Mechanistic investigation of the oxidation of hydrazides: implications for the activation of the TB drug isoniazid†
Ruth I. J.
Amos
ab,
Brendon S.
Gourlay
a,
Brian F.
Yates
a,
Carl H.
Schiesser
bc,
Trevor W.
Lewis
a and
Jason A.
Smith
*ab
aSchool of Chemistry, University of Tasmania, Hobart, Australia. E-mail: Jason.Smith@utas.edu.au; Fax: +613 6226 2858; Tel: +613 6226 2182
bAustralian Research Council Centre of Excellence for Free Radical Chemistry and Biotechnology, Australia
cSchool of Chemistry and Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Victoria, 3010, Australia
First published on 8th November 2012
Abstract
Aryl hydrazides are oxidised to acyl radicals through a mechanism involving diimide intermediates that are prone to nucleophilic acyl substitution. This oxidation occurs regardless of the oxidant involved, however there is no evidence that the acyl radical formed undergoes further oxidation to the corresponding acylium ion, even in the presence of strong oxidants. This study may provide insight into the mechanism of isoniazid resistance in Mycobacterium tuberculosis.
Introduction
Isoniazid, (1, Scheme 1) the hydrazide of isonicotinic acid, is a commonly used antibiotic against Mycobacterium tuberculosis.1 As resistance to the frontline antibiotics isoniazid, ethambutol, rifamycin and pyrazinamide increases, an understanding of the mode of action and mechanisms of resistance are key to the development of new antibiotics for multi-drug resistant tuberculosis (MDR-TB).2 Isoniazid acts by inhibiting the fatty acid synthesis pathway of Mycobacterium tuberculosis, effectively preventing the production of mycolic acid required for cell capsule synthesis.3 The target enzyme is InhA, an enoyl–acyl carrier protein.3,4 Sacchetini elucidated the structure of the inhibitor in the active site of the enzyme InhA and found it to be an isonicotinic acyl-NADH adduct (2, Scheme 1),5 confirming that a reactive intermediate from isoniazid and NAD+ (3, Scheme 1) combine to give the true inhibitor of the bacterium. As a consequence, isoniazid requires activation in order to react with NAD+ and be effective against the bacterium (Scheme 1).5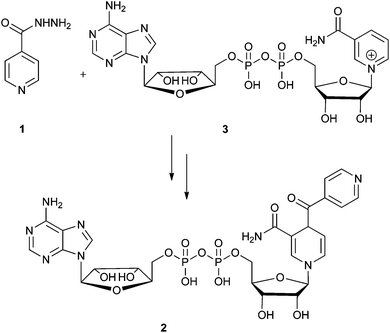 | ||
| Scheme 1 Isoniazid (1) combines with NAD+ (3) under oxidative conditions to form the true inhibitor of InhA 2. | ||
The activation of isoniazid was postulated to involve a free radical and this hypothesis was supported by electron spin resonance (ESR) spectroscopy studies.6,7 In the bacterium this activation is performed by the catalase-peroxidase enzyme KatG, therefore, oxidation of isoniazid plays an important role in this process.5,6 In many drug resistant strains of the bacterium, activation is inhibited as a result of a single amino acid residue change (S315T) in the KatG enzyme that appears to slightly contract the ligand access channel to the active site.8 Investigations into the mechanism of oxidation of isoniazid may give insight into drug resistance and provide new leads in the development of more effective antibiotics for MDR-TB.
Very little was known about the mechanism for the oxidation of hydrazides, although it was well established that an electrophilic species is generated and has been exploited for nucleophilic acyl substitution reactions.9,10 The oxidation of hydrazides to give carboxylic acids in water, and esters in the presence of alcohols is well known. However, there would appear to be some inconsistencies surrounding the mechanism of this transformation.9–12 Some studies invoke the presence of diimide intermediates (4) during the oxidation of hydrazides (5)11,12 and the possibility of nucleophilic acyl substitution of these intermediates (Scheme 2). However, it seems generally accepted in the literature that an acylium ion (6), or acyl cation, could be formed by oxidation, leading to carboxylic acids when the oxidation is carried out in water, and ester formation in the presence of alcohols.9,10 Recently it has been shown unequivocally that the oxidation of benzhydrazide forms acyl radicals (7, R = H).13 Preliminary investigations supported this observation and showed that isoniazid also forms an acyl radical14 but the involvement of an acylium ion in the oxidation of hydrazides is ambiguous.
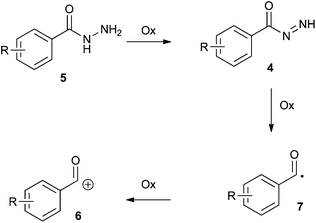 | ||
| Scheme 2 Proposed intermediates in the oxidation of hydrazides. | ||
Given the conflicting mechanisms previously proposed, an investigation into the oxidation of hydrazides was initiated to answer these questions. The results of this mechanistic study, involving both laboratory-based and computational techniques, provides some insight into possible mechanisms of drug resistance in S315T TB.
Results
Trapping of acyl radicals
Initial experiments on the oxidation of the phenyl derivative of isoniazid, benzhydrazide (8, Scheme 3), used the benzothiazolium ion reported by Minisci as a diagnostic probe for the presence of acyl radicals in the permanganate oxidation of alcohols to carboxylic acids.15 When the hydrazide 8 was oxidised with potassium permanganate in the presence of the benzothiazolium ion (formed by adding benzothiazole to 5 M H2SO4), 2-benzoyl benzothiazole 9 was obtained in 16% yield (Scheme 3).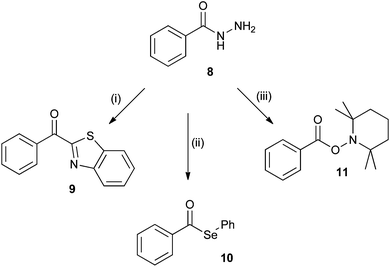 | ||
| Scheme 3 Reagents; (i) benzothiazole, AcOH, 5 M H2SO4, KMnO4; (ii) Ph2Se2, CH3CN/H2O, KMnO4; (iii) TEMPO, CH3CN/H2O, KMnO4. | ||
Diphenyldiselenide has also been used as a trap for acyl radicals16 and oxidation of benzhydrazide with potassium permanganate in the presence of diphenyldiselenide afforded phenyl selenobenzoate 10 in 34% yield (Scheme 3). The seleno ester was identified by the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch in the IR spectrum at 1683 cm−1.
O stretch in the IR spectrum at 1683 cm−1.
Oxidation of 8 with potassium permanganate in aqueous acetonitrile with TEMPO afforded the TEMPO adduct 11 (Scheme 3) in 41% yield. Importantly the analogous oxidation of isoniazid (1) gave the corresponding acyl radical trapped derivative 12 (Fig. 1) in 15% yield. The other products formed in these reactions were the corresponding carboxylic acids, benzoic and isonicotinic acid.
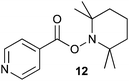 | ||
| Fig. 1 TEMPO adduct from isoniazid. | ||
Oxidations in the presence of nucleophiles
For further investigations, the manganese catalyst [MnIV–MnIV(μ-O)3L2](PF6)2 (L = 1,4,7-trimethyl-1,4,7-triazacyclononane) was utilised, as it was hypothesized to be a mimic for oxidation by the KatG enzyme, due to its catalase activity.17 This catalyst forms a MnVO species as the active oxidant.18 Under a nitrogen atmosphere in acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pyridine (9:1) with periodic acid as a co-oxidant, only a modest yield of 11 was observed (9–20%). However, when the solution was purged with nitrogen throughout the reaction, the yield was dramatically increased to 58% (see Table 1).
:pyridine (9:1) with periodic acid as a co-oxidant, only a modest yield of 11 was observed (9–20%). However, when the solution was purged with nitrogen throughout the reaction, the yield was dramatically increased to 58% (see Table 1).
When oxidations were performed on a range of hydrazides in methanol, instead of aqueous acetonitrile, in the presence of TEMPO, methyl esters (14, Scheme 4, and methyl isonicotinate) were formed in significant quantities. The reactions were then repeated in acetonitrile with only three equivalents of methanol to observe the effect of limiting the amount of nucleophile (Table 2, Fig. 2). The ratios of ester (14) to TEMPO adduct (15) resulting from the oxidation of various hydrazides in methanol were compared by 1H NMR spectroscopy of the crude reaction mixtures (Table 2).19
![Reagents; (i) TEMPO, [Mn2(μ-O)3L2(PF6)2/H5IO6], solvent: MeOH or MeCN with 3 equiv. MeOH.](/image/article/2013/OB/c2ob26419f/c2ob26419f-s4.gif)
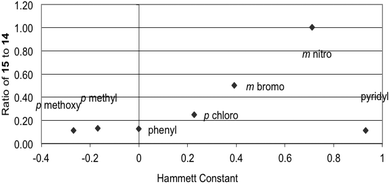 | ||
| Fig. 2 Hammett plot for ratio of products 15:14vs. Hammett constant for oxidation in the presence of 3 equiv. of MeOH. | ||
:15)
| Hydrazide | MeOH as solvent | 3 equiv. of MeOH |
|---|---|---|
| a The ratio of methyl isonicotinate to TEMPO adduct 12 was measured. | ||
| Isoniazid | 6:1a |
1:9a |
| Benzhydrazide | 1:1 |
1:8 |
| p-Methoxybenzhydrazide | 4:1 |
1:9 |
| p-Methylbenzhydrazide | 2:1 |
1:8 |
| p-Chlorobenzhydrazide | 2:1 |
1:4 |
| m-Bromobenzhydrazide | 4:1 |
1:2 |
| m-Nitrobenzhydrazide | 3:1 |
1:1 |
Attempted trapping of acylium ion
To investigate further the possibility of cation formation benzyhydrazide (8) and isoniazid (1) were oxidised in a non-nucleophilic solvent in the presence of anisole as a nucleophile. These reactions failed to yield any of the expected Friedel–Crafts product 16 (Scheme 6).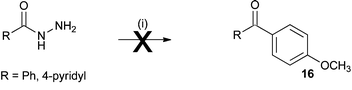 | ||
| Scheme 6 (i) Anisole (10 equiv.), ceric ammonium nitrate (4 equiv.), acetonitrile, N2 bubbled through reaction mixture. | ||
Computational analysis of acyl radical stability
Computational chemistry was utilised to investigate the bond dissociation enthalpy (BDE) of the various aryl radicals as a measure of radical stability. The calculations used the isodesmic reaction mechanism shown in Scheme 7 and were performed using the Gaussian0320 software package. The level of theory chosen after benchmarking was MPWB1K/6-311 + G(2d,2p)//MPWB1K/6-31G(d) (see ESI† for full details). The calculations were performed for 15 different aromatic groups including those in Table 2. The resulting BDEs were close in value, within 10 kJ mol−1. The highest BDE value was 65.2 kJ mol−1 for p-methylbenzaldehyde and the lowest 55.7 kJ mol−1 for the m-nitrobenzaldehyde (full tables of BDE values and benchmarking available in ESI†). | ||
| Scheme 7 Isodesmic scheme used for calculating Bond Dissociation Energies (BDE's) for acyl radical formation. | ||
Discussion
Acyl radical trapping
The formation of 2-benzoyl benzothiazole (9) from the oxidation of benzhydrazide (8) and trapping with the benzothiazolium ion (Scheme 3) supports the hypothesis that acyl radicals are formed from oxidation of hydrazides. The ketone would not be formed by nucleophilic acyl substitution. The hypothesis that the isoniazid-NADH adduct 2 is formed by radical trapping with NAD+3,21 is also supported by the formation of 9 as both the benzothiazolium ion and NAD+ are electron deficient heterocycles. However, no isonicotinoyl benzothiazole was observed in a similar oxidation of isoniazid. The formation of the phenyl selenobenzoate 10 upon oxidation of 8 in the presence of diphenyldiselenide is also consistent with formation of an acyl radical.16,22 The corresponding seleno ester was not observed upon oxidation of isoniazid. Therefore, the common radical trap TEMPO, which reacts with carbon centred radicals at near diffusion control with a rate constant of 1 × 109 M−1 s−1 at a temperature of 298 K,23 was employed.Oxidation of 8 with potassium permanganate and TEMPO afforded the TEMPO adduct 11 (Scheme 3). Importantly the oxidation of isoniazid gave the corresponding acyl radical trapped derivative 12. These products are the consequence of acyl radical generation (7, Scheme 2) and trapping, as supported by Braslau.13 It was noted that the yields appeared to vary depending on the strength of the oxidant (Table 1, 8–84%). Significantly, these results suggest that acyl radicals are formed when either typical single electron oxidants (such as potassium ferricyanide) or potential two electron oxidants were used.24
The combination of the different radical trapping results support the hypothesis that an acyl radical is formed upon oxidation of isoniazid by the catalase peroxidise enzyme KatG.
The corresponding carboxylic acids, benzoic and isonicotinic acid, formed in these trapping reactions were previously believed to result from acylium ion formation (6, Scheme 2) followed by reaction with water. The small yields of 12 (compared to the corresponding benzoyl derivative 11) (Table 1) could indicate that the acyl radical from isoniazid is much less stable than that derived from benzhydrazide and oxidised further to the cation (6). However, it is possible that the carboxylic acids could also be formed by the trapping of an acyl radical with O2.11,25
The manganese catalyst [MnIV–MnIV(μ-O)3L2](PF6)2 (L = 1,4,7-trimethyl-1,4,7-triazacyclononane) was utilised for the majority of our experiments and is a mimic for the oxygen evolving centre of photosystem II.17 Thus it could generate a low concentration of oxygen in the presence of periodic acid.18,26 When the reaction solution was purged with nitrogen throughout the reaction, the yield of the radical product 12 was dramatically increased from 9–20% to 58% (See Table 1). One reason for this increased yield may be that purging the reaction mixture with nitrogen gas removes traces of oxygen. As a consequence this reduces the competing trapping of an acyl radical with O2 to give the carboxylic acid (Scheme 4).25 A such, 12 was now observed as the major product. These experiments demonstrate that, under the conditions described above, the major oxidation pathway most likely involves formation of the acyl radical (7, Scheme 2).
Competing nucleophilic acyl substitution
When oxidations were performed in methanol, instead of aqueous acetonitrile, methyl esters (14 R = H, Scheme 4 and methyl isonicotinate) were significant products of reaction. It has been suggested in the literature that esters are formed from the trapping of an acylium ion (6, Scheme 2) by methanol.9,10 However, our preliminary research indicated that ester formation occurs predominantly by nucleophilic acyl substitution of the diimide 4 (Scheme 2), the precursor to the acyl radical.14The substitution of the diimide has been investigated recently using computational techniques and was found to proceed through a six centred cyclic intermediate involving at least two methanol molecules (Scheme 5). The transition state is substantially stabilised by the presence of additional solvent25 and is therefore a likely reaction pathway to ester formation.
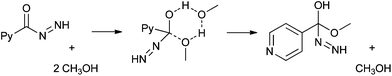 | ||
| Scheme 5 Six-centred cyclic intermediate formed during the nucleophilic acyl substitution of the diimide 4. | ||
While nucleophilic acyl substitution is clearly the dominant pathway in methanol, the amount of available nucleophile was reduced in repeat experiments to hinder this process in an attempt to favour acyl radical formation. As expected, acyl radical formation dominated with the reduced availability of methanol, and the TEMPO adducts 15 and 12 were then the major product. The apparent increase of radical formation when less nucleophile is available for reaction supports the hypothesis that esters are formed from a precursor to the acyl radical and not from further oxidation of the radical to an acylium ion.
The ratio of ester 14 to radical product 15 from the reduced methanol experiments increases with increasing electron withdrawing ability on the aryl ring. When the ratio is plotted against the corresponding Hammett constant the result is consistent with a nucleophilic acyl substitution mechanism for ester formation.28 Electron donation has little effect on the product ratio, with the methyl and methoxy benzhydrazides essentially having the same ratio of products as the parent benzhydrazide.
The Hammett constant for the pyridyl group predicts a 1:1 ratio similar to the nitro derivative but when methanol is limited, isoniazid (1) is less susceptible to nucleophilic acyl substitution than predicted. However, when isoniazid (1) is oxidised with methanol as the solvent, the methyl ester is the major product (Table 2). The contrasting experimental results are supported by calculations that predict that bulk solvent is required for the stabilisation of the transition state during nucleophilic acyl substitution of the diimide more for isoniazid than for other aryl hydrazides.27
Evidence against the formation of the acyl cation
Oxidation of benzyhydrazide or isoniazid in the presence of anisole as a nucleophile failed to yield any Friedel–Crafts product 16 that would be expected if an acylium ion were formed (Scheme 6). The acyl radical would have been formed under these conditions, as oxidation with ceric ammonium nitrate (Table 1) showed the presence of the acyl radical by the formation of the TEMPO adduct 11. The lack of addition product on the anisole ring is further supporting evidence that oxidation of the acyl radical to a cation is unlikely under these conditions.Cyclic voltammetry was utilised to investigate the oxidation of the radical to the cation, however the results were inconclusive.
Computational investigations into the stability of acyl radicals
To investigate the possibility that ester formation is related to radical stability, we turned to computational chemistry. Providing that the radicals are similar in nature, bond dissociation enthalpies (BDEs) can be used as a measure of radical stability, as they indicate the amount of energy required to homolyse the bond leading to the radicals in question.29 The calculated BDEs for 15 different aromatic groups were close in value, within 10 kJ mol−1, indicating that the stability of the radical is not affected by the substitution on the aromatic ring. As metioned in the results, the highest BDE value was 65.2 kJ mol−1 for p-methylbenzaldehyde and the lowest 55.7 kJ mol−1 for the m-nitrobenzaldehyde (see ESI†). The formation of the ester is favoured by electron withdrawing group substitution on the aromatic ring (vide supra). Therefore, the similarity of radical stabilities supports the hypothesis that interception of a precursor to the radical is a more likely mechanism for ester formation than generation of an acylium ion.Implications in understanding resistance
The determination of the mechanism of oxidation of isoniazid could assist in understanding why a single base pair mutation in the KatG enzyme in Mycobacterium tuberculosis confers resistance to isoniazid.8 The secondary alcohol moiety in the threonine side chain may act as a hydrogen donor to the acyl radical hindering the reaction with NAD+. In addition, resistance may be caused by the mutation favouring the nucleophilic acyl substitution pathway in preference to radical formation. This competition is particularly relevant for isoniazid activation in an aqueous medium, where nucleophilic acyl substitution could inhibit the formation of the acyl radical.Conclusions
The oxidation of hydrazides under different conditions has led to a greater understanding of the intermediates involved and their reactivity. We have demonstrated evidence for the formation of the acyl radical from the oxidation of hydrazides, in particular isoniazid, and that the diimide intermediate is susceptible to nucleophilic acyl substitution in polar aprotic media such as methanol. Isoniazid is particularly prone to this substitution pathway. These results are consistent with experimental details in the existing literature,9–12 however we find no evidence for the further oxidation of the acyl radical to the acylium ion. On the balance of evidence provided by this work, we suggest that nucleophilic acyl substitution is likely to be the major mechanism operating during the facile formation of esters by the oxidation of hydrazides in the presence of alcohols in this and previous studies.Experimental
See ESI† for general experimental details.Oxidation of benzhydrazide in the presence of protonated benzothiazole
A 3:2 ratio of glacial acetic acid and 5 M sulphuric acid (2.5 mL) at 0 °C was degassed by bubbling N2(g) through the solution for 15 min. To this was added 0.7 mmol benzothiazole (0.7 mmol) and benzhydrazide (4.5 mmol). Potassium permanganate (4.5 mmol) was added slowly and the solution stirred for 15 min. The reaction was quenched by the addition of water and sodium hydrogen carbonate and treated with potassium hydrogen sulphate, and sodium sulfite to reduce unreacted permanganate and manganese dioxide. The resulting solution was neutralized by the careful addition of sodium hydrogen carbonate and extracted with dichloromethane (3 × 15 mL). The combined extracts were dried and evaporated under reduced pressure to leave a yellow solid which was purified by column chromatography (silica gel; eluent 15% ethyl acetate in hexanes) to give a mixture of 2-benzoylbenzothiazole (9) and benzoic anhydride. This mixture was dissolved in ethanol and stirred overnight with 2 M sodium hydroxide. The resulting solution was extracted with dichloromethane (3 × 15 mL) and the combined extracts dried and evaporated under reduced pressure to give 9 (16% yield) as a white solid. The spectral data were consistent with that reported.30
Oxidation of benzhydrazide in the presence of diphenyldiselenide
A mixture of acetonitrile and water (4:1 ratio, 5 mL) was degassed by bubbling N2(g) through it for 10 min. Benzhydrazide (2.1 mmol) and diphenyl diselenide (0.3 mmol) were added followed by the slow addition of solid potassium permanganate (2.1 mmol). The resulting mixture was stirred for 15 min and the reaction quenched by the addition of water, acidification with saturated potassium hydrogen sulfate solution and addition of solid sodium sulfite until a clear solution was obtained. The resulting solution was neutralised by the careful addition of sodium hydrogen carbonate, extracted with dichloromethane (3 × 15 mL) and the combined extracts dried and evaporated under reduced pressure to yield a yellow solid which was purified by column chromatography (silica gel; eluent 10% ethyl acetate in hexanes) to give a mixture of phenylseleno ester 10 and diphenyl diselenide in a 4:1 ratio by 1H NMR. This data was extrapolated to give a 34% yield of 10.
Spectral data were consistent with that previously reported.31
General procedure for the oxidation of hydrazides in the presence of TEMPO
TEMPO (62.5 mg, 0.4 mmol) was added to a solution of isoniazid (1) (164.5 mg, 1.2 mmol) in degassed (with N2(g)) aqueous acetonitrile (4:1 MeCN:H2O, 5 mL) at 0 °C. Potassium permanganate (190 mg, 1.2 mmol) was added in small portions and the solution stirred for 5 min before being warmed to room temperature. The reaction was quenched by 0.5 M potassium hydrogen sulphate (1 mL) and saturated sodium sulfite solution (1 mL). The mixture was made alkaline with sodium carbonate and extracted with dichloromethane (3 × 20 mL). The organic extract was dried and evaporated to give a solid which was passed through a short plug of silica gel using 20% ethyl acetate in hexanes to give 2,2,6,6-tetramethylpiperidino isonicotinate (12) as a clear solid in 41% yield. mp. 60–61 °C. νMAX/cm−1 1755 (CO); δH(acetone-D6) 1.10 (6H, s), 1.27 (6H, s), 1.42–1.77 (6H, m), 7.90 (2H, m), 8.80 (2H, m); δC(acetone-D6) 17.0, 21.0, 32.1, 39.2, 60.9, 123.7, 138.1, 149.7, 164.7; m/z (EI) 262.1682 (M+ C15H22N2O2 requires 262.1681) 262 (M+, 1%), 247 (84), 156 (32), 139 (13), 25 (65), 123 (100), 106 (50), 98 (27), 83 (76), 77 (37), 69 (60), 55 (76), 51 (31), 41 (50).
General procedure for competitive trapping – methanol as solvent
To degassed (N2(g)) methanol (5 mL) was added TEMPO (62.5 mg, 0.4 mmol), isoniazid (54.9 mg, 0.4 mmol) and Mn catalyst ([MnIV–MnIV(μ-O)3L2](PF6)2 (L = 1,4,7-trimethyl-1,4,7-triazacyclononane)) (5 mg, 6 × 10−3 mmol, 1.6 mol%). Periodic acid (180 mg, 0.8 mmol) was added slowly and the reaction mixture stirred for a further 15 min. The solvent was removed and the reaction worked up as previously described. The resulting ratio of methyl ester 14 to TEMPO ester 15 was determined by 1H NMR integration.General procedure for competitive trapping – acetonitrile as solvent
To a solution of Mn catalyst ([MnIV–MnIV(μ-O)3L2](PF6)2 (L = 1,4,7-trimethyl-1,4,7-triazacyclononane)) (5 mg, 6 × 10−3 mmol, 1.6 mol%) in degassed (N2(g)) acetonitrile (4.5 mL) and pyridine (0.5 mL), was added TEMPO (62.5 mg, 0.4 mmol), methanol (50 μL, 1.2 mmol) and isoniazid (54.9 mg, 0.4 mmol). To this was added slowly periodic acid (180 mg, 0.8 mmol, in 1 mL 25% pyridine in acetonitrile) and the reaction mixture stirred for a further 15 min. The solvent was removed and the reaction worked up as previously described. The resulting ratio of methyl ester to TEMPO ester was found by 1H NMR integration.Procedure for attempted acyl cation trapping
A solution of anisole (500 mg, 4.6 mmol) in acetonitrile (4.5 mL) was degassed by bubbling N2(g) through the solution for 10 min. Light was excluded from the reaction and benzhydrazide (0.46 mmol) and ceric ammonium nitrate (1.62 mmol) were added and the mixture was stirred for 3.5 h. Water was added and the solution made alkaline by addition of solid sodium hydrogen carbonate followed by extraction with dichloromethane (3 × 20 mL). The extracts were dried and solvent removed under reduced pressure to give a red residue. GCMS analysis showed no ketone formation, only nitroanisole.Acknowledgements
Support of the Australian Research Council through the Centres of Excellence Program is gratefully acknowledged. JAS thanks the University of Tasmania for support through the IRGS program. RIJA thanks the University and the Australian Government for an APA and the ARC Centre of Excellence for Free Radical Chemistry and Biotechnology for a scholarship top-up and research support. We thank NCI and TPAC for computer time.Notes and references
- A. M. Rouhi, Chem. Eng. News, 1999, 77, 52–69 Search PubMed
.
- B. R. Bloom and C. J. Murray, Science, 1992, 257, 1055–1064 CAS
- A. Quemard, J. C. Sacchettini, A. Dessen, C. Vilcheze, R. Bittman, W. R. Jacobs Jr. and J. S. Blanchard, Biochemistry, 1995, 34, 8235–8241 CrossRef CAS
- A. Banerjee, E. Dubnau, A. Quemard, V. Balasubramanian, K. S. Um, T. Wilson, D. Collins, G. de Lisle and W. R. Jacobs Jr., Science, 1994, 263, 227–230 CrossRef CAS
- D. A. Rozwarski, G. A. Grant, D. H. R. Barton, W. R. Jacobs Jr. and J. C. Sacchettini, Science, 1998, 279, 98–102 CrossRef CAS
- N. Wengenack, H. M. Hoard and F. Rusnak, J. Am. Chem. Soc., 1999, 121, 9748–9749 CrossRef CAS
- N. Wengenack and F. Rusnak, Biochemistry, 2001, 40, 8990–8996 CrossRef CAS
- B. Saint-Joanis, H. Souchon, M. Wilming, K. Johnsson, P. M. Alzari and S. T. Cole, Biochem. J., 1999, 338, 753–760 CrossRef CAS
- B. Stefane, M. Kocevar and S. Polanc, Tetrahedron Lett., 1999, 40, 4429–4432 CrossRef CAS
- J. Tsuji, T. Nagashima, N. T. Qui and H. Takayanagi, Tetrahedron, 1980, 36, 1311–1315 CrossRef CAS
- M. Nguyen, C. Claparols, J. Bernadou and B. Meunier, C. R. Chim., 2002, 5, 325–330 Search PubMed
- R. S. Yalgurde and G. S. Gokavi, Ind. Eng. Chem. Res., 2012, 51, 5135–5140 Search PubMed
- R. Braslau, M. O. Anderson, F. Rivera, A. Jimenez, T. Haddad and J. R. Axon, Tetrahedron, 2002, 58, 5513–5523 CrossRef CAS
- R. I. J. Amos, B. S. Gourlay, C. H. Schiesser, J. A. Smith and B. F. Yates, Chem. Commun., 2008, 1695–1697 RSC
- T. Caronna, R. Galli, V. Malatesta and F. Minisci, J. Chem. Soc. C, 1971, 1747–1750 RSC
- C. Chen, D. Crich and A. Papadatos, J. Am. Chem. Soc., 1992, 114, 8313–8314 CrossRef CAS
- K. Wieghardt, U. Bossek, B. Nuber, J. Weiss, J. Bonvoisin, M. Corbella, S. E. Vitols and J. J. Girerd, J. Am. Chem. Soc., 1988, 110, 7398–7411 CrossRef CAS
- D. H. R. Barton, S. Y. Choi, B. Hu and J. A. Smith, Tetrahedron, 1998, 54, 3367–3337 CrossRef CAS
- Authentic samples of the TEMPO adducts were prepared by the general method for benzhydrazide and isoniazid.
-
M. J. Frisch, et al., Gaussian 03, Revision D.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed
- R. I. J. Amos, J. A. Smith, B. F. Yates and C. H. Schiesser, Tetrahedron, 2009, 65, 7643–7657 Search PubMed
- D. H. Brown, R. J. Cross and D. Millington, J. Chem., Dalton Trans.: Inorg. Chem. (1972–1999), 1977, 159–161 Search PubMed
- L. J. Johnston, J. C. Scaiano and K. U. Ingold, J. Am. Chem. Soc., 1984, 106, 4877–4881 CrossRef CAS
- While it is possible that TEMPO is oxidized in these reactions allowing substitution on the diimide intermediate, the case for the acyl radical is strong. It is supported by the evidence of the two other systems – protonated benzothiazole and diphenyl diselenide – and also by the evidence for radical trapping by TEMPO upon oxidation of hydrazides shown by Braslau et al.13.
- D. Crich and X. Hao, J. Org. Chem., 1997, 62, 5982–5988 CrossRef CAS
- Although it was reported that the manganese catalyst and periodic acid gave no detectable amount of oxygen by a change of the headspace volume, other activating agents such as oxone® do generate detectable levels.28.
- R. I. J. Amos, C. H. Schiesser, J. A. Smith and B. F. Yates, J. Org. Chem., 2009, 74, 5707–5710 Search PubMed
- I.-H. Um, J.-S. Min, J.-A. Ahn and H.-J. Hahn, J. Org. Chem., 2000, 65, 5659–5663 CrossRef CAS
- Y. Feng, H. Huang, L. Liu and Q.-X. Guo, Phys. Chem. Chem. Phys., 2003, 5, 685–690 RSC
- C. Boga, R. Stengel, R. Abdayem, E. Del Vecchio, L. Forlani and P. E. Todesco, J. Org. Chem., 2004, 69, 8903–8909 Search PubMed
- D. L. Boger and R. J. Mathvink, J. Org. Chem., 1992, 57, 1429–1443 CrossRef CAS
- J. Guin, S. De Sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727–8730 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ob26419f |
| This journal is © The Royal Society of Chemistry 2013 |