DOI:
10.1039/C2SC21163G
(Edge Article)
Chem. Sci., 2013,
4, 118-124
Photocatalytic generation of hydrogen from water using a cobalt pentapyridine complex in combination with molecular and semiconductor nanowire photosensitizers†
Received
3rd August 2012
, Accepted 5th September 2012
First published on 6th September 2012
Abstract
Recently, a family of cobalt pentapyridine complexes of the type [(R-PY5Me2)Co(H2O)])(CF3SO3)2, (R = CF3, H, or NMe2; PY5Me2 = 2,6-bis(1,1-di(pyridin-2-yl)ethyl)pyridine) were shown to catalyze the electrochemical generation of hydrogen from neutral aqueous solutions using a mercury electrode. We now report that the CF3 derivative of this series, [(CF3PY5Me2)Co(H2O)](CF3SO3)2 (1), can also operate in neutral water as an electrocatalyst for hydrogen generation under soluble, diffusion-limited conditions on a glassy carbon electrode, as well as a photocatalyst for hydrogen production using either molecular or semiconductor nanowire photosensitizers. Owing to its relatively low overpotential compared to other members of the PY5 family, complex 1 exhibits multiple redox features on glassy carbon, including a one-proton, one-electron coupled oxidative wave. Further, rotating disk electrode voltammetry measurements reveal the efficacy of 1 as a competent hydrogen evolution catalyst under soluble, diffusion-limited conditions. In addition, we establish that 1 can also generate hydrogen from neutral water under photocatalytic conditions with visible light irradiation (λirr ≥ 455 nm), using [Ru(bpy)3]2+ as a molecular inorganic chromophore and ascorbic acid as a sacrificial donor. Dynamic light scattering measurements show no evidence for nanoparticle formation for the duration of the photolytic hydrogen evolution experiments. Finally, we demonstrate that 1 is also able to enhance the hydrogen photolysis yield of GaP nanowires in water, showing that this catalyst is compatible with solid-state photosensitizers. Taken together, these data establish that the well-defined cobalt pentapyridine complex [(CF3PY5Me2)Co(H2O)]2+ is a versatile catalyst for hydrogen production from pure aqueous solutions using either solar or electrical input, providing a starting point for integrating molecular systems into sustainable energy generation devices.
Introduction
The combination of rising global energy demands, diminishing fossil fuel stores, and climate change has prompted intense interest in developing alternative carbon-neutral energy technologies. Harnessing solar energy to synthesize sustainable chemical fuels is a promising solution to the emerging energy challenge.1–5 An appealing approach to this ultimate goal is to drive chemical water splitting to hydrogen and oxygen using solar energy input,6 since the generation and combustion of hydrogen from water is carbon neutral and sunlight is a sustainable energy source.
A key challenge for water splitting is developing catalysts for the direct and efficient production of hydrogen from protons. Platinum and other heterogeneous precious metal catalysts have been studied for hydrogen generation for decades, but ultimately suffer from high cost and low abundance.7–9 Alternatively, solid-state catalysts composed of earth-abundant elements, such as metal alloys and molybdenum sulfides, have also been investigated.5,10–19 However, it remains a challenge to rationally assess precise structure–activity relationships in these heterogeneous systems. Enzymes like hydrogenases that utilize earth-abundant metals offer an attractive approach to hydrogen evolution catalysis with high catalytic activity and efficiency; however, it is difficult to integrate hydrogenases into solar water-splitting devices owing to their large size and relatively long-term instability. Enzyme mimics provide exquisite insight into biological systems and lay the groundwork for new catalyst design principles, but they often function in organic solvents with organic acids.20–23 As such, a growing number of abiotic molecular catalysts for electrocatalytic and photocatalytic hydrogen production featuring earth-abundant elements,24 including iron,25–27 cobalt,28–37 nickel,38–42 and molybdenum,43–45 are being reported.
An important step forward for the field of H2 catalysis is the development of systems that can operate in aqueous media, because using water as both the substrate and solvent increases substrate concentration while minimizing organic additives and waste by-products. As a molecular approach toward this end, we recently reported that a cobalt pentapyridine complex, [(PY5Me2)Co(H2O)](CF3SO3)2 (2; PY5Me2 = 2,6-bis(1,1-di(pyridin-2-yl)ethyl)pyridine; see Fig. 1) is a robust and efficient electrocatalyst for hydrogen evolution in pH 7 buffer on a mercury electrode, albeit at fairly high overpotential.46 However, the tunability of the PY5Me2 platform allowed us to modify the para-position of its central pyridine and synthesize the derivative [(CF3PY5Me2)Co(H2O)](CF3SO3)2 (1), which showed a positive shift in both the Co(II)/Co(I) reduction potential and the overpotential for H2 catalysis. To demonstrate that the performance of this catalyst is not restricted to electrochemistry on mercury, which can adsorb molecular species,28 we now report that 1 is indeed a versatile system for electro- and photochemical H2 generation in water. In particular, we show that it is a competent electrocatalyst under soluble, diffusion-limited conditions using a glassy carbon electrode, and, importantly, that it can function as a photocatalyst in combination with either a simple molecular chromophore such as [Ru(bpy)3]2+ or a semiconductor GaP nanowire photosensitizer system.47
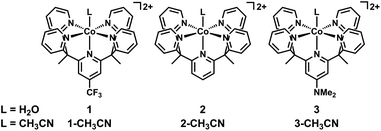 |
| | Fig. 1 Molecular cobalt pentapyridine complexes for catalytic H2 generation. | |
Results and discussion
Catalytic hydrogen generation on a glassy carbon electrode
Since the exogenous ligand L at the apical position of the PY5-cobalt complexes is exchangeable and sensitive to solvent, we chose to synthesize 1-CH3CN for electrochemical studies in acetonitrile, while using the aquo complex 1 for experiments conducted in aqueous media. Employing these complexes avoids possible solvent contamination in electrochemical studies. Similar to our previous report, metalation of CF3PY5Me2 with Co(CF3SO3)2(MeCN)2 in acetonitrile at room temperature results in the formation of 1-CH3CN, the crystal structure of which is shown in Fig. S1.†46 In agreement with the reported structure of 2-CH3CN, the Co(II) center in 1-CH3CN resides in a slightly distorted octahedral geometry with acetonitrile bound at the apical site. The structure of 1 has been reported previously.46
The cyclic voltammogram of 1-CH3CN in acetonitrile solution features two reversible redox processes at E1/2 = 0.98 V and −0.64 V vs. SHE, assigned to metal-based Co(III)/Co(II) and Co(II)/Co(I) couples, respectively, with another irreversible reduction peak at −1.57 V vs. SHE (Fig. S2†). Compared to the redox processes observed for parent PY5Me2 complex 2-CH3CN in acetonitrile, CF3 substitution on the para position of the central pyridine ring positively shifts the formal Co(III)/(II), Co(II)/(I), and Co(I)/(0) couples of 1-CH3CN by ca. 105, 140 and 120 mV, respectively. As the free ligand CF3PY5Me2 is redox-silent in the same potential region (see Fig. S3†), the data suggest that these observed features are metal-dependent.
Owing to the large overpotential of 2 for hydrogen evolution catalysis and the relatively small electrochemical window of the glassy carbon electrode in pH 7 aqueous media, no apparent reduction feature of 2 was observed before the rise of the glassy carbon background current. In contrast, the CF3 derivative 1 exhibits a well-resolved and irreversible cathodic peak at −0.89 V vs. SHE in 0.1 M phosphate buffered to pH 7, with a much more pronounced rise in current density compared to the background (Fig. 2). In the positive potential direction, a reversible redox feature at 0.35 V vs. SHE was also observed. The pH-dependent cyclic voltammograms of 1 in 0.1 M phosphate buffer with 0.1 M NaClO4 as the supporting electrolyte are shown in Fig. S4.† The oxidation wave shifts positively along the decrease of pH from 9 to 4, with a slope of 58.1 mV per pH (inset of Fig. 2), indicating a one-proton and one-electron redox process close to the ideal value of 59 mV per pH. This observation led us to assign the oxidation wave as a Co(II)–OH2/Co(III)–OH couple. Similar results have been reported for other cobalt complexes with pentadentate ligands in aqueous medium.35,48 The pH-dependence of the reduction feature is more complex and is a topic under current investigation.
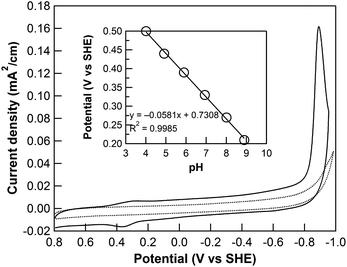 |
| | Fig. 2 Cyclic voltammograms of 0.1 mM 1 (solid line) and blank glassy carbon electrode (dotted line) in 0.1 M phosphate buffer at pH 7. Inset: pH dependence of the oxidation peak of 1 in 0.1 M buffered electrolytes at various pH values. Conditions: 0.1 M NaClO4 added as the supporting electrolyte, scan rate = 100 mV s−1, Ar atmosphere. | |
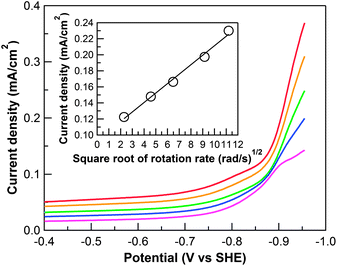 |
| | Fig. 3 Cathodic scans of 0.3 mM 1 in 0.1 M phosphate buffer and 0.1 M NaClO4 at pH 7 at different rotating rates: 100 (purple), 400 (blue), 800 (green), 1600 (orange), 2400 (red) rpm, (scan rate: 25 mV s−1). Inset: Levich plot of current density at overpotential = 500 mV versus the square root of rotating rate. | |
In order to compare the performance of 1 to that of other hydrogen catalysts in aqueous media, we sought to determine its apparent rate of proton reduction utilizing a method recently reported by Peters and co-workers.33 The parameter napp, defined as the apparent number of electrons delivered to the catalyst before it diffuses away from the electrode surface, can be calculated by the electrocatalytic current density normalized for the delivery of the catalyst to the surface as illustrated in eqn (1).
here,
jp is the plateau current density for the Co(
II)–aqua/Co(
III)–
hydroxide couple and
jc is the catalytic current density as shown in its
RDE voltammogram (
Fig. 4). The plot of
nappversus potential for
1 is included in the inset of
Fig. 4. The value of
napp increases dramatically after the onset of catalysis at
ca. −0.8 V
vs. SHE, consistent with the cyclic voltammogram in
Fig. 2, reaching nearly 16 at −0.900 V
vs. SHE (overpotential = −487 mV). Compared to the reported
napp values of 1–8 for a series of cobalt complexes at an overpotential of
ca. −500 mV in pH 2.2
buffers,
33 which were measured at the same scan rate and rotation rate as this present study,
1 exhibits better efficiency as a hydrogen evolution
catalyst in neutral pH aqueous solution at approximately the same overpotential. Similar results were obtained in 0.2 M NaClO
4 (Fig. S5 and S6
†), indicating that phosphate does not play a special role in this system. In addition, a Faradaic efficiency of 95 ± 10% was measured by
gas chromatography for a 3 h
bulk electrolysis of
1 at an applied potential of −0.963 V
vs. SHE (overpotential = −550 mV) in pH 7
buffer (
Fig. 5). The used glassy carbon plate was rinsed with
water and used as the working electrode in fresh
buffer for another blank
bulk electrolysis, which passed much less charge under the same condition (
Fig. 5). These results demonstrate the efficacy of
1 as an efficient and robust molecular electrocatalyst for the hydrogen evolution reaction.
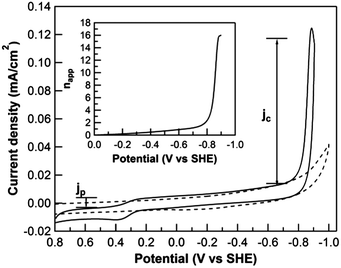 |
| | Fig. 4 Rotating disk electrode voltammograms of 0.1 mM 1 (solid) and blank glassy carbon electrode (dotted) in 0.1 M phosphate buffer at pH 7. Inset: napp plot of 1versus potential. Conditions: 0.1 M NaClO4 added as the supporting electrolyte, scan rate = 25 mV s−1, rotation rate = 400 rpm s−1, Ar atmosphere. | |
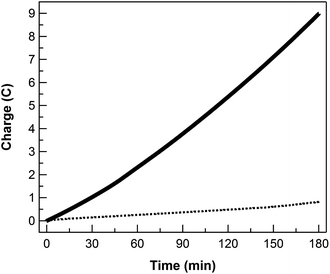 |
| | Fig. 5 Controlled potential electrolysis of 0.1 mM 1 (solid) and rinsed glassy plate after bulk electrolysis of 1 (dotted) in 0.1 M phosphate buffer and 0.1 M NaClO4 at pH 7, showing charge build-up versus time with an applied potential of −0.963 V vs. SHE (overpotential = −550 mV). | |
Photocatalytic hydrogen production using a molecular photosensitizer
After establishing that the cobalt pentapyridine complex 1 is a competent molecular electrocatalyst for hydrogen evolution in neutral water under diffusion-limited conditions, we next tested whether it could also be utilized as a photocatalyst under similar conditions. To this end, we carried out photocatalysis experiments in neutral aqueous solutions using [Ru(bpy)3]Cl2 as a soluble molecular inorganic photosensitizer and ascorbic acid as an electron donor. As shown in Fig. 6a and Fig. S7,† upon irradiation with light of wavelength λirr ≥ 455 nm at room temperature, a solution of 50 μM catalyst, 0.2 mM [Ru(bpy)3]Cl2, and 0.1 M ascorbic acid in 1.0 M phosphate buffer at pH 7 evolves hydrogen. The hydrogen evolution rate is initially linear in the first 2 h, followed by a slight deviation, until reaching the plateau of ca. 0.5 mL after 8 h of photolysis. In a separate experiment, we tested the stability of catalyst 1 and the chromophore during photolysis. As shown in Fig. S8,† after 10 h photolysis, further addition of 0.2 mM chromophore resumes nearly 40% activity of 1 under another 4 h illumination, indicating that although some of the catalyst may deactivate during the first 10 h photolysis, chromophore decomposition is the primary reason for the cease of hydrogen evolution after 8 h illumination.35 Importantly, control experiments without photosensitizer or ascorbic acid showed no H2 generation, and in the absence of catalyst, only negligible hydrogen was detected under the same conditions (Fig. 6a and S7†), establishing that all three components are necessary for the efficient photocatalytic evolution of hydrogen from water.35 Compared to the photocatalytic hydrogen evolution catalyzed by 2 and 3 (Fig. 6a), it is clear that 1 exhibits the highest catalytic performance under the conditions measured, which is consistent with its lower overpotential for electrocatalysis of hydrogen evolution in neutral water.46 During the first two hours of photolysis, an average quantum yield of 0.23% was obtained for 50 μM 1 in the presence of 0.2 mM [Ru(bpy)3]2+ and 0.1 M ascorbic acid. With increasing concentrations of catalyst, the average quantum yield during the first two hours of photolysis reached a plateau of ∼0.6% under the same conditions (Fig. S9†).
![(a) Photocatalytic hydrogen evolution over time as measured by gas chromatography for aqueous solutions containing 50 μM 1 (filled circles), 50 μM 2 (filled squares), 50 μM 3 (filled triangles), or no catalyst (open circles) with 0.2 mM [Ru(bpy)3]Cl2 and 0.1 M ascorbic acid in 1.0 M phosphate buffer of pH 7. (b) Photogenerated hydrogen volume after 2 h of illumination (λirr ≥ 455 nm) of 0.2 mM [Ru(bpy)3]Cl2 and 0.1 M ascorbic acid in 1.0 M phosphate buffer of pH 7 with various concentrations of 1. (c) Photogenerated hydrogen volume after 2 hour illumination (λirr ≥ 455 nm) of 50 μM 1 and 0.1 M ascorbic acid in 1.0 M phosphate buffer of pH 7 with various concentrations of [Ru(bpy)3]Cl2. The light source was a 150 W Xe lamp coupled to a 455 nm long-pass filter.](/image/article/2013/SC/c2sc21163g/c2sc21163g-f6.gif) |
| | Fig. 6 (a) Photocatalytic hydrogen evolution over time as measured by gas chromatography for aqueous solutions containing 50 μM 1 (filled circles), 50 μM 2 (filled squares), 50 μM 3 (filled triangles), or no catalyst (open circles) with 0.2 mM [Ru(bpy)3]Cl2 and 0.1 M ascorbic acid in 1.0 M phosphate buffer of pH 7. (b) Photogenerated hydrogen volume after 2 h of illumination (λirr ≥ 455 nm) of 0.2 mM [Ru(bpy)3]Cl2 and 0.1 M ascorbic acid in 1.0 M phosphate buffer of pH 7 with various concentrations of 1. (c) Photogenerated hydrogen volume after 2 hour illumination (λirr ≥ 455 nm) of 50 μM 1 and 0.1 M ascorbic acid in 1.0 M phosphate buffer of pH 7 with various concentrations of [Ru(bpy)3]Cl2. The light source was a 150 W Xe lamp coupled to a 455 nm long-pass filter. | |
To explore the effects of catalyst concentration on photochemical hydrogen generation, a series of 2 h photolysis experiments were carried out with various concentrations of 1. It is clear from Fig. 6b that, within the concentration range at less than 100 μM, the H2 evolution rate is first-order with catalyst concentration. An analogous observation has been made for other cobalt diglyoxime catalysts and a recent cobalt–ditholene catalyst, albeit in different solutions.36,49 Similarly, the effect of chromophore concentration on the system activity was also investigated. As shown in Fig. 6c, with the catalyst concentration kept constant at 50 μM and at relatively low concentrations of the photosensitizer (<60 μM), a linear relationship between hydrogen evolution rate and chromophore concentration was obtained. We note that at higher photosensitizer concentrations, the system activity is limited by the intrinsic efficiency of the catalyst.
Savéant and co-workers recently reported that in the presence of strong acids, the boron-capped tris(glyoximato) cobalt clathrochelate complexes decompose to form cobalt-containing nanoparticles that are actually responsible for the observed H2 generation activity.50,51 In addition, considering the concerns on the homogeneous or heterogeneous nature of some water oxidation cobalt catalysts published recently,52–54 we decided to investigate whether H2 evolution detected during photolysis may come from colloidal nanoparticle or other heterogeneous cobalt species. To this end, dynamic light scattering (DLS) measurements were conducted for photolysis solutions after 0 h, 0.5 h, and 3 h of irradiation (λirr ≥ 455 nm) at room temperature. As shown in Fig. 7, no species with a diameter larger than 1 nm could be detected in any of the three samples. Although we cannot unambiguously rule out any small particulate species, the different photocatalytic performance of the three cobalt catalysts with similar ligand platform, the first-order dependence on catalyst and photosensitizer, along with no evidence for particle formation at >1 nm sizes under the current photolysis conditions, strongly suggest the involvement of a molecular species.
![Particle size distribution of photolysis solutions containing 50 μM 1, 0.2 mM [Ru(bpy)3]Cl2, and 0.1 M ascorbic acid in 1.0 M phosphate buffer of pH 7 after illumination (λirr ≥ 455 nm) of 0 h (a), 0.5 h (b), and 3 h (c), determined by dynamic light scattering measurements.](/image/article/2013/SC/c2sc21163g/c2sc21163g-f7.gif) |
| | Fig. 7 Particle size distribution of photolysis solutions containing 50 μM 1, 0.2 mM [Ru(bpy)3]Cl2, and 0.1 M ascorbic acid in 1.0 M phosphate buffer of pH 7 after illumination (λirr ≥ 455 nm) of 0 h (a), 0.5 h (b), and 3 h (c), determined by dynamic light scattering measurements. | |
Photocatalytic hydrogen production using a semiconductor nanowire photosensitizer
Owing to the scarcity and hence high cost of ruthenium-based photosensitizers, it is desirable to replace [Ru(bpy)3]2+ with other chromophores composed of earth-abundant elements. As such, semiconductors possessing a high conduction band edge and visible light absorption offer a promising light-harvesting alternative. We recently reported the surfactant-free synthesis and characterization of GaP nanowires exhibiting hydrogen generation activity under visible light irradiation in water.47 To test whether our molecular cobalt catalyst could enhance H2 evolution performance, a photolysis experiment was conducted with 0.2 mM 1 and 1 mg of GaP nanowires in water, using methanol as a hole scavenger. The diameter and length of the nanowires were ∼40 nm and >1 μm, respectively. Indeed, as shown in Fig. 8, the hydrogen evolution rate is patently increased in the presence of catalyst 1, with a five-fold enhancement compared to the control sample during the first hour. Because hydrogen is quantified by head space gas sampling, we note that the hydrogen observed during the first 30 min is relatively low as a result of it being mainly dissolved in the electrolyte. Likewise, the decreased rate of hydrogen evolution after 2.5 h of photolysis may be due to the aggregation of GaP nanowires and/or decomposition of the catalysts. In addition, we observe that higher catalyst concentrations generally result in nanowire aggregation, which precludes quantitative analysis at this time. Since 1 is a relatively bulky molecular catalyst and the biomolecular electron-transfer rate between the GaP nanowires and cobalt complex 1 highly depends on their efficient collision, which is strikingly different from our reported Pt-coated GaP system,47 we believe that the hydrogen evolution performance can be improved upon by tuning electron transfer via covalent catalyst attachment or other means.
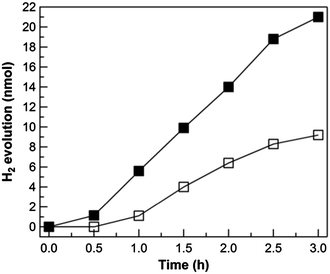 |
| | Fig. 8 Photocatalytic hydrogen evolution over time using a 0 mM (white square) and 0.2 mM (black square) solutions of 1 in the presence of 1 mg of GaP nanowires in 2.5 mL H2O with 0.5 mL of methanol added as the electron donor. The light source was a 450 W Xe lamp coupled to a 400 nm long-pass filter. | |
Conclusions
In summary, we have shown that the cobalt pentapyridine complex [(CF3PY5Me2)Co(H2O)]2+ is a competent molecular catalyst for the electrochemical production of hydrogen from neutral pH water under soluble, diffusion-limited conditions. Furthermore, the complex 1 can be employed as a photocatalyst for hydrogen evolution in neutral water, using [Ru(bpy)3]2+ as a molecular photosensitizer and ascorbic acid as the sacrificial electron donor. Dynamic light scattering measurements show no evidence for nanoparticle formation for the duration of the photolysis reactions, and photocatalysis is first-order in both catalyst and photosensitizer. We further demonstrated that 1 can operate in conjunction with a semiconductor photosensitizer, GaP nanowires, to enhance the hydrogen yield of photolysis upon visible light irradiation in water. These results present a starting point for constructing a molecular catalyst/semiconductor photosensitizer assembly using all earth-abundant components for H2 evolution catalysis in pure aqueous solution. Current lines of investigation include: performing further ligand modifications to decrease the overpotential and increase the rate of catalysis, strengthening the association between the molecular catalysts and the GaP nanowires to enhance the electron transfer, and coupling this reductive solar-driven process to oxidative oxygen evolution to generate a complete solar-to-fuel water-splitting system.
Experimental section
Materials
Ru(bpy)3Cl2, 4-trifluoromethyl-2,6-bis[(2-pyridyl)ethyl]pyridine (CF3PY5Me2), [(CF3PY5Me2)Co(H2O)](CF3SO3)2 (1), and GaP nanowires were synthesized and purified as previously reported.46,55,56 Glassy carbon rods (type 1) were purchased from Alfa Aesar for the electrochemical studies. Acetonitrile was dried over activated 4 Å molecular sieves, passed through a column of activated alumina, and stored over 3 Å molecular sieves under a nitrogen atmosphere. Water was deionized with the Millipore Milli-Q UF Plus system. All other chemical regents were purchased from commercial vendors and used without further purification. Unless noted otherwise, all manipulations were carried out at room temperature under a nitrogen atmosphere in a VAC glovebox or using high-vacuum Schlenk line techniques.
Syntheses
[(CF3PY5Me2)Co(NCCH3)](CF3SO3)2 (1-CH3CN) 1 eq. of Co(CF3SO3)2(CH3CN)2 (175 mg) was added to a 5 mL acetonitrile suspension of CF3PY5Me2 (177 mg). The mixture was stirred under nitrogen atmosphere at room temperature for 5 h. The orange solution was then concentrated to ∼1 mL under vacuum and diethyl ether vapor diffusion into this solution generated rod-shape light brown crystals suitable for X-ray crystallography. Yield: 312 mg (93%). LC-MS (M+) m/z calcd for C30H25CoF3N5O3S 651.0962, found 651.0947.
Physical methods
Carbon, hydrogen, and nitrogen analyses were obtained from the Microanalytical Laboratory of the University of California, Berkeley. Cyclic voltammetry experiments were carried out using BASI's Epsilon potentiostat and C-3 cell stand. A glassy carbon working electrode, a silver wire reference electrode, a platinum wire counter electrode were used for cyclic voltammetry experiments in CH3CN with 0.1 M Bu4NPF6 in glovebox. Ferrocene (E0 of Fc+/Fc = 0.64 V vs. SHE) was added during each experiment as an internal reference. For electrochemical studies conducted in aqueous media, a glassy carbon (3 mm diameter) was used as the working electrode and platinum wire as the auxiliary electrode. The reference electrode was a commercially available aqueous Ag/AgCl electrode, and the potentials were reported with respect to SHE by adding 0.195 V to the experimentally measured values. The electrolyte solution was thoroughly deaerated via bubbling argon 30 min prior to each electrochemical measurement in water and kept under positive argon pressure during the experiments. An Agilent 490-GC Micro-Gas Chromatograph with a molecular sieve column and heated syringe injector was used for hydrogen detection and quantification. The column was heated to 80 °C under Ar gas flow and an average sample volume of 200 nL was injected onto the column for each measurement. The ratio of the integrated areas of the hydrogen peak versus the internal standard methane peak was compared to a calibration curve (Fig. S10†) to calculate the hydrogen volume generated. A 150 W USHIO Xenon lamp with a 3 mm 455 nm optical glass filter was used to irradiate the photolysis sample solution which is kept in a water bath with cooling water circulating during the entire experiment.
Crystallographic structure determinations
The X-ray crystallographic data collection was carried out on a Bruker three-circle diffractometer mounted with an SMART 1000 detector using monochromated Mo Kα radiation (0.71073 Å) outfitted with a low-temperature, nitrogen-stream aperture, an APEXII CCD detector, and equipped with an Oxford Cryostream 700. The structure was solved using direct methods in conjunction with standard difference Fourier techniques and refined by full-matrix least-squares procedures.1 A semi-empirical absorption correction (SADABS) was applied to the diffraction data for all structures. All non-hydrogen atoms were refined anisotropically, and hydrogen atoms were treated as idealized contributions and refined isotropically. A summary of crystallographic data is given in Table S1.† All software used for diffraction data processing and crystal-structure solution and refinement are contained in the APEX2 program suite (Bruker AXS, Madison, WI).
Photolysis experiments
Samples of 4 mL solutions were prepared with known concentrations of catalyst, [Ru(bpy)3]Cl2, and ascorbic acid in 1.0 M phosphate buffer at pH 7 in a 30 mL schlenk flask sealed by a rubber septum with copper wire. The sample solutions were thoroughly deaerated by argon bubbling for 30 min prior to photolysis experiment. Each sample was irradiated by a USHIO 150 W Xenon lamp (λ ≥ 455 nm) or a 450 W Xenon lamp (λ ≥ 400 nm) at room temperature under constant stirring. The amounts of hydrogen produced during photolysis were determined by gas chromatography using methane as an internal standard. Quantum yields were determined following the procedure outlined in Handbook of Photochemistry, using ferrioxalate as the standard.57
Acknowledgements
The synthetic and catalytic portions of this work were supported by DOE/LBNL Grant 403801 (C.J.C.). The contributions of J.R.L. were supported by NSF grant CHE-1111900. The nanowire part of this study was supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division, of the US Department of Energy under Contract no. DE-AC02-05CH11231 (P.Y.). C.J.C. is an Investigator with the Howard Hughes Medical Institute.
Notes and references
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- D. G. Nocera, Chem. Soc. Rev., 2009, 38, 13–15 RSC.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- E. Aharon-Shalom and A. Heller, J. Electrochem. Soc., 1982, 129, 2865–2866 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef CAS.
- B. E. Conway, L. Bai and M. A. Sattar, Int. J. Hydrogen Energy, 1987, 12, 607–621 CrossRef CAS.
- O. Savadogo and H. Lavoie, Int. J. Hydrogen Energy, 1992, 17, 473–477 CrossRef CAS.
- O. Savadogo, J. Electrochem. Soc., 1992, 139, 1082–1087 CrossRef CAS.
- O. Savadogo, F. Carrier and E. Forget, Int. J. Hydrogen Energy, 1994, 19, 429–435 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jorgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Norskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS.
- T. F. Jaramillo, K. P. Jorgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS.
- Y. Li, H. Wang, L. Xie, Y. Liang, G. Hong and H. Dai, J. Am. Chem. Soc., 2011, 133, 7296–7299 CrossRef CAS.
- D. Merki, S. Fierro, H. Vrubel and X. Hu, Chem. Sci., 2011, 2, 1262–1267 RSC.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515–2525 RSC.
- V. Artero and M. Fontecave, Coord. Chem. Rev., 2005, 249, 1518–1535 CrossRef CAS.
- P. D. Tran, V. Artero and M. Fontecave, Energy Environ. Sci., 2010, 3, 727–747 CAS.
- A. M. Appel, D. L. DuBois and M. R. DuBois, J. Am. Chem. Soc., 2005, 127, 12717–12726 CrossRef CAS.
- B. E. Barton, C. M. Whaley, T. B. Rauchfuss and D. L. Gray, J. Am. Chem. Soc., 2009, 131, 6942–6943 CrossRef CAS.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 CAS.
- I. Bhugun, D. Lexa and J.-M. Saveant, J. Am. Chem. Soc., 1996, 118, 3982–3983 CrossRef CAS.
- J. W. Tye, J. Lee, H.-W. Wang, R. Mejia-Rodriguez, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Inorg. Chem., 2005, 44, 5550–5552 CrossRef CAS.
- M. J. Rose, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2012, 134, 8310–8313 CrossRef CAS.
- P. V. Bernhardt and L. A. Jones, Inorg. Chem., 1999, 38, 5086–5090 CrossRef CAS.
- J. P. Bigi, T. E. Hanna, W. H. Harman, A. Chang and C. J. Chang, Chem. Commun., 2010, 46, 958–960 RSC.
- V. Fourmond, P.-A. Jacques, M. Fontecave and V. Artero, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS.
- X. Hu, B. M. Cossairt, B. S. Brunschwig, N. S. Lewis and J. C. Peters, Chem. Commun., 2005, 4723–4725 RSC.
- X. Hu, B. S. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS.
- C. C. L. McCrory, C. Uyeda and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 3164–3170 CrossRef CAS.
- B. D. Stubbert, J. C. Peters and H. B. Gray, J. Am. Chem. Soc., 2012, 133, 18070–18073 CrossRef.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger, D. L. Baker, B. Yan, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 5941–5944 CrossRef CAS.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2012, 133, 15368–15371 CrossRef.
- W. R. McNamara, Z. Han, C.-J. M. Yin, W. W. Brennessel, P. L. Holland and R. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012 DOI:10.1073/pnas.1120757109.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, R. M. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS.
- U. J. Kilgore, J. A. S. Roberts, D. H. Pool, A. M. Appel, M. P. Stewart, M. R. DuBois, W. G. Dougherty, W. S. Kassel, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2012, 133, 5861–5872 CrossRef.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS.
- D. H. Pool, M. P. Stewart, M. O'Hagan, W. J. Shaw, J. A. S. Roberts, R. M. Bullock and D. L. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2012 DOI:10.1073/pnas.1120208109.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, Science, 2012, 335, 698–702 CrossRef CAS.
- V. S. Thoi, H. I. Karunadasa, Y. Surendranath, J. R. Long and C. J. Chang, Energy Environ. Sci., 2012, 5, 7762–7770 CAS.
- Y. Sun, J. P. Bigi, N. A. Piro, M. L. Tang, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 9212–9215 CrossRef CAS.
- J. Sun, C. Liu and P. Yang, J. Am. Chem. Soc., 2011, 133, 19306–19309 CrossRef CAS.
- D. J. Wasylenko, C. Ganesamoorthy, J. Borau-Garcia and C. P. Berlinguette, Chem. Commun., 2011, 47, 4249–4251 RSC.
- P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952–4962 CrossRef CAS.
- E. Anxolabéhère-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 6104–6107 CrossRef.
- Y. Z. Voloshin, A. V. Dolganov, O. A. Varzatskii and Y. N. Bubnov, Chem. Commun., 2011, 47, 7737–7739 RSC.
- Q. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle and C. L. Hill, Science, 2010, 328, 342–345 CrossRef CAS.
- J. J. Stracke and R. G. Finke, J. Am. Chem. Soc., 2011, 133, 14872–14875 CrossRef CAS.
- D. J. Wasylenko, R. D. Palmer, E. Schott and C. P. Berlinguette, Chem. Commun., 2012, 48, 2107–2109 RSC.
- Y. Sun, D. A. Lutterman and C. Turro, Inorg. Chem., 2008, 47, 6427–6434 CrossRef CAS.
- J. Sun, C. Liu and P. Yang, J. Am. Chem. Soc., 2011, 133, 19306–19309 CrossRef CAS.
-
S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, Marcel Dekker, New York, 1993 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallography data and structure of 2-CH3CN, electrochemistry data in CH3CN and NaClO4, additional photolysis data, gas chromatograms, and hydrogen calibration curves. CCDC 894807. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21163g |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.