Reactions of phosphorus/boron frustrated Lewis pairs with SO2†
Muhammad Sajida, Annika Klosea, Birgit Birkmannb, Liyuan Liangb, Birgitta Schirmer‡a, Thomas Wiegand§c, Hellmut Eckert§*c, Alan J. Lough¶b, Roland Fröhlich¶a, Constantin G. Daniliuc¶a, Stefan Grimme‡*d, Douglas W. Stephan*b, Gerald Kehra and Gerhard Erker*a
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität, Corrensstr. 40, 48149 Münster, Germany
bDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada
cInstitut für Physikalische Chemie and Graduate School of Chemistry, Westfälische Wilhelms-Universität, Corrensstr. 28/30, 48149 Münster, Germany
dMulliken Center for Theoretical Chemistry, Institut für Physikalische und Theoretische Chemie, Universität Bonn, Beringstr. 4, D-53115 Bonn, Germany
First published on 11th September 2012
Abstract
The frustrated Lewis pair tBu3P/B(C6F5)3 (1) readily adds SO2 to yield the zwitterionic adduct tBu3P+–S(O)–OB−(C6F5)3 (3). A series of intramolecular vicinal P/B FLPs Mes2P–(X)–B(C6F5)2 [X = –CH2–CH2– (2a), –CHMe–CH2– (2b), cyclo-C6H10 (5)] add SO2 at −78 °C to yield the corresponding six-membered addition products 4a, 4b, 6. The adducts contain a chiral sulfur center. The [B]–O–(O)S–[P] addition products 3, 4b and 6 were characterized by X-ray diffraction.
Introduction
The advent of frustrated Lewis pair (FLP) chemistry has provided a new strategy for small molecule binding and/or activation.1 Foremost in these efforts is the ability of FLPs to heterolytically split the dihydrogen molecule.2 There is an increasing scope of substrates that can be employed in FLP-mediated hydrogenations.3 In addition, this concept has been applied to effect the stoichiometric reduction of the aromatic rings of anilines to the corresponding cyclohexylamines.4 Small molecule binding is another aspect of FLP chemistry that has garnered much attention.5 FLP binding of alkenes, alkynes and unsaturated conjugated systems6 as well as a variety of carbonyl compounds7 have been explored. Perhaps most importantly, FLPs have been shown to bind to a number of greenhouse gases, such as carbon dioxide8 or nitrogen oxides.9 Sulfur dioxide is another environmentally problematic gas.10 Herein, we describe the ability of a small series of inter- and intramolecular FLPs to capture the gas sulfur dioxide.11 The dynamic and structural implications arising from the stereochemistry at sulfur are probed.Results and discussions
The intermolecular FLP 1 reacts with sulfur dioxide at room temperature in bromobenzene to give a new product 3 in 80% yield. This compound shows a 31P NMR resonance at δ = 67.8 ppm, typical of a phosphonium center. The corresponding 11B NMR signal is seen at δ = 0.3 ppm, suggestive of four-coordinate boron. Single crystals of compound 3 suitable for the X-ray crystal structure analysis were obtained by crystallization from CH2Cl2. It confirmed that the FLP 1 had undergone 1,2-addition to a S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of sulfur dioxide to form the zwitterion tBu3P(S(O)O)B(C6F5)33. The lengths of the newly formed P1–S1 and O1–B1 bonds are found to be 2.2753(6) and 1.565(2) Å, respectively, while the S1–O1–B1 angle is 118.2(1)°. The corresponding S1–O1 bond length is 1.572(1) Å while the corresponding terminal S1–O2 bond distance is 1.465(1) Å. The angles about S sum to 314.58° (O2–S1–O1 110.07(8)°, O2–S1–P1 105.89(6)°, O1–S1–P1 98.54(5)°) consistent with a distorted trigonal pyramidal geometry at sulfur (Scheme 1, Fig. 1).
O bond of sulfur dioxide to form the zwitterion tBu3P(S(O)O)B(C6F5)33. The lengths of the newly formed P1–S1 and O1–B1 bonds are found to be 2.2753(6) and 1.565(2) Å, respectively, while the S1–O1–B1 angle is 118.2(1)°. The corresponding S1–O1 bond length is 1.572(1) Å while the corresponding terminal S1–O2 bond distance is 1.465(1) Å. The angles about S sum to 314.58° (O2–S1–O1 110.07(8)°, O2–S1–P1 105.89(6)°, O1–S1–P1 98.54(5)°) consistent with a distorted trigonal pyramidal geometry at sulfur (Scheme 1, Fig. 1).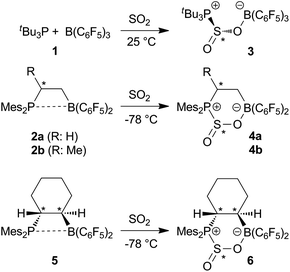 | ||
| Scheme 1 Reactions of inter- and intramolecular FLPs with SO2. | ||
 | ||
| Fig. 1 Molecular structure of compound 3 (thermal ellipsoids are shown with 50% probability). | ||
The intramolecular FLP 2a2b also cleanly adds sulfur dioxide in a rapid reaction in pentane solution at −78 °C to yield the product 4a. This was assigned the structure of the cyclic adduct (C6H2Me3)2PCH2CH2B(C6F5)2(S(O)O) (Scheme 1). The new compound shows typical heteronuclear NMR resonances at δ = 33.3 ppm (31P) and δ = 1.2 ppm (11B), respectively. Compound 4a is chiral due to the presence of the stereogenic sulfur atom. Consequently, NMR signals for the diastereotopic pair of C6F5 substituents on boron and a pair of diastereotopic mesityl groups at phosphorus are observed. In addition, the hydrogen atoms of the ethylene linker are also pairwise diastereotopic.
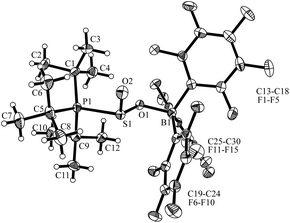
The overall structural features of this type of compounds were revealed by the X-ray crystal structure analysis of the methyl substituted derivative 4b that was formed from 2b and SO2 at −78 °C (for details see the ESI†). It features the rac-(R,SS)–4b diastereoisomer as different conformers (see Fig. 2), that shows B1A–O1A 1.574(3) Å [B1B–O1B 1.560(3) Å] and P1A–S1A 2.336(1) Å [P1B–S1B 2.347(1) Å] bond lengths inside the respective six-membered heterocyclic distorted half-chair conformations.
 | ||
| Fig. 2 Molecular structure of the two conformers of the rac-(1R,SS)-4b enantiomers found in the single crystal (thermal ellipsoids are shown with 30% probability; for clarity only the ipso-carbons of aryl ring at B and P are shown). | ||
Compound 4a shows temperature dependent dynamic NMR spectra. Coalescence of the pairs of o-, p- and m-19F NMR resonances of the B(C6F5)2 moiety was observed in the temperature range between 298 K and 339 K, indicating a rapid enantiomerization of the stereogenic sulfur atom on the NMR time scale (Scheme 2). From the dynamic 19F NMR spectra a Gibbs activation barrier of ΔG≠enant (333 K) = 15.4 ± 0.1 kcal mol−1 was calculated for this process.
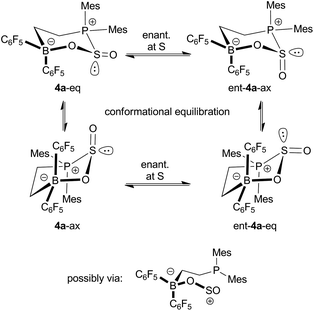 | ||
| Scheme 2 Enantiomerization and fluxional behavior of 4a. | ||
In addition a second distinct dynamic process of 4a was observed by monitoring the temperature dependent 31P NMR spectra. These spectra revealed the presence of a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture of two isomers at 183 K, giving rise to resonances at δ = 38.0 ppm and δ = 26.8 ppm. Warming the sample rapidly resulted in coalescence of the signals at 243 K. In view of the results of a theoretical study of this system (vide infra) it is likely that these spectral changes arise from the occurrence of two ring conformers, featuring the SO oxygen in the axial or equatorial position (Scheme 2). From the dynamic 31P NMR spectra an activation barrier of ΔG≠enant (223 K) = 10.0 ± 0.3 kcal mol−1 was estimated for this conformational equilibration process (see the ESI†).
:4 mixture of two isomers at 183 K, giving rise to resonances at δ = 38.0 ppm and δ = 26.8 ppm. Warming the sample rapidly resulted in coalescence of the signals at 243 K. In view of the results of a theoretical study of this system (vide infra) it is likely that these spectral changes arise from the occurrence of two ring conformers, featuring the SO oxygen in the axial or equatorial position (Scheme 2). From the dynamic 31P NMR spectra an activation barrier of ΔG≠enant (223 K) = 10.0 ± 0.3 kcal mol−1 was estimated for this conformational equilibration process (see the ESI†).
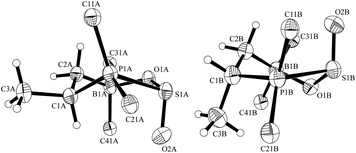
The results were augmented by a study of the reaction of the intramolecular FLP12 derived from the hydroboration of cyclohexenyldimesitylphosphane with Piers' borane [HB(C6F5)2].13 FLP 5 reacts readily with SO2 to give the cyclic adduct 6 (Scheme 1). In this case the FLP framework itself contains a pair of chiral carbon centers. The resulting stereochemistry from the hydroboration affords the single diastereoisomer that has the –PMes2 and –B(C6F5)2 groups trans-1,2-attached (rac-trans-5; this study involves only racemic compounds).14 Introduction of the stereogenic sulfur center in 6 leads to the formation of a pair of diastereomers of 6 in a ca. 1:1.3 ratio [31P NMR (193 K): δ = 60.5 ppm and δ = 41.1 ppm]. Each diastereoisomer exhibits conformational equilibria similar to those observed for the monocyclic FLP/SO2 adducts 4a/b (vide supra). At higher temperature (308 K) the reversible equilibration between the diastereoisomers of 6, by epimerization at sulfur, was evidenced by a 31P, 31P-NOESY experiment (see the ESI†). The X-ray crystal structure analysis of 6 confirmed that the phosphane component of the FLP 5 had added to the sulfur atom of sulfur dioxide. The diastereomers of 6 are present in a 3:1 ratio in the crystal (Fig. 3). The major adduct 6 shows typical bond lengths of P1–S1: 2.362(1) Å, S1–O(2A): 1.407(2) Å, S1–O1: 1.547(2) Å, O1–B1: 1.568(3) Å and bond angles C1–P1–S1: 102.80(7)°, P1–S1–O(2A): 114.82(11)°, O(2A)–S1–O1: 116.5(12)°, S1–O1–B1: 122.51(14)°, O1–B1–C6: 108.79(18)°.
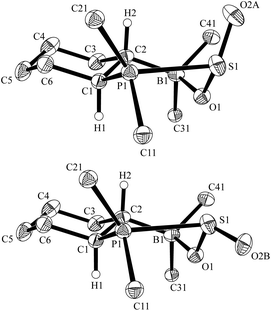 | ||
| Fig. 3 View of the molecular structures of the pair of diastereomers of the FLP-SO2 adduct 6 in the crystal (top 75%, bottom: 25%) (thermal ellipsoids are shown with 30% probability; For clarity only the ipso-carbons of aryl ring at B and P are shown). | ||
The FLP-SO2 adducts 4a and 6 were also characterized by 31P and 11B solid state NMR spectroscopy (see Fig. 4). The 31P{1H} CP MAS NMR spectrum of 4a shows a pair of resonances at 39.9 and 28.7 ppm in an intensity ratio of about 1:3 probably originating from the two [B]–O–(O)S–[P] ring conformers. The unusually broad line shape suggests some fluxional character in the solid state. The line shape simulation for the 11B MAS NMR spectrum of 4a reveals two resonances at about 1 ppm with quadrupolar coupling constants CQ of 1.6 and 1.9 MHz and nearly identical asymmetry parameters η of about 0.5. The experimental CQ and η values are much closer to those calculated for the [B]–O–(O)S–[P] isomer than to those calculated for the [B]–S(O)–O–[P] isomers 4a′-ax. and 4a′-eq. (see Table 1). Thus, the formation of the latter isomers can be ruled out for all the SO2 adducts studied here.
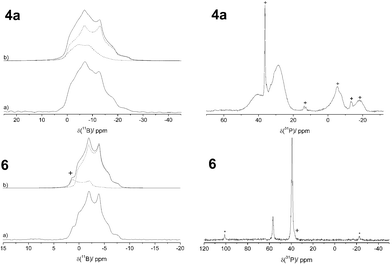 | ||
| Fig. 4 Left: 11B{1H} MAS NMR spectra of 4a and 6 (a) acquired at 7.1 T and 11.7 T, respectively, with their corresponding line shape simulations (b). Right: 31P{1H} CP MAS spectrum of 4a acquired at 7.1 T and 31P{1H} MAS NMR spectrum of 6 acquired at 9.4 T. * mark spinning sidebands, + impurities. | ||
| δisoCS/ppm ±0.5 | CQ/MHz ±10% | η ±0.1 | δisoCS/ppm (calc.) | CQ/MHz (calc.) | η (calc.) | |
|---|---|---|---|---|---|---|
| a Fully geometry optimized structure of 4a-ax.b Fully geometry optimized structure of 4a-eq.c Fully geometry optimized structure of 4a′-ax.d Fully geometry optimized structure of 4a′-eq.e Geometry of 6-ax. taken from the crystal structure.f Geometry of 6-eq. taken from the crystal structure.g Fully geometry optimized structure of 6′-ax.h Fully geometry optimized structure of 6′-eq.i Assigned to an impurity. | ||||||
| 4a | 1.0 | 1.62 | 0.46 | 1.6a | 2.21a | 0.35a |
| 1.0 | 1.92 | 0.48 | 2.2b | 2.15b | 0.37b | |
| −4.4c | 1.16c | 0.71c | ||||
| −5.4d | 1.00d | 0.56d | ||||
| 6 | 2.5i | 1.80i | 0.01i | −2.6e | 1.95e | 0.43e |
| 0.9 | 1.88 | 0.50 | 0.4f | 2.02f | 0.44f | |
| −2.8g | 1.05g | 0.74g | ||||
| −1.5h | 1.25h | 0.57h | ||||
Fig. 5 shows results from 11B{31P} Rotational Echo DOuble Resonance (REDOR)15,16 experiments for measuring the boron-phosphorus distances in 4a and 6. In contrast to previous results on non-reacted FLPs with much shorter B⋯P distances,16 no dipolar oscillation typical of spin pairs are seen in these curves. Most likely, these oscillations are damped out due to subtle differences in the B⋯P distances for the different conformers and/or slow dynamic processes as suspected for 4a. For the distance determination, we use the fact that at short evolution times (ΔS/S0 < 0.25) these curves are well approximated by parabolae, whose curvatures are proportional to the squared dipolar coupling constants.17 With the B⋯P internuclear distance for 6 known from the crystal structure (3.463(4) Å) we can use the REDOR curve of this compound (Fig. 5a) for calibrating analogous results obtained on 4a. We find a boron-phosphorus distance of 3.72 Å ± 0.2 Å in good agreement with the theoretically expected value (3.495 Å/3.506 Å) in the gas phase. Within the experimental error limit it also agrees well with the B⋯P internuclear distances of 3.501(2) Å and 3.537(6) Å for the two conformers determined from the crystal structure of 4b. The 31P{1H} MAS NMR spectrum of a sample of 6 reveals two resonances at 56.5 and 39.1 ppm, respectively, close to the respective 31P NMR chemical shifts found for the pair of diastereoisomers of 6 in solution (see above). In the solid state these two species were initially formed in a ca. 1:5 ratio which over a period of 25 days changed to 1:10. In contrast to 4a both diastereomers possess nearly identical 11B quadrupolar coupling parameters since only one resonance is detected in the 11B MAS NMR spectrum.
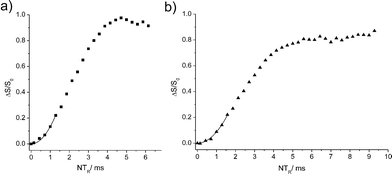 | ||
| Fig. 5 11B{31P} REDOR curves for 6 (a) and 4a (b) and corresponding parabolic approximations in the limit of short evolution times (for more details see ESI†). | ||
The FLP + SO2 reaction was investigated by state of the art DFT calculations [B2PLYP-D3/def2-TZVP//TPSS-D3/def2-QZVPP + COSMO-RS solvent corrections,18–23 estimated accuracy absolute about 1 kcal mol−1 for free enthalpies (for further details see the ESI†)]. As there were no X-ray data available for compound 4a several structural alternatives were tested. We investigated the observed [B]–O–(O)S–[P] conformational isomers 4a-eq. and 4a-ax. (see Table 2 and Fig. 6) as well as their hypothetical [B]–S(O)–O–[P] regioisomers (4a′-eq. and 4a′-ax.). The latter were found to be slightly less favourable than the observed 4a-eq./ax. pair (calculated for a bromobenzene solution). In addition the formation of the alternative five-membered isomer, [B]–S(O)2–[P], was computed to be highly thermodynamically unfavourable with reaction (free) enthalpies of ca. 7–14 kcal mol−1 higher than for any of the six-membered products.
| Conformer | 4a | rac-(R,R,SR) 6 |
|---|---|---|
| [B]–O–(O)S–[P], eq. | −7.9 | −6.2 |
| [B]–O–(O)S–[P], ax. | −7.7 | −6.7 |
| [B]–S(O)–O–[P], eq. | −3.6 | −5.1 |
| [B]–S(O)–O–[P], ax. | −7.1 | −4.5 |
| [B]–S(O)2–[P] | 6.7 | 2.5 |
![The DFT calculated [B]–O–(O)S–[P] isomers of compound 4a: (a) with Oexocycl. equatorial and (b) with Oexocycl. axial (color code: C grey, P orange, B purple, S yellow, O red, F green, H white).](/image/article/2013/SC/c2sc21161k/c2sc21161k-f6.gif) | ||
| Fig. 6 The DFT calculated [B]–O–(O)S–[P] isomers of compound 4a: (a) with Oexocycl. equatorial and (b) with Oexocycl. axial (color code: C grey, P orange, B purple, S yellow, O red, F green, H white). | ||
The relative free enthalpies of all four six-membered isomers of 4a (and similarly of 6 see Table 2) fall in a rather narrow range of about 4 kcal mol−1 for 4a and 2 kcal mol−1 for 6. The [B]–O–(O)S–[P] isomer 6-ax. is computed to be the thermodynamically most stable product. Although this would suggest the observed regioselectivity is under thermodynamic control, it should be noted that the error of the computations infer the first two isomers of 6 (and 1,2,4 of 4a) are more or less isoexergonic.
We investigated the possible pathways of the observed facile enantiomerization process at sulfur in 4a by DFT and found that a true inversion process at the sulfur atom would be far too high in energy (∼50 kcal mol−1). However, due to the calculations rapid reversible P–S bond rupture provides a viable pathway of enantiomerization at sulfur in such a system.
Conclusions
This study shows that FLPs react with remarkable ease with sulfur dioxide. The formation of the respective [B]–O–(O)S–[P] isomers has been observed by X-ray diffraction in three cases and by 11B solid state NMR/DFT calculations in two selected cases although the [B]–S(O)–O–[P] isomers are thermodynamically feasible according to the DFT calculations. FLP-SO2 adduct formation has remarkably low kinetic barriers, making these adducts readily available for further reactivity studies.Experimental
Synthesis of compound 3
A solution of B(C6F5)3 (25 mg, 0.049 mmol) and tBu3P (10 mg, 0.049 mmol) in C6H5Br (1 mL) was degassed and filled with SO2 (1 bar). The sample was sealed and allowed to react overnight to afford tBu3PS(O)OB(C6F5)3 (3) as a white powder (31 mg, 0.039 mmol, 80%). Crystals suitable for X-ray crystal structure analysis were obtained by crystallization from CH2Cl2. Anal. calcd (%) for C30H27BF15O2PS: C, 46.29; H, 3.50; found: C, 45.84; H, 3.40. 1H NMR (400 MHz, 298 K, [d2]-dichloromethane) δ = 1.43 (d, JPH = 14.1 Hz, 27H, tBu). 11B NMR (128 MHz) δ = 0.3 (ν1/2 ∼ 200 Hz). 31P NMR (162 MHz) δ = 67.8.X-ray crystal structure analysis of 3
Formula C30H27BF15O2PS, M = 778.36, colourless crystal, 0.24 × 0.22 × 0.19 mm, a = 9.4942(4), b = 29.0008(13), c = 11.5368(5) Å, β = 90.018(2)°, V = 3176.5(2) Å3, ρcalc = 1.628 g cm−3, μ = 0.270 mm−1, empirical absorption correction (0.937 ≤ T ≤ 0.950), Z = 4, monoclinic, space group P21/c (No. 14), λ = 0.71073 Å, T = 150(2) K, ω and φ scans, 30506 reflections collected (±h, ±k, ±l), 8222 independent (Rint = 0.040) and 6900 observed reflections [I > 2σ(I)], 452 refined parameters, R = 0.034, wR2 = 0.072, max. (min.) residual electron density 0.37 (−0.43) e Å−3, hydrogen atoms calculated and refined as riding atoms.Preparation of compound 4a
Compound 2a, was generated in situ from dimesitylvinylphosphane (100 mg, 0.34 mmol, 1.0 eq.) and bis(pentafluorophenyl)-borane (117 mg, 0.34 mmol, 1.0 eq.) in n-pentane. SO2 was introduced (−78 °C, 0.8 bar, 10 min.). After stirring the reaction mixture for 1.5 h workup gave compound 4a (198 mg, 83%) as a white solid. M.p.: 103 °C. Anal. calcd (%) for C32H26BF10O2PS: C, 54.41; H, 3.71; found C, 54.88; H, 3.91. 1H NMR (500 MHz, 298 K, [d2]-dichloromethane): δ = 7.00 (d, 4JPH = 3.6 Hz, 4H, m-MesA), 6.99 (d, 4JPH = 3.9 Hz, 4H, m-MesB), 3.75/2.93 (each m, each 1H, CH2P), 2.45 (br s, 6H, o-CH3MesA), 2.41 (br s, 6H, o-CH3MesB), 2.31 (s, 6H, p-CH3MesA,B), 1.88/1.23 (each m, each 1H, CH2B). 13C{1H} NMR (126 MHz): δ = 144.9, 144.8 (each s, p-MesA,B), 144.1 (d, 2JPC = 7.0 Hz, o-MesB), 143.4 (d, 2JPC = 5.6 Hz, o-MesA), 132.2 (d, 3JPC = 10.8 Hz, m-MesB), 131.6 (d, 3JPC = 9.8 Hz, m-MesA), 122.2 (d, 1JPC = 39.3 Hz, i-MesA), 118.3 (d, 1JPC = 52.5 Hz, i-MesB), 23.4 (br, o-CH3MesA), 23.1 (br, o-CH3MesB), 22.7 (d, 1JPC = 22.9 Hz, CH2P), 21.3, 21.2 (each s, p-CH3MesA,B), 11.4 (br, CH2B), [C6F5 not listed]. 11B{1H} NMR (160 MHz): δ = 1.2 (ν1/2 ≈ 400 Hz). 19F NMR (470 MHz): δ = −134.9 (m, 2F, o), −160.6 (m, 1F, p), −165.7 (m, 2F, m), Δδ19Fp,m = 5.1 [C6F5A]; −135.5 (m, 2F, o), −160.7 (m, 1F, p), −165.7 (m, 2F, m), Δδ19Fp,m = 4.7 [C6F5B]. 31P{1H} NMR (202 MHz): δ = 33.3 (ν1/2 ≈ 20 Hz).Synthesis of compound 4b
Compound 2b, was generated in situ from (1-methylethenyl)di-mesitylphosphane (24.8 mg, 0.08 mmol, 1.0 eq.) and bis(pentafluorophenyl)borane (27.6 mg, 0.08 mmol, 1.0 eq.) in toluene (2 mL). The solution was briefly exposed to sulfur dioxide at −78 °C (5 min, 1.8 bar, this provided for minimal condensation of SO2). The reaction mixture was stirred for 45 min. The excess SO2 was vented and the obtained reaction mixture was covered with precooled n-pentane (5 mL) and stored at −36 °C to finally get compound 4b as colorless crystals which were suitable for X-ray crystal structure analysis. Two diastereomers of 4b were found in [d2]-dichloromethane solution at 299 K (ratio: 84/16). M.p.: 126 °C. 11B{1H} NMR (160 MHz, CD2Cl2, 299 K): δ = 1.3 (ν1/2 ≈ 450 Hz). 31P{1H} NMR (202 MHz, CD2Cl2, 299 K): δ = 51.2 (ν1/2 ≈ 10 Hz, 16%), 39.5 (ν1/2 ≈ 10 Hz, 84%). 31P{1H} NMR (202 MHz, CD2Cl2, 198 K): δ = 55.4 (ν1/2 ≈ 15 Hz, 8%), 50.3 (ν1/2 ≈ 35 Hz, 3%), 39.0 (ν1/2 ≈ 25 Hz, 58%), 37.8 (ν1/2 ≈ 30 Hz, 31%). Major diastereomer [84%]:1H NMR (500 MHz, CD2Cl2, 299 K): δ = 7.02 (d, 4JPH = 3.6 Hz, 2H, m-MesA), 7.00 (d, 4JPH = 3.6 Hz, 2H, m-MesB), 4.14 (m, 1H, CHP), 2.38 (br, 6H, o-CH3MesA), 2.35 (br, 6H, o-CH3MesB), 2.325 (br m, 3H, p-CH3MesA), 2.318 (br m, 3H, p-CH3MesB), 1.82 (dd, 3JPH = 27.8 Hz, 3JHH = 16.4 Hz, 1H, CH2B), 1.41 (dd, 3JPH = 18.1 Hz, 3JHH = 7.0 Hz, 3H, CH3P), 1.30 (m, 1H, CH2B). 13C{1H} NMR (126 MHz): δ = 144.6 (d, 4JPC = 3.1 Hz, p-MesA), 144.3 (d, 4JPC = 3.8 Hz p-MesB), 144.0 (d, 2JPC = 5.6 Hz o-MesA), 143.6 (br d, 2JPC = 6.6 Hz o-MesB), 132.5 (br d, 3JPC = 11.4 Hz, m-MesB), 131.69 (d, 3JPC = 9.9 Hz, m-MesA), 120.4 (d, 1JPC = 49.6 Hz, i-MesB), 120.3 (d, 1JPC = 36.1 Hz, i-MesA) 31.1 (d, 1JPC = 15.0 Hz, CHP), 24.6 (br, o-CH3MesA)t, 23.3 (d, 3JPC = 4.1 Hz, o-CH3MesB)t, 22.3 (br d, 2JPC = 7.9 Hz, CH3P), 21.19, 21.18 (each d, each 5JPC = 1.8 Hz, p-CH3MesA,B), 21.1 (br, CH2B), [C6F5 not listed]. 19F NMR (470 MHz): δ = −135.4 (m, 2F, o), −160.6 (t, 1JFC = 20.4 Hz, 1F, p), −165.7 (m, 2F, m), Δδ19Fp,m = 5.1 [C6F5A]; −135.6 (m, 2F, o), −161.1 (t, 1JFC = 20.4 Hz, 1F, p), −165.5 (m, 2F, m), Δδ19Fp,m = 4.4 [C6F5B]. Minor diastereomer [16%]:1H NMR (500 MHz): δ = 7.04 (d, 4JPH = 3.7 Hz, 2H, m-MesA), 7.01 (d, 4JPH = 3.6 Hz, 2H, m-MesB), 3.36 (m, 1H, CHP), 2.37 (br, 6H, o-CH3MesA), 2.34 (br, 6H, p-CH3MesA,B), 2.27 (br, 6H, o-CH3MesB), 2.04 (m, 1H, CH2B), 1.68 (dd, 3JPH = 27.5 Hz, 3JHH = 16.4 Hz, 1H, CH2B), 1.406 (dd, 3JPH = 17.9 Hz, 3JHH = 7.1 Hz, 3H, CH3P). 13C{1H} NMR (126 MHz): δ = 144.4 (d, 4JPC = 3.3 Hz), 144.3t (d, 4JPC = 3.8 Hz) (p-MesA,B), 144.1 (d, 2JPC = 6.3 Hz, o-MesA), 142.9 (d, 2JPC = 6.3 Hz, o-MesB), 132.2 (d, 3JPC = 10.6 Hz, m-MesB), 131.67 (d, 3JPC = 10.8 Hz, m-MesA), 121.5 (d, 1JPC = 44.8 Hz, i-MesB), 121.1 (d, 1JPC = 34.5 Hz, i-MesA), 33.6 (d, 1JPC = 11.7 Hz, CHP), 24.4 (br, o-CH3MesA), 23.7 (d, 3JPC = 3.5 Hz, o-CH3MesB), 21.6 (d, 2JPC = 12.6 Hz, CH3P), n.o. (p-CH3Mes), 20.2 (br, CH2B), [C6F5 not listed; t tentative assignment]. 19F NMR (470 MHz): δ = −134.5 (m, 2F, o), −161.0 (t, 1JFC = 20.4 Hz, 1F, p), −165.9 (m, 2F, m), Δδ19Fp,m = 5.1 [C6F5A]; −134.7 (m, 2F, o), −159.9 (t, 1JFC = 20.4 Hz, 1F, p), −165.0 (m, 2F, m), Δδ19Fp,m = 4.4 [C6F5B].X-ray crystal structure analysis of 4b
Formula C33H28BF10O2PS, M = 720.39, colourless crystal, 0.20 × 0.13 × 0.05 mm, a = 13.2898(3), b = 13.3521(6), c = 22.5790(9) Å, α = 73.392(3), β = 73.073(3), γ = 83.328(4)°, V = 3670.7(2) Å3, ρcalc. = 1.304 g cm−3, μ = 1.903 mm−1, empirical absorption correction (0.702 ≤ T ≤ 0.910), Z = 4, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), λ = 1.54178 Å, T = 223(2) K, ω and φ scans, 53257 reflections collected (±h, ±k, ±l), 12647 independent (Rint = 0.043) and 10730 observed reflections [I > 2σ(I)], 877 refined parameters, R = 0.049, wR2 = 0.145, max. (min.) residual electron density 0.35 (−0.32) e.Å−3, hydrogen atoms calculated and refined as riding atoms.
(no. 2), λ = 1.54178 Å, T = 223(2) K, ω and φ scans, 53257 reflections collected (±h, ±k, ±l), 12647 independent (Rint = 0.043) and 10730 observed reflections [I > 2σ(I)], 877 refined parameters, R = 0.049, wR2 = 0.145, max. (min.) residual electron density 0.35 (−0.32) e.Å−3, hydrogen atoms calculated and refined as riding atoms.Synthesis of compound 6
Procedure A: Di(mesityl)cyclohexenylphosphane (120 mg, 0.34 mmol) and HB(C6F5)2 (119 mg, 0.34 mmol) dissolved in toluene (2.5 mL) and stirred for 30 min at r.t. to give a yellow solution of 5. The solution was cooled to −78 °C and SO2 gas was pressurized (2.0 bar) for 3 minutes. Then the solution was covered with n-pentane (−30 °C) to finally get compound 6 (136 mg, 52 %) as colourless crystals, which were suitable for X-ray crystal structure analysis. Procedure B: Di(mesityl)cyclohexenylphosphane (250 mg, 0.71 mmol) and HB(C6F5)2 (247 mg, 0.71 mmol) dissolved in n-pentane (15 mL) and stirred for 30 min at r.t. to give a yellow solution of 5. The solution was cooled to −78 °C and SO2 gas was pressurized (2.0 bar) for 5 minutes. The solvent was removed and the residue was dried at −78 °C. The colourless residue was dissolved in precooled CH2Cl2 and covered with precooled (−30 °C) n-pentane to finally get compound 6 (310 mg, 57 %) as colourless crystals. Anal. calcd (%) for C36H32PBSO2F10: C, 56.86; H, 4.24; found, C; 56.30; H, 4.06. The NMR resonances were not assigned to each diastereoisomer [1:1.3 (31P)] 1H NMR (600 MHz, CD2Cl2, 193 K) [all resonances are broad] δ = 7.18/6.97, 7.14/6.93, 7.09/6.80, 7.05/6.87 (each br s, each 1H, m,m'-Mes)1, 3.82, 3.47 (each br., each 1H, PCH), 2.79/2.24(p)/1.64, 2.74/2.30(p)/1.51, 2.67/2.31(p)/1.87, 2.54/2.25(p)/1.74 (each s, each 3H, CH3Mes)1, 2.25/1.23, 2.07/0.62, 1.92/0.61, 1.88/1.35, 1.60/1.22, 1.58/1.77, 1.56/1.28, 1.42/0.99 (each br, each 1H, CH2)2, 2.31, 1.85 (each br, each 1H, BH)2, [1 from the gcosy NMR experiment;2 from the ghsqc NMR experiment]. 11B{1H} NMR (192 MHz, CD2Cl2, 308 K): δ = 1.8 (ν1/2 ≈ 500 Hz). 31P{1H} NMR (243 MHz, CD2Cl2, 193 K): δ = 60.5 (ν1/2 ≈ 30 Hz, [42%]), 41.1 (ν1/2 ≈ 40 Hz, [58%]). 19F NMR (564 MHz, CD2Cl2, 193 K): δ = −128.6, −130.8, −131.7, −132.3, −132.47, −132.53, − 136.3, −139.0 (each br, each 1F, o-C6F5), −158.1, −159.3, −159.7, −160.1 (each br, each 1F, p-C6F5), −163.6 (1F), −163.9 (1F), −164.5 (5F), −164.9 (1F) (each br, m-C6F5).X-ray crystal structure analysis of 6
Formula C36H32BF10O2PS, M = 760.46, colourless crystal, 0.28 × 0.15 × 0.06 mm, a = 10.5993(4), b = 11.8650(4), c = 14.4165(9) Å, α = 92.700(3), β = 95.322(2), γ = 111.030(13)°, V = 1678.66(13) Å3, ρcalc. = 1.504 g cm−3, μ = 2.113 mm−1, empirical absorption correction (0.589 ≤ T ≤ 0.883), Z = 2, triclinic, space group P (no. 2), λ = 1.54178 Å, T = 223(2) K, ω and φ scans, 19387 reflections collected (±h, ±k, ±l), 5692 independent (Rint = 0.048) and 4822 observed reflections [I > 2σ(I)], 476 refined parameters, R = 0.043, wR2 = 0.116 max. (min.) residual electron density 0.24 (−0.29) e Å−3, hydrogen atoms calculated and refined as riding atoms.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft and the European Research Council is gratefully acknowledged. DWS gratefully acknowledges the financial support of NSERC of Canada, the award of a Canada Research Chair. TW thanks the Fonds der Chemischen Industrie for a research fellowship and acknowledges additional support by the NRW Forschungsschule Molecules and Materials.Notes and references
- D. W. Stephan and G. Erker, Angew. Chem., 2010, 122, 50 (Angew. Chem., Int. Ed., 2010, 49, 46) CrossRef.
- (a) G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS; (b) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC. See also: (c) S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., 2010, 122, 1444 (Angew. Chem., Int. Ed., 2010, 49, 1402) CrossRef.
- (a) P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich and G. Erker, Angew. Chem., 2008, 120, 7654 (Angew. Chem., Int. Ed., 2008, 47, 7543) CrossRef; (b) D. Chen and J. Klankermayer, Chem. Commun., 2008, 2130 RSC; (c) G. Erős, H. Mehdi, I. Pápai, T. A. Rokob, P. Király, G. Tárkányi and T. Soós, Angew. Chem., 2010, 122, 6709 (Angew. Chem., Int. Ed., 2010, 49, 6559) CrossRef; (d) B.-H. Xu, G. Kehr, R. Fröhlich, B. Wibbeling, B. Schirmer, S. Grimme and G. Erker, Angew. Chem., 2011, 123, 7321 (Angew. Chem., Int. Ed., 2011, 50, 7183) CrossRef; (e) S. Schwendemann, R. Fröhlich, G. Kehr and G. Erker, Chem. Sci., 2011, 2, 1842 RSC; (f) D. W. Stephan, S. Greenberg, T. W. Graham, P. Chase, J. J. Hastie, S. J. Geiger, J. M. Farrell, C. C. Brown, Z. M. Heiden, G. C. Welch and M. Ullrich, Inorg. Chem., 2011, 50, 12338 CrossRef CAS.
- T. Mahdi, Z. M. Heiden, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 4088 CrossRef CAS.
- (a) D. W. Stephan, Dalton Trans., 2009, 3129 RSC; (b) D. W. Stephan, Chem. Commun., 2010, 46, 8526 RSC; (c) G. Erker, C. R. Chim., 2011, 14, 831 CrossRef CAS.
- (a) M. Ullrich, K. Seto, A. J. Lough and D. W. Stephan, Chem. Commun., 2009, 2335 RSC; (b) A. Stirling, A. Hamza, T. A. Rokob and I. Pápai, Chem. Commun., 2008, 3148 RSC; (c) Y. Guo and S. Li, Eur. J. Inorg. Chem., 2008, 2501 CrossRef CAS; (d) C. M. Mömming, S. Frömel, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, J. Am. Chem. Soc., 2009, 131, 12280 CrossRef; (e) M. A. Doreen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396 CrossRef; (f) C. M. Mömming, G. Kehr, B. Wibbeling, R. Fröhlich, B. Schirmer, S. Grimme and G. Erker, Angew. Chem., 2010, 122, 2464 (Angew. Chem., Int. Ed., 2010, 49, 2414) CrossRef; (g) C. Chen, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2010, 46, 3580 RSC.
- (a) B. A. Arbuzov, G. N. Nikonov, A. S. Balueva, R. M. Kamalov, M. A. Pudovik, R. R. Shagidullin, A. K. Plyamovatyi and R. S. Khadiullin, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1991, 40, 2099 (Izv. Akad. Nauk SSSR, Ser. Khim., 1991, 2393) CrossRef; (b) A. S. Balueva, G. N. Nikonov, B. A. Arbuzov, R. Z. Musin and Yu. Ya. Ezremov, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1991, 40, 2103 (Izv. Akad. Nauk SSSR, Ser. Khim., 1991, 2397) CrossRef; (c) M. W. P. Bebbington, S. Boutemps, G. Bouhadir and D. Bourissou, Angew. Chem., 2007, 119, 3397 (Angew. Chem., Int. Ed., 2007, 64, 3333) CrossRef; (d) S. Moebs-Sanchez, G. Bouhadir, N. Saffon, L. Maron and D. Bourissou, Chem. Commun., 2008, 3435 RSC; (e) C. M. Mömming, G. Kehr, B. Wibbeling, R. Fröhlich and G. Erker, Dalton Trans., 2010, 39, 7556 RSC; (f) A. Stute, G. Kehr, R. Fröhlich and G. Erker, Chem. Commun., 2011, 47, 4288 RSC; (g) C. Rosorius, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, Organometallics, 2011, 30, 4211 CrossRef CAS.
- (a) C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., 2009, 121, 6770 (Angew. Chem., Int. Ed., 2009, 48, 6643) CrossRef; (b) I. Peuser, R. C. Neu, X. Zhao, M. Ulrich, B. Schirmer, J. Tannert, G. Kehr, R. Fröhlich, S. Grimme, G. Erker and D. W. Stephan, Chem.–Eur. J., 2011, 17, 9640 CrossRef CAS; (c) M. Harhausen, R. Fröhlich, G. Kehr and G. Erker, Organometallics, 2012, 31, 2801 CrossRef CAS.
- (a) E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918 CrossRef CAS; (b) A. J. P. Cardenas, B. J. Culotta, T. H. Warren, S. Grimme, A. Stute, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., 2011, 123, 7709 (Angew. Chem., Int. Ed., 2011, 50, 7567) CrossRef; (c) M. Sajid, A. Stute, A. J. P. Cardenas, B. J. Culotta, J. A. M. Hepperle, T. H. Warren, B. Schirmer, S. Grimme, A. Studer, C. G. Daniliuc, R. Fröhlich, J. L. Petersen, G. Kehr and G. Erker, J. Am. Chem. Soc., 2012, 134, 10156 CrossRef CAS.
- (a) D. H. Ehhalt, Phys. Chem. Chem. Phys., 1999, 1, 5401 RSC; (b) M. Vahedpour and F. Zolfaghari, Struct. Chem., 2011, 22, 1331 CrossRef CAS.
- For other trapping reactions of SO2 see e.g. (a) G. J. Kubas, Acc. Chem. Res., 1994, 27, 183 CrossRef CAS; (b) R. Mews, E. Lork, P. G. Watson and B. Görtler, Coord. Chem. Rev., 2000, 197, 277 CrossRef CAS; (c) M. K. Denk, K. Hatano and A. J. Lough, Eur. J. Inorg. Chem., 2003, 224 CrossRef CAS; (d) W. A. Schenk, Dalton Trans., 2011, 40, 1209 RSC; (e) F. Lavigne, E. Maerten, G. Alcaraz, V. Branchadell, N. Saffon-Merceron and A. Baceiredo, Angew. Chem., 2012, 124, 2539 (Angew. Chem., Int. Ed., 2012, 51, 2489) CrossRef; (f) P. Benndorf, S. Schmitt, R. Köppe, P. Oña-Burgos, A. Scheurer, K. Meyer and P. W. Roesky, Angew. Chem., 2012, 124, 5091 (Angew. Chem., Int. Ed., 2012, 51, 5006) CrossRef; (g) for amine/SO2 adducts see e.g. H. Woolven, C. González-Rodríguez, I. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876 CrossRef CAS and references cited therein.
- K. Axenov, C. M. Mömming, G. Kehr, R. Fröhlich and G. Erker, Chem.–Eur. J., 2010, 16, 14069 CrossRef CAS.
- (a) D. J. Parks, R. E. V. H. Spence and W. E. Piers, Angew. Chem., 1995, 107, 895 (Angew. Chem., Int. Ed. Engl., 1995, 34, 809) CrossRef; (b) R. E. V. H. Spence, D. J. Parks, W. E. Piers, M.-A. MacDonald, M. J. Zaworotko and S. J. Rettig, Angew. Chem., 1995, 107, 1337 (Angew. Chem., Int. Ed. Engl., 1995, 34, 1230) CrossRef; (c) W. E. Piers and T. Chivers, Chem. Soc. Rev., 1997, 26, 345 RSC; (d) R. E. V. H. Spence, W. E. Piers, Y. Sun, M. Parvez, L. R. MacGillivray and M. J. Zaworotko, Organometallics, 1998, 17, 2459 CrossRef CAS.
- V. Prelog and G. Helmchen, Angew. Chem., 1982, 94, 614 (Angew. Chem., Int. Ed. Engl., 1982, 21, 567) CrossRef CAS.
- T. Gullion and J. Schaefer, J. Magn. Reson., 1989, 81, 196 CAS.
- T. Wiegand, H. Eckert, O. Ekkert, R. Fröhlich, G. Kehr, G. Erker and S. Grimme, J. Am. Chem. Soc., 2012, 134, 4236 CrossRef CAS.
- Y. Pan, T. Gullion and J. Schaefer, J. Magn. Reson., 1990, 90, 330 CAS.
- (a) R. Ahlrichs, et al., TURBOMOLE, version 6.3, Universität Karlsruhe, 2011, see, http://www.turbomole.com Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef.
- A. Klamt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 699 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: For additional experimental details, further structural information, additional data from the solid state NMR and theoretical studies. CCDC 895193–895195. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21161k |
| ‡ Computational chemistry. |
| § Solid state NMR. |
| ¶ X-ray crystal analyses. |
| This journal is © The Royal Society of Chemistry 2013 |