particular elements, originating from the coating and/or electrolyte, as well as of their predominant forms (atomic, ionic, molecular) in the PEO plasma has been proven.
In another group of papers, the recorded emission spectra have been used to determine plasma parameters like temperature and electron density. Under assumption of a local thermal equilibrium (LTE), the spectral line intensity ratio method has been applied to obtain the (electron) temperature. For this purpose, different spectral lines can be used, as for example hydrogen Balmer,5 Al and Mg lines.6 In ref. 9 and 10, the plasma temperature has been obtained from the AlO and MgO emission molecular bands. The plasma electron density can be determined from the analysis of profiles of different spectral lines, as it was done for PEO plasmas in ref. 11 using hydrogen Balmer lines. Further, the recording of the PEO spectra can be carried out continuously in time, such that the so-obtained spectra can be correlated with the time evolution of the PEO process and can be used to monitor the PEO process, too, as for example in ref. 12.
The necessary condition for performing all the above-mentioned tasks is to record “good” spectra in the “proper” way. “Good” spectra means the spectra with a sufficiently large ratio of spectral lines/band intensity to noise, accompanied with well resolved spectral lines. The “proper” way means that we should pay attention to the properties of the detection device and choose the correct mode for collecting the signal in order, e.g., to avoid the saturation of signals.
In this paper we present the results of intensity measurements of D1 (589.5224 nm) and D2 (588.9950 nm) sodium doublet spectral lines emitted from a PEO plasma during the aluminum oxidation from a water solution of boric acid with sodium tetraborate decahydrate. We found that the sodium D2 to sodium D1 intensity ratios, D2/D1, were not constant, varying from 2 to 1.2. If the PEO plasma is in LTE this intensity ratio should be constant and equal to the ratio of the statistical weights of the atomic sublevels corresponding to the electronic transitions (D1: 32P1/2–32S1/2, D2: 32P3/2–32S1/2) i.e. 2.
Anomalous intensity ratios within spectral multiplets is a well known phenomenon. It has been observed in spectra of early type stars,13 in comet spectra,14 in the exosphere of the planet Mercury,15 in sodium fluorescence spectra (induced by a pulsed tunable dye laser) in flames,16 in high-density laser produced plasma17 but also in mesospheric night glow.18 Anomalous ratios within multiplet components may reflect the optical properties of particular plasma and can be a consequence of various mechanisms which cause the perturbation of the LTE. They are a typical characteristic of optically thick plasmas.19 In that case the effects of reabsorption take place, the radiation field influences the population of levels, etc., as a consequence the excited levels are not populated according to their statistical weights and the Boltzmann law. So, in some papers, the cause for the anomalous sodium D2 to D1 intensity ratio has been attributed to the fact that the particular plasma is optically thick.17 In ref. 13, the anomalous behavior of sodium doublet components emitted from early type stars has been explained by the inhomogeneity of large (interstellar) clouds where the sodium emission originates from. It has also been stressed that the sodium D2/D1 intensity ratio tends to be much closer to 2 for the weaker lines than for the stronger ones.
Experimental
The PEO process was performed on high purity cold-rolled aluminum (99.999%, obtained from Goodfellow) samples of 25 mm × 7 mm × 0.12 mm. The surface of aluminum was degreased in ethanol using an ultrasonic cleaner and dried in a warm air stream. The anodic oxidation process was carried out in a 100 ml volume electrolytic cell with flat glass windows. Platinum wires were used as cathodes and the electrolyte was thermostatted at 20 °C. During anodization the electrolyte circulated through the chamber–reservoir system, and the control temperature sensor was suited by the sample. The anodization of aluminum occurred in a water solution of boric acid with sodium tetraborate decahydrate (0.1 mole H3BO3 and 0.05 mole Na2B4O7·10H2O) as an electrolyte. The electrolyte was prepared with double distilled deionized water and PA grade chemical substances. The PEO process took place at the constant current density of 10 mA cm−2 achieved by a highly stabilized power supply.It is not a simple task to record optical emission spectra of PEO plasmas because of their random appearance across the anode surface, short lifetimes and usually low intensities. Besides, an anodizing process is time dependent and by recording spectra continuously in time, the evolution of the PEO process can be observed. Because of that, an optic-detection system with improved sensitivity has been developed.20 The emission spectra of PEO microdischarges have been recorded by projecting a small area of the anode surface onto the entrance slit of the spectrograph, in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Fig. 1. In order to collect and to focus the highest possible extent of the emitted light on the spectrometer slit, a collimating system consisting of a large-aperture achromatic spherical concave lens has been used. The collecting and focusing of the light on the grating have been performed using a set of flat and spherical concave mirrors, and a similar optics has been applied for the illumination of the Intensified Charge Coupled Device (ICCD) detector,21–24 as the most important part of this optic-detection system. A very sensitive PI-MAX ICCD cooled camera with high quantum efficiency, manufactured by Princeton Instruments, has been used. The CCD chip consists of 1024 × 256 pixels, approximately 26 μm × 26 μm each. To reduce the dark current, the CCD chip was cooled at −40 °C using Peltier devices. The real time images of the PEO plasmas have been taken with a video camera Sony DCR-DVD110 (800k pixels CCD, 40 × optical zoom and 40 mm lens filter). A reconstructed “Rank-Hilger” spectrometer with a diffraction grating of 1200 grooves per mm was used for spectra detection. Its linear dispersion is 0.07 nm per pixel, with spectral resolution in the first grating order of about 1200.
:1, Fig. 1. In order to collect and to focus the highest possible extent of the emitted light on the spectrometer slit, a collimating system consisting of a large-aperture achromatic spherical concave lens has been used. The collecting and focusing of the light on the grating have been performed using a set of flat and spherical concave mirrors, and a similar optics has been applied for the illumination of the Intensified Charge Coupled Device (ICCD) detector,21–24 as the most important part of this optic-detection system. A very sensitive PI-MAX ICCD cooled camera with high quantum efficiency, manufactured by Princeton Instruments, has been used. The CCD chip consists of 1024 × 256 pixels, approximately 26 μm × 26 μm each. To reduce the dark current, the CCD chip was cooled at −40 °C using Peltier devices. The real time images of the PEO plasmas have been taken with a video camera Sony DCR-DVD110 (800k pixels CCD, 40 × optical zoom and 40 mm lens filter). A reconstructed “Rank-Hilger” spectrometer with a diffraction grating of 1200 grooves per mm was used for spectra detection. Its linear dispersion is 0.07 nm per pixel, with spectral resolution in the first grating order of about 1200.
 | ||
| Fig. 1 Schematic diagram of the experimental set up: the electrolytic cell consists of two electrodes, platinum cathodes (−) and aluminum anode (+), in a glass container (GC) with a particular electrolyte solution; a very small area (0.07 mm × 6.7 mm) of the aluminum anode surface (S) is projected via spherical concave lens (CL) onto the slit of a spectrometer. Spectra have been detected using an intensified charge couple device (ICCD) without applying the microchannel plate (MCP). | ||
Because of the nature of the PEO plasma, the intensities of spectra are in general rather low. An increase in the spectral intensities can be achieved by choosing an appropriate combination of spectra-recording parameters like the slit width (d), exposure time (E), and by accumulation (A) of signals. The accumulation represents (computer) summing of A distinct signals obtained successively over a definite exposure time. The accumulation number, A, can be varied, from one (A = 1) to several hundreds. Thus the spectra can be recorded and presented in two manners. The first procedure (first mode) implies the recording of spectra over a certain longer exposure time. The other way (second mode, or accumulation mode) is to record spectra A times (usually A ≫ 1) over a certain shorter exposure time and after that to accumulate the results.
The intensity of the spectral lines measured in our experiments depended not only on the particular choice of technical parameters (like the time of recording with respect to the beginning of anodization, exposure time, slit width, measuring mode, possible filters applied to reduce the light intensity) but also showed stochastic behavior. For this reason we repeated each experiment with a fixed set of these parameters several (between five and few hundreds) times. (An analogous procedure was applied in our previous studies on Al and Mg.9,10,25) While generally being roughly linearly correlated with the exposure time and/or the accumulation number, the spectral line intensities showed relatively large dispersion with respect to the mean value of several measurements carried out under the same conditions. In extreme cases, appearing usually in one or two of ten experiments, particular peaks were up to one order of magnitude stronger or weaker than the mean value. In the following text we discuss our results primarily in terms of peak intensities (when working in the first mode) and the intensities divided by the number of accumulations (second mode).
All measurements described in the present study were performed without applying the microchannel plate (MCP) normally serving to intensify the signals, because the results of some previous studies26,27 had shown that this part of the device had been responsible for nonlinear response of ICCDs. To avoid confusion, from now on, we shall denote our detection device by CCD (instead of ICCD).
Results and discussion
In order to find an explanation for the anomalous sodium D2/D1 intensity ratio, found in a number of our spectra, we systematically investigated the influence of the detection parameters mentioned above on the sodium D1 and D2 spectral line intensities.At the beginning, we present in Fig. 2 the real time images of PEO microdischarges corresponding to different times of the anodizing process, to illustrate the main PEO-plasma features. The PEO plasmas become visible at about 20 s after the beginning of the anodizing process, their size becomes larger during the PEO process, while their number reduces. They have an average cross-section area of 0.05 mm2.
 | ||
| Fig. 2 Real time images of microdischarges appearing at different times t of the anodizing process: (a) 15 min; (b) 30 min; (c) 60 min; and (d) 120 min. | ||
In Fig. 3, six particular spectra involving sodium D1 and D2 spectral lines are presented. These spectra have been recorded at different times of the anodizing process e.g. at t1 = 5 min, t2 = 12 min etc. (t denotes the anodizing time; t = 0 is the beginning of anodization) with the exposure time of 1 s, with 50 accumulations and the slit width of 0.075 mm. The intensity of sodium spectral lines increases with increasing anodizing time. It is noticeable that the D2/D1 intensity ratios are not constant. They vary from the value close to 2 [cases (a and b)] to 1.2 [case (f)]. As it will be shown below, we ascribe this tendency to the continuously more expressed nonlinearity of the CCD response with increasing spectral line intensities.
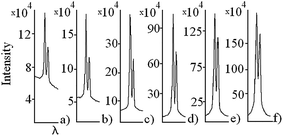 | ||
| Fig. 3 Sodium D2 and D1 emission spectral lines recorded at different times t of the anodizing process: (a) 5 min; (b) 12 min; (c) 43 min; (d) 94 min; (e) 142 min; and (f) 256 min. | ||
Particular difficulties in spectral measurements of the present kind are caused by some inherent characteristics of the PEO plasma, like its random appearance across the anode surface and its short lifetime. A consequence thereof is that the spectral features recorded are in general of low intensity (if they appear at all) and of poor reproducibility. Both of these drawbacks can be diminished by using longer exposure times and/or larger slit widths. This is illustrated in Fig. 4a and b. We present in Fig. 4a 70 records and in Fig. 4b 20 records (extracted of totally 300 ones) of the PEO plasma luminescence involving sodium D1 and D2 spectral lines. These spectra were recorded at t = 183 min after the beginning of the PEO without accumulation of signals (A = 1, mode one). In the first case (Fig. 4a) a narrow slit (d = 0.075 mm) and a relatively short exposure time (E = 0.05 s) were used, and in the second case (Fig. 4b) we chose a moderate slit width (d = 0.15 mm) and longer exposure time (E = 0.3 s). In the spectra presented in Fig. 4a sodium spectral lines are observed in only 6 records of 70 presented (an analysis of all 300 records gives a yield of 9%) and they are characterized by quite different intensities. The yield of observed sodium emission in spectra recorded with longer exposure time (Fig. 4b) is much higher (50–60%), and, naturally, higher are also the line intensities. We analyze now separately the influence of the slit width on the intensity and shape of the sodium spectral lines. The increase of the slit width results in a decrease of resolution of the spectral lines and in an increase of their intensities. In some cases the sodium lines presented in Fig. 4b are even saturated – they appear as peaks with flat “cut” maximum.
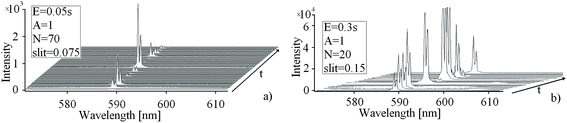 | ||
| Fig. 4 Successively recorded emission spectra of PEO plasma around sodium D1 and D2 spectral lines: (a) spectra taken with low exposure time (0.05 s) and narrow slit (0.075 mm) and (b) spectra taken with longer exposure time (0.3 s) and wider slit (0.15 mm). | ||
In Fig. 5a are presented D2 and D1 lines recorded with a relatively broad slit of 0.3 mm, obtained in the accumulation mode (A = 300) with short exposure times (E = 0.05 s). Let us first note that the background in our experiments is either the same for the both lines (because of their mutual vicinity) or is a very slowly varying linear function of the wavelength, and in general it is much less intense than the lines [1–2% in the first mode (without accumulation) and typically about 5% in the second (accumulation) mode]. Consequently, it can be easily subtracted from the measured spectral lines. The intensity ratio of the unresolved spectral lines (solid curve) equals (when the background is subtracted) 1.66. However, after applying a deconvolution, using Lorenzian fit, the intensity ratio of the resolved sodium lines nearly yields the theoretical value 2. However, if we deconvolute the sodium lines detected using a narrow slit (d = 0.075 mm) and longer exposure time (E = 1 s), Fig. 5b, the deviation of D2/D1 intensity ratios from 2 still remains (D2/D1 = 1.3). Note that the half-width of both lines is practically the same, with a consequence that the ratio of the areas below the lines (after deconvolution) equals the ratio of their intensities. Thus in order to avoid the artifacts in relative line intensities we carried out all the following experiments using a narrow slit, d = 0.075 mm.
 | ||
Fig. 5 Sodium D2 and D1 emission spectral lines: (a) recorded with a wider slit (0.3 mm) and short exposure time (0.05 s): ![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) before deconvolution, before deconvolution, ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) after deconvolution and (b) recorded with a narrow slit width (0.075 mm) and longer exposure time (1 s): before deconvolution, after deconvolution. after deconvolution and (b) recorded with a narrow slit width (0.075 mm) and longer exposure time (1 s): before deconvolution, after deconvolution. | ||
In order to investigate the influence of the exposure time on intensities of D1 and D2 spectral lines and particularly on D2/D1 intensity ratios, we recorded a series of spectra with longer exposure times (E = 60 s, 20 s, 10 s, 1 s, and 0.4 s) and without accumulation, and another series of spectra with shorter exposure times (0.2 s, 0.1 s and 0.05 s) with several hundred accumulations. The results of these measurements are presented in Fig. 6a–c. We show the correlation between the intensity of D2 spectral line and D1 line intensity multiplied by two. In the cases where the experiments are carried out in the second mode, the intensities are divided by the accumulation number A. Theoretically, all points should be embedded along the diagonal of that diagram.
 | ||
| Fig. 6 Correlation between the intensity of sodium D2 spectral line and that of D1 line multiplied by two. In the cases where the experiments are carried out in the second mode, the intensities are divided by the accumulation number A. (a) Intensity range 0–1000; (b) 0–10000; and (c) 0–100000. | ||
Although it could be expected that recording spectra at longer exposure time of, say 10 s, with only one accumulation (first mode) or using a short exposure time of 0.1 s with hundred accumulations (second mode) are equivalent procedures, both giving comparable spectral intensities, the right choice between these two procedures should be made, having in mind the properties of the excitation source whose spectra are recorded and the properties of the detection system. In a long exposure time mode, illumination of the detector over longer period of time and consequently the appearance of intense radiation can cause saturation effects or effects of approaching a saturation regime. As a result thereof the CCD may start operating outside the range of a linear relationship between the incident photon level and the output signal from CCD. Exceeding the saturation level is usually followed by visible signs like blooming23 or similar effects like those that can be seen in Fig. 4b. However, the CCD usually does not produce visible signs at the stage of approaching the saturation regime,24 although already operating outside the range of linear response. On the other hand, when the second (accumulation) mode of recording spectra is applied, spectra of sufficient intensities are obtained due to computer summing of particular signals obtained over a short period of time. In that way the saturation or (pre)saturation effects can be avoided.
The content of Fig. 6 speaks in favor of our belief that the anomaly in the D2/D1 ratio observed in a number of our experiments should be attributed to the presaturation effects. At small intensities of the spectral lines (Fig. 6a) the deviation of the measured D2/D1 ratio from 2 is negligible. It becomes continuously larger with increasing spectral line intensities. At highest intensities measured in this series of experiments, corresponding to the exposure time of E = 60 s, the D2/D1 ratio becomes approximately 1.6 (Fig. 6c). Note, however, that in the majority of experiments with such a long exposure time the signals are beyond the saturation point and thus are not presented in Fig. 6c. On the other hand, the lines of the same intensity obtained in the second mode (say with E = 0.2 s and A = 300) show nearly normal D2/D1 ratio, close to 2. We obtained the same effect (quasi-normal D2/D1) in long-exposure measurement using the filters that transmitted 2.4% and 0.6% of initial radiation.
An important question is whether there is another explanation for the anomaly of the D2/D1 ratio. A condition for obtaining the theoretical ratio of 2 is the existence of the LTE in our plasma. There are several arguments speaking in favor of reliability of the assumption that there is LTE. The first one is that an ratio very close to 2 is always obtained when the intensities of spectral lines are low, i.e. at short exposure times. If we did not have the LTE, this would be reflected in both short- and long-exposure time regimes. Further, in our previous studies on AlO9 and MgO10 we showed that the plasma of the present type is in the LTE, at least concerning the vibrational motion modes. Finally, in an extensive study Dunleavy et al.5 concluded that a similar plasma is in its high-temperature core region in LTE, while in the colder zones at least partial LTE takes place.
Conclusions
A series of spectra involving D1 and D2 sodium doublet spectral lines, emitted from the PEO plasma during the aluminum oxidation, have been recorded by varying different detection parameters, like the exposure time, slit width, detection mode, number of accumulations, etc. These detailed investigations have been performed in order to find an explanation for the anomalous sodium D2/D1 spectral line intensity ratio observed in a number of our spectra. The largest deviations of D2/D1 intensity ratios from the theoretical value of 2 have been found in spectra recorded with long exposure time (60 s, 20 s, etc.). A correlation between the deviation of the D2/D1 ratio from 2 and the overall spectral line intensities has been established. Based on these findings we attribute the anomalous D2/D1 intensity ratio to the effect of CCD's approaching a saturation regime and thus to an early indication of saturation occurrence. Let us stress that at the stage of approaching the saturation regime CCD usually does not produce visible signs like blooming or evidently saturated signals, although already operating outside the range of linear response.Anomalous intensity ratios within doublet components can also be a consequence of complex physical and chemical processes that cause the perturbation of the LTE and of the energy level population. Having in mind that the plasma arising during the electrolytic oxidation process of different metals has not been explored enough in spite of a great number of papers dealing with it, at the beginning of our investigations we did not rule out the possibility that some complex physical and chemical processes induced the anomalous D2/D1 intensity ratio. But, at last, we came to the conclusion that the anomaly we observed is due to a more trivial cause connected with our detection system and CCD characteristics, i.e. that it represents a manifestation of the CCD's presaturation effect. This presaturation effect can be successfully avoided by recording spectra in the accumulation mode. In this mode successive scans of shorter exposure time are summed to achieve sufficiently high effective intensities without long exposure time, overcoming in that way the risk of CCD overcharging. The same effect could be, of course, achieved by the use of appropriate filters, but a consequence of that would be inconvenient lowering of the intensity of other inherently weak spectral features, particularly of molecular spectral bands.
Acknowledgements
This work is supported by the Ministry of Education and Science of the Republic of Serbia under projects no. 171035 and 172040. We thank Prof. Miljenko Perić for precious contribution to this paper.References
- A. L. Yerokhin, X. Nie, A. Lezland, A. Mathews and S. J. Dowey, Surf. Coat. Technol., 1999, 122, 73–93 CrossRef CAS.
- L. Wang, L. Chen, Z. Yan and W. Fu, Surf. Coat. Technol., 2010, 205, 1651–1658 CrossRef CAS.
- R. O. Hussein, X. Nie and D. O. Northwood, Surf. Coat. Technol., 2010, 205, 1659–1667 CrossRef CAS.
- M. Petković, S. Stojadinović, R. Vasilić, I. Belča, B. Kasalica and L. Zeković, Serb. J. Electr. Eng., 2012, 9, 81–94 CrossRef.
- C. S. Dunleavy, L. O. Golosnoy, J. A. Curran and T. W. Clyne, Surf. Coat. Technol., 2009, 203, 3410–3419 CrossRef CAS.
- R. O. Hussein, X. Nie, D. O. Northwood, A. Yerokhin and A. Mathews, J. Phys. D: Appl. Phys., 2010, 43, 1–13 CrossRef.
- B. Kasalica, M. Petković, I. Belča, S. Stojadinović and Lj. Zeković, Surf. Coat. Technol., 2009, 203, 3000–3004 CrossRef CAS.
- S. Stojadinović, J. Jovović, M. Petković, R. Vasilić and N. Konjević, Surf. Coat. Technol., 2011, 205, 5406–5413 CrossRef.
- S. Stojadinović, M. Perić, M. Petković, R. Vasilić, B. Kasalica, I. Belča and J. Radić-Perić, Electrochim. Acta, 2011, 56, 10122–10129 CrossRef.
- S. Stojadinović, M. Perić, J. Radić-Perić, R. Vasilić, M. Petković and Lj. Zeković, Surf. Coat. Technol., 2012, 206, 2905–2913 CrossRef.
- J. Jovović, S. Stojadinović, N. M. Šišović and N. Konjević, Surf. Coat. Technol., 2011, 206, 24–28 CrossRef.
- S. Stojadinović, R. Vasilić, I. Belča, M. Petković, B. Kasalica, Z. Nedić and Lj. Zeković, Corros. Sci., 2010, 52, 3258–3265 CrossRef.
- O. C. Wilson and P. W. Merril, Ap. J., 1937, 86, 44–69 CrossRef CAS.
- N. S. Kovar and R. P. Kovar, Sol. Phys., 1968, 3, 611–617 CrossRef CAS.
- C. Barbieri, S. Verani, G. Cremonese, A. Sprague, M. Mendillo, R. Consentino and D. Hunten, Planet. Space Sci., 2004, 52, 1169–1175 CrossRef CAS.
- R. A. Van Calcar, M. J. M. Van de Ven, B. K. Van Uitert, K. J. Biewenga, Tj. Hollander and C. Th. J. Alkemade, J. Quant. Spectrosc. Radiat. Transfer, 1979, 21, 11–18 CrossRef CAS.
- F. E. Irons, Aust. J. Phys., 1980, 33, 283–301 CAS.
- T. G. Slanger, P. C. Cosby, D. L. Huestis, A. Saiz Lopez, B. J. Murray, D. A. O'Sullivan, J. M. C. Plane, C. Allende Prieto, F. J. Martin-Torres and P. Jenniskens, J. Geophys. Res., 2005, 110, 1–8 CrossRef.
- Plasma Diagnostics, ed. W. Lochte-Holtgreven, North Holland, Amsterdam, 1968 Search PubMed.
- S. Stojadinović, M. Tadić, I. Belča, B. Kasalica and Lj. Zeković, Electrochim. Acta, 2007, 52, 7166–7170 CrossRef.
- W. S. Boyle and G. E. Smith, Bell Syst. Tech. J., 1970, 49, 587–593 Search PubMed.
- G. E. Smith, Nobel Lecture: The Invention and Early History of the CCD, 2009 Search PubMed.
- J. R. Janesick, Scientific Charge-Coupled Devices, SPIE Press, Monograph Vol. PM83, 2001 Search PubMed.
- T. J. Fellers and M. W. Davidson, Hamamatsu articles: Concepts in Digital Imaging Technology: CCD Saturation and Blooming, http://hamamatsu.com/articles, 2009 Search PubMed.
- M. Sarvan, M. Perić, Lj. Zeković, S. Stojadinović, I. Belča, M. Petković and B. Kasalica, Spectrochim. Acta, Part A, 2011, 81, 672–678 CrossRef CAS.
- T. C. Williams and C. R. Shaddix, Rev. Sci. Instrum., 2007, 78, 123702–123707 CrossRef.
- J. Lindén, C. Knappe, M. Richter and M. Aldén, Meas. Sci. Tecnol., 2012, 23, 035201–035208 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |