Photochemically induced radical alkynylation of C(sp3)–H bonds†
Tamaki
Hoshikawa
,
Shin
Kamijo‡
and
Masayuki
Inoue
*
Graduate School of Pharmaceutical Sciences, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: inoue@mol.f.u-tokyo.ac.jp; Fax: (+81) 3-5841-0568
First published on 16th October 2012
Abstract
A general strategy for photochemical alkynylation of unreactive C(sp3)–H bonds has been developed. After C–H abstraction by the photo-excited benzophenone, a two-carbon unit was efficiently transferred to the generated radical from 1-tosyl-2-(trimethylsilyl)acetylene to afford the alkynylated product. The present reaction enables construction of various tri- and tetra-substituted carbons from heteroatom-substituted methylenes, methines and alkanes in a highly chemoselective fashion, and would serve as a new synthetic strategy for rapid construction of complex structures.
Introduction
Carbon–carbon (C–C) bond formation plays a central role in chemical syntheses, and innovations in these types of reactions profoundly improve the overall synthetic efficiency. Among the various C–C forming strategies, the direct transformation of C(sp3)–H bonds into C(sp3)–C bonds has attracted much interest in recent years, since it eliminates prior functional group manipulations for substrate activation, resulting in simpler and shorter synthetic schemes.1,2Typically, such a direct transformation is a challenge, because the reagents are required to cleave the requisite strong C(sp3)–H bond selectively without affecting other C–H bonds in the organic molecules. Recently, we employed photochemically-generated highly reactive oxyl radicals to induce C(sp3)–H functionalizations,3 and developed chemoselective acylation,4a carbamoylation,4b and cyanation strategies.4c These studies prompted us to apply the photochemical reaction system for attachment of an alkyne, a more versatile building block.
The carbon–carbon triple bond is one of the most important functional groups in organic chemistry due to its unique physicochemical properties, as well as the wide range of available methods for its functionalization.5 Therefore, direct transfer strategies of acetylene to organic molecules are highly desirable in the syntheses of functional materials, pharmaceuticals and natural products. Here we report direct alkynylation of C(sp3)–H bonds under photo-irradiation conditions (Scheme 1).6–8 The present reaction enables the construction of various tri- and tetra-substituted carbons from heteroatom-substituted methylenes, methines and alkanes, and provides a new synthetic strategy for rapid construction of architecturally complex molecules.
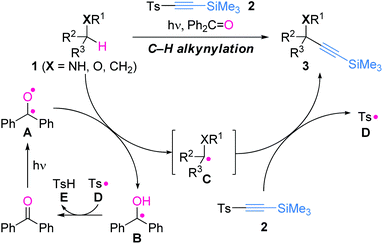 | ||
| Scheme 1 Direct alkynylation of C(sp3)–H bonds and proposed reaction mechanism. | ||
Results and discussion
Our plan for the direct C(sp3)–H alkynylation is illustrated in Scheme 1. We employed benzophenone (Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) as an oxyl radical precursor and 1-tosyl-2-(trimethylsilyl)acetylene 2 as an alkynylating agent (Scheme 1).9,10 The photochemically formed A is an electrophilic oxyl radical, and thus would chemoselectively abstract the hydrogen of an electron-rich C–H bond of 1 to furnish carbon radical C.11 Upon reaction with the electron-deficient alkyne 2, C is expected to preferentially add at the α-position than the β-position of the sulfonyl group, due to the unfavorable steric interaction with the bulky trimethylsilyl group. Subsequent release of the tosyl radical D from the produced vinyl radical intermediate would result in formation of the alkynylated product 3, while D abstracts a hydrogen from the ketyl radical B to regenerate Ph2CO. Importantly, high-yielding transformation from 1 to 3 is realized only when all the radical species properly follows the series of reactions depicted in Scheme 1.
O) as an oxyl radical precursor and 1-tosyl-2-(trimethylsilyl)acetylene 2 as an alkynylating agent (Scheme 1).9,10 The photochemically formed A is an electrophilic oxyl radical, and thus would chemoselectively abstract the hydrogen of an electron-rich C–H bond of 1 to furnish carbon radical C.11 Upon reaction with the electron-deficient alkyne 2, C is expected to preferentially add at the α-position than the β-position of the sulfonyl group, due to the unfavorable steric interaction with the bulky trimethylsilyl group. Subsequent release of the tosyl radical D from the produced vinyl radical intermediate would result in formation of the alkynylated product 3, while D abstracts a hydrogen from the ketyl radical B to regenerate Ph2CO. Importantly, high-yielding transformation from 1 to 3 is realized only when all the radical species properly follows the series of reactions depicted in Scheme 1.
We first established the optimum photochemical conditions for an efficient C–H alkynylation (Table 1). Pyrrolidinone 1a was selected as a substrate based on the expectation that the nitrogen functionality would secure the selective functionalization of its electron-rich nitrogen-substituted methylene. In fact, irradiation of 1a (3 equiv.), 2 (1 equiv.) and Ph2CO (1 equiv.) in MeCN with a medium-pressure mercury lamp successfully provided the adduct 3a in 53% yield (entry 1). While the reaction proceeded in benzene and t-BuOH in similar yields (entries 2 and 3), conversion in t-BuOH was apparently faster in comparison to other solvents. The modest yields in entries 1–3 appeared to originate from the undesired reaction involving the reactive propargylic tertiary C–H bond of 3a, since disappearance of 3a was observed upon irradiation of 3a just in the presence of Ph2CO.12 Consequently, a significant improvement in the yield of 3a was attained by applying 8 equiv. of 1a in t-BuOH (83%, entry 4).13
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1, equiv. | 2, equiv. | Solvent | T (h) | Yieldb (%) | Recoveryb (%) |
|
a Reaction conditions: 1a, 2, Ph2CO (1 equiv.), solvent (0.04 M), rt, photo-irradiation using a Riko 100 W medium pressure mercury lamp.
b Yield was determined by NMR analysis.
c Isolated yield.
|
||||||
| 1 | 3 | 1 | MeCN | 4 | 53 | 31 |
| 2 | 3 | 1 | Benzene | 4 | 52 | 9 |
| 3 | 3 | 1 | t-BuOH | 1 | 57 | 13 |
| 4 | 8 | 1 | t-BuOH | 1 | 83c | 0 |

The established conditions were next applied to a variety of electron-rich secondary C–H bonds adjacent to nitrogen-based functional groups (Table 2). Similar to alkynylation of the five-membered lactam 1a (entry 1), both the six- and seven-membered lactams 1b and 1c were chemoselectively functionalized to afford the corresponding adducts 3b and 3c, respectively, in high yields (entries 2 and 3). The reactions of the protected piperidines, bearing Boc 1d, Ac 1e, and Troc 1f, all efficiently provided the corresponding products 3d–3f (entries 4–6). The Boc-substituted alkylamine 1g and the Ph-substituted diethylamine 1h were also converted to the non-cyclic products 3g and 3h, respectively (entries 7 and 8). In the case of N-Boc morpholine 1i, C–H functionalization chemoselectively occurred at the methylene proximal to the N-Boc group to generate 3i (entry 9), clearly indicating that the C–H bond attached to the nitrogen atom is more reactive than that attached to the oxygen atom. When the substrates with preexisting stereocenters were used, high diastereoselectivity was observed (entries 10 and 11). C–H alkynylations of the cyclic carbamate 1j and the proline derivative 1k stereoselectively produced the 1,2-trans-disubstituted 3j and the 1,3-trans-disubstituted 3k, respectively, in a completely chemo- and stereoselective fashion.
| Entry | Starting material | t (h) | Product | Yieldb (%) |
|---|---|---|---|---|
a Reaction conditions: 1/2/Ph2CO = 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, t-BuOH (0.04 M), rt, photo-irradiation.
b Isolated yield.
c
1l (16 equiv.) was used.
d Mixture of diastereomers (dr = 3:4). :1:1, t-BuOH (0.04 M), rt, photo-irradiation.
b Isolated yield.
c
1l (16 equiv.) was used.
d Mixture of diastereomers (dr = 3:4).
|
||||

|

|
|||
| 1 | 1a: n = 1 | 1 | 3a | 83 |
| 2 | 1b: n = 2 | 1.5 | 3b | 81 |
| 3 | 1c: n = 3 | 2 | 3c | 73 |

|

|
|||
| 4 | 1d: R = Boc | 1 | 3d | 82 |
| 5 | 1e: R = Ac | 1.5 | 3e | 89 |
| 6 | 1f: R = Troc | 1.5 | 3f | 87 |
| 7 |

|
2.5 |

|
62 |
| 8 |

|
1.5 |

|
78 |
| 9 |

|
1 |

|
92 |
| 10 |

|
3 |

|
77 |
| 11 |

|
2 |

|
94 |
| 12c |

|
2 |

|
75 |
| 13 |

|
1 |

|
82 |
| 14 |

|
1 |

|
74d |
| 15 |

|
2 |

|
73 |
The present protocol realized high-yielding functionalizations of oxygen-substituted methylenes (Table 2, entries 12–15). Dioxane 1l (entry 12) and 15-crown-5 ether 1m (entry 13) underwent mono-alkynylation to produce the adducts 3l and 3m, respectively, while (−)-ambroxide 1n was chemoselectively alkynylated at the ethereal C–H bond among other potentially reactive C–H bonds to provide 3n (entry 14). Moreover, two carbon-elongation of the non-protected primary alcohol 1o was possible, leading to the propargylic alcohol 3o (entry 15). The order of the favorable reactive sites is clarified from Table 2, and determined to be nitrogen- > oxygen- > non-heteroatom-substituted carbons.
Despite their more sterically hindered nature, tertiary C–H bonds adjacent to nitrogen- and oxygen-based functional groups were efficiently transformed (Table 3). Alkynylation of 5-methylpyrrolidin-2-one 1p under the optimized conditions in Table 1 (conditions A: 1p/2/Ph2CO = 8:1:1) resulted in formation of the alkyne-attached tetrasubstituted carbon of 3p in quantitative yield within 1 h (entry 1). Because the product 3p does not have a reactive propargylic C–H bond, increasing the irradiation time did not cause over-reactions of 3p, and thus reducing the reagent amount was possible without decreasing the yield. Namely, 3p was obtained in 75% yield even upon irradiating a limited amount of 1p (1 equiv.) with 1.5 equiv. of 2 and 0.5 equiv. of Ph2CO (conditions B: 1p/2/Ph2CO = 1:1.5:0.5) for 10 h (entry 2). Significantly, conditions B were applicable for construction of nitrogen- and oxygen-substituted tetrasubstituted carbons in high yields (entries 3–6). The bicyclic lactam 1q14 was chemoselectively alkynylated at the methine position in the presence of the less hindered oxygen-substituted methylene, providing 3q in 81% yield (entry 3).15,16 Mono-functionalization of both the cis- and trans-diaminocyclohexane derivatives 1r and 1s17 took place stereoselectively, providing the same cis-fused bicyclic compound 3r as a sole product (entries 4 and 5). The configurational change of the bicyclic system between 1s and 3r supported the intermediacy of an α-amino carbon radical.4,18 Alkynylation of the ethereal tertiary C–H bond in the cyclohexanediol derivative 1t stereoselectively furnished the cis-fused ring system 3t in 70% yield (entry 6).15,19 Since the secondary alcohol 1u exhibited lower reactivity, conditions A were again adopted to generate the propargylic alcohol 3u in 57% yield (entry 7).
| Entry | Starting material | Cond., t (h) | Product | Yieldb (%) |
|---|---|---|---|---|
|
a Conditions A: 1/2/Ph2CO = 8:1:1, t-BuOH (0.04 M), rt, photo-irradiation; conditions B: 1–2–Ph2CO = 1:1.5:0.5, t-BuOH (0.04 M), rt, photo-irradiation.
b Isolated yield.
c Reaction was conducted in the presence of 2,6-di(t-butyl)pyridine (1 equiv.).
d Recovery of 1 was observed in 10% yield.
e Ph2CO (1 equiv.) was used.
|
||||
| 1 |

|
A, 1 |

|
99 |
| 2 | B, 10 | 75 | ||
| 3c |

|
B, 7 |

|
81 |

|

|
|||
| 4 | 1r: cis | B, 10 | 3r | 80 |
| 5 | 1s: trans | B, 20 | 3r | 52d |
| 6c,d |

|
B, 12 |

|
70e |
| 7 |

|
A, 4 |

|
57 |
The protocol was then utilized for the direct alkynylation of C–H bonds of alkanes, which are known to be less reactive than those of heteroatom-substituted compounds (Table 4). Cyclooctane 1v was converted to the alkyne-branched carbocycle 3v in 70% yield (entry 1). When the alcohol was capped with an electron-withdrawing Ac group, alkynylation did not occur at the α-oxy carbon, but the most electron-rich and hindered tertiary C–H bond was selectively functionalized in the presence of less electron-rich primary and secondary C–H bonds, giving rise to 3w in 54% yield (entry 2). In the case of the adamantane structures 1x and 1y, the reaction also proceeded exclusively at the methine positions to install the quaternary carbons. Consequently, compound 3x was obtained in 86% yield (entry 3), and 3y, which is a two carbon homolog of the antiviral drug, amantadine, was isolated in 73% yield after removal of the Boc group (entry 4). In particular, constructions of the hindered tetrasubstituted centers from a variety of structures (Tables 3 and 4, entries 2–4) demonstrated the power and generality of the present methodology.








The synthetic utility of the introduced C–C triple bond was exemplified by the two functional group transformations from the proline derivative 3k (Scheme 2). Treatment of 3k with RuO2 and Oxone20 transformed the alkyne moiety to the carboxylic acid via oxidative C–C bond cleavage, giving rise to the differentially protected C2-symmetric pyrrolidine 4 in 88% yield.21 On the other hand, the Sonogashira-type reaction22 using the combination of Pd0 and AgI catalysts directly coupled TMS-protected acetylene 3k and phenyl iodide to provide phenylacetylene 5 in 89% yield.
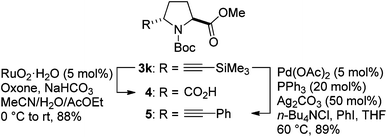 | ||
| Scheme 2 Transformations of the TMS-protected acetylene. | ||
Conclusions
In conclusion, we developed a direct photochemical alkynylation of unreactive C(sp3)–H bonds using Ph2CO as the precursor reagent of the C–H bond abstraction and 1-tosyl-2-(trimethylsilyl)acetylene as the alkynylating agent. The present transformation proceeds at ambient temperature with wide applicability of starting substrates, including amine derivatives, ethers, alcohols, and alkanes, and enables one-step construction of tetrasubstituted carbon centers. The sequence of the preferred reaction sites was established to be nitrogen- > oxygen-substituted carbons > non-substituted methines > non-substituted methylenes. The simple procedure, mild conditions, and predictable chemoselectivity make this protocol a unique and powerful tool for carbon–carbon bond formation. Since transformations of the introduced C–C triple bonds allow further attachments of a variety of carbon units at the alkyne terminus as well as the conversion to carboxylic acids, the newly developed C–H alkynylation strategy should be highly suitable for streamlined construction of pharmaceuticals and natural products.
Acknowledgements
This research was financially supported by the Funding Program for Next Generation World-Leading Researchers (JSPS) to M.I.Notes and references
- For recent reviews on direct C–H transformations, see: (a) Handbook of C–H Transformations, ed. G. Dyker, Wiley-VCH, Weinheim, 2005, vol. 1 and 2 Search PubMed; (b) Handbook of Reagents for Organic Synthesis: Reagents for Direct Functionalization of C–H Bonds, ed. L. A. Paquette and P. L. Fuchs, Wiley, Chichester, 2007 Search PubMed; (c) Chem. Soc. Rev., 2011, 40(4) Search PubMed, special issue on C–H Functionalizations in Organic Synthesis.
- For recent reviews on direct C(sp3)–H transformation to form C–C bonds, see: (a) Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393 CrossRef CAS; (b) A. A. Fokin and P. R. Schreiner, Adv. Synth. Catal., 2003, 345, 1035 CrossRef CAS; (c) R. Knorr, Chem. Rev., 2004, 104, 3795 CrossRef CAS; (d) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS; (e) F. Kakiuchi and T. Kochi, Synthesis, 2008, 3013 CrossRef CAS; (f) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (g) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS; (h) T. Akindele, K. Yamada and K. Tomioka, Acc. Chem. Res., 2009, 42, 345 CrossRef CAS; (i) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS; (j) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761 RSC; (k) M. Klussmann and D. Sureshkumar, Synthesis, 2011, 353 CrossRef CAS; (l) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS.
- For recent reviews on photochemical reactions, see: (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS; (b) N. Hoffmann, Chem. Rev., 2008, 108, 1052 CrossRef CAS.
- (a) S. Kamijo, T. Hoshikawa and M. Inoue, Tetrahedron Lett., 2010, 51, 872 CrossRef CAS; (b) S. Kamijo, T. Hoshikawa and M. Inoue, Tetrahedron Lett., 2011, 52, 2885 CrossRef CAS; (c) S. Kamijo, T. Hoshikawa and M. Inoue, Org. Lett., 2011, 13, 5928 CrossRef CAS.
- Acetylene Chemistry, ed. F. Diederich, P. J. Stang and R. R. Tykwinski, Wiley-VCH, Weinheim, 2005 Search PubMed.
- For pioneering works of the photo-induced direct C(sp3)–H alkynylation, see: (a) J. Gong and P. L. Fuchs, J. Am. Chem. Soc., 1996, 118, 4486 CrossRef CAS; (b) J. S. Xiang and P. L. Fuchs, Tetrahedron Lett., 1996, 37, 5269 CrossRef CAS; (c) J. Gong and P. L. Fuchs, Tetrahedron Lett., 1997, 38, 787 CrossRef CAS; (d) J. Xiang, W. Jiang and P. L. Fuchs, Tetrahedron Lett., 1997, 38, 6635 CrossRef CAS.
- For representative examples of the transition metal-catalyzed direct C(sp3)–H alkynylation, see: (a) Z. Li and C.-J. Ji, J. Am. Chem. Soc., 2004, 126, 11810 CrossRef CAS; (b) Z. Li, D. S. Bohle and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8928 CrossRef CAS; (c) Y. Ano, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2011, 133, 12984 CrossRef CAS; (d) M. Niu, Z. Yin, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2008, 73, 3961 CrossRef CAS; (e) X. Xu and X. Li, Org. Lett., 2009, 11, 1027 CrossRef CAS; (f) D. B. Freeman, L. Furst, A. G. Condie and C. R. Stephenson, Org. Lett., 2012, 14, 94 CrossRef CAS; (g) M. Rueping, R. M. Koenigs, K. Poscharny, D. C. Fabry, D. Leonori and C. Vila, Chem.–Eur. J., 2012, 18, 5170 CrossRef CAS.
- For recent examples of direct C(sp3)–H alkenylation using alkynes, see: (a) Y. Zhang and C.-J. Li, Tetrahedron Lett., 2004, 45, 7581 CrossRef CAS; (b) R. A. Doohan, J. J. Hannan and N. W. A. Geraghty, Org. Biomol. Chem., 2006, 4, 942 RSC.
- For the chemistry of acetylenic sulfones, see: T. G. Back, Tetrahedron, 2001, 57, 5263 CrossRef CAS.
- For representative examples of radical reactions using tosyl acetylene as an alkynylating agent, see: (a) A.-P. Schaffner, V. Darmency and P. Renaud, Angew. Chem., Int. Ed., 2006, 45, 5847 CrossRef CAS; (b) V. Liautard, F. Robert and Y. Landais, Org. Lett., 2011, 13, 2658 CrossRef CAS.
- Generally, the more electron rich C–H bonds are more reactive toward C(sp3)–H bond functionalizations when using electrophilic reactants. See, for examples: (a) R. Mello, M. Fiorentino, C. Fusco and R. Curci, J. Am. Chem. Soc., 1989, 111, 6749 CrossRef CAS; (b) M. S. Chen and M. C. White, Science, 2007, 318, 783 CrossRef CAS; (c) K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042 CrossRef CAS; (d) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS . See also ref. 4c.
- Compound 3a was recovered only in 24% yield after photo-irradiation of only 3a for 2 h at rt in the presence of Ph2CO (1 equiv.) in t-BuOH (0.04 M).
- No product was obtained in the absence of Ph2CO. Use of other oxyl radical precursors instead of Ph2CO (e.g. 4,4′-dimethoxybenzophenone, acetophenone and xanthone) gave alkynylated product 3a in lower yields (<25%).
- Compound 1q was synthesized from L-pyroglutamic acid. S. G. Davies, D. J. Dixon, G. J.-M. Doisneau, J. C. Prodger and H. J. Sanganee, Tetrahedron: Asymmetry, 2002, 13, 647 CrossRef CAS.
- Because of the acid sensitive nature of the acetal moiety, 2,6-di(t-butyl)pyridine was added for neutralization of the generated sulfinic acid during the reaction.
- The stereochemical information of the methine carbon center in 1q appeared to be lost in 3q ([α]25D −0.2).
- Á. L. Fuentes de Arriba, D. G. Seisdedos, L. Simón, V. Alcázar, C. Raposo and J. R. Morán, J. Org. Chem., 2010, 75, 8303 CrossRef.
- The steric hindrance around the reaction sites as well as the stereoelectronic effect of the abstracting C–H bonds is responsible for the difference in yields between 1r and 1s.
- Alkynylation of 1t under conditions A gave the adduct 3t within 1 h in 88% yield.
- D. Yang, F. Chen, Z.-M. Dong and D.-W. Zhang, J. Org. Chem., 2004, 69, 2221 CrossRef CAS.
- The stereochemistry of the ester-bearing carbon was confirmed to be retained. For the structural determination, see ESI.†.
- (a) Y. Hatanaka, K. Matsui and T. Hiyama, Tetrahedron Lett., 1989, 30, 2403 CrossRef CAS; (b) Y. Nishihara, K. Ikegashira, A. Mori and T. Hiyama, Chem. Lett., 1997, 1233 CrossRef CAS; (c) Y. Koseki, K. Omino, S. Anzai and T. Nagasaka, Tetrahedron Lett., 2000, 41, 2377 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ob26785c |
| ‡ Current address: Graduate School of Science and Engineering, Yamaguchi University, Yoshida, Yamaguchi 753-8511, Japan. |
| This journal is © The Royal Society of Chemistry 2013 |