Calciomics: integrative studies of Ca2+-binding proteins and their interactomes in biological systems†
Yubin
Zhou
ab,
Shenghui
Xue
c and
Jenny J.
Yang
*b
aCenter for Translational Cancer Research, Institute of Biosciences and Technology, Texas A&M University System Health Science Center, Houston, TX 77030, USA
bDepartment of Chemistry, Georgia State University, Atlanta, GA 30303, USA
cDepartment of Biology, Georgia State University, Atlanta, GA 30303, USA. E-mail: jenny@gsu.edu
First published on 26th November 2012
Abstract
Calcium ion (Ca2+), the fifth most common chemical element in the earth's crust, represents the most abundant mineral in the human body. By binding to a myriad of proteins distributed in different cellular organelles, Ca2+ impacts nearly every aspect of cellular life. In prokaryotes, Ca2+ plays an important role in bacterial movement, chemotaxis, survival reactions and sporulation. In eukaryotes, Ca2+ has been chosen through evolution to function as a universal and versatile intracellular signal. Viruses, as obligate intracellular parasites, also develop smart strategies to manipulate the host Ca2+ signaling machinery to benefit their own life cycles. This review focuses on recent advances in applying both bioinformatic and experimental approaches to predict and validate Ca2+-binding proteins and their interactomes in biological systems on a genome-wide scale (termed “calciomics”). Calmodulin is used as an example of Ca2+-binding protein (CaBP) to demonstrate the role of CaBPs on the regulation of biological functions. This review is anticipated to rekindle interest in investigating Ca2+-binding proteins and Ca2+-modulated functions at the systems level in the post-genomic era.
 Yubin Zhou | Dr Yubin Zhou is currently an assistant professor at the Center for Translational Cancer Research, Institute of Biosciences & Technology at Texas A&M University System Health Science Center. Dr Zhou obtained his PhD and MS degrees in Biochemistry from Georgia State University and received postdoctoral training at Harvard Medical School and La Jolla Institute for Allergy & Immunology. He also holds a Bachelor’s degree in Medicine from Zhejiang University School of Medicine. His research is mainly focused on understanding how calcium ions are transported across membrane, how ion channels can be engineered to achieve tailored function, and elucidating the structure–function relationships and physiological roles of calcium channels in health and disease. |
 Shenghui Xue | Shenghui Xue obtained his bachelor’s degree major in Biotechnology from the School of Life Science in Lanzhou University in 2005. Now he is a PhD student in the Department of Biology of Georgia State University. His current research interests include the role of calcium in virus and design of novel protein–metal complexes for disease diagnostics and therapeutics. |
 Jenny J. Yang | Dr Jenny J. Yang obtained her BS and MS in Chemistry and Analytic Chemistry at Xiangtan University. She obtained her PhD in Biochemistry at Florida State University in 1992. She then conducted her postdoctoral training at the University of Oxford and Yale University. Currently, she is a full professor at the Department of Chemistry at Georgia State University and the associate director of the Center for Diagnostics & Therapeutics. Her research interests are mainly focused on the chemical, biological, material and environmental role of calcium and calcium binding proteins (termed calciomics) and development of novel tools, methods and reagents for research, diagnostics and therapeutics using protein design and engineering approaches. |
1. Introduction
Ca2+, the fifth most abundant element on earth, constitutes approximately 3.6% of the earth's crust. Ca2+ has a flexible coordination number of 4–8, an irregular coordination geometry and rapid binding kinetics, which maximizes its potential to interact with various ligands. Ca2+ tends to precipitate both inorganic and organic anions (at ∼mM range) so that high Ca2+ is incompatible with life and it has to be extruded, sequestered or compartmentalized.1 These unique physio-chemical properties of Ca2+ make it the only available candidate in the environment to perform both micro (as a messenger) and macro (e.g., biomineralization) functions in biological systems. During the evolution from prokaryotes to eukaryotes that began over 2 billion years ago, the Fe2+–H2S buffering system in the sea was progressively exhausted, along with the increase of oxygen supply and prevalence of oxidative reactions.1 Ca2+ played an important role in facilitating the adaptive changes of prokaryotes to an oxidative environment, and contributed to the evolution of more advanced organisms and land colonization.2 For example, Ca2+ is closely involved in crucial evolutionary events such as the evolution of cytoskeleton, cell motility, the formation of skeletons and hard-shelled eggs (biomineralization), and the ability to supply milk.By mass, Ca2+ represents the most abundant mineral (∼1 kg) in the adult human body, with 99% in the form of calcium phosphates in bones. In mammals, a specific Ca2+ homeostatic system is evolved to set both the extracellular and intracellular Ca2+ concentrations at proper levels, and produces a steep Ca2+ gradient (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times) across the plasma membrane.3 The external Ca2+, sensed by the extracellular Ca2+ sensing receptor, is maintained at 1.1–1.4 mM through the coordinated actions of three major hormones (i.e., parathyroid hormone, calcitonin and 1,25-dihydroxyvitamin D3).4 Compared to the extracellular Ca2+ concentration of about 10−3 M, the internal Ca2+ concentration is maintained within the narrow range of 10−7 to 10−8 M in nearly all living cells. In multi-cellular eukaryotes, Ca2+ levels in different cellular compartments or organelles are tightly controlled through the molecular choreography of a repertoire of Ca2+ signaling molecules, including Ca2+ channels, Ca2+ transporters, receptors, Ca2+ buffers, and Ca2+-responsive proteins (e.g., Ca2+-sensitive enzymes and transcription factors) that are distributed in cytoplasm, endoplasmic/sarcoplasmic reticulum (ER/SR), Golgi complex, mitochondria, nucleus or the extracellular matrix (Fig. 1).5 Even in the most primitive prokaryotes, the free cytosolic Ca2+ is maintained at ∼10−7 M by using a variety of prokaryotic ion transporters (e.g., Ca2+/H+ antiporter, Ca2+/Na+ antiporter, and Ca2+-ATPase) or channels.6 Although bacteria does not possess complex intracellular organelles and Ca2+ is not extensively used as an intracellular messenger, accumulating evidence indicates that Ca2+ plays an essential role in the regulation of bacterial movements, chemotaxis, survival reactions and sporulation.3b,6c,d,7
000 times) across the plasma membrane.3 The external Ca2+, sensed by the extracellular Ca2+ sensing receptor, is maintained at 1.1–1.4 mM through the coordinated actions of three major hormones (i.e., parathyroid hormone, calcitonin and 1,25-dihydroxyvitamin D3).4 Compared to the extracellular Ca2+ concentration of about 10−3 M, the internal Ca2+ concentration is maintained within the narrow range of 10−7 to 10−8 M in nearly all living cells. In multi-cellular eukaryotes, Ca2+ levels in different cellular compartments or organelles are tightly controlled through the molecular choreography of a repertoire of Ca2+ signaling molecules, including Ca2+ channels, Ca2+ transporters, receptors, Ca2+ buffers, and Ca2+-responsive proteins (e.g., Ca2+-sensitive enzymes and transcription factors) that are distributed in cytoplasm, endoplasmic/sarcoplasmic reticulum (ER/SR), Golgi complex, mitochondria, nucleus or the extracellular matrix (Fig. 1).5 Even in the most primitive prokaryotes, the free cytosolic Ca2+ is maintained at ∼10−7 M by using a variety of prokaryotic ion transporters (e.g., Ca2+/H+ antiporter, Ca2+/Na+ antiporter, and Ca2+-ATPase) or channels.6 Although bacteria does not possess complex intracellular organelles and Ca2+ is not extensively used as an intracellular messenger, accumulating evidence indicates that Ca2+ plays an essential role in the regulation of bacterial movements, chemotaxis, survival reactions and sporulation.3b,6c,d,7
![Schematics of the Ca2+ signaling machinery, the range of Ca2+-binding affinities and the timescale of Ca2+ modulated activities. The extracellular Ca2+ homeostasis is maintained by the coordinated actions of hormones, bone cells and balanced uptake and excretion of Ca2+ in the intestine and kidney. The internal Ca2+ homeostasis is achieved through the exquisite choreography of the Ca2+ signaling toolkits. Under resting conditions, cytosolic Ca2+ is maintained at a submicromolar range by extruding Ca2+ outside of the plasma membrane via plasma membrane Ca2+-ATPase (PMCA) and Na+/Ca2+ exchanger (NCX), or by pumping Ca2+ back into internal stores through sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) or secretory pathway Ca2+-ATPase (SPCA). Upon extracellular stimulation, the free cytosolic Ca2+ rapidly increases by the entry of extracellular Ca2+ across the plasma membrane via Ca2+ channels, including voltage-operated channels (VOC), receptor-operated channels (ROC), transient receptor potential ion-channel (TRP) and store-operated channels (SOC), or by the release of Ca2+ from internal stores (e.g., endoplasmic reticulum (ER) and Golgi complex) through inositol-1,4,5-triphosphate receptors (IP3R) and ryanodine receptors (RyR) due to activation of membrane receptors (G protein coupled receptors (GPCRs) and receptor tyrosine kinase [RTK]) and the subsequent synthesis of IP3. In the mitochondria, Ca2+ can easily pass through outer mitochondrial membrane pores and cross the inner mitochondrial membrane through the membrane-embedded Ca2+ uniporter (MCU). Ca2+ exits mitochondria through the opening of a nonselective high-conductance channel permeability transition pore (PTP) in the inner mitochondrial membrane and the Na+/Ca2+ exchanger (NCX). The Ca2+ signals are delivered by affecting the activity of Ca2+ buffers, Ca2+ effectors and Ca2+-regulated enzymes. Ca2+-binding proteins have Ca2+ affinities that vary by 106-fold or more depending upon their locations and functions (left panel). Ca2+ can exert short-term effects by triggering neurotransmitter release within microseconds. The signals can also elicit “long-term” effects by modulating gene expression (right panel).](/image/article/2013/MT/c2mt20009k/c2mt20009k-f1.gif) | ||
| Fig. 1 Schematics of the Ca2+ signaling machinery, the range of Ca2+-binding affinities and the timescale of Ca2+ modulated activities. The extracellular Ca2+ homeostasis is maintained by the coordinated actions of hormones, bone cells and balanced uptake and excretion of Ca2+ in the intestine and kidney. The internal Ca2+ homeostasis is achieved through the exquisite choreography of the Ca2+ signaling toolkits. Under resting conditions, cytosolic Ca2+ is maintained at a submicromolar range by extruding Ca2+ outside of the plasma membrane via plasma membrane Ca2+-ATPase (PMCA) and Na+/Ca2+ exchanger (NCX), or by pumping Ca2+ back into internal stores through sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) or secretory pathway Ca2+-ATPase (SPCA). Upon extracellular stimulation, the free cytosolic Ca2+ rapidly increases by the entry of extracellular Ca2+ across the plasma membrane via Ca2+ channels, including voltage-operated channels (VOC), receptor-operated channels (ROC), transient receptor potential ion-channel (TRP) and store-operated channels (SOC), or by the release of Ca2+ from internal stores (e.g., endoplasmic reticulum (ER) and Golgi complex) through inositol-1,4,5-triphosphate receptors (IP3R) and ryanodine receptors (RyR) due to activation of membrane receptors (G protein coupled receptors (GPCRs) and receptor tyrosine kinase [RTK]) and the subsequent synthesis of IP3. In the mitochondria, Ca2+ can easily pass through outer mitochondrial membrane pores and cross the inner mitochondrial membrane through the membrane-embedded Ca2+ uniporter (MCU). Ca2+ exits mitochondria through the opening of a nonselective high-conductance channel permeability transition pore (PTP) in the inner mitochondrial membrane and the Na+/Ca2+ exchanger (NCX). The Ca2+ signals are delivered by affecting the activity of Ca2+ buffers, Ca2+ effectors and Ca2+-regulated enzymes. Ca2+-binding proteins have Ca2+ affinities that vary by 106-fold or more depending upon their locations and functions (left panel). Ca2+ can exert short-term effects by triggering neurotransmitter release within microseconds. The signals can also elicit “long-term” effects by modulating gene expression (right panel). | ||
In eukaryotic cells, Ca2+ functions as a universal and versatile signal by interacting with hundreds of proteins over a 106-fold range of affinities (nM to mM) (Fig. 1, left panel). Inside cells, Ca2+-binding proteins, such as calmodulin (CaM) or parvalbumin, possess Ca2+-binding affinities in the submicromolar range corresponding to the resting cytosolic Ca2+ concentrations at ∼100 nM. Ca2+ concentrations in organelles such as the ER, Golgi, and endocytic vacuoles are in the hundreds of μM. By modulating the activities of these proteins, Ca2+ participates in various biological processes (e.g., hormonal secretion, muscle contraction, neurotransmission and memory formation) over a wide range of timescale, ranging from milliseconds up to days (Fig. 1, right panel). Overall, the Ca2+ signaling system exhibits great versatility in speed, amplitude and spatial–temporal patterns. In this review, we focus on how integrative studies can be carried out to probe the sophisticated Ca2+ signaling network in biological systems (termed calciomics). First, we describe bioinformatics approaches tailored for predicting Ca2+-binding sites and Ca2+-modulated activities from genomic and structural information. Second, we discuss how experimental approaches can be applied to probe Ca2+-binding properties (stoichiometry, selectivity, affinity, cooperativity etc.). Finally, we present herein recent progress in identifying the interactomes of CaBPs by using CaM–target interactions as an example.
2. Bioinformatics approaches to predict Ca2+-binding proteins
In the last decade we have witnessed an explosion of genomic information that far exceeds the capability of experimental characterization on each gene product. Nearly 30% of all proteins bind various metals. This has created unprecedented need for the Ca2+ signaling field to predict Ca2+-binding sites in proteins based on the amino acid sequences translated from nucleotide sequences, or from low-resolution or modeled structures. The prediction of a protein's capability to bind Ca2+ may provide important insights into its biological function and guide experimental designs.82.1 Prediction from primary sequences
Ca2+-binding sites in proteins can be divided into two types: continuous and discontinuous ones. Continuous Ca2+-binding sites are formed by a stretch of continuous amino acids in a linear sequence with at least some of the binding ligands well conserved. Thus, a pattern (or motif signature) representing the conserved features can be generated, and used to determine whether a protein contains a given Ca2+-binding pattern with simple string matching algorithms. A major class of Ca2+-binding proteins (CaBPs) contains continuous Ca2+-binding sites with an EF-hand structure (Fig. 2).9 A typical canonical EF hand (Fig. 2, middle panel) contains a helix–loop–helix topology with the Ca2+-binding pocket adopting a pentagonal bipyramidal geometry. Another continuous Ca2+-binding motif is called “pseudo” EF-hands that are exclusively found in the N-termini of S100 and S100-like proteins (Fig. 2, lower panel).9 The major difference between these two types of EF-hands lies in the Ca2+-binding loop (Fig. 2): the 12-residue canonical EF-hand loop binds Ca2+ mainly via sidechain carboxylates or carbonyls (loop sequence positions 1, 3, 5, 12), whereas the 14-residue pseudo EF-hand loop chelates Ca2+ primarily via backbone carbonyls (positions 1, 4, 6, 9) and the sidechain carboxylates (position 14). The residue at position 9 in a canonical EF-hand, or position 11 in a pseudo EF-hand, coordinates the Ca2+ ion through a bridged water molecule. While the majority of the known EF-hand CaBPs contain paired EF-hand motifs, CaBP's with single EF hands have also been discovered in prokaryotes and viruses.10 In addition, “EF-hand-like motifs” (e.g., Excalibur as shown in Fig. 2, upper panel) have been found in a number of bacterial proteins.10a,11 Although their coordination properties remain similar to the canonical 29-residue helix–loop–helix EF-hand motif, the EF-hand-like motifs differ from EF-hands in that they contain deviations in the secondary structure of the flanking sequences and/or variation in the length of the Ca2+ coordinating loop. A series of patterns reflecting these features have been generated with satisfactory predictive accuracy and sensitivity. We have used these patterns to detect putative EF-hand or EF-hand like Ca2+-binding motifs in both viral and prokaryotic genomes.10 A list of all the predicted CaBPs in these primitive genomes can be found in Tables S1 and S2 (ESI†). Aside from our contribution, several other groups have developed webservers or libraries using their own patterns or published data to predict and catalog EF-hand CaBPs (see Table 1).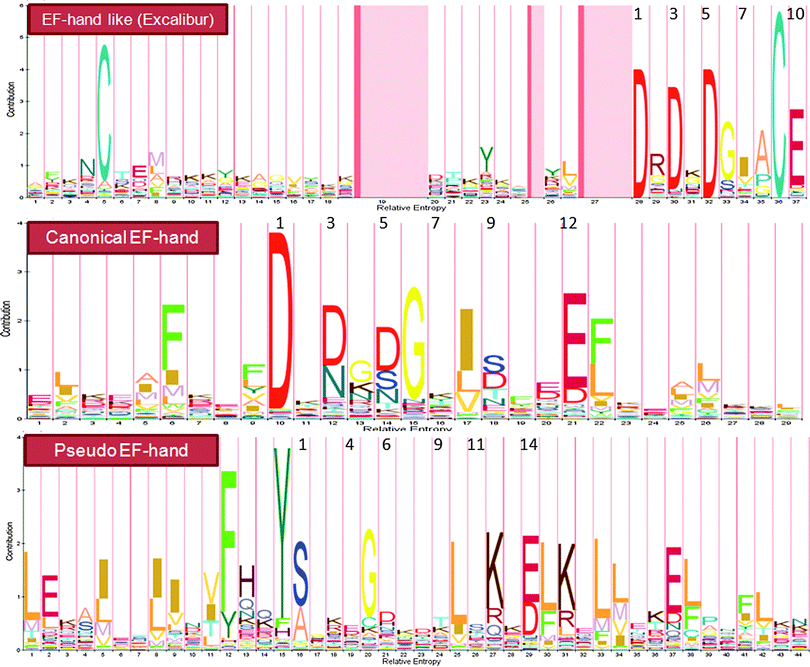 | ||
| Fig. 2 Profile HMMs for three representative continuous Ca2+-binding motifs. The numbers on top of the amino acids indicate the positions of ligands in the Ca2+-binding loop. The Pfam entries for EF-hand like (Excalibur), canonical EF-hand and pseudo EF-hand motifs are PF05901, PF00036 and PF01023, respectively. The letter size is positively correlated to the distribution probability of amino acids at each position. See the text for detailed discussions (Section 2.1 Prediction from primary sequences). | ||
| Resource (reference) | Type of resource | Web address |
|---|---|---|
| Predicting Ca2+-binding sites based on protein primary sequences | ||
| CaPS10a | Webserver for prediction of EF-hand or EF-hand like Ca2+-binding motifs | http://chemistry.gsu.edu/faculty/Yang/Calciomics.htm |
| EF-hand CaBPs data library | Database of sequence and structural information on EF-hand CaBPs | http://structbio.vanderbilt.edu/cabp_database/cabp.html |
| EF-Handome58 | A collection of gene, mRNA and protein information of EF-hand proteins in humans, rat and mouse | Available from the authors |
| MFSCa59 | A meta-functional signature algorithm for Ca2+-binding residue prediction | Available from the authors |
| MetalloPred60 | Webserver for prediction of metal-binding sites using a cascade of neural networks from sequence derived features | http://www.juit.ac.in/assets/Metallopred/ |
| CalPred | Web tools for EF-hand CaBP prediction and calcium binding region identification | http://www.bioinformatics.org/calpred/index.html |
| Predicting Ca2+-binding sites based on protein structures (PDB entries or modeled structures) | ||
| GG,23 MUG,24 MUGSR28 | Tools for predicting Ca2+-binding sites based on graph theory and geometric analyses | http://chemistry.gsu.edu/faculty/Yang/Calciomics.htm |
| Fold-X18 | A computations algorithm based on empirical force field to predict the position of metal ions in protein | http://foldx.crg.es/ |
| SVMProt20 | Webserver for assigning protein functions (including Ca2+-binding) with support vector machine learning | http://bidd.cz3.nus.edu.sg/cgi-bin/svmprot.cgi |
| WebFEATURE26 | Webserver for automated function prediction (including Ca2+-binding sites) in protein structures with machine learning | http://feature.stanford.edu/webfeature/ |
| SitePredict21 | Webserver for predicting metal ion binding sites with the Random Forest machine learning method | http://sitepredict.org/index.php |
| FunFOLD61 | An integrated web resource for ligand binding site prediction | http://www.reading.ac.uk/bioinf/FunFOLD/FunFOLD_form.html |
| FINDSITE-metal22 | A threading-based method to detect metal-binding sites in modeled structures by integrating evolutionary information and machine learning | http://cssb.biology.gatech.edu/findsite-metal |
| MetaPocket62 | A consensus method to predict ligand binding sites by integrating LIGSITE, PASS, Q-SiteFinder and SURFNET, Fpocket, GHECOM, ConCavity and POCASA | http://projects.biotec.tu-dresden.de/metapocket/index.php |
| MetSite63 | An automatic approach for detecting metal-binding residues in low-resolution 3D models with neural network classifiers | http://bioinf.cs.ucl.ac.uk/metsite |
| Predicting Ca2+-modulated functions | ||
| Calmodulin Target Database30 | Webserver for predicting calmodulin binding sites from protein sequences | http://calcium.uhnres.utoronto.ca/ctdb/ctdb/home.html |
| MeTaDor64 | Webserver for predicting membrane targeting domains (e.g., Ca2+-dependent C2 domain) | http://proteomics.bioengr.uic.edu/metador/predict.html |
| ORBIT65 | De novo design of protein sequence based on a desired backbone fold | Available from the authors |
| DEZYMER66 | Design of metal binding sites by selecting suitable ligands | Available from the authors |
Given that the pattern search method simply looks for Ca2+-binding motif signatures in proteins without considering the similarity to known domains, it tends to generate false positives. Another convenient method to predict CaBPs is to search for conserved Ca2+-binding domains (CaBDs) by online resources such as the Pfam or InterPro databases.12 The Pfam database has a comprehensive collection of profile hidden Markov models (HMM),13 which contain probability-based consensus information for protein domains. The profile HMMs can be visualized by HMM Logos14 (Fig. 2) and are compatible with HMMER,15 a free and effective software package used to assign sequence similarity to a query protein sequence. This prediction strategy is based on sequence homology alignment using a known Ca2+-binding protein (such as C2 domain) against a genome. It is extremely useful for identifying non-continuous CaBPs as it allows for the identification of any homologous proteins without the restriction of any gaps in sequences. An excellent introduction to using profile HMMs to predict metalloproteomes has been recently described in a tutorial review in Metallomics.16 As a common limitation of sequence-based predictive computational approaches, one has to be aware that both methods are unlikely to detect potential CaBPs that do not contain either a known Ca2+-binding pattern or a well-established CaBD.
All these prediction methods enabled us to make a phylogenetic distribution of typical Ca2+-binding motifs or domains found in nature (Table 2). Interestingly, EF-hands or EF-hand like motifs (particularly the Excalibur motif11) are frequently present in prokaryotes in a single unit, which raises the hypothesis that the prokaryotic single EF-hand motif is likely the precursor for the paired EF-hands found in eukaryotes.9,10 In contrast, the pseudo EF-hand is almost exclusively found in vertebrates. Thus, it is reasonable to postulate that pseudo EF-hands are phylogenetically younger and have a shorter history than canonical EF-hands. Likewise, calreticulin and calsequestrin, two signature CaBPs inside ER/SR, are only found in eukaryotes given that endo-membranous structures evolved later and that the ER structure is absent in prokaryotes.
| Motifs/CaBPs (Pfam entry) | Locations and functions | Organisms | ||||||
|---|---|---|---|---|---|---|---|---|
| Viruses | Archaea | Bacteria | Fungi | Yeast | Plants | Metazoa | ||
| a Note: the values indicate the numbers of predicted Ca2+-binding motifs in protein sequences annotated by Pfam (http://pfam.sanger.ac.uk/) as 2011. One protein sequence may contain multiple predicted motifs. b The number includes both EF-hand and EF-hand like motifs according to Zhou et al.10b | ||||||||
| EF-hand like (e.g., Excalibur (PF05901)) | External: enzymes | 93b | — | 662 | — | — | — | — |
| EF-hand (PF13499) | Internal: triggers, buffers, enzymes, messengers | 6 | 509 | 2303 | 192 | 3540 | 6471 | |
| S100 (PF01023) | Internal or external: triggers, buffers, messengers | — | — | — | — | — | — | 393 |
| C2 domains (PF00168) | Internal: membrane-linked enzymes | — | 1 | 8 | 1435 | 257 | 2180 | 6700 |
| Annexins (PF00168) | Internal: binds to membrane phospholipids; triggers | — | — | 3 | 336 | — | 823 | 2765 |
| Calreticulin (PF00262) | Calcium stores in reticula; chaperone | — | — | — | 106 | 20 | 158 | 352 |
| Calsequestrin (PF01216) | Calcium stores in reticula | — | — | — | — | — | — | 105 |
2.2 Prediction from 3D structures
Ca2+-binding sites do not always follow a set of linear amino acid sequences. Approximately 90% of known Ca2+-binding sites in Protein Data Bank (PDB) are non-linear, which means that the Ca2+-coordinating ligands may come from different loops in a protein, or different subunits, or even several different proteins. Prediction of those Ca2+-binding sites requires three-dimensional structural information from either determined structures deposited in PDB or from homology modeling. Fortunately, key structural factors governing Ca2+-binding share some common features with defined ligand types, charge distribution, Ca2+–ligand distances, bond angles and geometric configurations.17 This enables the development of a number of computational methods to predict non-continuous Ca2+-binding sites based on 3D structures (summarized in Table 1). Those prediction algorithms employ structural parameters as distinguishing factors between Ca2+-binding and non-binding sites. Frequently, multiple algorithms are combined, typically using some types of scoring function with a grid system or other methods of spatial analysis (e.g., graph theory) to evaluate physicochemical features systematically and identify either binding ligands or binding sites based on comparative scoring. It is worthy of note that these functions are dependent on a priori statistical analyses to define or constrain function parameters based on structural, chemical or thermodynamic data. Selected examples are briefly discussed below.An earlier example of using combinatory methods to predict Ca2+-binding sites is the Fold-X algorithm.18 This method combines geometric pattern matching and energy calculations based on Fold-X empirical force field, where Ca2+-binding sites are constrained to four to six ligand oxygen atoms.18 The Fold-X force field is used for energy calculation and for the optimization of the position of the predicted Ca2+. This algorithm can also coarsely predict binding affinity based on energy-optimized placement of Ca2+ to distinguish between low and high affinity sites, as well as high affinity Mg2+-binding sites. Although these results indicate higher prediction rates than previous efforts,19 this method was less successful at identifying either multiple binding sites or sites with lower coordination numbers (CN < 4).
Another commonly used predicting strategy is based on the machine learning method (e.g., SVMProt,20 SitePredict21 and FINDSITE-METAL22). Machine learning is particularly effective at classifying metal-binding ligands and determining whether the target atom is close enough for chelating the ion. In this method, a window including neighboring residues slides through the sequence so that each successive residue is viewed as a target residue in turn, to identify binding residues. The training procedures include applying the same learning machine to a training dataset consisting of sequences with known metal-binding residues. Classification features include evolutionary information, location in the primary sequence, secondary structure, metal-binding propensity, and solvent accessibility. Additional information on machine learning methods for predicting CaBPs can be found through the webservers listed in Table 1.
Toward the goal of predicting Ca2+-binding sites with improved accuracy and sensitivity, we have developed several prediction algorithms during the recent five years. The first-generation program GG (Graph theory and Geometry) presented a new approach to prediction with the combination of graph theory and geometric parameters.23 Based on the a priori statistical parameters that govern Ca2+-binding, the GG algorithm identifies oxygen clusters as the basis for predicting Ca2+-binding sites (Fig. 4). In this method, a graph G (V, E) was first constructed with oxygen atoms as its vertices and edges between two vertices if their spatial distance was no more than 6.0 Å. Oxygen clusters corresponding to cliques in G (V, E) consisting of exactly four vertices were identified, and the Ca2+ center was determined at an equidistant center within each cluster if the distance ranged from 1.8 to 3.0 Å. This algorithm did not include a procedure to merge overlapping, predicted Ca2+ locations, resulting in multiple, adjacent predicted sites near the documented Ca2+-binding site. Nonetheless, this algorithm produces rapid results, mainly due to the absence of a grid algorithm, and achieves approximately 90% site sensitivity and 80% site selectivity on testing datasets. To fully utilize the Ca2+-binding geometric properties, we have also developed the next-generation MUG (MUltiple Geometries) program, which is capable of predicting Ca2+-binding sites with different coordination numbers in proteins at atomic resolution.24 MUG requires that potential Ca2+-binding sites consisting of at least four oxygen atoms although MUG further allows oxygen atoms from either water or cofactors in addition to amino acid residues. Spatial analysis is achieved based on graph theory modeling as well as imposition of a grid system. MUG also has the capability of predicting both low coordination sites and multiple Ca2+-binding sites that share a common ligand. Very recently, we further developed the third-generation MUGC algorithm25 to predict Ca2+-binding sites in both X-ray and NMR structures with flexibility in the binding region or low occupancy in a Ca2+-binding site. Using second shell carbon atoms, and without explicit reference to side-chain oxygen ligand coordinates, MUGC is capable of achieving 94% sensitivity with 76% selectivity on a dataset of X-ray structures comprised of 43 Ca2+-binding proteins. Additionally, prediction of Ca2+-binding sites in NMR structures was obtained by MUGC using a different set of parameters determined by analysis of both Ca2+-constrained and unconstrained Ca2+-loaded structures derived from NMR data. The geometric arrangement of the second-shell carbon cluster is sufficient for both accurate identification of Ca2+-binding sites in NMR and X-ray structures, and for selective differentiation between Ca2+ and other relevant divalent cations.
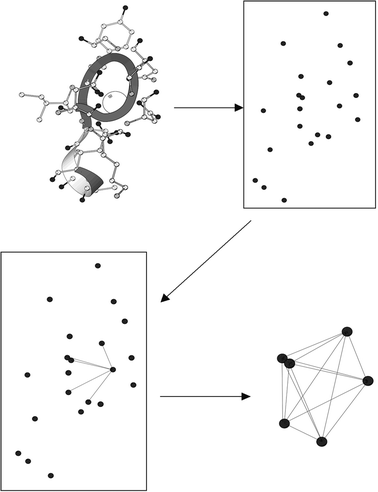 | ||
| Fig. 4 Prediction of Ca2+-binding sites by the geometry and graph (GG) algorithm program. To predict Ca2+-binding sites in proteins, oxygen ions (black) from protein 3D-structure are extracted, while other types of ions are excluded. Next, the distance between any of the two oxygen ions is calculated, and an edge will be assigned if the calculated oxygen distance is below a cutoff value. A potential Ca2+-binding site contains oxygen clusters in which every oxygen ion is linked to each other with an O–O distance less than the cutoff and every oxygen ion is linked to all other oxygen atoms by the assigned edges in this oxygen cluster. The Ca2+ center is determined at an equidistant center within each oxygen cluster if the distance ranges from 1.8 to 3.0 Å. Adapted from Deng et al.,23 with permission from John Wiley and Sons. | ||
While the majority of the computational algorithms are tailored for predicting CaBPs in their metal-bound (holo) forms, predicting Ca2+-binding sites from apo-structures is equally important but much more challenging due to two facts: first, a large number of protein structures are determined in the absence of metal ions; second, the binding of Ca2+ may trigger conformational changes and cause the rearrangement of the binding pocket. Tremendous efforts have been directed to this end with steady progress. A FEATURE program, with machine learning that utilizes a Bayesian approach to classify potential Ca2+-binding sites, has been recently reported.26 Following the identification of distributions of predefined Ca2+-binding characteristics, the query protein structure was embedded in a 3D-grid of 2 Å grid cubes, and a score was computed for each grid (e.g., query site) based on a scoring function that uses Bayes' rule to combine prior known distributions and observed frequencies (e.g., derived from a training dataset and the query site) to indicate the likelihood of Ca2+-binding in the queried site. Besides being applied to static X-ray crystal structures, FEATURE has recently been applied to multiple conformations of the protein parvalbumin β generated from molecular dynamics (MD) simulation.27 For MD generated conformations of both holo and apo forms, FEATURE correctly identifies the Ca2+-binding sites and non-sites with some interesting fluctuations in score. The combination of machine learning techniques such as FEATURE with MD simulations may provide new insights into the Ca2+-binding affinity, persistence and the stability of Ca2+-binding sites. More recently, we have improved the MUG algorithm (MUGSR) to accurately predict CaBPs with multiple-binding sites that undergo local conformational change or side chain rotations upon Ca2+-binding.28 Given an apo (unbound) protein structure, this new approach conducts a side chain rotation procedure according to a rotamer library to find binding sites with possible side-chain movements (local conformational changes) caused by Ca2+-binding. Given an apo protein structure, MUGSR is capable of capturing Ca2+-binding sites that may undergo local conformational changes, which is validated by comparison with the corresponding holo protein structure sharing more than 98% sequence similarity with the apo protein.
2.3 Toward the prediction of the CaBP–target interactomes
Upon the stimulation of signals or alteration in the membrane potential, the cytosolic free Ca2+ concentration ([Ca2+]c) could be elevated by 100 folds from 10−8 M to 10−6 M.5 The elevation of [Ca2+]c can be effectively sensed by Ca2+ sensors inside cells. Ca2+ signaling is a fast process and takes place in milliseconds. To convert these transient Ca2+ signals into more sustained physiological processes, CaBPs, such as calmodulin (CaM), undergo conformational changes and subsequently interact with a variety of protein kinases (e.g., CaM-dependent kinase I) and phosphatase (e.g., calcineurin), both of which are capable of covalently modifying downstream effectors by adding or removing a phosphate group.29 The time scale of this process usually lasts for seconds or even minutes. In addition, to more efficiently modulate the signaling cascades, Ca2+-CaM (sometimes apo-CaM) targets enzymes or proteins (e.g., adenylate cyclases, phosphodiesterase, ion channels, and GPCRs) involved in the conversion of other second messengers, such as cAMP and cGMP. These enzymes or proteins themselves do not respond to changes in Ca2+ concentration. However, CaBPs, such as CaM, can sense the intracellular Ca2+ concentration changes. Binding or releasing of Ca2+ induces conformational changes of these intracellular “Ca2+ sensor proteins” (e.g., CaM), which further alter the binding affinity of “Ca2+ sensor proteins” to these enzyme and proteins. The reciprocal interactions between CaBP and signaling proteins will regulate the function of these proteins. Thus, CaBPs like CaM relay the Ca2+ signaling to a bewildering number of target enzymes or proteins to carry out corresponding cellular functions. More importantly, the Ca2+ signal itself can even be modulated by CaM through CaM's interaction with membrane receptors, ion channels and pumps. CaM resides in almost every subcellular compartment and targets a wide range of proteins (Fig. 5). The complex CaBP–target interactions lead to the divergent cellular responses to transient Ca2+ signals in different cell types. Thus, the prediction of CaBP–target interactomes would not only provide a testable hypothesis and functional information in Ca2+-regulated biological processes, but would also lay the cornerstone for the ultimate computational modeling of the sophisticated Ca2+ signaling network.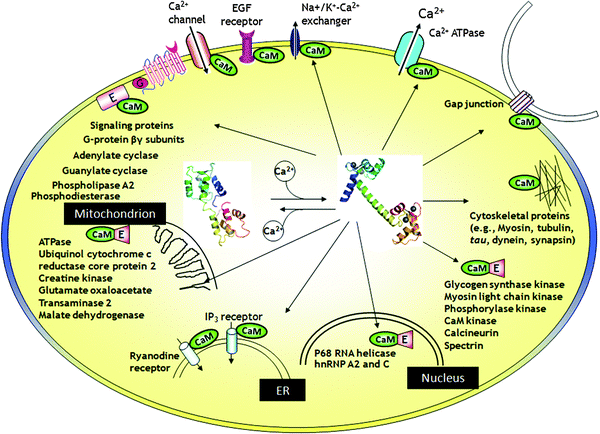 | ||
| Fig. 5 The interaction of Ca2+-CaM with target proteins. Temporal and spatial changes of the Ca2+ concentration in different compartments of cells affect the regulation of cellular signaling, by modulating the activity of a large body of CaBPs. Upon binding to Ca2+, CaBP such as calmodulin (CaM) undergoes substantial conformational changes and subsequently activate or inactivate over 100 functional enzymes, cellular receptors and ion channels through direct protein–protein interactions. | ||
The most successful example of predicting the CaBP–target interactome is the CaM target database.30 Although low sequence identity has been observed among different sequences, CaM target peptides share some key features: the hydrophobic residues with bulky sidechains that anchor the target sequence to the hydrophobic cleft of CaM are arranged in particular spacing patterns with 2–6 positively charged residues interspersed in between. According to the distribution of anchoring residues in the continuous polypeptide sequence, the binding motifs can be divided into five classes: 1–10 class, 1–14 class, 1–16 class, IQ class and others.30,31 Among them, the first three classes have been found to interact with CaM almost exclusively in a Ca2+-dependent way. Additional binding modes of 1–7 and 1–17 for CaM interaction of the ryanodine receptor (RyR) and the NMDA receptor were identified, respectively, and were also shown to be Ca2+-dependent.32 The IQ class motifs, however, are activated or inhibited by CaM in a Ca2+-independent fashion and CaM seems to be a constitutive part of its functional unit. Recent works by Shea and Chazin's groups have shown that the C-domain of apo-CaM interacts with several voltage dependent sodium channels via binding to IQ motifs.33 In addition, there are several unclassified CaM binding motifs that interact with CaM when the four Ca2+-binding sites are only half occupied. All of these common features enable one to gain more confidence when confronted by the following three questions: (1) are the proteins under investigation capable of interacting with CaM? If so, (2) is it Ca2+-dependent or independent? and (3) how the CaM-mediated function is affected by intracellular Ca2+ concentration change? An online server dedicated to predicting CaM binding sites has been generated by Ikura's group.30 We have successful experience using this prediction algorithm to predict putative CaM target sequences in a viral protease domain,34 the gap junction protein connexins35 and the intracellular domain of the Ca2+-sensing receptor,34 and to further demonstrate the functional importance of such CaM–target interactions. CaM has been shown to use either N or C-domain to sense local or global Ca2+ concentration change to regulate a number of Ca2+ permeable channels.36 If only CaM variants with removed/decreased Ca2+-binding affinities are used, it is difficult to unambiguously distinguish between the effects of Ca2+ directly on the channel and the indirect effect of Ca2+ on its target mediated by CaM. Hence, we engineered a series of mutant CaMs to individually tune the Ca2+ affinity of each of CaM's EF-hands by increasing the number of acidic residues in Ca2+ chelating positions.37 Domain-specific Ca2+ affinities of each CaM variant were determined by equilibrium fluorescence titration. Mutations in site I (T26 > D) or II (N60 > D) in CaM's N-terminal domain had little effect on CaM Ca2+ affinity and regulation of RyR1. However, the site III mutation (N97 > D) increased the Ca2+-binding affinity of CaM's C-terminal domain and caused CaM to inhibit RyR1 at a lower Ca2+ concentration than wild type CaM. Conversely, the site IV mutation (Q135 > D) decreased the Ca2+-binding affinity of CaM's C-terminal domain and caused CaM to inhibit RyR1 at higher Ca2+ concentrations. These results support the hypothesis that Ca2+-binding to CaM's C-terminal acts as the switch converting CaM from a RyR1 activator to a channel inhibitor. These results further indicate that targeting CaM's Ca2+ affinity may be a valid strategy to tune the activation profile of CaM-regulated ion channels.37
Overall, the rapid progress in bioinformatics has led to the constant improvement and expanding of prediction algorithms, databases and tools that are rapidly becoming indispensable to the metallomics field. The availability of genomic information (both 1D sequences and 3D structures), prediction algorithms and software for modeling structural and energetic characteristics has facilitated an important new phase in calciomic research, allowing for pre-experimental evaluation of hypotheses and the rational design of experimental plans (Fig. 3).
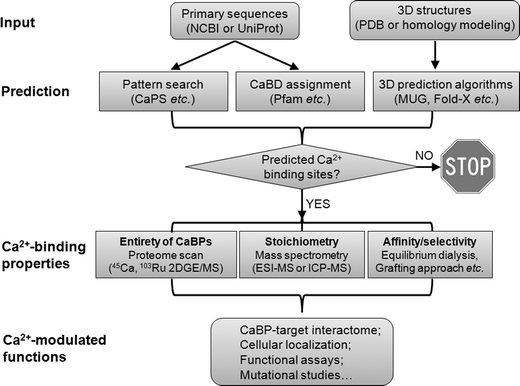 | ||
| Fig. 3 Schematic diagram of approaches used to study calciomics. Starting from protein amino acid sequences, continuous Ca2+-binding sites can be simply detected by pattern search, whereas some non-continuous Ca2+-binding domains (CaBDs) can be predicted by domain assignment using Pfam. Complementary to these methods, prediction can be made from 3D structural information using several online webservers (e.g., MUG, as listed in Table 1). The proteome for Ca2+-binding proteins (CaBPs) can be obtained by standard proteomic approaches coupled with the use of radioactive metals (45Ca or 103Ru). Quantitative mass spectrometry techniques (e.g., ESI-MS and ICP-MS) can be further applied to characterize the metal–protein stoichiometry of CaBPs. To determine the Ca2+-binding affinities, one can use equilibrium dialysis coupled with ICP-MS (or 45Ca) under circumstances that the predicted CaBP can be readily produced to high purity while retaining its function. For continuous Ca2+-binding sites, a grafting approach can be used to probe metal binding properties without the need to purify the predicted protein. An important aspect of calciomics is to define the CaBP–target interactome by combining both computational and high-throughput experimental approaches. Next, the cellular localization of putative CaBPs can be traced by tagging the CaBP of interest with fluorescent proteins. Lastly, to establish possible functional correlations of Ca2+-modulated activities, loss-of-function experiments should be carried out by removing the predicted Ca2+-binding ligands. | ||
3. Experimental approaches to detect Ca2+-binding proteins
To understand the molecular mechanism of Ca2+ modulated biological processes, we have to answer at least three important questions with experimental evidence: (i) What is the entirety of Ca2+-binding proteins? (ii) What is the exact Ca2+–protein stoichiometry? (iii) What are the site-specific Ca2+-binding affinities for Ca2+-binding proteins, particularly those that utilize multiple coupled Ca2+-binding sites to respond to sharp changes in cellular Ca2+ concentration? However, finding answers to these questions is often hampered by challenges listed below. First, while X-ray analysis has been the “gold standard” for revealing the metal-binding sites, Ca2+ has often been missing in the determined structure, particularly for those proteins with weak Ca2+-binding affinities. This is possibly due to a rapid off rate owing to the low affinity of the Ca2+-binding and the existence of multiple conformations that are in equilibrium with one another. Second, Ca2+ is spectroscopically silent, making it impossible to trace by fluorescence or other spectroscopic techniques that are amenable to study other metal ions. Fortunately, this could be partially overcome by using radioactive 45Ca, [103Ruthenium] azido ruthenium (Section 3.1), or Ca2+ analogs that are spectroscopically visible (e.g., Eu3+ and Tb3+) (Section 3.3). Third, most experimental methods such as dialysis coupled with mass spectrometry (Section 3.2) are only sensitive to the total Ca2+ content, instead of the protein-bound fraction of Ca2+. Fourth, overcoming the persistent background contamination of Ca2+ during the preparation of a Ca2+-free sample for proteins with strong Ca2+-binding affinities is not a trivial task. Last, obtaining information for site-specific Ca2+-binding is difficult due to complexities encountered in cooperative, multi-site systems, and the use of Ca2+-binding energy to bring about conformational changes. Unfortunately, the use of peptide models has been limited by lack of well-defined conformation of peptides in solution. To circumvent these limitations, we have developed a grafting approach coupled with luminescence resonance energy transfer (LRET) to probe intrinsic metal binding properties of continuous Ca2+-binding sites (Section 3.3).3.1 Proteomic approaches based on gel electrophoresis
The first question regarding the entirety of CaBPs can be partially answered by taking proteomic approaches (two dimensional gel electrophoresis (2D-GE) coupled with mass spectrometry) and the additional use of radioactive metal ions, such as 45Ca or 103Ruthenium.The 45Ca overlay method, first described by Maruyama et al.,38 has been widely used for detecting CaBPs from crude mixtures for almost three decades. Proteins from various sources are separated by means of either one- (1D) or two-dimensional (2D) gel electrophoresis, transferred to a membrane (nitrocellulose or PVDF), and overlaid with radioactive 45Ca. The “Stains-all” method described by Campbell et al. provides a simple and efficient way to probe CaBPs.39 CaBPs, such as troponin C, S-100 and calsequestrin, resolved in SDS-PAGE gel can be stained as blue and purple color. The dye has a maximum absorbance at 615 nm, indicating that this dye is in the anionic sites when it associates with Ca2+-binding proteins and then forms a J complex. Interestingly, “stains-all” also interacts with native proteins with a maximal absorbance around 600 nm. Further studies showed that the “stains-all” dye could cause changes in both the UV/vis absorbance and the circular dichroism signals when it interacts with Ca2+-binding motifs.40
The advent of powerful mass spectrometry techniques further extends the applicability of this methodology to efficiently identify unknown CaBPs. This method has been recently adapted to investigate the repertoire of CaBPs associated with the plasma membrane of Arabidopsis thaliana41 or human sperm.42 These techniques have also been used to characterize CaBPs such as p95, calreticulin, synaptophysin and γ-crystallin.43 When coupled with equilibrium dialysis, the 45Ca overlay method can be further used to determine the stoichiometry and binding affinity of a Ca2+-binding protein.
The recent use of azido ruthenium (AzRu or PhotoCal) represents a new avenue of studying Ca2+-dependent processes.44 AzRu was shown to specifically interact with Ca2+-binding proteins and remain covalently bound after ultraviolet irradiation. By taking advantage of this unique feature, Israelson et al. generated an AzRu-cellulose column to purify CaBPs with affinity chromatography, and developed AzRu-based biosensor chips for identifying previously unknown CaBPs.44 Moreover, radioactive [103Ru] AzRu can be used to detect CaBPs in biological samples after SDS-PAGE and autoradiography. It is anticipated that the wide use of this reagent will pave the way for generating a comprehensive Ca2+-targeted protein database and detecting defective CaBPs implicated in various disorders and diseases associated with dysregulated Ca2+ signaling. Nonetheless, this method has two potential caveats: it has not been fully established that AzRu mimics the coordination chemistry of all known CaBPs and the metal selectivity (except for Mg2+) has not been rigorously tested.
In summary, the gel electrophoresis and autoradiography-based method is well-known for its high sensitivity and accuracy. However, its disadvantages are also obvious: first, it requires the handling of radioactive products and involves relatively labor-intensive and time-consuming procedures. Second, the amount of proteins encoded by rare genes is far from sufficient to be detected by SDS-PAGE. Third, the demanding gel electrophoresis step is likely to disrupt the native Ca2+-binding sites in protein samples. Last but critically, CaBPs with weak affinities for Ca2+ may not be readily detected with this method.
3.2 Mass spectrometry approaches
The determination of Ca2+–protein stoichiometry and metal selectivity in protein samples can be routinely performed using electrospray ionization mass spectrometry (ESI-MS) or inductively coupled plasma mass spectrometry (ICP-MS). ESI-MS is widely used to capture non-covalent interactions as seen in Ca2+:protein complexes. Metal-bound biological samples can be mildly ionized and transferred to the gas-phase while maintaining their native states. This technique is very sensitive to CaBPs with a strong affinity to Ca2+. The comparison of the mass/charge peaks corresponding to the same sample under native or denatured conditions gives directly the mass of bound metal. Nonetheless, ESI-MS has a poor tolerance toward nonvolatile salt buffers and solubilizing agents, which are often necessary to maintain the structural integrity and functionality of CaBPs. Furthermore, ESI-MS often cannot provide quantitative information on the amount of bound metals. Complementary to ESI-MS, ICP-MS can quantitatively detect the total metal contents in a biological sample with high sensitivity regardless of its molecular environment, and therefore, is often used in speciation analysis coupled with liquid chromatographic techniques.45 A detailed description of the application of these mass spectrometry techniques in bioinorganic analytical chemistry can be found in a comprehensive review by Lobinski et al.45a
3.3 A grafting approach to probe isolated continuous Ca2+-binding sites
To answer the third question listed above, we have developed a grafting approach to dissect the key structural factors that control the Ca2+-binding affinity, conformational change and cooperativity.46 In principle, the key determinants for Ca2+ affinity can be systematically introduced into a stable host protein frame and evaluated by eliminating or minimizing the contribution of conformational change. We have shown that domain 1 of CD2 (CD2.D1) is an excellent scaffold protein.47 It retains its native structure following insertion of the EF-hand motif with proper flanking linkers. This provides the foundation for measuring the intrinsic metal-binding affinity with minimized contribution of protein conformational change. More importantly, the aromatic residues (particularly Trp32) in CD2, which is juxta-positioned to the grafted Ca2+-binding loop, enable us to obtain Tb3+ binding affinity with LRET using fluorescence spectroscopy. Tb3+ is commonly used to probe Ca2+-binding because of their similar ionic radii (Ca2+, 1.00 Å; Tb3+, 0.92 Å) and metal coordination chemistry.48 A Ca2+ competition assay can be further performed to obtain the Ca2+-binding affinity. This approach has been successfully applied to determine the site-specific metal-binding affinities of calmodulin, a ubiquitous Ca2+-sensing protein with four EF-hands. Based on the energetics relationship, the cooperativity of coupled Ca2+-binding sites can also be estimated after obtaining the intrinsic Ca2+-binding affinity for each site.46a Another advantage of this approach lies in the fact that it circumvents the expression and purification of the original proteins that harbor the predicted Ca2+-binding sites. The same method has also been used to probe the site-specific metal-binding properties of putative EF-hand motifs in viral and bacterial proteins,10,49 as well as predicted continuous Ca2+-binding sites in the family C of G-protein coupled receptors (GPCRs)35a,50 and the ER-resident stromal interaction molecule 1 (STIM1).51 By taking advantage of the sensitive LRET signal and the dropping cost of mini-gene synthesis, we foresee ample opportunities to further extend this approach to systematically validate putative linear Ca2+-binding sites in a high-throughput manner with fluorescence microplate readers. For example, GST-tagged engineered proteins containing various putative Ca2+-binding sites can be enriched from E. coli cell lysates with agarose beads conjugated with glutathione and subjected to LRET measurement in a 96-well format.3.4 Approaches to map the CaBP–target interactome
We use the most extensively studied CaM–target interactions as an example here. Several robust methods have been developed to screen CaM-binding proteins (CaMBPs) on the proteome-wide scale. A CaM binding overlay (CAMBOT) technique that requires labeling of CaM with 35S and the use of SDS-PAGE was established by O'Day.52 In this technique, the bacteria-expressed 35S-CaM was used as the probe to detect CaMBPs and compare the CaMBPs expression level under different conditions. Under Ca2+-saturated and Ca2+-free conditions, it is possible, in theory, to detect both Ca2+-dependent and Ca2+-independent CaMBPs. This method can be potentially applied to verify and profile the CaMBPs, to assess the effects of drugs on CaM–CaMBP networks, and to evaluate the effects of heavy metal ions in the environment on the CaM–CaMBP network.An alternative way to screen CaMBP is explored by Shen et al.53 This large-scale screening method utilizes the mRNA display technique. A mRNA-displayed enriched proteome library can be selected by biotinylated CaM, which will be further captured by streptavidin–agarose beads. Another round of selection involving six steps (PCR, in vitro transcription, crosslinking, cell-free translation and fusion, reverse transcription and affinity chromatography) can be repeated to diminish unspecific interactions. Screening of CaMBPs in the human and C. elegans proteome with this method reveals a large body of previously undetected proteins or uncharacterized proteins, such as ribosomal proteins, proteasome 26S subunits, deubiquitinating enzymes, and leucine zipper proteins.
In another method, combining affinity chromatography and tandem mass spectrometry, Berggard et al. detected 140 putative CaM binding proteins in the mouse brain.54 Recently, a protein microarray based method was used to map plant proteins that interact with Ca2+-CaM or CaM-like (CaML) protein in Arabidopsis thaliana.55 The protein array strategy overcomes at least two limitations of the above-mentioned methods. First, all the target proteins are presented in distinct locations and identification of secondary targets is minimized. Second, the protein array platform is compatible with transmembrane proteins, including GPCRs and ion channels, which are often closely implicated in Ca2+ signaling. A total of 1133 high-quality Arabidopsis proteins were screened for their capability to interact with CaM that is conjugated with Alexa Fluor 647 or Alexa Fluor 594. This medium-content array identified 173 novel CaM/CaML targets in plants. A high-content protein array has been applied to profile CaM–target interactions in the human proteome.56 This protein array contains 37200 redundant proteins and over 10000 unique human proteins expressed from a human brain cDNA library. One of the novel findings from this study is the identification of a high-affinity interaction between CaM and the ER-resident single-pass transmembrane protein, stromal interaction molecule (STIM), which plays instrumental roles in the store operated Ca2+ entry process by translocating from the ER toward the plasma membrane to open the ORAI1 store operated Ca2+ channel.57
4. Conclusions
The Ca2+ signaling field has been constantly reinvigorated with the continuous discovery of new components and expansion of the Ca2+-signaling toolkits. The combination of powerful bioinformatics tools and innovative methodologies is expected to enable us to explore a growing list of Ca2+-binding proteins and Ca2+-modulated interactomes that are currently underrepresented. Our ability to investigate the structure–function correlations of CaBPs has advanced in leaps with the evolution of algorithms capable of recognizing or predicting Ca2+-binding sites. It is optimistically anticipated that the development of tools to predict Ca2+-binding sites in apo-structures may become routine in the near future. Moving toward rational protein design based on computational prediction, it is highly expected that the engineering of metal binding proteins with precise affinity and metal selectivity will significantly improve our ability to provide proteins with tailored functions for diagnostic or therapeutic purposes.Acknowledgements
We appreciate the critical review by Natalie White and Katheryn Meenach. This work is supported in part by research from NIH GM081749 to JJY and a Special Fellow Award (3013-12) from the Leukemia & Lymphoma Society to YZ.References
- R. J. Williams, The evolution of calcium biochemistry, Biochim. Biophys. Acta, 2006, 1763, 1139–1146 CrossRef CAS.
- M. D. Ross, The influence of gravity on structure and function of animals, Adv. Space Res., 1984, 4, 305–314 CrossRef CAS.
- (a) W. A. Stini, Calcium homeostasis and human evolution, Coll. Antropol., 1998, 22, 411–425 CAS; (b) R. M. Case, D. Eisner, A. Gurney, O. Jones, S. Muallem and A. Verkhratsky, Evolution of calcium homeostasis: from birth of the first cell to an omnipresent signalling system, Cell Calcium, 2007, 42, 345–350 CrossRef CAS.
- E. M. Brown, Clinical lessons from the calcium-sensing receptor, Nat. Clin. Pract. Endocrinol. Metab., 2007, 3, 122–133 CrossRef CAS.
- M. J. Berridge, M. D. Bootman and H. L. Roderick, Calcium signalling: dynamics, homeostasis and remodelling, Nat. Rev. Mol. Cell Biol., 2003, 4, 517–529 CrossRef CAS.
- (a) P. Gangola and B. P. Rosen, Maintenance of intracellular calcium in Escherichia coli, J. Biol. Chem., 1987, 262, 12570–12574 CAS; (b) B. P. Rosen, Bacterial calcium transport, Biochim. Biophys. Acta, 1987, 906, 101–110 CrossRef CAS; (c) N. K. Vyas, M. N. Vyas and F. A. Quiocho, A novel calcium binding site in the galactose-binding protein of bacterial transport and chemotaxis, Nature, 1987, 327, 635–638 CrossRef CAS; (d) N. J. Watkins, M. R. Knight, A. J. Trewavas and A. K. Campbell, Free calcium transients in chemotactic and non-chemotactic strains of Escherichia coli determined by using recombinant aequorin, Biochem. J., 1995, 306(Pt 3), 865–869 CAS; (e) R. N. Reusch, R. Huang and L. L. Bramble, Poly-3-hydroxybutyrate/polyphosphate complexes form voltage-activated Ca2+ channels in the plasma membranes of Escherichia coli, Biophys. J., 1995, 69, 754–766 CrossRef CAS; (f) S. R. Durell and H. R. Guy, A putative prokaryote voltage-gated Ca(2+) channel with only one 6TM motif per subunit, Biochem. Biophys. Res. Commun., 2001, 281, 741–746 CrossRef CAS.
- F. Kippert, Endocytobiotic coordination, intracellular calcium signaling, and the origin of endogenous rhythms, Ann. N. Y. Acad. Sci., 1987, 503, 476–495 CrossRef CAS.
- M. Kirberger, X. Wang, K. Zhao, S. Tang, G. Chen and J. J. Yang, Integration of Diverse Research Methods to Analyze and Engineer Ca-Binding Proteins: From Prediction to Production, Curr. Bioinf., 2010, 5, 68–80 CrossRef CAS.
- H. Kawasaki, S. Nakayama and R. H. Kretsinger, Classification and evolution of EF-hand proteins, Biometals, 1998, 11, 277–295 CrossRef CAS.
- (a) Y. Zhou, W. Yang, M. Kirberger, H. W. Lee, G. Ayalasomayajula and J. J. Yang, Prediction of EF-hand calcium-binding proteins and analysis of bacterial EF-hand proteins, Proteins, 2006, 65, 643–655 CrossRef CAS; (b) Y. Zhou, T. K. Frey and J. J. Yang, Viral calciomics: interplays between Ca2+ and virus, Cell Calcium, 2009, 46, 1–17 CrossRef CAS.
- (a) D. J. Rigden, D. D. Woodhead, P. W. Wong and M. Y. Galperin, New structural and functional contexts of the Dx[DN]xDG linear motif: insights into evolution of calcium-binding proteins, PLoS One, 2011, 6, e21507 CAS; (b) D. J. Rigden, M. J. Jedrzejas and M. Y. Galperin, An extracellular calcium-binding domain in bacteria with a distant relationship to EF-hands, FEMS Microbiol. Lett., 2003, 221, 103–110 CrossRef CAS; (c) J. Michiels, C. Xi, J. Verhaert and J. Vanderleyden, The functions of Ca(2+) in bacteria: a role for EF-hand proteins?, Trends Microbiol., 2002, 10, 87–93 CrossRef CAS.
- (a) S. Hunter, P. Jones, A. Mitchell, R. Apweiler, T. K. Attwood, A. Bateman, T. Bernard, D. Binns, P. Bork, S. Burge, E. de Castro, P. Coggill, M. Corbett, U. Das, L. Daugherty, L. Duquenne, R. D. Finn, M. Fraser, J. Gough, D. Haft, N. Hulo, D. Kahn, E. Kelly, I. Letunic, D. Lonsdale, R. Lopez, M. Madera, J. Maslen, C. McAnulla, J. McDowall, C. McMenamin, H. Mi, P. Mutowo-Muellenet, N. Mulder, D. Natale, C. Orengo, S. Pesseat, M. Punta, A. F. Quinn, C. Rivoire, A. Sangrador-Vegas, J. D. Selengut, C. J. Sigrist, M. Scheremetjew, J. Tate, M. Thimmajanarthanan, P. D. Thomas, C. H. Wu, C. Yeats and S. Y. Yong, InterPro in 2011: new developments in the family and domain prediction database, Nucleic Acids Res., 2012, 40, D306–D312 CrossRef CAS; (b) M. Punta, P. C. Coggill, R. Y. Eberhardt, J. Mistry, J. Tate, C. Boursnell, N. Pang, K. Forslund, G. Ceric, J. Clements, A. Heger, L. Holm, E. L. Sonnhammer, S. R. Eddy, A. Bateman and R. D. Finn, The Pfam protein families database, Nucleic Acids Res., 2012, 40, D290–D301 CrossRef CAS.
- A. Krogh, M. Brown, I. S. Mian, K. Sjolander and D. Haussler, Hidden Markov models in computational biology. Applications to protein modeling, J. Mol. Biol., 1994, 235, 1501–1531 CrossRef CAS.
- B. Schuster-Bockler, J. Schultz and S. Rahmann, HMM Logos for visualization of protein families, BMC Bioinf., 2004, 5, 7 CrossRef.
- R. D. Finn, J. Clements and S. R. Eddy, HMMER web server: interactive sequence similarity searching, Nucleic Acids Res., 2011, 39, W29–W37 CrossRef CAS.
- I. Bertini and G. Cavallaro, Bioinformatics in bioinorganic chemistry, Metallomics, 2010, 2, 39–51 RSC.
- (a) M. Kirberger, X. Wang, H. Deng, W. Yang, G. Chen and J. J. Yang, Statistical analysis of structural characteristics of protein Ca2+-binding sites, J. Biol. Inorg. Chem., 2008, 13, 1169–1181 CrossRef CAS; (b) W. Yang, H. W. Lee, H. Hellinga and J. J. Yang, Structural analysis, identification, and design of calcium-binding sites in proteins, Proteins, 2002, 47, 344–356 CrossRef CAS; (c) E. Pidcock and G. R. Moore, Structural characteristics of protein binding sites for calcium and lanthanide ions, J. Biol. Inorg. Chem., 2001, 6, 479–489 CrossRef CAS.
- J. W. Schymkowitz, F. Rousseau, I. C. Martins, J. Ferkinghoff-Borg, F. Stricher and L. Serrano, Prediction of water and metal binding sites and their affinities by using the Fold-X force field, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10147–10152 CrossRef CAS.
- (a) M. Nayal and E. Di Cera, Predicting Ca(2+)-binding sites in proteins, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 817–821 CrossRef CAS; (b) M. M. Yamashita, L. Wesson, G. Eisenman and D. Eisenberg, Where metal ions bind in proteins, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5648–5652 CrossRef CAS.
- H. H. Lin, L. Y. Han, H. L. Zhang, C. J. Zheng, B. Xie, Z. W. Cao and Y. Z. Chen, Prediction of the functional class of metal-binding proteins from sequence derived physicochemical properties by support vector machine approach, BMC Bioinf., 2006, 7(Suppl 5), S13 CrossRef.
- A. J. Bordner, Predicting small ligand binding sites in proteins using backbone structure, Bioinformatics, 2008, 24, 2865–2871 CrossRef CAS.
- M. Brylinski and J. Skolnick, FINDSITE-metal: integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level, Proteins, 2011, 79, 735–751 CrossRef CAS.
- H. Deng, G. Chen, W. Yang and J. J. Yang, Predicting calcium-binding sites in proteins – a graph theory and geometry approach, Proteins, 2006, 64, 34–42 CrossRef CAS.
- X. Wang, M. Kirberger, F. Qiu, G. Chen and J. J. Yang, Towards predicting Ca2+-binding sites with different coordination numbers in proteins with atomic resolution, Proteins, 2009, 75, 787–798 CrossRef CAS.
- K. Zhao, X. Wang, H. C. Wong, R. Wohlhueter, M. P. Kirberger, G. Chen and J. J. Yang, Predicting Ca(2+)-binding sites using refined carbon clusters, Proteins, 2012, 80, 2666–2679 CAS.
- M. P. Liang, D. R. Banatao, T. E. Klein, D. L. Brutlag and R. B. Altman, WebFEATURE: An interactive web tool for identifying and visualizing functional sites on macromolecular structures, Nucleic Acids Res., 2003, 31, 3324–3327 CrossRef CAS.
- D. S. Glazer, R. J. Radmer and R. B. Altman, Combining molecular dynamics and machine learning to improve protein function recognition, Pac. Symp. Biocomput., 2008, 332–343 CAS.
- X. Wang, K. Zhao, M. Kirberger, H. Wong, G. Chen and J. J. Yang, Analysis and prediction of calcium-binding pockets from apo-protein structures exhibiting calcium-induced localized conformational changes, Protein Sci., 2010, 19, 1180–1190 CrossRef CAS.
- K. P. Hoeflich and M. Ikura, Calmodulin in action: diversity in target recognition and activation mechanisms, Cell (Cambridge, Mass.), 2002, 108, 739–742 CrossRef CAS.
- K. L. Yap, J. Kim, K. Truong, M. Sherman, T. Yuan and M. Ikura, Calmodulin target database, J. Struct. Funct. Genomics, 2000, 1, 8–14 CrossRef CAS.
- P. Radivojac, S. Vucetic, T. R. O'Connor, V. N. Uversky, Z. Obradovic and A. K. Dunker, Calmodulin signaling: analysis and prediction of a disorder-dependent molecular recognition, Proteins, 2006, 63, 398–410 CrossRef CAS.
- (a) Z. A. Ataman, L. Gakhar, B. R. Sorensen, J. W. Hell and M. A. Shea, The NMDA receptor NR1 C1 region bound to calmodulin: structural insights into functional differences between homologous domains, Structure, 2007, 15, 1603–1617 CrossRef CAS; (b) A. A. Maximciuc, J. A. Putkey, Y. Shamoo and K. R. Mackenzie, Complex of calmodulin with a ryanodine receptor target reveals a novel, flexible binding mode, Structure, 2006, 14, 1547–1556 CrossRef CAS.
- (a) M. D. Feldkamp, L. Yu and M. A. Shea, Structural and energetic determinants of apo calmodulin binding to the IQ motif of the Na(V)1.2 voltage-dependent sodium channel, Structure, 2011, 19, 733–747 CrossRef CAS; (b) B. Chagot and W. J. Chazin, Solution NMR structure of Apo-calmodulin in complex with the IQ motif of human cardiac sodium channel NaV1.5, J. Mol. Biol., 2011, 406, 106–119 CrossRef CAS.
- Y. Zhou, W. P. Tzeng, H. C. Wong, Y. Ye, J. Jiang, Y. Chen, Y. Huang, S. Suppiah, T. K. Frey and J. J. Yang, Calcium-dependent association of calmodulin with the rubella virus nonstructural protease domain, J. Biol. Chem., 2010, 285, 8855–8868 CrossRef CAS.
- (a) Y. Zhou, W. Yang, M. M. Lurtz, Y. Ye, Y. Huang, H. W. Lee, Y. Chen, C. F. Louis and J. J. Yang, Identification of the calmodulin binding domain of connexin 43, J. Biol. Chem., 2007, 282, 35005–35017 CrossRef CAS; (b) Y. Zhou, W. Yang, M. M. Lurtz, Y. Chen, J. Jiang, Y. Huang, C. F. Louis and J. J. Yang, Calmodulin mediates the Ca2+-dependent regulation of Cx44 gap junctions, Biophys. J., 2009, 96, 2832–2848 CrossRef CAS; (c) Y. Chen, Y. Zhou, X. Lin, H. C. Wong, Q. Xu, J. Jiang, S. Wang, M. M. Lurtz, C. F. Louis, R. D. Veenstra and J. J. Yang, Molecular interaction and functional regulation of connexin50 gap junctions by calmodulin, Biochem. J., 2011, 435, 711–722 CrossRef CAS.
- (a) M. R. Tadross, I. E. Dick and D. T. Yue, Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel, Cell (Cambridge, Mass.), 2008, 133, 1228–1240 CrossRef CAS; (b) I. E. Dick, M. R. Tadross, H. Liang, L. H. Tay, W. Yang and D. T. Yue, A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels, Nature, 2008, 451, 830–834 CrossRef CAS.
- J. Jiang, Y. Zhou, J. Zou, Y. Chen, P. Patel, J. J. Yang and E. M. Balog, Site-specific modification of calmodulin Ca(2)(+) affinity tunes the skeletal muscle ryanodine receptor activation profile, Biochem. J., 2010, 432, 89–99 CrossRef CAS.
- K. Maruyama, T. Mikawa and S. Ebashi, Detection of calcium binding proteins by 45Ca autoradiography on nitrocellulose membrane after sodium dodecyl sulfate gel electrophoresis, J. Biochem., 1984, 95, 511–519 CAS.
- K. P. Campbell, D. H. MacLennan and A. O. Jorgensen, Staining of the Ca2+-binding proteins, calsequestrin, calmodulin, troponin C, and S-100, with the cationic carbocyanine dye “Stains-all”, J. Biol. Chem., 1983, 258, 11267–11273 CAS.
- Y. Sharma, A. Gopalakrishna, D. Balasubramanian, T. Fairwell and G. Krishna, Studies on the interaction of the dye, stains-all, with individual calcium-binding domains of calmodulin, FEBS Lett., 1993, 326, 59–64 CrossRef CAS.
- Y. Ide, N. Nagasaki, R. Tomioka, M. Suito, T. Kamiya and M. Maeshima, Molecular properties of a novel, hydrophilic cation-binding protein associated with the plasma membrane, J. Exp. Bot., 2007, 58, 1173–1183 CrossRef CAS.
- S. Naaby-Hansen, A. Diekman, J. Shetty, C. J. Flickinger, A. Westbrook and J. C. Herr, Identification of calcium-binding proteins associated with the human sperm plasma membrane, Reprod. Biol. Endocrinol., 2010, 8, 6 CrossRef.
- (a) J. S. Gilchrist and G. N. Pierce, Identification and purification of a calcium-binding protein in hepatic nuclear membranes, J. Biol. Chem., 1993, 268, 4291–4299 CAS; (b) R. E. Milner, S. Baksh, C. Shemanko, M. R. Carpenter, L. Smillie, J. E. Vance, M. Opas and M. Michalak, Calreticulin, and not calsequestrin, is the major calcium binding protein of smooth muscle sarcoplasmic reticulum and liver endoplasmic reticulum, J. Biol. Chem., 1991, 266, 7155–7165 CAS; (c) B. Rajini, P. Shridas, C. S. Sundari, D. Muralidhar, S. Chandani, F. Thomas and Y. Sharma, Calcium binding properties of gamma-crystallin: calcium ion binds at the Greek key beta gamma-crystallin fold, J. Biol. Chem., 2001, 276, 38464–38471 CrossRef CAS; (d) H. Rehm, B. Wiedenmann and H. Betz, Molecular characterization of synaptophysin, a major calcium-binding protein of the synaptic vesicle membrane, EMBO J., 1986, 5, 535–541 CAS.
- (a) A. Israelson, N. Zilberberg and V. Shoshan-Barmatz, Azido ruthenium: a new photoreactive probe for calcium-binding proteins, Nat. Protoc., 2006, 1, 111–117 CrossRef CAS; (b) A. Israelson, L. Arzoine, S. Abu-hamad, V. Khodorkovsky and V. Shoshan-Barmatz, A photoactivable probe for calcium binding proteins, Chem. Biol., 2005, 12, 1169–1178 CrossRef CAS.
- (a) R. Lobinski, D. Schaumloffel and J. Szpunar, Mass spectrometry in bioinorganic analytical chemistry, Mass Spectrom. Rev., 2006, 25, 255–289 CrossRef CAS; (b) M. Montes-Bayon, K. DeNicola and J. A. Caruso, Liquid chromatography-inductively coupled plasma mass spectrometry, J. Chromatogr., A, 2003, 1000, 457–476 CrossRef CAS.
- (a) Y. Ye, H. W. Lee, W. Yang, S. Shealy and J. J. Yang, Probing site-specific calmodulin calcium and lanthanide affinity by grafting, J. Am. Chem. Soc., 2005, 127, 3743–3750 CrossRef CAS; (b) J. J. Yang, A. Gawthrop and Y. Ye, Obtaining site-specific calcium-binding affinities of calmodulin, Protein Pept. Lett., 2003, 10, 331–345 CrossRef CAS; (c) Y. Ye, S. Shealy, H. W. Lee, I. Torshin, R. Harrison and J. J. Yang, A grafting approach to obtain site-specific metal-binding properties of EF-hand proteins, Protein Eng., 2003, 16, 429–434 CrossRef CAS.
- Y. Ye, H. W. Lee, W. Yang, S. J. Shealy, A. L. Wilkins, Z. R. Liu, I. Torshin, R. Harrison, R. Wohlhueter and J. J. Yang, Metal binding affinity and structural properties of an isolated EF-loop in a scaffold protein, Protein Eng., 2001, 14, 1001–1013 CrossRef CAS.
- W. D. Horrocks, Jr., Luminescence spectroscopy, Methods Enzymol., 1993, 226, 495–538 Search PubMed.
- (a) Y. Zhou, W. P. Tzeng, W. Yang, Y. Ye, H. W. Lee, T. K. Frey and J. Yang, Identification of a Ca2+-binding domain in the rubella virus nonstructural protease, J. Virol., 2007, 81, 7517–7528 CrossRef CAS; (b) C. Yanyi, X. Shenghui, Z. Yubin and Y. J. Jie, Calciomics: prediction and analysis of EF-hand calcium binding proteins by protein engineering, Sci. China: Chem., 2010, 53, 52–60 CrossRef.
- Y. Jiang, Y. Huang, H. C. Wong, Y. Zhou, X. Wang, J. Yang, R. A. Hall, E. M. Brown and J. J. Yang, Elucidation of a novel extracellular calcium-binding site on metabotropic glutamate receptor 1alpha (mGluR1) that controls receptor activation, J. Biol. Chem., 2010, 285, 33463–33474 CrossRef CAS.
- Y. Huang, Y. Zhou, H. C. Wong, Y. Chen, S. Wang, A. Castiblanco, A. Liu and J. J. Yang, A single EF-hand isolated from STIM1 forms dimer in the absence and presence of Ca2+, FEBS J., 2009, 276, 5589–5597 CrossRef CAS.
- D. H. O'Day, CaMBOT: profiling and characterizing calmodulin-binding proteins, Cell. Signalling, 2003, 15, 347–354 CrossRef CAS.
- (a) X. Shen, C. A. Valencia, W. Gao, S. W. Cotten, B. Dong, B. C. Huang and R. Liu, Ca(2+)/Calmodulin-binding proteins from the C. elegans proteome, Cell Calcium, 2008, 43, 444–456 CrossRef CAS; (b) X. Shen, C. A. Valencia, J. W. Szostak, B. Dong and R. Liu, Scanning the human proteome for calmodulin-binding proteins, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5969–5974 CrossRef CAS.
- T. Berggard, G. Arrigoni, O. Olsson, M. Fex, S. Linse and P. James, 140 mouse brain proteins identified by Ca2+-calmodulin affinity chromatography and tandem mass spectrometry, J. Proteome Res., 2006, 5, 669–687 CrossRef.
- S. C. Popescu, G. V. Popescu, S. Bachan, Z. Zhang, M. Seay, M. Gerstein, M. Snyder and S. P. Dinesh-Kumar, Differential binding of calmodulin-related proteins to their targets revealed through high-density Arabidopsis protein microarrays, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4730–4735 CrossRef CAS.
- D. J. O'Connell, M. Bauer, S. Linse and D. J. Cahill, Probing calmodulin protein–protein interactions using high-content protein arrays, Methods Mol. Biol., 2011, 785, 289–303 CAS.
- (a) P. G. Hogan, R. S. Lewis and A. Rao, Molecular basis of calcium signaling in lymphocytes: STIM and ORAI, Annu. Rev. Immunol., 2010, 28, 491–533 CrossRef CAS; (b) Y. Zhou, P. Meraner, H. T. Kwon, D. Machnes, M. Oh-hora, J. Zimmer, Y. Huang, A. Stura, A. Rao and P. G. Hogan, STIM1 gates the store-operated calcium channel ORAI1 in vitro, Nat. Struct. Mol. Biol., 2010, 17, 112–116 CrossRef CAS.
- J. Haiech, S. B. Moulhaye and M. C. Kilhoffer, The EF-Handome: combining comparative genomic study using FamDBtool, a new bioinformatics tool, and the network of expertise of the European Calcium Society, Biochim. Biophys. Acta, 2004, 1742, 179–183 CrossRef CAS.
- J. A. Horst and R. Samudrala, A protein sequence meta-functional signature for calcium binding residue prediction, Pattern Recognit. Lett., 2010, 31, 2103–2112 CrossRef.
- K. N. Pradeep, R. Piyush and K. Pooja, MetalloPred: A tool for hierarchical prediction of metal ion binding proteins using cluster of neural networks and sequence derived features, J. Biophys. Chem., 2011, 2, 112–123 CrossRef.
- D. B. Roche, S. J. Tetchner and L. J. McGuffin, FunFOLD: an improved automated method for the prediction of ligand binding residues using 3D models of proteins, BMC Bioinf., 2011, 12, 160 CrossRef CAS.
- B. Huang, MetaPocket: a meta approach to improve protein ligand binding site prediction, Omics, 2009, 13, 325–330 CrossRef CAS.
- J. S. Sodhi, K. Bryson, L. J. McGuffin, J. J. Ward, L. Wernisch and D. T. Jones, Predicting metal-binding site residues in low-resolution structural models, J. Mol. Biol., 2004, 342, 307–320 CrossRef CAS.
- N. Bhardwaj, R. V. Stahelin, R. E. Langlois, W. Cho and H. Lu, Structural bioinformatics prediction of membrane-binding proteins, J. Mol. Biol., 2006, 359, 486–495 CrossRef CAS.
- B. I. Dahiyat and S. L. Mayo, De novo protein design: fully automated sequence selection, Science, 1997, 278, 82–87 CrossRef CAS.
- H. W. Hellinga and F. M. Richards, Construction of new ligand binding sites in proteins of known structure. I. Computer-aided modeling of sites with pre-defined geometry, J. Mol. Biol., 1991, 222, 763–785 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2mt20009k |
| This journal is © The Royal Society of Chemistry 2013 |