A novel strategy for surface treatment on hematite photoanode for efficient water oxidation†
Lifei
Xi
a,
Sing Yang
Chiam
b,
Wai Fatt
Mak
a,
Phong D.
Tran
c,
James
Barber
*ade,
Say Chye Joachim
Loo
*a and
Lydia Helena
Wong
*a
aSchool of Materials Science and Engineering, Nanyang Technological University, Singapore, 639798. E-mail: j.barber@imperial.ac.uk; Joachimloo@ntu.edu.sg; Lydiawong@ntu.edu.sg
bInstitute of Materials Research and Engineering(IMRE), Agency of Science, Technology, and Research (A* Star), 3 Research Link, 117602, Singapore
cEnergy Research Institute @ NTU, Nanyang Technological University, 50 Nanyang Drive, Research Techno Plaza, X-Frontier Block, Level 5, Singapore, 637553
dDivision of Molecular Biosciences Imperial College London, London, SW7 2AZ, UK
eBioSolar Laboratory, Department of Material Sciences and Chemical Engineering, Polytechnic of Torino, Corso Duca degli Abruzzi, 24, 10129, Torino, Italy
First published on 12th September 2012
Abstract
In this paper, we report a novel strategy for surface treatment of hematite nanorods for efficient photo-driven water oxidation. This is the first report describing the growth of Sn treated hematite from α-FeOOH nanorod arrays in one step without substantially altering morphologies. With this treatment the photocurrent density increased from 1.24 for pristine hematite nanorods to 2.25 mA cm−2 at 1.23 V vs. RHE (i.e. 81% improvement). The increase in photocurrent density was also accompanied by improved incident-photon-to-current efficiencies and oxygen evolution. The photocurrent improvement is mainly attributed to a reduced electron–hole recombination at the hematite–electrolyte interface through the formation of FexSn1−xO4 layer at the hematite nanorod surface as shown by XPS, HRTEM, EDAX line scan analyses and PEC measurements.
Introduction
Photoelectrochemical (PEC) cells offer the ability to convert solar energy to stored chemical energy through the splitting of water into molecular oxygen and hydrogen.1–3 Hematite (α-Fe2O3) has recently emerged as a promising photoanode material for the generation of dioxygen from water due to its favorable optical band gap (Eg = 2.2 eV), excellent chemical stability in aqueous environments, ample abundance and low cost.4 Hematite has been theoretically predicted to achieve a water splitting efficiency of 12.4%.5 However, the reported efficiencies of hematite are lower than this predicted value, mainly due to the very short lifetime of photo-generated charge carriers (<10 ps) and short hole diffusion length (2–4 nm), which gives rise to a high recombination rate of photo-generated carriers in the bulk.6 Another fundamental limitation of hematite system is the need for externally applied bias because the energy levels of the conduction band of hematite are lower than the potential required to reduce protons to hydrogen (in the vacuum scale).7,8 Currently, various nanostructuring approaches have been proposed to overcome the very short lifetime and short hole diffusion length of hematite by putting the hematite in high proximity to the semiconductor–liquid junction (SCLJ) while maintaining a sufficient amount of material for complete light harvesting.7,9,10 These approaches, which involve the manipulation of the dimensions and morphology of hematite at the nanometer scale, include the deposition of porous thin films using solution-based colloidal methods,9 growth of nanowire arrays on conducting substrates,10,11 electrodeposition,12 spray pyrolysis13,14 and atmospheric pressure chemical vapor deposition (APCVD).15 In addition, chemical composition modification by doping was also reported to improve the optical absorption coefficient, the electron donor density and the flat band potential of hematite.4,16,17 Hematite has been doped with different elements such as Al,18,19 Cd,20 Mg,19 Mo,21 Si,22,23 Sn,6,9,19,24 Ti25 and Zn.26 For example, dopants such as Si4+ in the place of Fe3+ have been found to improve electrical conductivity of hematite.22 Ling et al. attributed their performance enhancement of hematite to Sn doping.9 Sn ions have a similar ionic radius and Pauling electronegativity to Fe ions and have been used to improve the electronic properties of hematite.27 They found that the morphology of hematite nanorods changed substantially when Sn was initially added.However, even with these strategies and improvements, water oxidation efficiencies of the various hematite systems are still far below expectation, partly because of surface recombination of trapped holes. Surface treatment was previously found to be an effective way to prevent electron–hole recombination with inorganic materials, such as Al2O3,7,28 CoF3,16 Ga2O328 and ZnO,13 providing improvements in the magnitude of the photocurrents and advantageous shifts of the photocurrent onset potentials. For example, Hu et al. found that the photocurrent onset potential was negatively shifted by about 200 mV for Ti-doped hematite modified with CoF3.16 Recently, Spray et al. treated hematite nanoparticle films with metal ions and found that a solid-state reaction can be induced at the surface of nanostructured electrodes.29 They found that their surface treated samples showed significantly enhanced photocurrents. Very recently we found that the photocurrent onset potential of hematite photoanodes was improved by 170 mV after 3 cycles of ZnAc treatment, while the photocurrent was increased from 0.75 to 1.08 mA cm−2 at 1.23 V vs. RHE.13 We proposed that the ZnO overlayer changes the flat band potential of hematite and reduces surface defects. Indeed, our study showed that advantageous surface treatments of hematite can be achieved using safe, inexpensive materials and simple processing techniques.
Here, we report a novel strategy for surface treatment of hematite to produce a photoanode for efficient water oxidation without any substantial changes in morphology of the electrode. This has been achieved by treating hydrothermally grown iron oxyhydroxide (FeOOH) nanorod arrays with Sn(IV) aqueous solution before subsequent annealing in air at 750 °C for 30 min (see ESI†). Since Sn(IV) was introduced as a solution phase it could easily penetrate FeOOH nanorods and uniformly wet the surface and the annealing temperature of 750°C was sufficient to induce reaction.29 During annealing, the solvent was evaporated and a very thin layer of SnO2 was formed coating the hematite nanorods. Following a solid-state diffusion reaction, Sn(IV) is gradually incorporated into hematite.29 Any SnO2 remaining on the surface was later removed by NaOH treatment and the very thin layer of FexSn1−xO4 formed on the surface was found to be unaffected by repeating this treatment.29,30 The presence of this layer was proposed to passivate surface defects as well as suppress the tunneling of electrons from the hematite core. These effects of Sn(IV) treatment are supported by experimental data obtained by XPS, HRTEM, EDAX line scan analyses and PEC measurements. To the best of our knowledge, this study is the first report describing a simple Sn(IV) treatment on α-FeOOH nanorod array without substantially changing their morphologies. This method showed significant improvement in PEC performance and can be applied to a broader range of nanostructured metal oxyhydroxide prepared by other methods such as electrochemical deposition and sol–gel methods.
Results and discussion
The top view SEM image of the pristine hematite nanorods is shown in Fig. 1a. The diameter and length of round nanorods were around 50 nm and 500 nm (see Fig. 1a inset). After treatment with 5 mM Sn(IV), there is no apparent residue as observed by SEM (not shown) and this is due to the low Sn(IV) concentration. However when the pristine hematite nanorods were treated with 20 mM Sn(IV), some residue appeared on the surface (Fig. 1b). With further increase of Sn(IV) concentration to 60 mM, the hematite nanorods are observed to be fully covered in a SnO2 matrix (see Fig. 1c). It can be seen that the initial square-like shape of original FeOOH nanorods is retained (refer to Fig. S1a, ESI†). This is probably due to the confinement effect of the SnO2 matrix during annealing. Agglomerated nanorods are subjected to stress during annealing, thereby causing cracks and formation of islands. When the concentration of Sn(IV) was raised to 90 mM, significant cracking in the nanorod arrays was observed (see Fig. S1b, ESI†). After soaking in 1 M NaOH solution for 12 h, it can be seen that the SnO2 matrix was removed (see Fig. 1d) and a slight increase in diameter was also observed when compared to the pristine hematite (see Fig. 1a and d). This increase can be attributed to the incorporation of Sn into the hematite structure and to the formation of a thin interfacial layer of amorphous FexSn1−xO4, which is chemically stable in NaOH, on the surface as detected by HRTEM (see Fig. 1e). The thickness of this amorphous layer is around 1–2 nm. A slight growth in particle size was also observed when Al(III) was used to treat Fe2O3 nanoparticles.29 XRD studies showed that after annealing, there was no peak shift or broadening and no additional peaks appeared (see Fig. S2, ESI†). The absence of any indication for Sn doping or additional peaks in the XRD spectrum implies that the modifications due to Sn treatment were limited to the surface of the hematite nanorods. Indeed, images derived from Scanning TEM (STEM) and elemental mapping of the hematite showed a good distribution of Sn on the surface (see Fig. S3, ESI†). It was clearly observed that Sn concentration in the shell is higher than that in the core and vice versa for Fe and O (Fig. S3e, ESI†). This is further evidence of the formation of FexSn1−xO4 at the surface. A diagrammatic representation of Sn(IV) treated hematite nanorod arrays is shown in Fig. 1f.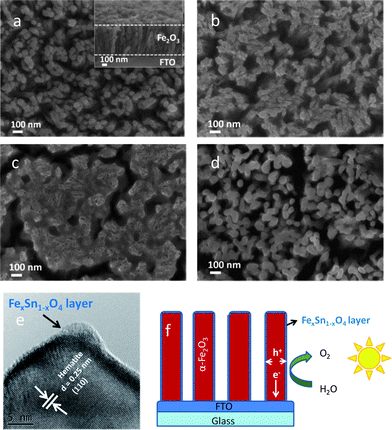 | ||
| Fig. 1 SEM image of (a) the pristine hematite after annealed at 750 °C for 30 min (inset: cross-section image), (b) and (c) hematite after treatment with different concentrations of Sn(IV) aqueous solution: 20 and 60 mM and annealed at 750 °C for 30 min. (d) Film shown in (b) after removing residual SnO2 in 1 M NaOH solution. (e) HRTEM image of hematite after treatment with 20 mM Sn(IV) solution. (f) Schematic effect of Sn(IV) treated hematite nanorod arrays for efficient water oxidation. | ||
The composition of the Sn treated samples was studied by XPS. Fig. 2a shows the spectra of the pristine hematite and hematite treated with 5 mM and 20 mM Sn(IV). It can be seen that with increasing Sn(IV) concentration, more Sn is incorporated as shown by the increasing intensity of Sn 3d5/2 peaks (centered at 486.5 and 494.9 eV). The fitted high resolution Sn 3d scans is shown in Fig. 2b. The origin of the initial Sn detected in the pristine sample is contamination from the FTO substrate. Increasing Sn(IV) concentration leads to the enhancement of the Sn 3d5/2 peak but at a lower binding energy of 486.5 eV. This lower binding energy peak is attributed to the Sn that is incorporated in the FeO structure. This is consistent with the expected binding energy shifts based on the fact that electronegativity of Sn (1.96) is higher than that of Fe (1.83). The second nearest neighbour effect of substituting Fe with Sn in O–Fe–O structure will thereby increase the relative transfer of electrons to the incorporated Sn atoms leading to a lower binding energy.20 When Sn ions were incorporated into the hematite structure, the same oxidation state between SnO2 and Sn(IV) means that we should only expect slight shifts in the O 1s spectra (see Fig. 2c).29 The spectra of O 1s shows that with increasing Sn(IV) concentration the binding energy centered around 530.1 eV for the pristine hematite, is shifted to a slightly higher binding energy, which is consistent with a lower electron density around O after Sn incorporation.31,32 On the other hand the Fe 2p scans looks comparable in all cases and their features match well with those reported for hematite (see Fig. 2a and d).21,25,29 With increasing Sn(IV) concentration, an additional peak at 715.8 eV becomes evident. This peak belongs to the Sn 3p3/2 spin-orbit split. Fitted Fe 2p scan for 20 mM Sn(IV) treated sample was shown in Fig. S4a (ESI†). It can be fitted with the different oxidation states of Fe along with a satellite state at higher energy level (Fig. S4a, ESI†). The molar ratio of Sn and Fe obtained from quantitative analysis of XPS data gives an atomic percentage (Sn/(Sn + Fe)) of 6.99, 13.63 and 34.87% for the pristine, 5 mM and 20 mM Sn(IV) treatments (see Fig. S4b, ESI†). The presence of Sn in the pristine sample comes from the FTO as mentioned above. The relative high concentration of Sn found for the 5 and 20 mM treated samples supports the contention that there is a significant level of Sn ions present mainly at the surface of hematite. Diffuse reflectance UV-Vis spectra and corresponding Tauc-Plots of samples with and without Sn(IV) treatment are shown in Fig. S5 (ESI†). The absorption properties of the sample were affected by the Sn(IV) treatment. The bandgap obtained from Tauc-Plots slightly increased from 2.05 eV for the pristine sample to 2.08 and 2.09 eV for samples treated with 5 and 20 mM Sn(IV) solution. A slight change of bandgap implies that doping level in the nanorods is low or negligible. A possible reason for the absorption decrease is the presence of the FexSn1−xO4 layer which may absorb light.30 Other effects of surface treatment from PEC and EIS measurements are presented below.
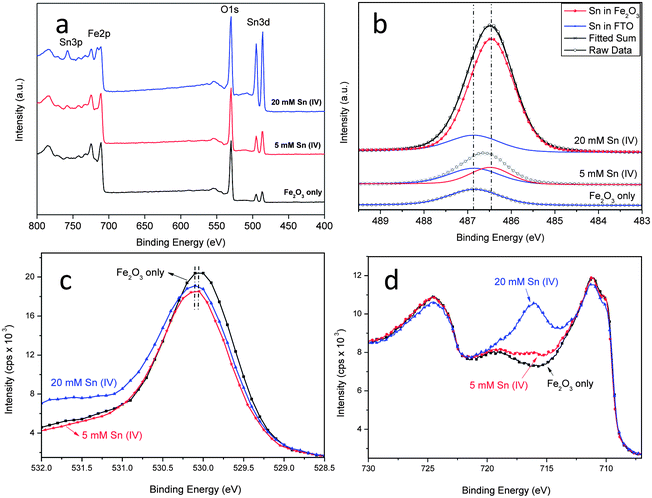 | ||
| Fig. 2 XPS spectra of (a) survey, (b) fitted high resolution Sn 3d5/2 scans, (c) O 1s scans for the pristine and hematite treated with Sn(IV) and (d) XPS spectra of Fe 2p scans for the pristine hematite and hematite treated with 5 and 20 mM Sn(IV) solution. | ||
Fig. 3a shows photocurrent-potential (I–V) curves of the pristine hematite and hematite treated with different concentration of Sn(IV). It was found that the addition of Sn(IV) gave rise to a strong effect on the I–V characteristics. The photocurrent densities increased from 1.24 for the pristine nanorods to 1.80, 2.25 and 1.16 mA cm−2 at 1.23 V vs. RHE with 5, 20 and 60 mM Sn(IV) treatments, respectively. In this study, the potentials used are referred to RHE. With an initial photocurrent onset potential of 0.62 V in 1 M NaOH electrolyte (pH 13.6), the photo-induced current density generated by the hematite photoanode with 20 mM Sn(IV) increased rapidly, attaining approximately 2.25 mA cm−2 at 1.23 V and reached a plateau of about 2.66 mA cm−2 at 1.4–1.6 V before the current increased exponentially. It can also be seen that the plateau of Sn(IV) treated samples is quite broad, which indicates a large active surface area and provides good evidence of surface modification.7,16 Thus the photocurrent of hematite treated with 20 mM Sn(IV) at 1.23 V increased by 81% as compared to that of the pristine nanorods. It should be noted that this value is comparable to the highest reported photocurrent of 2.4 mA cm−2 at 1.23 V for hematite without catalyst added prepared by APCVD15 and is the highest value generated by hematite prepared by the hydrothermal method.6,11,33 This pronounced effect of Sn(IV) treatment was found to be highly reproducible; in more than 30 photoanodes tested (with initial photocurrents of 0.9–1.25 mA cm−2), the photocurrents have been enhanced to 70–100% after 20 mM Sn(IV) treatment. Further increasing Sn(IV) concentration to 60 mM or even higher resulted in a decrease of the photocurrent. This was probably due to the formation of a thick FexSn1−xO4 layer on the surface of hematite nanorods (see Fig. 1e), which may then block both electron and hole transport. A moderate thickness of FexSn1−xO4 interfacial layer is therefore preferred since it can passivate surface defects as well as suppress the tunneling of electrons from the hematite core.29 This is believed to be major contributing factor for the pronounced photocurrent improvement. In our hands, 20 mM Sn(IV) treatment was found to be optimum. In previous studies, Dotan and Hisatomi et al. reported that recombination losses in the bulk and the surface of hematite can be distinguished by using H2O2.34,35 In this study, H2O2 was added to 1 M NaOH solution during chopped light I–V test (Fig. S6a–c, ESI†). It can be seen that surface recombination gives rise to photocurrent spikes under chopped light I–V curves (Fig. S6a, ESI†). These positive photocurrent spikes upon turning the light on represent the accumulation of holes at the hematite–electrolyte interface; similarly negative current spikes upon turning the light off represent the back reaction of electrons from the conduction band with the accumulated holes.34 It can be seen that in the high potential range (>1.0 V vs. RHE), a steady state photocurrent of the Sn treated hematite was much higher than that of pristine hematite which implies less recombination events in the Sn treated hematite. It can also be seen that these photocurrent spikes decrease with increasing potentials until they nearly disappear at 1.6 V for the Sn treated hematite or 1.8 V vs. RHE for the pristine hematite which means that the photogenerated holes were efficiently injected to the electrolyte and so drive the water oxidation reaction.34 The photocurrent spikes in the Sn treated hematite disappear at a lower potential than observed with the pristine sample, which could be an indication of a more efficient photogenerated hole transport to the electrolyte. Once H2O2 was added to the electrolyte, these photocurrent spikes were suppressed because photogenerated holes that reach the semiconductor–electrolyte interface are injected to the electrolyte at a faster rate, thus reducing recombination (Fig. S6b and c, ESI†). Charge separation yield is the fraction of the photo-generated holes that do not recombine with electrons in the bulk while charge injection yield is the fraction of those holes that do not recombine with electrons at surface traps.34,35 The charge separation yield of hematite treated with Sn(IV) was slightly higher than that of the pristine hematite over the entire potential range in this study (see Fig. S6d, ESI†). On the other hand, the charge injection yield of the hematite treated with Sn was also higher than that of the pristine hematite at the potential >1.05 V which is the main photoresponse region. For instance, the charge injection yields of the pristine and Sn(IV) treated hematite at 1.2 V were 48 and 65%, respectively. It can be seen that the charge injection yield was improved 35% after Sn treatment. This means that after Sn(IV) treatment, there is a higher fraction of photogenerated holes injected from the nanorods to electrolyte for water oxidation, resulting in less recombination events at the hematite–electrolyte interface.35 It seems likely that the higher charge injection yield is a consequence of the FexSn1−xO4 layer at the hematite–electrolyte interface.
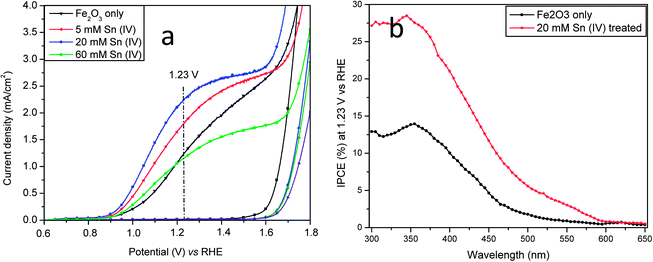 | ||
| Fig. 3 (a) I–V curves and (b) IPCE spectra of hematite photoanode with and without Sn(IV) treatment. IPCE measurements were carried out at an applied potential of 1.23 V vs. RHE in a 1 M NaOH electrolyte. | ||
We also found it to be vital that Sn(IV) treatment of FeOOH nanorods occurred during the course of annealing to obtain maximum photocurrents. This is probably because of the presence of defects and dangling bonds within hematite. When post-treatments are used, the defects and dangling bonds on the surface of hematite were only partially passivated and not at all for those buried within the core of hematite nanorods.17 In addition, Sn incorporation into hematite is expected to be more difficult than into FeOOH generated nanorods. These reasons probably explain the poor performance for the other approaches reported.7,29,30 Interestingly, a negative onset potential shift was also observed for 5, 20 and 60 mM Sn(IV) treated samples, but the largest shift was 100 mV for hematite treated with 20 mM Sn(IV). The enhancement of the photocurrent and photocurrent onset potential shift can be attributed to the effect of surface passivation as suggested by other groups.7,16,28 The photocurrent improvements were further verified by measuring the photoanode incident-photon-to-current efficiency (IPCE) at 1.23 V as a function of the wavelength of the incident light (see Fig. 3b). It can be seen that the hematite treated with 20 mM Sn(IV) showed substantially enhanced IPCE values as compared to the pristine samples. This is therefore indicative of an efficient charge injection, since they have a similar charge separation and absorption, and is consistent with their I–V characteristics. The IPCE drops substantially at wavelengths longer than 590 nm, as expected from the measurements of the hematite band gap.6 The IPCE was 28.4% at 345 nm for 20 mM Sn(IV) treated sample, a value comparable to that of Ti- and Sn-doped hematite.6,25 The pristine samples showed lower IPCE (13.8%), which further strengthens the argument that surface treatment is important in improving the photoelectrochemical efficiency of hematite.6,9,27,36 To compare the IPCE spectra with the photocurrent under AM1.5 illumination, the IPCE data were integrated over the standard AM1.5 solar spectrum.15,23 The integrated photocurrent for 20 mM Sn(IV) treated sample is close to 1.3 mA cm−2 which is lower than the measured photocurrent. This discrepancy is previously observed in PEC devices.37,38 The main reason for the discrepancy is presumably caused by the second order diffraction from the monochromator which may underestimate the IPCE result. In addition, the light distribution variation with different light source and filter used may be other reasons.
In order to study the role of Sn(IV) treatment on the electronic properties of hematite in electrolyte solution, EIS measurements were carried out. In general, the Mott–Schottky plots of nanorod array electrodes should be used with caution because the development of the space charge regions in nanorod arrays may not be same as that for planar electrodes.6,29 The Mott–Schottky plots (see Fig. 4a), which were collected from hematite nanorod arrays at a frequency of 1 KHz, exhibit a linear behaviour for the pristine hematite. Fig. 4a showed that the flat band (VFB) potential of our untreated hematite was around 0.38 V vs. RHE, which is in line with previous reported values for hematite nanowires.6,11,33 After surface treatment, a nonlinear behaviour, especially for 20 mM Sn treated hematite, appeared probably due to the change in surface state39 and therefore the flat band potentials could not be estimated. However, they could be used to estimate the electron donor densities (ND). It can be seen that Sn(IV) treatment decreased the measured slope in the potential range of 0.4–0.9 V (see Fig. 4a).6 A smaller slope means a bigger electron donor density. With a dielectric constant (ε) of 80 for hematite6,7 and a geometry area of 1 cm2 (an active surface area is 10 times higher than test area due to cylindrical structure of nanorod, see ESI†), ND was calculated to be 6 × 1020 cm−3, 12 × 1020 cm−3 and 29 × 1020 cm−3 for the pristine hematite, 5 and 20 mM Sn(IV) treated samples, respectively. Clearly, both samples treated with Sn showed increased donor density compared to the pristine hematite. We noted that the increase in donor density results in a decrease of space charge region width (W) and thus does not favour charge separation at the space charge region (ESI†). The space charge region widths were calculated to be 3.3, 2.1 and 1.5 nm for the pristine, 5 mM and 20 mM Sn treated samples which were far smaller than the radius of nanorods (25 nm). Considering that the charge separation yield of hematite treated with Sn(IV) was slightly higher than that of the pristine hematite as discussed above, the probable reduction in charge separation yield due to the reduction of the space charge region width is negligible and is compensated by the improvement in charge injection yield due to the formation of FexSn1−xO4 layer at the surface. This FexSn1−xO4 layer can reduce electron–hole recombination at hematite nanorods–electrolyte interface and is believed to be the major contributing factor for the pronounced photocurrent improvement in this study.
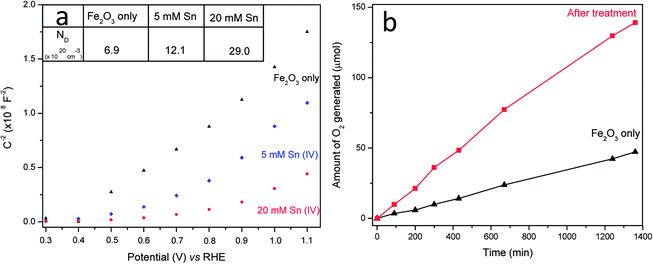 | ||
| Fig. 4 (a) Mott–Schottky plots in dark. Inset: ND is donor density with a unit of 1020 cm−3. (b) Time course of oxygen evolution of hematite photoanodes with and without 20 mM Sn(IV) treatment at an applied potential of 1.23 V vs. RHE in a 1 M NaOH electrolyte based on equal levels of hematite. | ||
In addition, the effect of surface passivation was also examined through the ideality factors extracted from the slope of the dark ln(I) vs. V curves under forward bias (see Fig. S7, ESI†).13,39,40 The ideality factor measurements of the pristine hematite and Sn treated hematite were carried out in a reversible redox couple-[Fe(CN)6]3−/4−. The decrease of ideality factor from 4.6 for the pristine hematite rods to 2.3 after 20 mM Sn(IV) treatment may be an indication of a reduction of recombination events as a consequence of FexSn1−xO4 surface passivation. It should also be noted that the surface passivation might also alter the surface catalytic properties and series resistances, which altogether would result in photocurrent improvement. Finally, the oxygen generation was recorded by gas chromatography as shown in Fig. 4b. It can be seen that the amount of oxygen generated from 20 mM Sn(IV) treated hematite photoanode was stable and nearly three times larger than that of the pristine sample after 22 h illumination (see Fig. S8, ESI†). The Faradaic efficiencies of both the pristine hematite and hematite treated with 20 mM Sn(IV) were calculated equal to 93 and 98% respectively, which indicates that the amount of O2 evolved is slightly less than expected. This is probably because of gas leakage during manual injection.11
Conclusions
In summary, a novel strategy for surface treatment on hematite nanorods for efficient water oxidation is reported. It was found that the growth of Sn treated hematite from α-FeOOH nanorod arrays can be readily achieved in a simple one step process without substantially changing the morphology of the nanorods. After treatment with 1 M NaOH for 12 h, XPS, HRTEM, EDAX line scan analyses confirmed the formation of FexSn1−xO4 layer at the surface of the hematite photoanode while PEC measurements showed improvements in surface passivation. It was found that the highest photocurrent increase and photocurrent onset potential shift was observed with 20 mM Sn(IV) treatment. The photocurrent density increases from 1.24 to 2.25 mA cm−2 (i.e. 81% improvement) at 1.23 V for the pristine Fe2O3 nanorod and the 20 mM Sn(IV) treated sample respectively. Concomitant with these improvements were a shift in the photocurrent onset potential by 100 mV, improvements in IPCE and increase in the efficiency of oxygen evolution. The reasons for the photocurrent improvement are attributed to surface passivation effect through the formation of a very thin FexSn1−xO4 layer. This layer can reduce electron–hole recombination at hematite nanorods–electrolyte interface and increases the charge injection efficiency.Acknowledgements
We appreciated Prof. M. Gratzel (EPFL, Switzerland) and Dr Oh Jihun (NREL, US) for their discussion and suggestions on IPCE measurements. Financial supports from the Centre of Artificial Photosynthesis and MOE Tier 1 are also gratefully acknowledged.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425 CrossRef CAS.
- M. Gratzel, Nature, 2001, 414, 338 CrossRef CAS.
- K. Sivula, F. Le Formal and M. Gratzel, ChemSusChem, 2011, 4, 432 CrossRef CAS.
- A. B. Murphy, P. R. F. Barnes, L. K. Randeniya, I. C. Plumb, I. E. Grey, M. D. Horne and J. A. Glasscock, Int. J. Hydrogen Energy, 2006, 31, 1999 CrossRef CAS.
- Y. Ling, G. Wang, D. A. Wheeler, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 2119 CrossRef CAS.
- F. Le Formal, N. Tetreault, M. Cornuz, T. Modehl, M. Gratzel and K. Sivula, Chem. Sci., 2011, 2, 737 RSC.
- J. Brillet, M. Cornuz, F. Le Formal, J.-H. Yum, M. Gratzel and K. Sivula, J. Mater. Res., 2010, 25, 17 CrossRef CAS.
- K. Sivula, R. Zboril, F. Le Formal, R. Robert, A. Weidenkaff, J. Tucek, J. Frydrych and M. Gratzel, J. Am. Chem. Soc., 2010, 132, 7436 CrossRef CAS.
- N. Beermann, L. Vayssieres, S. E. Lindquist and A. Hagfeldt, J. Electrochem. Soc., 2000, 147, 2456 CrossRef CAS.
- L. F. Xi, P. D. Tran, S. Y. Chia, P. S. Bassi, W. F. Mak, J. Barber, J. S. C. Loo and L. H. Wong, J. Phys. Chem. C, 2012, 116, 13884 CAS.
- Y. S. Hu, A. Kleiman-Shwarsctein, A. J. Forman, D. Hazen, J. N. Park and E. W. McFarland, Chem. Mater., 2008, 20, 3803 CrossRef CAS.
- L. F. Xi, P. S. Bassi, S. Y. Chia, W. F. Mak, P. D. Tran, J. Barber, J. S. C. Loo and L. H. Wong, Nanoscale, 2012, 4, 4430 RSC.
- A. S. N. Murthy and K. S. Reddy, Mater. Res. Bull., 1984, 19, 241 CrossRef CAS.
- S. Tilley, M. Cornuz, K. Sivula and M. Gratzel, Angew. Chem., Int. Ed., 2010, 49, 6405 CrossRef CAS.
- Y. S. Hu, A. Kleiman-Shwarsctein, G. D. Stucky and E. W. McFarland, Chem. Commun., 2009, 2652 RSC.
- P. Liao, M. C. Toroker and E. A. Carter, Nano Lett., 2011, 11, 1775 CrossRef CAS.
- C. J. Sartoretti, B. D. Alexander, R. Solarska, W. A. Rutkowska, J. Augustynski and R. Cerny, J. Phys. Chem. B, 2005, 109, 13685 CrossRef CAS.
- J. S. Jang, J. Lee, H. Ye, F. R. F. Fan and A. J. Bard, J. Phys. Chem. C, 2009, 113, 6719 CAS.
- A. Bak, W. Choi and H. Park, Appl. Catal., B, 2011, 110, 207 CrossRef CAS.
- A. Kleiman-Shwarsctein, Y. S. Hu, A. J. Forman, G. D. Stucky and E. W. McFarland, J. Phys. Chem. C, 2008, 112, 15900 CAS.
- J. A. Glasscock, P. R. F. Barnes, I. C. Plumb and N. Savvides, J. Phys. Chem. C, 2007, 111, 16477 CAS.
- Y. Q. Liang, C. S. Enache and R. van de Krol, Int. J. Photoenergy, 2008, 739864 Search PubMed.
- M. Gaudon, N. Pailhe, J. Majimel, A. Wattiaux, J. Abel and A. Demourgues, J. Solid State Chem., 2010, 183, 2101 CrossRef CAS.
- G. Wang, Y. Ling, D. A. Wheeler, K. E. N. George, K. Horsley, C. Heske, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3503 CrossRef CAS.
- S. Kumari, C. Tripathi, A. P. Singh, D. Chauhan, R. Shrivastav, S. Dass and V. R. Satsangi, Curr. Sci., 2006, 91, 1062 CAS.
- W. W. Wang and J. L. Yao, Mater. Res. Bull., 2012, 47, 1762 CrossRef CAS.
- T. Hisatomi, F. Le Formal, M. Cornuz, J. Brillet, N. Tetreault, K. Sivula and M. Gratzel, Energy Environ. Sci., 2011, 4, 2512 CAS.
- R. L. Spray, K. J. McDonald and K. S. Choi, J. Phys. Chem. C, 2011, 115, 3497 CAS.
- K. J. McDonald and K. S. Choi, Chem. Mater., 2011, 23, 4863 CrossRef CAS.
- V. Muller, M. Rasp, G. Stefanic, J. H. Ba, S. Gunther, J. Rathousky, M. Niederberger and D. Fattakhova-Rohlfing, Chem. Mater., 2009, 21, 5229 CrossRef CAS.
- K. L. Purvis, G. Lu, J. Schwartz and S. L. Bernasek, J. Am. Chem. Soc., 2000, 122, 1808 CrossRef CAS.
- Y. R. Hong, Z. L. Liu, S. F. B. S. A. Al-Bukhari, C. J. J. Lee, D. L. Yung, D. Z. Chi and T. S. A. Hor, Chem. Commun., 2011, 47, 10653 RSC.
- H. Dotan, K. Sivula, M. Gratzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958 CAS.
- T. Hisatomi, H. Dotan, M. Stefik, K. Sivula, A. Rothschild, M. Gratzel and N. Mathews, Adv. Mater., 2012, 24, 2699 CrossRef CAS.
- D. D. Qin, C. L. Tao, S. I. In, Z. Y. Yang, T. E. Mallouk, N. Z. Bao and C. A. Grimes, Energy Fuels, 2011, 25, 5257 CrossRef.
- G. M. Wang, H. Y. Wang, Y. C. Ling, Y. C. Tang, X. Y. Yang, R. C. Fitzmorris, C. C. Wang, J. Z. Zhang and Y. Li, Nano Lett., 2011, 11, 3026 CrossRef CAS.
- I. Cesar, K. Sivula, A. Kay, R. Zboril and M. Gratzel, J. Phys. Chem. C, 2009, 113, 772 CAS.
- Photoelectrochemical Hydrogen Production, ed. R. van de Krol and M. Gratzel, Springer, LLC, 2012, vol. VIII Search PubMed.
- T. Okumura and C. Kaneshiro, Electron. Comm. Jpn., 1999, 82, 13 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterizations and supporting figures. See DOI: 10.1039/c2sc20881d |
| This journal is © The Royal Society of Chemistry 2013 |