Expanding DNAzyme functionality through enzyme cascades with applications in single nucleotide repair and tunable DNA-directed assembly of nanomaterials†
Yu
Xiang
a,
Zidong
Wang
b,
Hang
Xing
a and
Yi
Lu
*ab
aDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
bDepartment of Material Science, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: yi-lu@illinois.edu; Fax: +1 217-244-3186; Tel: +1 217-333-2619
First published on 21st September 2012
Abstract
Many biological functions require two or more enzymes working together in cascades. While many examples of protein and RNA enzyme cascades are known, few enzyme cascades containing solely DNAzymes have been reported. Herein we demonstrate the combination of an 8–17 DNAzyme with RNA cleavage activity and an E47 DNAzyme with DNA ligation activity to achieve a new function of single ribonucleotide repair in DNA while maintaining the integrity of the original DNA sequence, which is difficult for a single DNAzyme to achieve. In addition, this method is applied to modify the sequences of DNA strands immobilized on the surface of nanoparticles to control the DNA-directed assembly selectively and sequentially. Such an approach can be applied to other DNAzymes with different activities to expand the functions of DNAzymes and the scope of their applications.
Introduction
The discovery of deoxyribozymes (DNAzymes) with enzymatic activity in the 1990s1,2 has demonstrated that DNA molecules are not simply inert biopolymers for genetic storage; they can be active catalysts as well.3–8 Since then, many DNAzymes have been obtained with catalytic functions such as cleavage,2,9–13 ligation,14–16 phosphorylation,17 adenylation18 or depurination19 of nucleic acids, as well as other reactions including porphyrin metallation, C–C bond formation, nucleopeptide linkage formation, oxygen transfer and thymine dimer repair.20–26 Because DNAzymes are facile to synthesize and more stable than protein and RNA enzymes, they have been widely used in applications such as nanomaterial assembly,27,28 biosensing,29–31 logical computing,32 nanomachine engineering,33 antiviral or gene therapy,34 and in vitro RNA manipulation.35 Despite these successes, the application of DNAzymes is limited by the narrower range of catalytic functionality compared to protein enzymes. One possible approach to addressing this issue would be to combine enzymes with different reactivities to form a cascade of successive enzymatic reactions, which together create new functionality. Indeed, many such examples exist in biology, since nearly all important biological functions, such as the pathways involved in DNA repair and protein synthesis, require a cascade of multiple protein enzymes to carry out their full function. In contrast, little has been reported about the use of DNAzyme cascades to realize enhanced functionality. Such a strategy could expand the functionality of DNAzymes to a level more on par with protein and RNA enzymes, which should greatly increase the range of possible applications.One such application is single nucleotide repair, i.e., excision of a misincorporated ribonucleotide in single-stranded DNA and subsequent insertion of the corresponding deoxyribonucleotide at the excision site. The misincorporation of ribonucleotides into DNA strands can occur from exposure to external oxidizing agents or ionizing radiation,36 or spontaneously during DNA replication.37 Misincorporation of ribonucleotide can distort the structure of DNA,38 reduce its stability,39 and interfere with the normal interaction between DNA and DNA polymerases.40 In fact, the overexpression of DNA polymerases that are prone to ribonucleotide misincorporation has been linked to many cancers, including ovarian, prostate, breast and colon cancers.41 In nature, protein enzymes such as RNase H and FEN-1 can efficiently excise misincorporated ribonucleotides in DNA by cleaving the DNA at the ribonucleotide site and then restoring the correct deoxyribonucleotides by DNA polymerases,42,43 which is an example of an enzyme cascade. It would be interesting to find out if a similar function could be achieved through DNAzyme cascades.
Another potential application is in tuning the properties of DNA-functionalized nanomaterials. For example, DNA-functionalized gold nanoparticles27 have emerged as an attractive platform for biosensing,32,44–50 nanomedicine,45 and as building blocks for controlled nanoassemblies.51–55 Although much research has been focused on the surface modification of gold nanoparticles with DNA for various applications, there are still limited methods to modify the sequences of DNA already immobilized on gold nanoparticles in order to make the properties of the DNA-modified nanomaterials tunable after fabrication. The use of DNAzymes is a promising approach for DNA modification on nanomaterials56 due to the excellent stability of DNAzymes and their smaller size compared to protein enzymes, thereby minimizing steric effects between the enzyme and the DNA in order to avoid reduction in reaction efficiency. However, it is still very challenging to modify a specific DNA sequence on multiple-DNA-functionalized nanomaterials to tune their functions in a selective and sequential fashion.
Herein, we demonstrate a cascade of two DNAzymes with RNA cleavage and DNA ligation activities, respectively, in order to carry out single nucleotide repair or selective sequence modification of DNA. In a one-pot reaction, a single misincorporated ribonucleotide in a DNA strand was converted to the corresponding deoxyribonucleotide while maintaining sequence integrity. Furthermore, the sequences of DNA strands immobilized on multiple functional nanoparticles were successfully modified in order to control and alter the DNA-directed assembly of nanoparticles in a stepwise and selective fashion.
Results and discussion
To demonstrate that single nucleotide repair in DNA can be achieved by the cascade of two DNAzymes, we used a 26-nt DNA strand (O1) containing a misincorporated cytidine (rC) ribonucleotide as an example. The goal was to convert the rC in O1 into a deoxycytidine (C), as seen in O4 (Fig. 1a), while maintaining the integrity of the DNA sequence. The DNAzymes 17Em1 (Fig. 1a, blue) with RNA cleavage activity2,57–59 and E47 (Fig. 1a, red) with DNA ligation activity14,60 were chosen as the cascade pair in this study. The 17Em1 DNAzyme catalyzes the hydrolysis of the 3′ phosphodiester linkage of the internal rC in the DNA strand when metal ion cofactors such as Pb2+ and Zn2+ are present (Fig. S1a in ESI†). On the other hand, the E47 DNAzyme can induce the catalytic ligation of the 5′ –OH of the DNA substrate with another 3′-phosphorylated DNA strand (activated by imidazole)14 in the presence of Cu2+ or Zn2+ as the metal cofactor (Fig. S1b in ESI†). Therefore, by sequential cleavage and ligation reactions catalyzed by these two DNAzymes on O1 containing rC, O1 could first be cleaved at the 3′ phosphodiester of the rC by 17Em1 and then undergo ligation at the cleavage site with another 3′-phosphorylated DNA strand of an identical sequence (except with deoxyribonucleotide C in place of ribonucleotide rC) by E47. The product O4 has a sequence identical to the starting strand O1, with the rC replaced with C.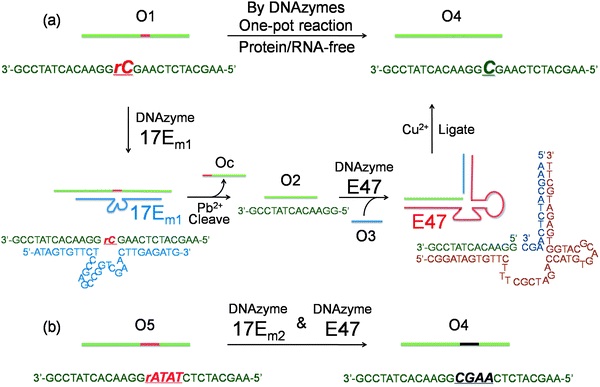 | ||
| Fig. 1 (a) Conversion of a single ribonucleotide (rC) in a DNA strand O1 to a deoxyribonucleotide (C) by the cascade of DNAzymes 17Em1 and E47: O1 is cleaved by 17Em1 to afford products of O2 and Oc; O2 is then ligated with O3 (activated by imidazole) to form O4 by E47. (b) Sequence modification of a DNA strand O5 to O4 through a similar protocol by the cascade of DNAzymes 17Em2 and E47. | ||
Initially, 3′-fluorescein-labeled O1 was treated with 17Em1 to form DNA duplex O1-17Em1via 18 matched base pairs (9 on each binding arm). In the presence of Pb2+, O1 was efficiently cleaved by 17Em1 into fragments O2 and Oc, resulting in the dehybridization of the duplex because the melting temperature of the duplex between 17Em1 and O2 or between 17Em1 and Oc is below room temperature (Fig. 1a). The fluorescence image after polyacrylamide gel electrophoresis (PAGE) suggested the complete cleavage of O1 and formation of O2 (Fig. 2a, lane 1 and 2 for O1 and O2, respectively), while Oc was not visible on the gel due to the lack of a fluorescein label. The cleavage reaction product O2 was also confirmed by the result from matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrum (Table 1 and Fig. S2 in ESI†). Control experiments using a DNAzyme of a different sequence (17Em2) or without Pb2+ showed negligible cleavage of the substrate O1 (Fig. S3 in ESI†) due to the specificity of the DNAzyme and the essential role of the metal ion cofactor.2,57–59 Subsequently, without any purification of O2 from the mixture solution after the previous cleavage step, E47 and 3′-phosphorylated O3 (imidazole-activated) were added into the solution to generate another DNA complex O2–O3-E47, which gave O4 as the product after the E47-catalyzed ligation reaction in the presence of Cu2+ (Fig. 1a).14,60 The formation of O4 was confirmed by both fluorescent PAGE (Fig. 2a, the upper band of lane 3) and MALDI-TOF MS (Table 1 and Fig. S2 in ESI†), while some unreacted O2 was also observed on the gel (Fig. 2a, the lower band of lane 3). Here, O3 was invisible due to the lack of a fluorescein label. Considerably lower levels of ligation between O2 and O3 were observed if either E47 or Cu2+ was absent (Fig. S3 in ESI†). Together these results indicate that the reactions catalyzed by the DNAzyme cascade were achieved through a one-pot reaction without isolation and purification of the intermediate O2.
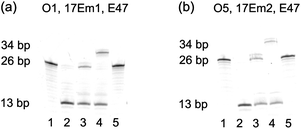 | ||
| Fig. 2 (a) Fluorescent PAGE (20% denaturing gel) images of the transformation from O1 to O4 by DNAzymes 17Em1 and E47. Lanes in (a): 1, O1; 2, 1 after cleavage by 17Em1 to yield O2 and Oc in the presence of Pb2+; 3, 2 after ligation to yield O4 by E47 in the presence of O3 and Cu2+; 4, 2 after ligation to yield O4 + 8A by E47 in the presence of O3 + 8A and Cu2+; 5, O4 in the presence of Pb2+ and 17Em1. (b) Fluorescent PAGE images of the transformation from O5 to O4 by 17Em2 and E47: Lanes in (b): 1, O5; 2, 1 after cleavage by 17Em2 to yield O2 in the presence of Pb2+; 3, 2 after ligation to yield O4 by E47 in the presence of O3 and Cu2+; 4, 2 after ligation to yield O4 + 8A by E47 in the presence of O3 + 8A and Cu2+; 5, O4 in the presence of Pb2+ and 17Em2. | ||
| DNA | O1 | O2 | O4 | O5 |
|---|---|---|---|---|
| Measured | 8831.6 | 4824.1 | 8812.3 | 8819.1 |
| Calculated | 8831.9 | 4826.3 | 8815.9 | 8821.9 |
To provide further confirmation of the above successful conversion of rC in O1 to C in O4, while keeping other sequences identical, a longer O3 + 8A (O3 extended by A8 at 5′) was used in place of O3 (Fig. 1a). Under the same conditions, a longer product O4 + 8A was obtained (Fig. 2a, the upper band of lane 4) with slower gel migration compared to O4 (Fig. 2a, the upper band of lane 3), suggesting that the ligation reaction occurred mostly between O2 and imidazole-activated O3, and not between O2 and un-activated Oc (Oc is the product from the previous cleavage reaction of O1 and 17Em1), in which case a band with the same migration as O4 would have been observed. The presence of C rather than rC in the product O4 was supported by the lower molecular weight of O4 in the MALDI-TOF mass spectrum as compared to that of O1 (Table 1), as well as the increased resistance to hydrolysis of O4 even in the presence of Pb2+ and 17Em1 (Fig. 2a, lane 5), which can catalyze the cleavage of a substrate containing an internal ribonucleotide linkage (O1),2,57 but not a substrate containing entirely deoxyribonucleotides (O4).
In addition to the single nucleotide repair functionality, it is also possible to use this methodology to edit the sequence of a DNA strand, which was used to convert the DNA strand O5 into O4 using the same cascade and conditions as before (Fig. 1b and S4 in ESI†). The product O4 was confirmed by PAGE (Fig. 2b) and MALDI-TOF MS (Table 1 and Fig. S2 in ESI†) and found to be identical to that obtained from the method in Fig. 1a.
Encouraged by the above results, we applied this method to modify the sequence of DNA immobilized on gold nanoparticles27 (AuNPs) to control the DNA-directed assembly of the AuNPs in a selective manner. DNA-functionalized gold AuNPs have been used in a variety of applications due to both their unique properties and the sequence-dependent hybridization of ssDNA immobilized on the AuNPs for controlled assembly.51–55 As shown in Fig. 3, when AuNPs are functionalized by complementary DNAs, the AuNPs can assemble into an “aggregated” state via DNA hybridization, which shows red-shifted and broadened absorption spectra compared to AuNPs functionalized by non-complementary DNAs. By modifying the sequences of DNA on the AuNPs, the assembly of the particles can be effectively controlled. Although methods for fabrication of DNA-functionalized AuNPs have been developed,27 there are still limited methods to modify the DNA sequences already immobilized on AuNPs in order to tune their functions. It is even more challenging to modify a specific DNA sequence on multiply-functionalized AuNPs with different DNA sequences on each nanoparticle. Selective modification can allow each different function of the AuNP to be controlled in a selective fashion for potential applications.
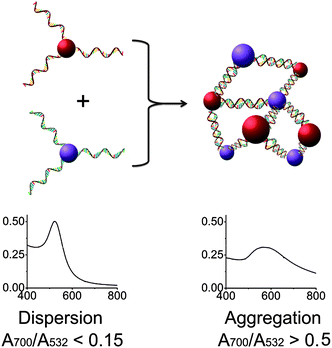 | ||
| Fig. 3 Assembly of two types of DNA-functionalized gold nanoparticles. If the sequences of the two DNAs are not complementary, the gold nanoparticles are in a “dispersed” state and exhibit a red color with a sharp absorption band peaked around 532 nm (left). In contrast, the assembly of the gold nanoparticles with complementary DNAs causes the formation of an “aggregated” state with a broad absorption band around 600 nm (right). | ||
AuNPs of 13 nm diameter were functionalized with DNA molecules via 3′-end thiols and used for this study. The formation of the AuNP assembly was confirmed by TEM images (Fig. S5 in ESI†) and characterized by large changes in absorption spectra27 (A700/A532 changed from <0.15 to >0.50 as illustrated in Fig. 5 and Table S1 in ESI†). As shown in Fig. 4, O6-functionalized AuNPs (red) were found to be able to form aggregates with O9-functionalized AuNPs (blue) via DNA-directed assembly through 12 complementary base pairs (Fig. 5 and S5 and Table S1†), but not with O10-functionalized AuNPs (purple), because of the 4 mismatched base pairs in the middle of the binding arm (Fig. 5 and S5 and Table S1†). After being treated with 17Em2 and Pb2+, the O6 on the surface of AuNPs was cleaved and converted to O8, which could not hybridize with either O9 or O10 efficiently. Thus no DNA-directed assembly was observed between the resulting O8-functionalized AuNPs and either O9- or O10-functionalized AuNPs (Fig. 5 and S5 and Table S1†). However, after a subsequent ligation reaction catalyzed by E47 in the presence of imidazole-activated O3 and Cu2+, O8 on the surface of AuNPs could be extended to O7, making the AuNPs capable of assembling with O10-, but not O9-functionalized AuNPs (Fig. 5 and S5 and Table S1†).
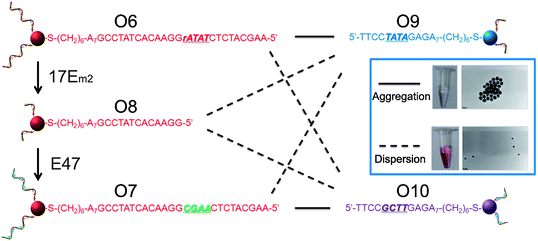 | ||
| Fig. 4 Controlling the assembly of DNA-functionalized gold nanoparticles via cascade-mediated modification of the DNA sequences. The solid and dashed lines indicate the successful and unsuccessful formation of aggregates, respectively. The inset shows the assembly of AuNPs modified with the complementary strands O6 and O9 (top) and the lack of assembly between AuNPs modified with noncomplementary strands O9 and O7 (obtained by treating O6-functionalized AuNPs with 17Em2 and E47) (bottom). | ||
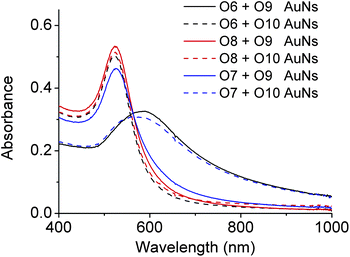 | ||
| Fig. 5 Absorption spectra of O6-, O7- or O8-functionalized AuNPs in the presence of O9- and O10-functionalized AuNPs, respectively. The red-shift of the peak indicates the formation of AuNP aggregations due to the hybridization of complementary DNAs on the AuNPs.27 For the ratios of absorbance (A700/A532), see Table S1 in ESI.† | ||
Interestingly, the product, O7-functionalized AuNPs, showed inverse characteristics in the formation of DNA-directed assembly with O10- and O9-functionalized AuNPs, compared to the original O6-functionalized AuNPs. TEM images of O6-functionalized AuNPs, either with or without treatment with 17Em2/E47, mixed with O10-functionalized AuNPs, are displayed in the inset of Fig. 4. These results clearly demonstrate that the ability to edit and replace DNA on AuNPs allows for exquisite programmable control over the assembly of nanoparticles.
Taking advantage of the specificity of DNAzyme to its nucleic acid substrates by complementary base pairing in the binding arms, selective modification of DNA sequences on surface of multiple functional AuNPs was also achieved in this work. As depicted in Fig. 6, O6 (red) and O11 (blue) bi-functional AuNPs could be modified by 17Em1, 17Em2 and E47 selectively and sequentially. As shown in Fig. 6, AuNPs capable of forming DNA-directed assembly with both (A and E), either (B, C and F), or neither (D) of the O9- and O10-functionalized AuNPs could be obtained by monitoring the significant increase of A700/A532 as indication of assembly formation (Fig. 7 and Table S2 in ESI†). Such a result, which is challenging to achieve by other techniques, can be used for the construction of tunable nanoassemblies for various applications.
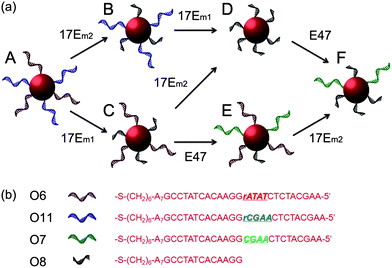 | ||
| Fig. 6 (a) Scheme showing stepwise modification of DNA sequences on multiply-functional gold nanoparticles by the collaboration of 17Em1 or 17Em2 and E47. (b) DNA sequences of O6–O8 and O11. | ||
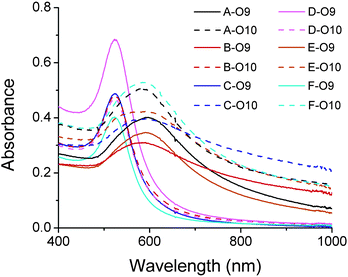 | ||
| Fig. 7 Absorption spectra of DNA-functionalized gold nanoparticles (A–F) (Fig. 6a) in the presence of O9- and O10-functionalized AuNPs, respectively. The red-shift of the peak indicates the formation of AuNP aggregations due to the hybridization of complementary DNAs on the AuNPs.27 For the ratios of absorbance (A700/A532), see Table S2 in ESI.† | ||
Conclusions
In summary, by putting together a cascade of two DNAzymes with cleaving and ligating activities, we have generated a new functionality for effective DNA modification. This function was applied in the conversion of a single misincorporated ribonucleotide into the corresponding dexoyribonucleotide in DNA and the modification of DNA sequences on the surface of gold nanoparticles to modify and control their self-assembly through DNA hybridization. The results suggest that combining DNAzymes with different catalytic activities may achieve more interesting functions and thus broaden the applications of DNAzymes.Experimental section
Materials
All DNA samples were purchased from Integrated DNA Technologies Inc. (Coralville, IA). Substrate and enzyme strands of the DNAzyme were purified by HPLC; thiol-modified and phosphorylated DNAs underwent standard desalting. Other chemicals were purchased from Sigma-Aldrich Inc.The sequences of DNA used in this work were as follows:
Cleaving DNAzyme 17Em1: 5′-ATAGTGTTCT CCGAGCCGGTCGAA ATAGAGATG-3′
Cleaving DNAzyme 17Em2: 5′-ATAGTGTTCT CCGAGCCGGTCGAA CTTGAGATG-3′
Ligating DNAzyme E47: 5′-CGGATAGTGTTCTTTCGCTAGACCATGTGACGCATGG TGAGATGCTT-3′
Fluorescein-labeled substrate O1: 3′-FAM-GCCTATCACAAGGrATATCTCTACGAA-5′
3′-phosphorylated O3: 3′-PO3-CGAACTCTACGAA-5′
3′-phosphorylated O3 + 8A: 3′-PO3-CGAACTCTACGAAAAAAAAAA-5′
Fluorescein-labeled substrate O5: 3′-FAM-GCCTATCACAAGGrCGAACTCTACGAA-5′
3′-thiol-modified O6: 3′-HS-AAAAAAAGCCTATCACAAGGrATATCTCTACGAA-5′
3′-thiol-modified O9: 3′-HS-AAAAAAAGAGATATCCTT-5′
3′-thiol-modified O10: 3′-HS-AAAAAAAGAGTTCGCCTT-5′
3′-thiol-modified O11: 3′-HS-AAAAAAAGCCTATCACAAGGrCGAACTCTACGAA-5′
Experimental procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with a stop solution containing 100 mM EDTA, to quench the cleavage and ligation reactions. This solution was mixed 1:1 with glycerol and transferred to a denaturing 20% acrylamide gel. The resulting gel was documented by a fluorescence image scanner (model FLA-3000G, Fuji). DNA isolation from the gel was performed by soaking the cut-out gel band in 1 mL Millipore water for 1 day, then centrifuging at 10000 rpm for 15 minutes. Afterwards, the fluorescent supernatant was concentrated to about 20 μL and purified by PD-10 column. The fraction with fluorescence at 520 nm was collected (total volume approximately 0.5–1.5 mL), concentrated to 20 μL, and then analyzed by MALDI-TOF MS.
000 rpm for 15 minutes. The supernatant was removed and nanoparticles were re-dispersed in buffer containing 100 mM NaCl and 25 mM tris-acetate (pH 8.2). This centrifugation process was repeated once more to remove free DNA in solution.
:1 ratio with a stop solution containing 100 mM EDTA, to quench the cleavage and ligation reactions. This solution was mixed 1:1 with glycerol and transferred to a denaturing 20% acrylamide gel. The resulting gel was documented by a fluorescence image scanner (model FLA-3000G, Fuji). DNA isolation from the gel was performed by soaking the cut-out gel band in 1 mL Millipore water for 1 day, then centrifuging at 10000 rpm for 15 minutes. Afterwards, the fluorescent supernatant was concentrated to about 20 μL and purified by PD-10 column. The fraction with fluorescence at 520 nm was collected (total volume approximately 0.5–1.5 mL), concentrated to 20 μL, and then analyzed by MALDI-TOF MS.
000 rpm for 15 minutes. The supernatant was removed and nanoparticles were re-dispersed in buffer containing 100 mM NaCl and 25 mM tris-acetate (pH 8.2). This centrifugation process was repeated once more to remove free DNA in solution.
For nanoparticles functionalized with both O6 and O11 in sequential tuning experiments, the molar ratio of O6 and O11 in loading was 4:1, with a total loading amount of 25 μL of 1 mM DNA. Other procedures were the same as above.
000 rpm for 15 minutes. The supernatant was removed and nanoparticles were re-dispersed in a buffer containing 100 mM NaCl and 25 mM HEPES (pH 7.0) to prepare a 3 nM solution. Then DNAzyme 17Em1 or 17Em2 (3 μM) and Pb(NO3)2 (4 μM) were added to the solution. The mixture was incubated at room temperature for 10 hours, then centrifuged at 13000 rpm for 15 minutes. The supernatant was removed and nanoparticles were re-dispersed in 1 mM HEPES (pH 7.0) without NaCl for 15 min to denature DNA hybridization. This centrifugation process was repeated once to remove free DNA in solution. At last, gold nanoparticles were re-dispersed in 100 mM NaCl and 25 mM HEPES (pH 7.0) to prepare a 3 nM solution.
000 rpm for 15 minutes. The supernatant was removed and nanoparticles were re-dispersed in 1 mM HEPES (pH 7.0) without NaCl for 15 min to denature DNA hybridization. This centrifugation process was repeated twice to remove free DNA in solution. Finally, the nanoparticles were re-dispersed in 100 mM NaCl and 25 mM HEPES (pH 7.0) to a final concentration of 3 nM.
Acknowledgements
We wish to thank the US National Institute of Health (ES016865) and Department of Energy (DE-FG02-08ER64568) for financial support.Notes and references
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344, 467–468 CrossRef CAS.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 CrossRef CAS.
- D. Sen and C. R. Geyer, Curr. Opin. Chem. Biol., 1998, 2, 680–687 CrossRef CAS.
- Y. F. Li and R. R. Breaker, Curr. Opin. Struct. Biol., 1999, 9, 315–323 CrossRef CAS.
- Y. Lu, Chem.–Eur. J., 2002, 8, 4588–4596 CrossRef CAS.
- R. R. Breaker, Nature, 2004, 432, 838–845 CrossRef CAS.
- K. Schlosser and Y. F. Li, Chem. Biol., 2009, 16, 311–322 CrossRef CAS.
- S. K. Silverman, Acc. Chem. Res., 2009, 42, 1521–1531 CrossRef CAS.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1995, 2, 655–660 CrossRef CAS.
- N. Carmi, S. R. Balkhi and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2233–2237 CrossRef CAS.
- A. R. Feldman and D. Sen, J. Mol. Biol., 2001, 313, 283–294 CrossRef CAS.
- J. W. Liu, A. K. Brown, X. L. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056–2061 CrossRef CAS.
- M. Chandra, A. Sachdeva and S. K. Silverman, Nat. Chem. Biol., 2009, 5, 718–720 CrossRef CAS.
- B. Cuenoud and J. W. Szostak, Nature, 1995, 375, 611–614 CrossRef CAS.
- A. Sreedhara, Y. F. Li and R. R. Breaker, J. Am. Chem. Soc., 2004, 126, 3454–3460 CrossRef CAS.
- W. E. Purtha, R. L. Coppins, M. K. Smalley and S. K. Silverman, J. Am. Chem. Soc., 2005, 127, 13124–13125 CrossRef CAS.
- Y. F. Li and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2746–2751 CrossRef CAS.
- Y. F. Li, Y. Liu and R. R. Breaker, Biochemistry, 2000, 39, 3106–3114 CrossRef CAS.
- T. L. Sheppard, P. Ordoukhanian and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7802–7807 CrossRef CAS.
- Y. F. Li and D. Sen, Nat. Struct. Biol., 1996, 3, 743–747 CrossRef CAS.
- D. Sen and L. C. H. Poon, Crit. Rev. Biochem. Mol. Biol., 2011, 46, 478–492 Search PubMed.
- M. Chandra and S. K. Silverman, J. Am. Chem. Soc., 2008, 130, 2936–2937 CrossRef CAS.
- P. I. Pradeepkumar, C. Hobartner, D. A. Baum and S. K. Silverman, Angew. Chem., Int. Ed., 2008, 47, 1753–1757 CrossRef CAS.
- P. Travascio, Y. F. Li and D. Sen, Chem. Biol., 1998, 5, 505–517 CrossRef.
- L. C. H. Poon, S. P. Methot, W. Morabi-Pazooki, F. Pio, A. J. Bennet and D. Sen, J. Am. Chem. Soc., 2011, 133, 1877–1884 CrossRef CAS.
- D. J. F. Chinnapen and D. Sen, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 65–69 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- J. W. Liu and Y. Lu, J. Am. Chem. Soc., 2003, 125, 6642–6643 CrossRef CAS.
- Y. Li and Y. Lu, Functional Nucleic Acids for Sensing and Other Analytical Applications, Springer, New York, 2009 Search PubMed.
- J. W. Liu, Z. H. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS.
- M. M. Ali, S. D. Aguirre, H. Lazim and Y. F. Li, Angew. Chem., Int. Ed., 2011, 50, 3751–3754 Search PubMed.
- I. Willner, B. Shlyahovsky, M. Zayats and B. Willner, Chem. Soc. Rev., 2008, 37, 1153–1165 RSC.
- Y. Chen, M. S. Wang and C. D. Mao, Angew. Chem., Int. Ed., 2004, 43, 3554–3557 CrossRef CAS.
- A. Peracchi, Rev. Med. Virol., 2004, 14, 47–64 Search PubMed.
- A. M. Pyle, V. T. Chu, E. Jankowsky and H. Boudvillain, Methods Enzymol., 2000, 317, 140–146 Search PubMed.
- M. Isildar, M. N. Schuchmann, D. Schultefrohlinde and C. Vonsonntag, Int. J. Radiat. Biol., 1981, 40, 347–354 Search PubMed.
- P. S. Eder, R. Y. Walder and J. A. Walder, Biochimie, 1993, 75, 123–126 CrossRef CAS.
- N. C. Horton and B. C. Finzel, J. Mol. Biol., 1996, 264, 521–533 CrossRef CAS.
- S. Nakano, T. Kanzaki and N. Sugimoto, J. Am. Chem. Soc., 2004, 126, 1088–1095 CrossRef CAS.
- C. M. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1619–1622 CrossRef CAS.
- K. J. Scanlon, M. Kashanisabet and H. Miyachi, Cancer Invest., 1989, 7, 581–587 Search PubMed.
- B. Rydberg and J. Game, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16654–16659 CrossRef CAS.
- N. Kim, S. Y. N. Huang, J. S. Williams, Y. C. Li, A. B. Clark, J. E. Cho, T. A. Kunkel, Y. Pommier and S. Jinks-Robertson, Science, 2011, 332, 1561–1564 Search PubMed.
- S. J. Park, T. A. Taton and C. A. Mirkin, Science, 2002, 295, 1503–1506 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS.
- C. S. Thaxton, D. G. Georganopoulou and C. A. Mirkin, Clin. Chim. Acta, 2006, 363, 120–126 CrossRef CAS.
- Z. D. Wang and Y. Lu, J. Mater. Chem., 2009, 19, 1788–1798 RSC.
- H. Wang, R. H. Yang, L. Yang and W. H. Tan, ACS Nano, 2009, 3, 2451–2460 CrossRef CAS.
- W. Xu, X. J. Xue, T. H. Li, H. Q. Zeng and X. G. Liu, Angew. Chem., Int. Ed., 2009, 48, 6849–6852 CrossRef CAS.
- F. Wang, Y. Han, C. S. Lim, Y. H. Lu, J. Wang, J. Xu, H. Y. Chen, C. Zhang, M. H. Hong and X. G. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS.
- J. Sharma, R. Chhabra, A. Cheng, J. Brownell, Y. Liu and H. Yan, Science, 2009, 323, 112–116 CrossRef CAS.
- Y. Wang, G. Chen, M. X. Yang, G. Silber, S. X. Xing, L. H. Tan, F. Wang, Y. H. Feng, X. G. Liu, S. Z. Li and H. Y. Chen, Nat. Commun., 2010 DOI:10.1038/ncomms1089.
- A. V. Pinheiro, D. R. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS.
- Z. Zhao, E. L. Jacovetty, Y. Liu and H. Yan, Angew. Chem., Int. Ed., 2011, 50, 2041–2044 CrossRef CAS.
- W. A. Zhao, J. C. F. Lam, W. Chiuman, M. A. Brook and Y. F. Li, Small, 2008, 4, 810–816 CrossRef CAS.
- J. Li and Y. Lu, J. Am. Chem. Soc., 2000, 122, 10466–10467 CrossRef.
- Y. Okumoto, Y. Tanabe and N. Sugimoto, Biochemistry, 2003, 42, 2158–2165 CrossRef CAS.
- S. Nakano, H. T. Karimata, Y. Kitagawa and N. Sugimoto, J. Am. Chem. Soc., 2009, 131, 16881–16888 CrossRef CAS.
- J. W. Liu and Y. Lu, Chem. Commun., 2007, 4872–4874 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2sc20763j |
| This journal is © The Royal Society of Chemistry 2013 |