Access to formally Ni(I) states in a heterobimetallic NiZn system†
Christopher Uyeda and Jonas C. Peters*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: jpeters@caltech.edu
First published on 12th September 2012
Abstract
Heterobimetallic NiZn complexes featuring metal centers in distinct coordination environments have been synthesized using diimine–dioxime ligands as binucleating scaffolds. A tetramethylfuran-containing ligand derivative enables a stable one-electron-reduced S = 1/2 species to be accessed using Cp2Co as a chemical reductant. The resulting pseudo-square planar complex exhibits spectroscopic and crystallographic characteristics of a ligand-centered radical bound to a Ni(II) center. Upon coordination of a π-acidic ligand such as PPh3, however, a five-coordinate Ni(I) metalloradical is formed. The electronic structures of these reduced species provide insight into the subtle effects of ligand structure on the potential and reversibility of the NiII/I couple for complexes of redox-active tetraazamacrocycles.
Introduction
Metalloenzymes that mediate multiproton and multielectron redox processes, such as the reduction of H+,1 O2,2 N2,3 and CO24 commonly utilize polymetallic reaction sites, a characteristic that engenders greater redox flexibility than is possible at a single metal center, both in the range of accessible potentials and the number of stored reducing or oxidizing equivalents. Additionally, the binding and activation of small molecule substrates may be facilitated by simultaneous interaction with multiple transition metals. For example, the C-cluster of NiFe CO dehydrogenase, which catalyzes the reversible oxidation of CO to form CO2, has been crystallographically characterized with CO2 bound simultaneously through carbon to a reduced nickel center and through oxygen to a Lewis-acidic iron site.4 The development of well-defined synthetic systems that accommodate two metal centers in different coordination environments can facilitate the study of these cooperativity effects.5Tetraazamacrocyclic complexes of nickel have been studied extensively as electroreduction catalysts for a variety of substrates including H+,6 CO2,7 and alkyl electrophiles.8 A prototypical example is the nickel tetrapyrrole cofactor F430, which is the site at which S-methyl coenzyme M is catalytically reduced to generate methane.9 Key intermediates in these processes are paramagnetic reduced states, formulated either as Ni(I) metalloradicals or Ni(II)-stabilized ligand-centered radicals. The nature of these species dictates the potentials at which they are accessible as well as their reactivity and stability under catalytic conditions. Complexes featuring conjugated π-systems in the ligand framework, including diimine–dioxime ligands (Fig. 1),10 have attracted interest due to their relatively anodic reduction potentials. Nevertheless, while a number of solution-phase electrochemical6 and spectroscopic studies11 have been reported, isolable and structurally characterized reduced nickel N4-macrocycles are relatively uncommon,12 particularly for those cases in which the ligand is redox active.
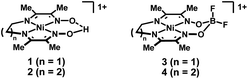 | ||
| Fig. 1 Diimine–dioxime complexes of nickel containing either a bridging proton (1 and 2) or BF2 (3 and 4) group. | ||
An understanding of the specific factors that influence the potential of the NiII/I couple and the stability of various intermediates in catalytic systems is predicated on a detailed description of the reduced state. We, therefore, have pursued the synthesis of S = 1/2 formally Ni(I) species in a series of heterobimetallic NiZn complexes supported by diimine–dioxime ligands. Significant spin density localized in π-systems of these ligands causes hydrogen-atom abstraction pathways to be facile for the parent, methyl-substituted complexes; however, a modified tetramethylfuran-containing ligand framework is described that sufficiently stabilizes these species to allow for isolation and characterization by a variety of spectroscopic and crystallographic techniques. Insights from these studies are used to provide a rationale for the impact of subtle changes in macrocycle structure on the observed potential and reversibility of the NiII/I redox couples.
Results and discussion
Synthesis and structural characterization of NiZn complexes
Based on precedence by Chaudhuri et al. for the synthesis of related copper complexes,13 substitution of the bridging proton in [Ni(MedoenH)]ClO4 (1) and [Ni(MedopnH)]ClO4 (2) for a Zn2+ ion was accomplished by deprotonation with excess Et3N in the presence of [Zn(H2O)6](ClO4)2 (Scheme 1). In order to fill out the Zn coordination sphere and prevent the formation of dimeric species, 1,4,7-trimethyl-1,4,7-triazacyclononane (Me3TACN) was employed as a capping ligand. The resulting NiZn complexes, 5 and 6, are diamagnetic, and the 1H-NMR resonances corresponding to the methyl and methylene groups of the Me3TACN ligand appear as two singlets at room temperature in CD3CN, suggestive of rapid fluxional processes. Positive ions corresponding to [Ni(Medoen/Medopn)Zn(Me3TACN)(ClO4)]+ were observed by ESI-MS, confirming the presence of one nickel and one zinc atom.![Preparation of [Ni(Medoen)Zn]2+ (5) and [Ni(Medopn)Zn]2+ (6).](/image/article/2013/SC/c2sc21231e/c2sc21231e-s1.gif) | ||
| Scheme 1 Preparation of [Ni(Medoen)Zn]2+ (5) and [Ni(Medopn)Zn]2+ (6). | ||
Orange-red crystals of 5 and 6 grown from MeCN solutions were analysed by X-Ray diffraction (XRD). Solid state structures reveal a square planar coordination environment for nickel, consistent with these complexes being diamagnetic in solution (Fig. 2). In both cases, zinc is six-coordinate, associating a molecule of MeCN. In order to accommodate the zinc ion, the separation between the oxygen atoms of the oximato groups increases by 0.35 to 0.4 Å relative to the corresponding proton-bridged complexes. Zinc is displaced approximately 1 Å above the mean NiN4 plane, and the distances between the Ni and Zn are 3.6 Å. The short N–O bond lengths of the oximato groups, ranging from 1.309(3) to 1.316(3) Å, suggest a significant degree of π-bonding. By comparison, N–O distances of 1.340–1.367 Å were observed for the BF2-bridged complex 4.14 The proton-bridged complex 1, which features a non-symmetrical hydrogen-bond, has one long and one short N–O distance at 1.361(1) and 1.308(1) Å respectively. A summary of some of the key metrical parameters is shown in Table 1.
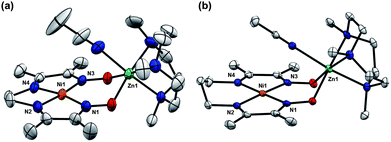 | ||
| Fig. 2 Solid-state structures of (a) 5 and (b) 6 excluding ClO4− counterions and non-coordinated solvent molecules (thermal ellipsoids at 50% probability). Selected bond distances (Å) and angles (°) for 5: Ni1–N1, 1.884(2); Ni1–N2, 1.835(3); Ni1–N3, 1.885(2); Ni1–N4, 1.836(2); N1–Ni–N3, 106.40(7); N2–Ni–N4, 86.3(1). Selected bond distances (Å) and angles (°) for 6: Ni1–N1, 1.923(2); Ni1–N2, 1.905(2); Ni1–N3, 1.911(2); Ni1–N4, 1.917(2); N1–Ni–N3, 99.49(9); N2–Ni–N4, 96.5(1). | ||
| Mean Ni–N | O–O | N–O | Ni–Zn | Zn–mean N4 plane | |
|---|---|---|---|---|---|
| a From ref. 6b.b Bond metrics for one of two crystallographically distinct molecules in the asymmetric unit are shown. | |||||
| 1 | 1.833 | 2.613(1) | 1.361(1), 1.308(1) | — | — |
| 2a | 1.884 | 2.420 | 1.339, 1.333 | — | — |
| 5 | 1.860 | 3.035(2) | 1.312(3), 1.318(3) | 3.5919(4) | 0.916 |
| 6 | 1.914 | 2.758(3) | 1.309(3), 1.316(3) | 3.6150(4) | 1.266 |
| 7 | 1.857 | 3.089(3) | 1.320(3), 1.303(4) | 3.4755(8) | 1.023 |
| 8b | 1.867 | 3.032(4) | 1.327(5), 1.319(4) | 3.4467(6) | 1.276 |
As compared to complexes derived from 1,3-diaminopropane, the two-carbon tether in the 1,2-diaminoethane-derived complexes enforces shorter Ni–N distances—by approximately 0.05 Å on average. Additional effects of the shorter tether length are a compressed N2–Ni–N4 angle and expanded N1–Ni–N3 angle, which result in an increase in the distance between the oximato oxygen atoms. These structural differences between 5 and 6 are manifested in their relative susceptibility toward protonolysis. When treated with 10 equivalents of AcOH, complex 6 is completely converted to the proton-bridged species 2, while complex 5 is stable under these conditions. This observation can be rationalized by the relative strength of the intramolecular hydrogen-bonding interactions in 1 and 2, with greater O–O separation resulting in weaker hydrogen-bonds. Consistent with this hypothesis, the bridging proton in complex 1 is in rapid exchange with residual water in CD3CN on the 1H-NMR chemical shift timescale at room temperature, whereas a sharp downfield resonance at 18.3 ppm is observed for complex 2 under the same conditions.
In order to explore whether these bimetallic platforms are capable of supporting ligands that span the two metal centers, complex 5 was treated with n-Bu4N+ salts of OAc− or NO2− to yield the corresponding dark-brown monocationic complexes. Complex 7 exhibits strong IR bands at 1569 and 1400 cm−1, assigned to the antisymmetric and symmetric stretching vibrations of the acetate group. The Δυ of 169 cm−1 falls within the typical range for syn–syn bridging carboxylates.15 This coordination mode was confirmed by XRD (Fig. 3). While multiple bridging geometries have been observed in bimetallic μ-NO2 complexes,16 the μ-(η1-N:η1-O) motif is favored in the solid-state for 8. Coordination of one of the nitrite oxygen atoms to zinc induces a moderate asymmetry in the N–O bond distances (Δ ≈ 0.05 Å).
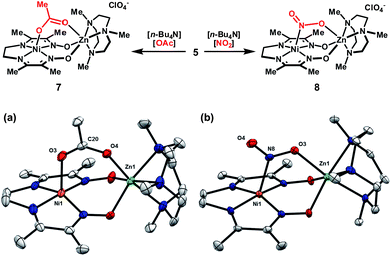 | ||
| Fig. 3 Association of bridging anions to complex 5 and solid-state structures of (a) 7 and (b) 8 excluding ClO4− counterions (thermal ellipsoids at 50% probability). Only one of two crystallographically distinct molecules in the asymmetric unit is shown for 8. Selected bond distances (Å) for 7: Ni1–O3, 2.173(3); Zn1–O4, 2.060(2); O3–C20, 1.239(4); O4–C20, 1.260(4). Selected bond distances (Å) for 8: Ni1–N8, 2.209(3); Zn1–O3, 2.148(3); N8–O3, 1.266(5); N8–O4, 1.236(4). | ||
Electrochemical and chemical reduction
In order to compare the electrochemistry of complexes 5 and 6 to the corresponding proton and BF2-bridged complexes, cyclic voltammetry (CV) studies were conducted in MeCN. Complex 5 exhibits a reversible reduction (100 mV s−1 scan rate) at −0.98 V vs. SCE, followed by a quasi-reversible reduction at −1.42 V (Fig. 5). Despite the fact that the Zn2+ ion causes complex 5 to be overall dicationic, the E° for the first reduction is cathodically shifted by 270 mV relative to the proton-bridged, monocationic complex 1. The shift observed for 6 relative to 2 is of a similar magnitude. This effect may reflect the relatively modest Lewis acidity of pseudo-octahedral Zn2+ ions, particularly when coordinated to multiple amine donors. This hypothesis is supported by the relatively short N–O distances in the NiZn complexes. The expanded macrocycle core size upon substitution of a proton for a larger metal ion may also contribute to this shift. It is noteworthy in this context that the complexes derived from the longer diaminopropane tether are reduced at more negative potentials than those derived from diaminoethane (Table 2).| E° ([Ni]n/n−1) | E° ([Ni]n−1/n−2) | |
|---|---|---|
| a 0.5 mM concentration of the complex in MeCN; 0.1 M [n-Bu4N][ClO4] supporting electrolyte; glassy carbon working electrode; N2 atmosphere; 100 mV s−1 scan rate; internally referenced to the Fc/Fc+ redox couple at +0.38 V vs. SCE.b Reduction waves are quasi-reversible. | ||
| 1 | −0.71 | −1.42 |
| 2 | −0.86 | −1.39 |
| 5 | −0.98 | −1.42b |
| 6 | −1.08 | −1.43b |
| 10 | −0.76 | −1.21 |
When a chemical reduction of complex 5 was attempted using one equivalent of Cp2Co, a green, diamagnetic product was instead observed to form within 15 min at room temperature. The 1H-NMR spectrum of this product is consistent with the enamide structure, 9, shown in Scheme 2: the methylene groups of the two-carbon tether appear as non-equivalent triplets, and two singlets are observed, at 4.01 and 3.75 ppm, in a region that is typical for olefinic protons.
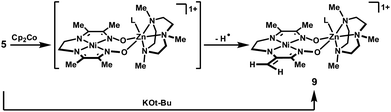 | ||
| Scheme 2 Hydrogen-atom and proton abstraction reactions. | ||
The enamide product 9, formed in 71% yield by 1H-NMR integration against an internal standard, is presumed to arise by hydrogen-atom abstraction from the putative reduced species. Dihydrogen was not detected by gas chromatography as a byproduct of this process, suggesting that the fraction of metal-containing product not observed by NMR is the hydrogen-atom acceptor in this reaction. When reduction with Cp2Co was conducted in the presence of TEMPO, the formation of 9 was nearly quantitative, and an equimolar amount of TEMPO–H was generated. This result suggests that the C–H bond dissociation energies for the methyl substituents are lowered significantly in the reduced complex, a likely indication of significant radical character in the ligand. Compound 9 can alternatively be accessed directly by treatment of the Ni(II) complex 5 with one equivalent of KOt-Bu.
The observation of this ligand-based reactivity is significant in the context of H+ reduction catalysis and constitutes a Faradaic process that does not generate H2. While cobalt complexes of diimine–dioxime ligands evolve H2 in high yield under a variety of conditions, the corresponding nickel complexes generally exhibit comparatively low Faradaic efficiency.6b
Synthesis and electrochemistry of [Ni(TMFdoen)Zn]2+ (10)
In order to circumvent hydrogen-atom transfer pathways from the ligand, the tetramethylfuran-derived17 NiZn complex 10, lacking hydrogen-atoms adjacent to the imines, was prepared. While many of the major features of the solid-state structure are similar to complex 5, a noteworthy difference is that zinc is nearly coplanar with the NiN4 unit, and the separation between the oximato oxygen atoms is correspondingly greater by 0.1 Å (Fig. 4).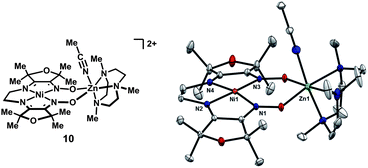 | ||
| Fig. 4 Solid-state structure of 10 excluding ClO4− and BPh4− counterions and non-coordinated solvent molecules (thermal ellipsoids at 50% probability). | ||
In contrast to complexes 5 and 6, both the first and second reduction waves for 10 are electrochemically reversible (100 mV s−1 scan rate) with E° values of −0.76 and −1.21 V vs. SCE (Fig. 5). These potentials are approximately 200 mV more positive than for the methyl-substituted complex 5. One possible explanation for this shift is an inductive electron-withdrawing effect of the furan oxygens,18 which may be particularly significant in cases where reduction is ligand based. Alternatively, the shorter C–C bond distance between the imine and oxime groups enforced by the five-membered ring may lower the energy of the ligand-based π* orbital, allowing it to be populated at less reducing potentials.
![Cyclic voltammograms for 0.5 mM (a) 5 and (b) 10 in MeCN. Scans that extend past the second reduction wave are in black, and a scan that extends only past the first reduction wave is in red. (0.1 M [n-Bu4N][ClO4] supporting electrolyte; glassy carbon working electrode; N2 atmosphere; 100 mV s−1 scan rate; internally referenced to the Fc/Fc+ redox couple at +0.38 V vs. SCE).](/image/article/2013/SC/c2sc21231e/c2sc21231e-f5.gif) | ||
| Fig. 5 Cyclic voltammograms for 0.5 mM (a) 5 and (b) 10 in MeCN. Scans that extend past the second reduction wave are in black, and a scan that extends only past the first reduction wave is in red. (0.1 M [n-Bu4N][ClO4] supporting electrolyte; glassy carbon working electrode; N2 atmosphere; 100 mV s−1 scan rate; internally referenced to the Fc/Fc+ redox couple at +0.38 V vs. SCE). | ||
Chemical reduction of [Ni(TMFdoen)Zn]2+ and PPh3 coordination
Reduction of complex 10 using Cp2Co yielded a dark green, paramagnetic species that was isolated in 81% yield (Scheme 3). The reduced complex, 11, exhibits a sharp, nearly isotropic EPR signal at g = 2.02. This spectrum is detectable in 2-MeTHF/THF solution at room temperature and in a frozen glass at 77 K. The narrow line width, lack of anisotropy, and g-factor near the free electron value are suggestive of a ligand-centered radical with minimal spin population at the nickel center. | ||
| Scheme 3 Chemical reduction and coordination of PPh3. | ||
Room temperature diffusion of pentane into concentrated solutions of complex 11 in THF produced plate-like, green-brown crystals. Characterization by XRD revealed a structure in which zinc is five-coordinate. Combustion analysis of bulk samples isolated at room temperature also indicates a formula lacking associated solvent molecules. Although crystals obtained in this manner were sufficient to establish connectivity between atoms, reliable metrical parameters could not be obtained. In order to further characterize the reduced complex, crystallizations were conducted at −30 °C under otherwise identical conditions, yielding the THF adduct shown in Fig. 6.
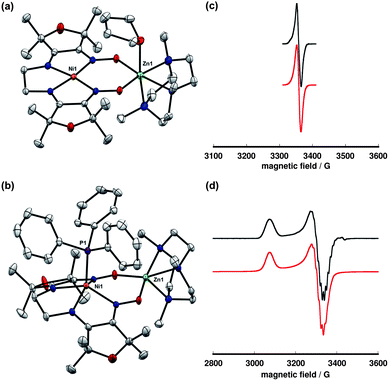 | ||
| Fig. 6 Solid-state structures of (a) 11 and (b) 12 excluding BPh4− counterions and non-coordinated solvent molecules (thermal ellipsoids at 50% probability). Selected bond lengths (Å) and angles (°) for 11: Ni1–mean N4-plane, 0.021; Σ N–Ni1–N, 360.0. Selected bond lengths (Å) and angles (°) for 12: Ni1–P1, 2.2263(5); Ni1–mean N4-plane, 0.666; Σ N–Ni1–N, 334.4. Experimental (black, top) and simulated (red, bottom) X-band EPR spectra for (c) 11 (2-MeTHF/THF frozen solution, 77 K) and (d) 12 (2-MeTHF/THF frozen solution, 50 K). Simulated parameters for 11: giso = 2.020. Simulated parameters for 12: g∥ = 2.208, g⊥ = 2.044. | ||
The metrical parameters associated with the solid-state structure of 11 support the electronic configuration suggested by the EPR data. The Ni–N distances are contracted relative to the more oxidized species, 10, consistent with greater electrostatic attraction between the metal cation and the increased negative charge on the ligand framework. This propensity for shorter Ni–N bonds in the reduced state provides an explanation for the observed effect of macrocycle core size on the reduction potential of Ni(II) diimine–dioxime complexes: the diaminoethane-derived complex 5 exhibits both shorter Ni–N distances and an anodically-shifted reduction potential than for 6. This trend additionally extends to the proton and BF2-bridged complexes.
Distortions in the ligand geometry are also consistent with a delocalized organic radical: as compared to complex 10, C–N distances for the imine and oxime groups in 11 are elongated, and the intervening C–C bonds are contracted. No significant differences were observed in the bond metrics for the two halves of the macrocycle suggesting that the unpaired spin is distributed over both ligand π-systems. The reduced C–N bond order of the imine group is also apparent in the IR spectrum: the imine C–N stretching band at 1657 cm−1 for complex 10 is shifted to 1560 cm−1 in complex 11.
Gagne has reported EPR studies of a series of reduced nickel tetraazamacrocycle complexes.11a For ligands containing conjugated π-systems, reduction was observed to be primarily ligand-centered. By contrast, complexes containing amine and isolated imine donors exhibited axial EPR spectra that were attributed to Ni(I) species. Consistent with this assignment, structural characterization of four-coordinate diimine–diamine and tetraamine Ni(I) complexes by Fujita revealed significantly elongated Ni–N distances relative to the Ni(II) complexes of the same ligand.12b,12c This structural change can be readily interpreted as a consequence of an electron residing in a σ-anti-bonding orbital of dx2−y2 parentage. Accordingly, tetraamine complexes of nickel are reduced at more positive potentials as the macrocycle core size is increased19—an opposing trend to what is observed for diimine–dioxime complexes.
Nickel K-edge data (Fig. 7) are also consistent with the reduction of 10 being primarily ligand-based. For related square planar tetraazamacrocycle20 and dithiolene/dithiolato21 complexes of nickel, the pre-edge feature, assigned to a 1s → 4pz transition, has been used as a measure of the effective nuclear charge at the metal center. The energies for this transition are nearly coincident at 8333 eV for complexes 10 and 11. By comparison, a Ni(I) tetramethylcyclam complex investigated by Fujita et al.20exhibited a significantly lower energy for both the pre-edge feature (approximately 8328 eV) and the subsequent rising edge.
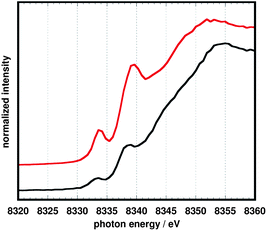 | ||
| Fig. 7 Nickel K-edge spectra of 10 (top, red) and 11 (bottom, black). | ||
The addition of PPh3 to solutions of complex 11 in THF caused a subtle color change to green-blue. A frozen THF/2-MeTHF solution of the resulting adduct exhibited an axial EPR signal (g∥ = 2.208, g⊥ = 2.044) with a 12-line hyperfine coupling pattern, attributed to the four roughly equivalent equatorial nitrogen atoms (Fig. 6). In the solid-state structure of 12, nickel is five-coordinate and positioned 0.67 Å above the N4 basal plane. The Ni–N distances are significantly elongated relative to the four-coordinate complexes in either oxidation state, consistent with population of a dx2−y2 orbital. The relevant C–N and C–C distances are similar to complex 10 and indicate that the oxidation state of the ligand is largely unchanged.
Computational electronic structure model
More detailed insight into the electronic structure of the S = 1/2 reduced complex 11 and the PPh3 adduct 12 was obtained by calculating the singly occupied molecular orbitals (SOMO) and spin densities using DFT methods (B3LYP/6-31G(d)). Tetramethylfuran groups were truncated to methyl substituents and outer-sphere anions were excluded in these calculations. The computationally optimized geometries accurately reproduce the decrease in Ni–N distances upon reduction as well as the changes in the C–N, C–C, and N–O bond distances of the ligand π-systems. A more detailed comparison between experimental and computational values for key metrical parameters is shown in Fig. 8.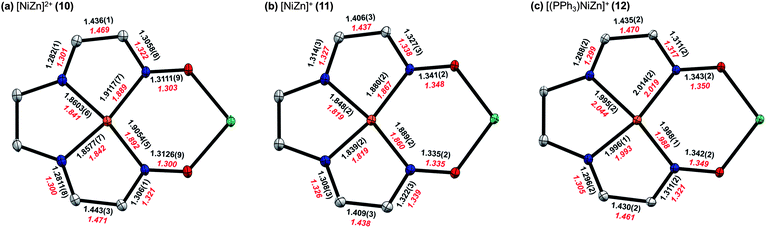 | ||
| Fig. 8 Comparison of selected bond lengths (Å) from solid-state structures (black) for (a) 10, (b) 11, and (c) 12. For 11, metrical parameters are shown for only one of the two crystallographically distinct molecules in the asymmetric unit. Calculated values from computationally optimized structures (red) at the B3LYP/6-31G(d) level of DFT are shown for comparison. | ||
For complex 11, the Mulliken spin population on nickel was calculated to be 0.02, indicating negligible metalloradical character (Fig. 9). The SOMO has π-anti-bonding character between the carbon and nitrogen atoms of the imine and oxime groups, and π-bonding character between the intervening C–C bond, accounting for the observed geometric changes upon reduction. The square pyramidal PPh3 adduct, by contrast, was calculated to have a 1.12 spin population on nickel. The SOMO is derived from a dx2−y2 orbital and is σ-anti-bonding with respect to the Ni–N bonds. For both reduced complexes, elongation of the oximato N–O bonds can be rationalized by the π-anti-bonding character in the calculated SOMOs.
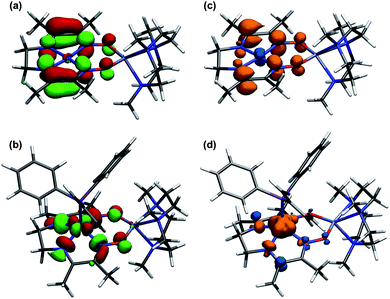 | ||
| Fig. 9 Calculated SOMO for (a) 11 and (b) 12, and spin density plots for (c) 11 and (d) 12. Geometries were optimized at the B3LYP/6-31G(d) level of DFT and verified by frequency analysis. | ||
Hydrogen-atom abstraction reactivity of [Ni(TMFdoen)Zn]+
In order to assess the stability of complex 11 toward hydrogen-atom abstraction, reaction with TEMPO was conducted in CD3CN. The formation of TEMPO–H was observed by 1H-NMR, and two diamagnetic products were detected in 55% and 13% yield (Scheme 4). For the major product, the presence of seven well-resolved peaks corresponding to the furan methyl substituents indicates a non-symmetrical structure, and the two singlets at 4.85 and 5.35 ppm suggest unsaturation in the ligand. The identity of this species was established by XRD analysis of crystals obtained from concentrated THF solutions (Fig. 10). The unusual cyclopropane motif presumably arises by H-atom abstraction from a methyl substituent and cyclization onto the adjacent imine carbon. The short C17–C18 distance of 1.36 Å is consistent with a double bond. By comparison, the corresponding C–C distance in complex 10 is 1.54 Å. While the identity of the minor product has not been definitively established, a plausible structure based on 1H-NMR data and analogy to complex 13 is the cyclopropane-containing product with a saturated two-carbon tether. | ||
| Scheme 4 Hydrogen-atom abstraction from the reduced complex. | ||
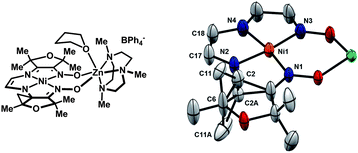 | ||
| Fig. 10 Solid-state structure of 13 highlighting parts of the ligand relevant to the hydrogen-atom abstraction reactivity (thermal ellipsoids at 50% probability). The occupancy of C2, C2A, C11, and C11A refined to a value of 50%. Selected bond lengths (Å): Ni1–N1, 1.883(3); Ni1–N2, 1.820(3); Ni1–N3, 1.921(3); Ni1–N4, 1.881(4); N2–C2, 1.43(1); N2–C2A, 1.48(1); C17–C18, 1.357(6). | ||
Conclusions
Bimetallic complexes containing a low-valent, reduced nickel center in combination with a Lewis-acidic zinc site can be synthetically accessed using the dianionic form of diimine–dioxime ligands. These scaffolds afford sufficient geometric flexibility to accommodate a variety of bridging and non-bridging ligands, with metal–metal distances ranging from 3.45 to 3.78 Å. The incorporation of zinc in the place of a bridging proton results in a significant cathodic shift in the NiII/I couple, illustrating the utility of varying this substituent in order to tune the overall redox potential of the complex.Reduced states of nickel diimine–dioxime complexes are accessible at modest potentials owing to the presence of low-lying ligand-based π-orbitals. The resulting four-coordinate, S = 1/2 species exhibits characteristic features of a ligand-centered radical bound to Ni(II), yet is capable of associating π-acidic ligands such as PPh3 to form five-coordinate Ni(I) metalloradicals. The ability of a single platform to reversibly access multiple oxidation states and conformations at minimal energetic cost is a key characteristic of redox catalysts that are capable of operating without the need for large thermodynamic or kinetic driving forces. The application of the complexes described here as bifunctional electrocatalysts is an ongoing area of research.
Acknowledgements
This work was supported by the NSF Center for Chemical Innovation: Powering the Planet grant CHE-0802907, and by the Gordon and Betty Moore Foundation. We thank Larry Henling and Charlene Tsay for assistance with crystallography, Dr Angelo Di Bilio for EPR measurements, and Dr Limei Zhang and Dr Jens Kaiser for XAS measurements. We acknowledge the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi-Aventis BRP at Caltech for their generous support of the Molecular Observatory at Caltech. SSRL is operated for the DOE and supported by its Office of Biological and Environmental Research, and by the NIH, NIGMS (including P41GM103393) and the NCRR (P41RR001209).Notes and references
- (a) A. Volbeda, M.-H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580–587 CrossRef CAS; (b) J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS; (c) Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS.
- (a) E. I. Solomon, P. Chen, M. Metz, S.-K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570–4590 CrossRef CAS; (b) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1046 CrossRef CAS.
- (a) J. Kim and D. C. Rees, Science, 1992, 257, 1677–1682 CAS; (b) M. Chan, J. Kim and D. Rees, Science, 1993, 260, 792–794 CAS.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS.
- (a) Z. Li, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2005, 127, 8950–8951 CrossRef CAS; (b) B. E. Barton, C. M. Whaley, T. B. Rauchfuss and D. L. Gray, J. Am. Chem. Soc., 2009, 131, 6942–6943 CrossRef CAS; (c) Y. J. Park, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2011, 133, 9258–9261 CrossRef CAS; (d) G. Fachinetti, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1978, 100, 7405–7407 CrossRef CAS; (e) S. Gambarotta, F. Arena, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1982, 104, 5082–5092 CrossRef CAS; (f) J. P. Krogman, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582–14585 CrossRef CAS.
- (a) L. L. Efros, H. H. Thorp, G. W. Brudvig and R. H. Crabtree, Inorg. Chem., 1992, 31, 1722–1724 CrossRef CAS; (b) P.-A. Jacques, V. Artero, J. Pecaut and M. Fontecave, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20627–20632 CrossRef CAS.
- (a) B. J. Fisher and R. Eisenberg, J. Am. Chem. Soc., 1980, 102, 7361–7363 CrossRef CAS; (b) C. M. Lieber and N. S. Lewis, J. Am. Chem. Soc., 1984, 106, 5033–5034 CrossRef CAS; (c) J. P. Collin and J. P. Sauvage, Coord. Chem. Rev., 1989, 93, 245–268 CrossRef CAS; (d) E. Fujita, J. Haff, R. Sanzenbacher and H. Elias, Inorg. Chem., 1994, 33, 4627–4628 CrossRef CAS.
- (a) M. C. Helvenston and C. E. Castro, J. Am. Chem. Soc., 1992, 114, 8490–8496 CrossRef CAS; (b) G. K. Lahiri, L. J. Schussel and A. M. Stolzenberg, Inorg. Chem., 1992, 31, 4991–5000 CrossRef CAS; (c) A. Bakac and J. H. Espenson, J. Am. Chem. Soc., 1986, 108, 713–719 CrossRef CAS.
- (a) W. L. Ellefson, W. B. Whitman and R. S. Wolfe, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 3707–3710 CrossRef CAS; (b) A. Pfaltz, B. Jaun, A. Fassler, A. Eschenmoser, R. Jaenchen, H. H. Gilles, G. Diekert and R. K. Thauer, Helv. Chim. Acta, 1982, 65, 828–865 CrossRef CAS; (c) G. Färber, W. Keller, C. Kratky, B. Jaun, A. Pfaltz, C. Spinner, A. Kobelt and A. Eschenmoser, Helv. Chim. Acta, 1991, 74, 697–716 CrossRef.
- E. Uhlig and M. Friedrich, Z. Anorg. Allg. Chem., 1966, 343, 299–307 CrossRef.
- (a) R. R. Gagne and D. M. Ingle, Inorg. Chem., 1981, 20, 420–425 CrossRef CAS; (b) K. M. Kadish, M. M. Franzen, B. C. Han, C. Araullo-McAdams and D. Sazou, Inorg. Chem., 1992, 31, 4399–4403 CrossRef CAS; (c) S. P. J. Albracht, D. Ankel-Fuchs, J. W. Van der Zwaan, R. D. Fontijn and R. K. Thauer, Biochim. Biophys. Acta, 1986, 870, 50–57 CrossRef CAS; (d) B. Jaun and A. Pfaltz, J. Chem. Soc., Chem. Commun., 1986, 1327–1329 RSC.
- (a) L. Latos-Grazynski, M. M. Olmstead and A. L. Balch, Inorg. Chem., 1989, 28, 4065–4066 CrossRef CAS; (b) L. R. Furenlid, M. W. Renner, D. J. Szalda and E. Fujita, J. Am. Chem. Soc., 1991, 113, 883–892 CrossRef CAS; (c) D. J. Szalda, E. Fujita, R. Sanzenbacher, H. Paulus and H. Elias, Inorg. Chem., 1994, 33, 5855–5863 CrossRef CAS; (d) M. P. Suh, H. K. Kim, M. J. Kim and K. Y. Oh, Inorg. Chem., 1992, 31, 3620–3625 CrossRef CAS; (e) V. S. Thoi and C. J. Chang, Chem. Commun., 2011, 47, 6578–6580 RSC.
- F. Birkelbach, M. Winter, U. Floerke, H.-J. Haupt, C. Butzlaff, M. Lengen, E. Bill, A. X. Trautwein, K. Wieghardt and P. Chaudhuri, Inorg. Chem., 1994, 33, 3990–4001 CrossRef CAS.
- M. Summers, A. L. Rheingold, M. B. Allen, 1996, private communication to the CSD (CSD entry ZOHTIZ).
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- M. A. Hitchman and G. L. Rowbottom, Coord. Chem. Rev., 1982, 42, 55–132 CrossRef CAS.
- S. Kiani, R. J. Staples, S. Ted Treves and A. B. Packard, Polyhedron, 2009, 28, 775–781 CrossRef CAS.
- J. D. Roberts and W. T. Moreland, J. Am. Chem. Soc., 1953, 75, 2167–2173 CrossRef CAS.
- F. V. Lovecchio, E. S. Gore and D. H. Busch, J. Am. Chem. Soc., 1974, 96, 3109–3118 CrossRef CAS.
- L. R. Furenlid, M. W. Renner and E. Fujita, Phys. B, 1995, 208–209, 739–742 CrossRef.
- (a) K. Ray, S. DeBeer George, E. I. Solomon, K. Wieghardt and F. Neese, Chem.–Eur. J., 2007, 13, 2783–2797 CrossRef CAS; (b) R. K. Szilagyi, B. S. Lim, T. Glaser, R. H. Holm, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 9158–9169 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic and crystallographic details, and calculated geometries. CCDC 895612–895620. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21231e |
| This journal is © The Royal Society of Chemistry 2013 |