Chloride, carboxylate and carbonate transport by ortho-phenylenediamine-based bisureas†‡
Stephen J.
Moore
a,
Cally J. E.
Haynes
a,
Jorge
González
ab,
Jennifer L.
Sutton
a,
Simon J.
Brooks
a,
Mark E.
Light
a,
Julie
Herniman
a,
G. John
Langley
a,
Vanessa
Soto-Cerrato
c,
Ricardo
Pérez-Tomás
c,
Igor
Marques
d,
Paulo J.
Costa
d,
Vítor
Félix
d and
Philip A.
Gale
*a
aChemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: philip.gale@soton.ac.uk; Tel: +44 (0)23 8059 3332
bMolecular Science Institute (ICMol), Inorganic Chemistry Department, University of Valencia, Edificio de Institutos de Paterna, Apartado de Correos 22085, 46071, Valencia, Spain
cDepartment of Pathology and Experimental Therapeutics, Cancer Cell Biology Research Group, University of Barcelona, Barcelona, Spain
dDepartamento de Química, CICECO and Secção Autónoma de Ciências da Saúde, Universidade de Aveiro, 3810-193 Aveiro, Portugal. E-mail: vitor.felix@ua.pt
First published on 29th August 2012
Abstract
Highly potent but structurally simple transmembrane anion transporters are reported that function at receptor to lipid ratios as low as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000000. The compounds, based on the simple ortho-phenylenediamine-based bisurea scaffold, have been studied for their ability to facilitate chloride/nitrate and chloride/bicarbonate antiport, and HCl symport processes using a combination of ion selective electrode and fluorescence techniques. In addition, the transmembrane transport of dicarboxylate anions (maleate and fumarate) by the compounds was examined. Molecular dynamics simulations showed that these compounds permeate the membrane more easily than other promising receptors corroborating the experimental efflux data. Moreover, cell based assays revealed that the majority of the compounds showed cytotoxicity in cancer cells, which may be linked to their ability to function as ion transporters.
:1000000. The compounds, based on the simple ortho-phenylenediamine-based bisurea scaffold, have been studied for their ability to facilitate chloride/nitrate and chloride/bicarbonate antiport, and HCl symport processes using a combination of ion selective electrode and fluorescence techniques. In addition, the transmembrane transport of dicarboxylate anions (maleate and fumarate) by the compounds was examined. Molecular dynamics simulations showed that these compounds permeate the membrane more easily than other promising receptors corroborating the experimental efflux data. Moreover, cell based assays revealed that the majority of the compounds showed cytotoxicity in cancer cells, which may be linked to their ability to function as ion transporters.
Introduction
The regulation of cellular ion concentration by complex membrane spanning proteins is critical for a range of biological processes.1 Misregulation of chloride transport has been associated with myotonia, nephrolithiasis (kidney stones), Bartter's syndrome and cystic fibrosis.2 Similarly, bicarbonate transport misregulation is linked to a range of disease states,3 and has been linked with pathogenic mucus production in cystic fibrosis.4 By synthesising molecules that can facilitate the transmembrane transport of chloride and bicarbonate it may be possible to develop new treatments for this type of disease. Furthermore, compounds that can facilitate H+/Cl− symport or Cl−/HCO3− antiport processes may function as anticancer agents since the deacidification of acidic organelles leads to cytoplasmic acidification, an early event in apoptosis.5 Indeed, there are a number of anticancer agents that have been reported to modulate intracellular pH. The prodiginines are one example, with the anticancer activity of these molecules linked to their ability to facilitate both H+/Cl− symport and Cl−/HCO3− antiport processes.6The synthesis of small molecules that function as membrane resident, mobile carriers for anions is an expanding field of research. By incorporating hydrogen-bond donor groups into a suitably lipophilic scaffold it is possible to transport anions through the hydrophobic interior of a lipid bilayer. Recent examples of such scaffolds include the steroid-based cholapods,7 calix[4]pyrrole and its fluorinated or triazole-strapped analogues,8 amphiphilic catechols and monoacylglycerols,9 preorganised isophthalamides and squaramides,10 and tripodal tris(aminoethyl)amine (tren) thioureas.11
Interest in this area has grown to encompass transporters (carriers or otherwise) that show activity in biological systems.6,12 To improve the likelihood of new transmembrane transporter motifs exhibiting biological activity it is important to design compounds that have suitable absorption, distribution, metabolism, excretion and toxicity (ADMET) characteristics.13 There are guidelines, such as Lipinski's rule of five, that specify ranges of ‘ideal’ molecular properties such as molecular weight, partition coefficient and the number of hydrogen bond donors and acceptors.14 By considering these guidelines we generated transporters that exhibit biological activity in vivo, such as fluorinated molecules based on indolylurea/thiourea15 scaffolds that show promise as anticancer agents. By synthesising transporters that obey the rules of thumb defined by Lipinski and others, the possibility that these compounds will be biologically compatible is greater, i.e. have acceptable absorption, distribution, metabolism and excretion properties.
We have previously shown that bisurea receptors in which the two urea groups are linked by alkyl chains can promote chloride transport across phospholipid bilayers.16 In other work we have shown that ortho-phenylenediamine-based bisureas are effective anion receptors.17 We decided to explore the anion transport properties of ortho-phenylenediamine-based bisureas as promising candidates for biologically active ion transporters that may conform to the rules of thumb defined by Lipinski and others.14
Molecules developed from this scaffold have found extensive use in the field of anion complexation; we have previously reported that ortho-phenylenediamine-based bisureas function as selective carboxylate complexation agents,17 whilst the analogous 4,5-dimethyl-1,2-phenylenediamine scaffold has been used to construct colorimetric anion sensors.18 These motifs can be extended with additional hydrogen bond donor groups to yield receptors with enhanced anion affinity.19 Trisureas and tetraureas developed in this way show good selectivity for phosphate and sulfate,20 whilst a tripodal tren-based hexylurea functions as a sulfate extractant.21 Extended amidourea macrocycles based on these scaffolds exhibit good selectivity towards carboxylates.22 Replacement of the phenylene core with anthraquinone yields colorimetric sensors for the detection of Hg2+ and H2PO4−,23 whilst replacement with a cyclohexyl core has been used for the chiral recognition of phosphate ions24 and the colorimetric sensing of cyanide.25 Further, these molecules have found other applications in crystal engineering,26 as gelators,27 and in catalysis.28
In this paper we report the transmembrane transport properties of ortho-phenylenediamine-based bisureas 1–8, and compare them to monoureas 9–11; the latter being analogues of the three most active bisurea transporters. The anion binding properties of this series of compounds were determined using 1H NMR titration techniques in DMSO-d6/0.5% H2O mixtures. Anion transport was studied using a combination of ion selective electrode assays and fluorescence techniques in phospholipid vesicles. In vitro viability and fluorescence assays have been performed in order to evaluate the anticancer properties of these compounds. We show that this hydrogen bonding array is a highly potent anion transport motif with the most active compound studied showing transport at 1:1000000 receptor:lipid ratios. Effective transport at very low concentrations is a desirable feature allowing the potential use of these compounds in biological systems at low dose. Finally, in silico studies were also performed to bring further insights into the interactions of 5 and 6 with a lipid bilayer model.
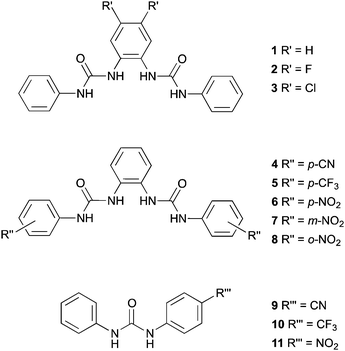
Results and discussion
Synthesis
We have previously reported the synthesis of compounds 1,17a3,17b6,17b and 8.17b Reaction of the appropriate isocyanate with ortho-phenylenediamine in dichloromethane/pyridine gave compounds 4, 5 and 7 in 39%, 84% and 82% yields respectively. Compound 2 was prepared by a similar method, using 4,5-difluoro-2-nitroaniline as the starting material. Hydrogenation using Pd/C in methanol, followed by reaction with phenylisocyanate in dichloromethane/pyridine yielded compound 2 in 37% yield.The syntheses of monoureas 9,2910,30 and 11 (ref. 31) have been previously reported.32 In all cases aniline was added to the corresponding isocyanate in dichloromethane/pyridine to produce compounds 9, 10, and 11 in 56%, 58% and 75% yields respectively. These compounds are analogues of the three most active bisurea transporters 4, 5 and 6.
Anion binding in solution
The ability of compounds 1–11 to bind anions in solution was investigated using 1H NMR titration techniques in DMSO-d6/0.5% water (with the anions added as tetrabutylammonium (TBA) or tetraethylammonium (TEA) salts) both to provide comparability with earlier studies33 and for solubility reasons. The binding studies were performed for anions relevant to both our transmembrane transport assays and biological systems. Where possible the change in chemical shift of the most downfield NH signal was fitted to a 1:1 binding model using WinEQNMR2 software.34 The results are summarised in Table 1. Previously reported stability constants are included for comparison.17 Fitted curves and selected Job plots can be found in the ESI.†
| Chloride | Bicarbonate | Nitrate | |
|---|---|---|---|
|
a All errors <15%. Data fitted to a 1:1 binding model. Binding constant obtained by following the most downfield urea NH unless stated otherwise.
b No significant interaction observed.
c Sigmoidal curve could not be fitted to a suitable binding model.
d Significant broadening of urea NHs observed.
e Reliable binding constant could not be obtained due to significant broadening of urea NHs and significant overlap of aromatic CHs.
f Dramatic colour change observed upon addition of bicarbonate.
|
|||
| 1 (ref. 17a) | 43 | 1370 | b |
| 2 | 73 | c , d | b |
| 3 (ref. 17b) | 67 | 2570d | b |
| 4 | 74 | e | b |
| 5 | 78 | 2090d | b |
| 6 (ref. 17b) | 78 | 3770d,f | b |
| 7 | 81 | 3140d,f | b |
| 8 (ref. 17b) | <10 | 826e | b |
| 9 | 41 | 2580 | b |
| 10 | 61 | 1380 | b |
| 11 | 57 | 1630d,f | b |
In general, compounds 1–11 exhibit strong 1:1 binding with tetraethylammonium bicarbonate, moderate 1:1 binding with tetrabutylammonium chloride and no significant interaction with tetrabutylammonium nitrate under the conditions of the NMR experiments. A sigmoidal binding curve was obtained for compound 2 with tetraethylammonium bicarbonate that could not be fitted to a binding model.35 Job plot analysis suggested 1:1 binding, however the plot was an unusual shape, that may be due to more complex solution phase behaviour such as deprotonation of the bound oxoanion.33b Significant urea NH peak broadening was observed upon addition of tetraethylammonium bicarbonate to compounds 2, 3, 4, 5, 6, 7, 8 and 11. A stability constant could not be determined for compound 4 with tetraethylammonium bicarbonate due to this broadening effect, however Job plot analysis confirmed 1:1 binding.
Comparing functionalised receptors 2–7 with receptor 1, it can be observed that addition of electron withdrawing substituents to either the central core or the peripheral phenyl groups increases anion affinity. In addition, it has been suggested that halogenation of the phenylene core (2 and 3) can aid receptor preorganisation by increasing the acidity of the phenylene CH groups, strengthening intramolecular hydrogen bonding interactions.17b For compounds 2–7 the binding constants obtained with tetrabutylammonium chloride are of similar magnitude whilst no significant interaction was previously reported between compound 8 and tetrabutylammonium chloride.17b This lack of interaction was attributed to intramolecular hydrogen bonding. The ortho-nitro groups may also provide a steric constraint on the binding site. Similarly, compound 8 has a significantly lower binding constant with tetraethylammonium bicarbonate than compounds 6 and 7.
Colour changes were observed upon addition of tetraethylammonium bicarbonate to nitro-functionalised compounds 6, 7, 8 and 11. Such behaviour has been reported previously in the presence of basic anions and has been attributed to deprotonation in analogous systems.17b,18,25,36
Interestingly, the two least active ion transporters (1 and 8) display the lowest affinity for both chloride and bicarbonate. The most active transporter 6 has the highest affinity of the series for bicarbonate. In addition, the monourea compounds 9, 10 and 11 show reduced anion affinity compared to their bisurea analogues, demonstrating the impact of incorporating additional convergent hydrogen bond donor groups into the receptor scaffold.
To complement the transport studies, we attempted to titrate compounds 4, 5 and 6 with tetrabutylammonium maleate and tetrabutylammonium fumarate in DMSO-d6/0.5% H2O.37 Addition of the tetrabutylammonium dicarboxylate to the receptor solution resulted in the formation of a precipitate in both instances, preventing determination of a binding constant. Instead ESI mass spectrometry was used to examine the interaction between bisureas and dicarboxylate anions. Compound 5 was studied owing to both its good solubility and high transport activity. Negative ESI experiments with solutions of 5 and either maleic or fumaric acid revealed the formation of 1:1 bisurea–carboxylate complexes in the gas phase. Further, by analysing samples containing different ratios of bisurea host and carboxylate anion it was possible to determine that compound 5 binds maleate more strongly than fumarate.38,39
Solid state
The solid-state behaviour of compound 7 was examined using single-crystal X-ray diffraction (Fig. 1). Tables of hydrogen bonds, data collection and refinement details, and thermal ellipsoid plots can be found in the ESI.†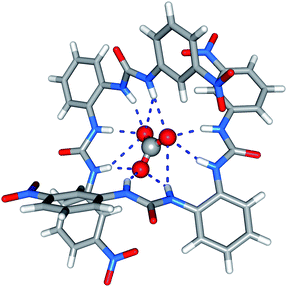 | ||
| Fig. 1 X-ray crystal structure of 7 (tetraethylammonium carbonate complex). Counterions are omitted for clarity. | ||
Crystals were obtained by slow evaporation of a methanol–water solution of compound 7 in the presence of tetraethylammonium bicarbonate. Analysis revealed that the meta-nitro functionalised bisurea forms a 2:1 (receptor:anion) complex with carbonate, the encapsulated carbonate anion bound by eleven hydrogen-bonding interactions. All the available urea NH groups were found to be involved in hydrogen bonding the carbonate (N–O distances 2.726(2)–3.368(2) Å, N–H⋯O angles 139.0–166.9°). It appears that in the solid state the meta-nitro group does not hinder guest inclusion.
Transport studies
We assessed the ability of compounds 1–11 to transport anions across phospholipid bilayers using a combination of ion selective electrode and fluorescence techniques. In a typical assay unilamellar, 1-palmitoyl-2-oleoylphosphatidyl-choline (POPC) vesicles (200 nm diameter) were prepared containing sodium chloride (488 mM with 5 mM phosphate buffer at pH 7.2) and suspended in a solution of sodium nitrate (488 mM with 5 mM phosphate buffer at pH 7.2).40 Compounds 1–11 were added as solutions in DMSO and the resulting chloride efflux from vesicles monitored using a chloride selective electrode (Accumet). At the end of the experiment detergent (octaethylene glycol monododecyl ether) was added to lyse the vesicles. The final reading was used to calibrate the electrode to 100% chloride release.In these assays ion transport occurs by a passive process. To maintain charge balance, ion transport must occur by either a symport or an antiport mechanism.40 The chloride efflux in Fig. 2 could therefore be a consequence of chloride/nitrate antiport, chloride/sodium symport or HCl symport processes.
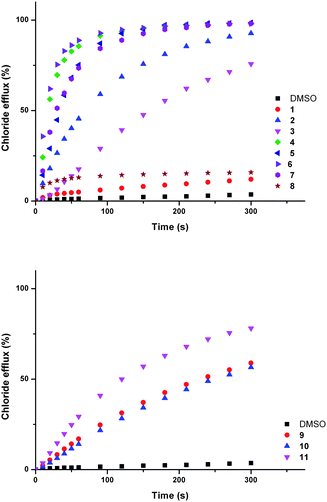 | ||
| Fig. 2 Chloride efflux promoted by a DMSO solution of compounds 1–11 (2 mol% carrier to lipid) from unilamellar POPC vesicles loaded with 488 mM NaCl buffered to pH 7.2 with 5 mM sodium phosphate salts. The vesicles were dispersed in 488 mM NaNO3 buffered to pH 7.2 with 5 mM sodium phosphate salts. At the end of the experiment detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents an average of three trials. DMSO was used as a control. See Fig. S44 and S45† for versions of this figure with error bars. | ||
To examine the possibility of sodium/chloride co-transport, an ISE assay was performed using vesicles containing caesium chloride buffered to pH 7.2 with 5 mM phosphate buffer. The vesicles were suspended in a solution of sodium nitrate buffered to pH 7.2 with 5 mM phosphate buffer. In the event of metal chloride co-transport we would expect the rate of chloride release to be dependent on the nature of the metal cation.8,41 The rates of chloride release for vesicles containing sodium chloride were within error of those observed for vesicles containing caesium chloride, implying that metal/chloride symport is not facilitated by these compounds (see ESI† for comparative plots).
Chloride/bicarbonate antiport is a biologically significant process.3,6e,42 Compounds 1–11 were tested for ability to facilitate chloride exchange using a chloride selective electrode assay with vesicles containing sodium chloride (451 mM with 20 mM phosphate buffer at pH 7.2) suspended in a solution of sodium sulphate (150 mM with 20 mM phosphate buffer at pH 7.2). Compounds 1–11 were added as solutions in DMSO. After two minutes a sodium bicarbonate ‘pulse’ was added such that the external concentration of bicarbonate was 40 mM.
The external sulfate is more challenging to transport through a lipid bilayer than bicarbonate, due to its higher hydrophilicity.43 Consequently, chloride efflux observed in the first two minutes of the assay (before the bicarbonate ‘pulse’) is not expected to be the result of an anion antiport mechanism. Upon addition of the ‘bicarbonate pulse’ a chloride/bicarbonate antiport mechanism becomes possible. A marked increase in transport activity was observed for compounds 2–7 at this point (Fig. 3).
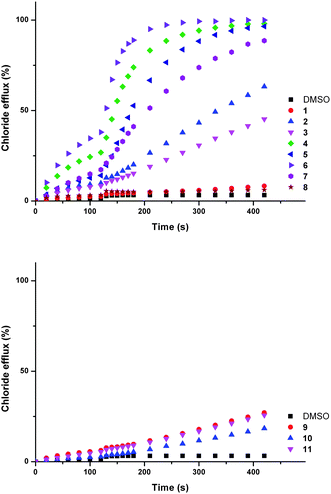 | ||
| Fig. 3 Chloride efflux promoted by a DMSO solution of compounds 1–12 (2 mol% carrier to lipid) from unilamellar POPC vesicles loaded with 451 mM NaCl buffered to pH 7.2 with 20 mM sodium phosphate salts. The vesicles were dispersed in 150 mM Na2SO4 buffered to pH 7.2 with 20 mM sodium phosphate salts. At t = 120 s a solution of sodium bicarbonate was added such that the external concentration of bicarbonate was 40 mM. At the end of the experiment, detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents an average of three trials. DMSO was used as a control. See Fig. S57 and S58† for versions of this figure with error bars. | ||
Low levels of chloride efflux were observed for some of the compounds in the two minutes before the bicarbonate pulse in the aforementioned assay. To investigate this effect further, the experiment was repeated without the bicarbonate ‘pulse’. Significant chloride efflux was observed for some of the compounds (see ESI, Fig. S59†).
Evidence in support of transmembrane sulfate transport has been observed previously for fluorinated tripodal ureas and thioureas.44 To test if the chloride efflux observed in the aforementioned assay was linked to the transport of sulfate, unilamellar POPC vesicles were prepared containing 100 mM NaCl and 2 mM lucigenin buffered to pH 7.2 with 20 mM phosphate buffer. These vesicles were suspended in 100 mM NaCl solution buffered to pH 7.2 with 20 mM phosphate salts. A solution of sodium sulfate was added such that the external concentration of sulfate was 40 mM, and after one minute compounds 2–7 were added as solutions in methanol (2 mol% carrier to lipid). No evidence of sulfate transport was observed using this assay (see ESI†).
Compound 6 facilitated the highest chloride efflux in the sulfate ‘blank’ assay and was used to explore the possibility of HCl co-transport using a pH gradient assay.45 Vesicles containing 451 mM sodium chloride buffered to pH 4.0 with 20 mM citric acid buffer were suspended in a solution of 150 mM sodium sulfate buffered to pH 7.2 with 20 mM sodium phosphate salts. Fig. 4 shows that the rate of chloride transport facilitated by compound 6 under gradient conditions is greater than in the absence of a pH gradient, implying that this molecule can facilitate HCl symport in addition to anion–anion antiport.
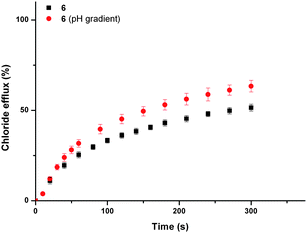 | ||
| Fig. 4 Chloride efflux promoted by a DMSO solution of compound 6 (2 mol% carrier to lipid) from unilamellar POPC vesicles loaded with either 451 mM NaCl buffered to pH 7.2 with 20 mM sodium phosphate salts or 451 mM NaCl buffered to pH 4.0 with 20 mM sodium citrate salts. The vesicles were dispersed in 150 mM Na2SO4 buffered to pH 7.2 with 20 mM sodium phosphate salts. At the end of the experiment, detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents an average of three trials. DMSO was used as a control. | ||
Proton transport was confirmed using POPC vesicles loaded with NaCl (488 mM) and 1 mM 8-hydroxy-1,3,6-pyrenetrisulfonate (HPTS), a pH sensitive fluorescent dye.46 These vesicles were suspended in a solution of Na2SO4 (150 mM) and the HPTS fluorescence measured upon addition of a DMSO solution of compound 6. An increase in intravesicular pH was observed, corresponding to deacidification of the vesicles via either H+/Cl− co-transport or an equivalent Cl−/OH− antiport pathway (see ESI†).
Three modes of operation are known for synthetic transmembrane ion transporters; a mobile carrier mechanism, channel formation, or a relay mechanism.47 We probed the transport mechanism of these systems using an ion selective electrode assay with POPC vesicles containing cholesterol (7:3 molar ratio). The vesicles were prepared containing sodium chloride (488 mM with 5 mM phosphate buffer at pH 7.2) and suspended in a solution of sodium nitrate (488 mM with 5 mM phosphate buffer at pH 7.2). By comparing the chloride efflux observed in this assay with that observed in the absence of cholesterol we can deduce if these molecules are mobile carriers. Cholesterol decreases membrane fluidity and thus transport that relies on a diffusion mechanism (i.e. mobile carrier), is expected to be slower in the presence of cholesterol.48 Compounds 2–7 all showed reduced chloride efflux in the cholesterol containing vesicles (see ESI†).
Further evidence for a carrier mechanism was obtained from U-tube experiments.15,44 A membrane is modelled by two aqueous phases separated by a nitrobenzene organic phase. The source phase was loaded with sodium chloride (488 mM buffered to pH 7.2 with 5 mM sodium phosphate salts) and the receiving phase was loaded with sodium nitrate (488 mM buffered to pH 7.2 with 5 mM sodium phosphate salts). The carrier (1 mM) was dissolved in the nitrobenzene phase and chloride transport into the receiving phase was monitored using an ion selective electrode. The separation of the two aqueous phases rules out the possibility of channel formation.49 For solubility reasons only compounds 2, 4, 5 and 7 could be tested in this way. All yielded a higher concentration of chloride in the receiving phase than in the control (see ESI†). These results are further evidence that the compounds operate via a mobile carrier mechanism.
To quantify the transport activity of compounds 1–11 Hill analyses50 for the chloride/nitrate and chloride/bicarbonate antiport assays were performed (see ESI†). Hill analysis enables determination of an EC50,270s value; the concentration of carrier (mol% with respect to lipid) required to afford 50% chloride efflux 270 s after addition of the carrier (or after the bicarbonate ‘pulse’), enabling us to compare the transport activity of the compounds. These values are summarised in Table 2, together with the Hill coefficients (which provide supporting evidence for a mobile carrier mechanism), the calculated values for polar surface area (PSA) and total surface area (TSA) and log P.51 The PSA and TSA parameters were estimated as described in the ESI† from molecular dynamic (MD) simulations carried out with compounds 1–11 in water solution.
| clogPa | PSAb (Å2) | TSAc (Å2) | EC50,270sd (Cl−/NO3−) | n e (Cl−/NO3−) | EC50,270sd (Cl−/HCO3−) | n e (Cl−/HCO3−) | |
|---|---|---|---|---|---|---|---|
| a clogP calculated using Fieldview version 2.0.2 for Macintosh (Wildman–Crippen model). b Polar surface area (PSA). c Total surface area (TSA). Both PSA and TSA values were calculated with MD simulations of 10 ns length (see ESI for details1). d EC50, 270 s defined as the concentration (mol% carrier to lipid) needed to obtain 50% chloride efflux after 270 s. e Hill coefficient. f Some of the transport activity observed may be the consequence of H+/Cl− co-transport. | |||||||
| 1 | 4.97 | 89.9 ± 7.1 | 482.7 ± 18.0 | na | na | na | na |
| 2 f | 5.80 | 155.5 ± 6.0 | 484.6 ± 17.9 | 0.14 | 0.9 | 1.40 | 0.7 |
| 3 f | 6.33 | 198.9 ± 6.7 | 530.0 ± 17.9 | 0.39 | 0.7 | >5 | 0.9 |
| 4 f | 4.72 | 203.6 ± 8.4 | 564.3 ± 23.2 | 0.011 | 0.7 | 0.073 | 1.0 |
| 5 f | 7.01 | 271.8 ± 11.9 | 601.8 ± 28.4 | 0.018 | 1.2 | 0.15 | 0.9 |
| 6 f | 4.58 | 286.0 ± 12.4 | 565.0 ± 27.1 | 0.0048 | 0.9 | 0.038 | 1.1 |
| 7 f | 4.58 | 263.4 ± 27.6 | 544.6 ± 40.4 | 0.084 | 1.0 | 0.21 | 0.8 |
| 8 | 4.58 | 203.0 ± 21.7 | 520.1 ± 21.8 | na | na | na | na |
| 9 | 3.20 | 114.1 ± 1.3 | 392.9 ± 2.9 | 1.76 | 1.8 | na | na |
| 10 | 4.35 | 150.2 ± 1.5 | 415.1 ± 3.0 | 1.80 | 2.1 | na | na |
| 11 | 3.13 | 157.5 ± 1.5 | 396.4 ± 2.9 | 1.05 | 1.5 | na | na |
In general, we observed that incorporation of electron withdrawing functionalities into the bisurea scaffold affords compounds capable of facilitating the transmembrane transport of anions. Parent compound 1 shows poor transport activity. Halogenation of the phenyl core in the 4- and 5-positions improved transporter activity, but to a less dramatic extent than the addition of electron withdrawing functionalities to the 4-position of the peripheral phenyl groups. In these instances transporter activity was observed to increase with increasing electron withdrawing strength of the substituents (represented by the Hammett constants shown in Table 3); H < F ∼ Cl < CF3 < CN < NO2. This trend is consequently reflected in the binding constants, with transport inactive receptor 1 exhibiting relatively low binding constants with chloride and bicarbonate (43 M−1 and 1270 M−1 respectively), in comparison to the most active transporter 6, (78 M−1 and 3770 M−1 respectively), reflecting the strong electron withdrawing effect of the nitro functionality. Indeed, para-nitro functionalised compound 6 displays very high transport activity, facilitating chloride efflux at receptor:lipid ratios as low as 1:1000000 (Fig. 5). To the best of our knowledge we believe that this is the lowest loading level of transporter observed that can facilitate transport to date.
| Compound | Substituent | σ meta | σ para |
|---|---|---|---|
| 1 | H | 0.00 | 0.00 |
| 2 | F | 0.34 | 0.06 |
| 3 | Cl | 0.37 | 0.23 |
| 4 | CN | — | 0.66 |
| 5 | CF3 | — | 0.54 |
| 6 | NO2 | — | 0.78 |
| 7 | NO2 | 0.71 | — |
| 8 | NO2 | — | — |
| 9 | CN | — | 0.66 |
| 10 | CF3 | — | 0.54 |
| 11 | NO2 | — | 0.78 |
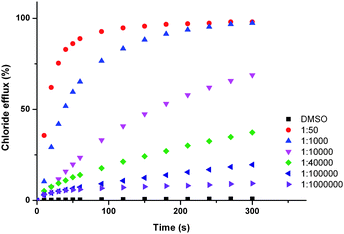 | ||
| Fig. 5 Chloride efflux promoted by a DMSO solution of compounds 6 at various loadings from unilamellar POPC vesicles loaded with 488 mM NaCl buffered to pH 7.2 with 5 mM sodium phosphate salts. The vesicles were dispersed in 488 mM NaNO3 buffered to pH 7.2 with 5 mM sodium phosphate salts. At the end of the experiment detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents an average of three trials. DMSO was used as a control. | ||
We have also examined the effect of varying substituent position on the peripheral phenyl rings (compounds 6, 7, and 8). The nitro functionality is most electron withdrawing in the ortho- and para-positions.52para-Nitro functionalised compound 6 shows greater transporter activity than meta-nitro functionalised compound 7. The ortho-nitro functionalised compound 8 displays poor transport activity, likely a consequence of the involvement of the nitro group in intramolecular hydrogen bonding and also sterically hindering the binding site.17b This is reflected in the stability constants for compound 8 with chloride and bicarbonate (<10 M−1 and 826 M−1 respectively) which are significantly lower than those observed for compounds 6 and 7.
The transport activity of the monourea analogues of the three most active bisurea compounds (4, 5 and 6) were tested using vesicles containing sodium chloride (488 mM with 5 mM phosphate buffer at pH 7.2) and suspended in a solution of sodium nitrate (488 mM with 5 mM phosphate buffer at pH 7.2). Compounds 9, 10 and 11 were tested at loadings of 2 mol% (with respect to lipid), and the results compared to the efflux measured upon addition of compounds 4, 5 and 6 at loadings of 1 mol% (with respect to lipid). This ensured that the concentration of urea groups was the same. The results (Fig. 6) show that the bisurea compounds are more active than their monourea analogues.
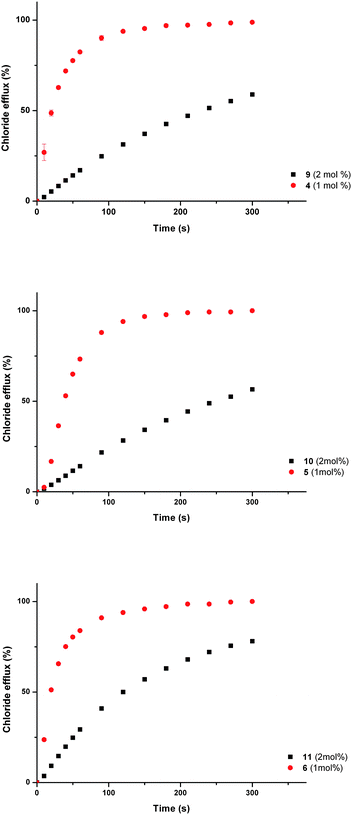 | ||
| Fig. 6 Chloride efflux promoted by a DMSO solution of monourea compounds 9–11 (2 mol% carrier to lipid) and their analogous bisureas 4–6 (1 mol% carrier to lipid) from unilamellar POPC vesicles loaded with 488 mM NaCl buffered to pH 7.2 with 5 mM sodium phosphate salts. The vesicles were dispersed in 488 mM NaNO3 buffered to pH 7.2 with 5 mM sodium phosphate salts. At the end of the experiment detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents an average of three trials. | ||
Carboxylate transport
Recently, interest has grown in the construction of molecules capable of discriminating between small isomeric dicarboxylates.53 Several examples of bisurea compounds with structural similarity to compounds 1–8, have been shown to selectively bind carboxylates.54 Owing to this similarity, we postulated that out most active transporters (4, 5 and 6) might be able to facilitate the transmembrane transport of carboxylate anions. The transmembrane transport of carboxylates is biologically important; failure of glutamate transporters is linked neurodegenerative disorders including Alzheimer's and Huntingdon's disease,55 whilst disruption of oxalate transport can result in renal disorders.56We decided to focus on the transmembrane transport of fumarate and its stereoisomer, maleate. Fumarate has biological importance as a key intermediate in the citric acid cycle.57 Carboxylate transport was examined using vesicles containing sodium chloride buffered to pH 7.2, suspended in a solution of sodium sulphate buffered at pH 7.2. Compounds 4, 5 and 6 were added as solutions in DMSO (2 mol% with respect to lipid). After two minutes a sodium maleate or sodium fumarate ‘pulse’ was added such that the external concentration of the carboxylate was 40 mM. At pH 7.2 significant additional chloride efflux was observed on addition of a sodium maleate ‘pulse’ with all three compounds, whilst no significant enhancement in chloride efflux was seen with the sodium fumarate ‘pulse’. The experiment was repeated at pH 4.0, whereupon an enhancement in chloride efflux was observed upon addition of both maleate and fumarate pulses (Fig. 7 shows compound 4 as a representative example).
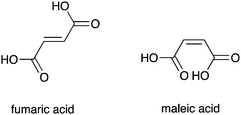
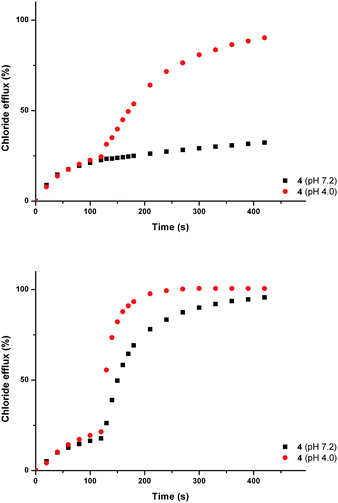 | ||
| Fig. 7 Chloride efflux promoted by a DMSO solution of compound 4 (2 mol% carrier to lipid) from unilamellar POPC vesicles loaded with either; (i) 451 mM NaCl buffered to pH 7.2 with 20 mM sodium phosphate salts and dispersed in 150 mM Na2SO4 buffered to pH 7.2 with 20 mM sodium phosphate salts, or (ii) 451 mM NaCl buffered to pH 4.0 with 20 mM sodium citrate salts and dispersed in 150 mM Na2SO4 buffered to pH 4.0 with 20 mM sodium citrate salts. At t = 120 s a solution of either sodium fumarate (top) or sodium maleate (bottom) was added such that the external concentration of carboxylate was 40 mM. At the end of the experiment, detergent was added to lyse the vesicles and calibrate the ISE to 100% chloride efflux. Each point represents an average of three trials. DMSO was used as a control. | ||
We have already shown that pre-pulse chloride efflux is the consequence of HCl symport and not sulfate transport or NaCl symport. We can rationalise the pH dependant nature of the transport by considering the pKa values for maleate (pKa1 = 1.92, pKa2 = 6.23) and fumarate (pKa1 = 3.02, pKa2 = 4.38).52 Thus, at pH 7.2 a higher proportion of maleate exists as the monoanion, whilst fumarate will predominantly exist as the dianion. Dianions are more hydrophilic than their monoanionic counterparts and will consequently be more difficult to transport.43 Hence, at pH 7.2 we observe an increase in chloride transport upon addition of maleate, but little enhancement upon addition of a fumarate. At pH 4.0 the monoionic forms of both maleate and fumarate will predominate, and an increase in chloride efflux was observed upon addition of both maleate and fumarate, corresponding to chloride/carboxylate exchange.
In summary, we have observed that ortho-phenylenediamine-based bisureas functionalised with electron withdrawing substituents are potent ion transporters, facilitating chloride/nitrate and chloride/bicarbonate antiport. In some instances we have observed carboxylate/chloride antiport and HCl symport. We have demonstrated that these compounds operate via a mobile carrier mechanism and that sulphate transport and metal/chloride symport processes are not facilitated.
Cell based assays
The potent anion transport activity of some members of the series prompted examination of the in vitro cytotoxicity towards a range of cancerous cell lines. A single point MTT cell viability assay was performed with compounds 1–8 (10 μM) on a selection of cancer cell lines of diverse origin (human small-cell lung carcinoma GLC4, human alveolar adenocarcinoma A549, human colon adenocarcinoma SW480, human melanoma A375 and human oral adenosquamos carcinoma CAL27). Cell viability was assessed 48 hours after exposure to each of the compounds. Fig. 8 shows that compounds 2, 5 and 8 display significant cytotoxicity towards the GLC4, A549, SW480 and A375 cell lines. In comparison compounds 3, 6 and 7 are less toxic, whilst compounds 1 and 4 are the least toxic in the series (Fig. 8).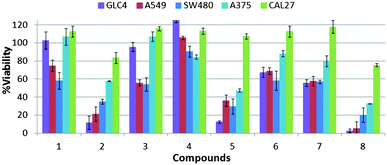 | ||
| Fig. 8 Cell viability after 48 h of compound treatment measured by MTT assay. Single-point screening of compounds 1–8 (10 μM) on a range of cancer cell lines, from left to right, GLC4, A549, SW480, A375 and CAL27. | ||
Dose–response curve experiments were performed and IC50 values (Inhibitory Concentration of 50% of cell population) were calculated for the most cytotoxic compounds (fluorinated compounds 2 and 5, and nitro functionalised compound 8) in the most sensitive cancerous cell lines (Table 4). These results corroborate the potency of these cytotoxic receptors, showing IC50 values around 10 μM in GLC4 cells.
| GLC4 | A549 | SW480 | |
|---|---|---|---|
| 2 | 8.0 ± 1.1 | 10.8 ± 0.7 | 10.6 ± 0.7 |
| 5 | 5.2 ± 0.1 | 6.0 ± 0.3 | 11.1 ± 2.0 |
| 8 | 5.0 ± 1.2 | 9.2 ± 2.2 | 3.3 ± 1.2 |
We have previously reported that fluorination is an effective strategy for the production of compounds that can function as cytotoxic agents in cancerous cell lines.15,44 In contrast, the nitrophenyl group is a known toxicophore.58 The disruption of intracellular pH (pHi) has been suggested as a strategy for cancer treatment.59 Modulation of pHi is essential for control of the cell cycle (amongst other cellular processes) and is regulated by several mechanisms, including Na+/H+ co-transport, Cl−/HCO3− antiport, Na+/HCO3− symport and H+ uniport.60 To determine if the observed cytotoxicity was caused by sustained changes in pHi compounds 1–8 were studied in A375 melanoma cells stained with acridine orange (AO) (Fig. 9). Upon protonation this cell-permeable dye displays an orange fluorescence and accumulates within acidic intracellular compartments. Green fluorescence is observed under the more basic conditions of the cystol.61 Cells in basal conditions were stained with AO and showed typical granular orange fluorescence, corresponding to cellular acidic compartments such as lysosomes (Fig. 9 Ct). A complete loss of orange fluorescence was observed on exposure to compounds 5, 6 and 7 and a significant reduction in orange fluorescence was observed with compounds 2 and 3. This corresponds to an increase in the pH of the intracellular organelles, achieved through acidification of the cytoplasm. Granular orange fluorescence was retained with compounds 1, 4 and 8, indicating that exposure to these compounds did not facilitate changes in pHi. This finding, combined with the inactivity of compound 8 in the vesicle based ion transport assays, suggests that the cytotoxicity in this instance is the result of a mechanism other than transmembrane ion transport.58
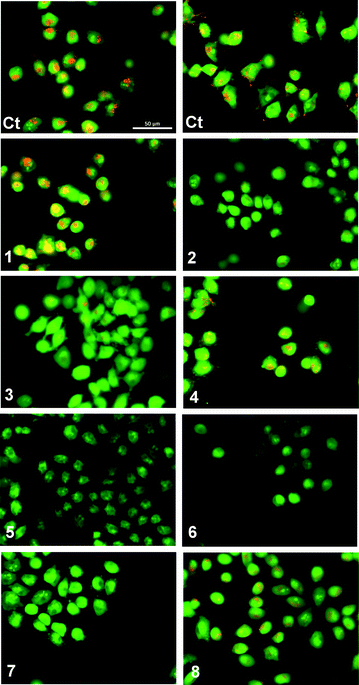 | ||
| Fig. 9 Acridine Orange staining of melanoma A375 cells after 1 hour exposure to compounds 1–8 (50 μM): (Ct) control (untreated cells). Cells with cytoplasmic granular orange fluorescence (Ct, 1, 4 and 8); cells with a significant reduction in cytoplasmic orange fluorescence (2 and 3); cells with complete disappearance of cytoplasmic orange fluorescence (5, 6 and 7). | ||
Cancer cell death can be induced by sustained changes in pHi.62 Molecules capable of facilitating HCl symport and Cl−/HCO3− antiport have been shown to induce apoptosis by facilitating ion transport processes that lower pHi.15,44,63 All of the compounds that facilitated a reduction in pHi function as Cl−/NO3− and Cl−/HCO3− antiport agents in vesicle based assays. In addition, the most active transporter (compound 6) has been shown to facilitate HCl co-transport. The biological activity observed in the AO assay could therefore be the direct result of the transmembrane transport of HCl or bicarbonate, or the result of ion transport coupled to the uniport of chloride.64
Apoptosis is a form of programmed cell death that facilitates the removal of damaged cells without causing inflammation and can be induced by sustained changes in pHi. Nuclear condensation, fragmentation and the formation of apoptotic bodies are characteristic of this type of cell death.65 The type of cell death induced by compounds 1–8 was examined using Hoechst 33342 staining in A375 cells. Hoechst 33342 is a fluorescent nuclear dye, permitting changes in nuclear morphology to be monitored. In Fig. 10 untreated A375 cells have typical rounded nuclei (Ct). Following treatment with the compounds (10 μM for 48 hours), the most cytotoxic compounds 2, 5 and 8 showed nuclear condensation (2a, 5a, 8a), and at 50 μM the formation of apoptotic bodies was observed, confirming that these compounds induce apoptotic cell death (2b, 5b and 8b).
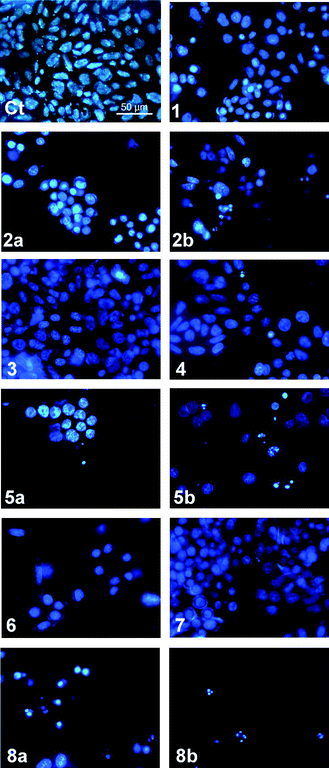 | ||
| Fig. 10 Hoechst 33342 staining of A375 cells after 48 hours exposure to compounds 1–8 (10 μM or 10 μM (a) and 50 μM (b)): (Ct) control (untreated cells). Cells with typical nuclear morphology (Ct, 1, 3, 4 and 7), cells with nuclear condensation and apoptotic bodies (2a, 2b, 5a, 5b, 6, 8a, 8b). | ||
In summary, compounds 2, 3, 5, 6, 7 and 8 have been shown to be toxic in a range of cancer cell lines. With the exception of compound 8, all of the cytotoxic compounds were found to facilitate Cl−/NO3− and Cl−/HCO3− antiport in vesicle based ion transport assays. In an AO assay compounds 2, 3, 5, 6 and 7 were found to alter pHi in A375 cells and the formation of apoptotic bodies was clearly observed with the most cytotoxic compounds (2, 5 and 8) using Hoechst 33342 staining in A375 cells. Compounds 2 and 5 facilitated apoptotic cell death in A375 cells, likely through sustained changes in pHi as a consequence of in vitro ion transport. In contrast, compound 8 induced apoptotic cell death but organelle deacidification was not observed in the AO assay. The cytotoxicity of this compound thus appears to be unrelated to its ion transport activity (compound 8 was found to be inactive in vesicle based ion transport assays) and may therefore be the result of an alternative mechanism.58 Compound 5 was shown to reduce the viability of the cell lines to a greater extent than compound 6 whilst compound 6 is the most effective anion transporter of the two compounds. This could be due to differences between the model membrane used for the vesicle studies and the biological membranes present in the cells. We are continuing to study the effects of these compounds on cells.
Molecular dynamics simulations on POPC bilayer model
The ability of 5 and 6 to permeate a POPC bilayer model, being the first step towards the anion transport, was also investigated by molecular dynamics (MD) simulations using the AMBER12 software.66 These mobile carriers were selected taking into account that, among the ortho-phenylendimine-based bisureas, 5 with two p-CF3 substituents is significantly the more lipophilic one, while 6 functionalized with two p-NO2 withdrawing electron substituents is the most active Cl−/HCO3− anion exchanger.These modelling studies were carried out with 5·Cl−, 6·Cl− and 5·HCO3− complexes and using a bilayer system (square in the xy plane), composed of 128 POPC lipids and 6500 TIP3P water molecules,67 as model of a POPC vesicle. The compounds 5 and 6 and bicarbonate anion were described with GAFF68 and RESP atomic charges (see ESI for details†), while the LIPID11 force field parameters69 were used for the lipid molecules. The chloride anion and sodium counterion with respective net charges of −1 and +1 were described with van der Waals parameters developed for the TIP3P water model.70 The membrane simulations were performed under periodic boundary conditions in a NPT ensemble at 303 K with a surface tension of 17 dyn cm−1 and an 8 Å cut-off for van der Waals and non-bonded electrostatic interactions following the protocol detailed in ESI.†
The starting binding arrangements of the anionic complexes were determined in gas phase by quenched molecular dynamics as stated in ESI.† In the lowest energy structures of the halide complexes, the two-bisurea binding sites adopt a syn configuration, establishing four N–H⋯Cl− hydrogen bonds. In the 5·HCO3− complex, three N–H binding sites form three N–H⋯O hydrogen bonds with an oxygen atom from the –CO2− group while the remaining N–H binding site is involved in a N–H⋯O single hydrogen bond with the other oxygen atom from this group. These two binding scenarios are illustrated in Fig. 11 with the structures of 5·Cl− and 5·HCO3− complexes, along with hydrogen bonds lengths.
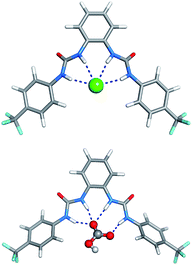 | ||
| Fig. 11 Molecular mechanics lowest energy structures of 5·Cl− (top) and 5·HCO3− (bottom) complexes, with N⋯Cl− distances ranging from 3.367 to 3.378 Å, and N⋯O distances ranging from 2.748 to 2.873 Å. | ||
Afterwards, 5·Cl−, 6·Cl− and 5·HCO3− complexes were initially positioned in the water slab of a pre-equilibrated POPC bilayer at distances larger than 8 Å away from the water–lipid interface (see Fig. S95 in ESI†) and subject to a production run of 80 ns simulation length, preceded by a multi-stage equilibration process (see ESI†). Two independent replicates (R1 and R2) were performed for each system using different initial velocities and comparable sequences of events were observed as described below. The chloride and bicarbonate anions are promptly solvated by water molecules and released in the water phase before either 5 or 6 reach the water–lipid interface, which occurs within the first 20 ns of the production runs as depicted in Fig. 12 for all anion complexes. This first insight indicates that in the water phase, 5 displays equivalent binding behaviours towards both anions and henceforth the simulations carried out with 5·Cl− and 5·HCO3− are analysed together. Afterwards, the free receptors interact with the membrane interface in a comparable fashion. Indeed, 5 is able to either permeate the membrane or to remain at the interface during almost of the subsequent simulation time, as also evident in Fig. 12 (top and middle plots). Surprisingly, the two replicates performed with 6·Cl− reveal that this less lipophilic receptor (see Table 2) permeates the membrane more easily, which can be correlated with the population of N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P hydrogen bonds established between the amide binding sites and phosphate head groups, as discussed below.
P hydrogen bonds established between the amide binding sites and phosphate head groups, as discussed below.
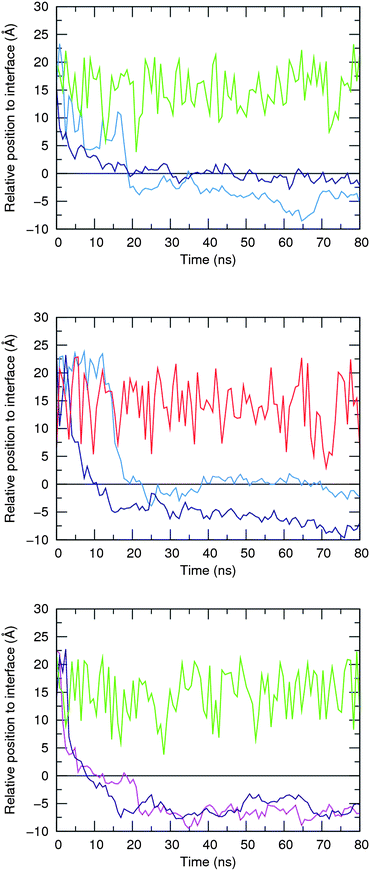 | ||
| Fig. 12 Relative positions of 5, 6, Cl− and HCO3− species defined by the distances between their centre of mass and the closest water–lipid interface, which results from the average position of the corresponding phosphorus atoms in the z coordinate. Initial complexes per plot: 5·Cl− – top; 5·HCO3− – middle; and 6·Cl− – bottom. The following line colour scheme was used: light blue for R1 and dark blue for R2 of 5; purple for R1 and magenta for R2 of 6; green for Cl−; and red for HCO3−. The water–lipid interface is represented as a black line at z = 0 Å. Only the position of the anion along the replicate R1 is represented. | ||
The events’ sequences associated with the passive diffusion of 5 and 6 towards POPC, described above, are illustrated in Fig. 13 and 14 with three consecutive snapshots taken from the MD simulations of 5·Cl− and 6·Cl− complexes, respectively.
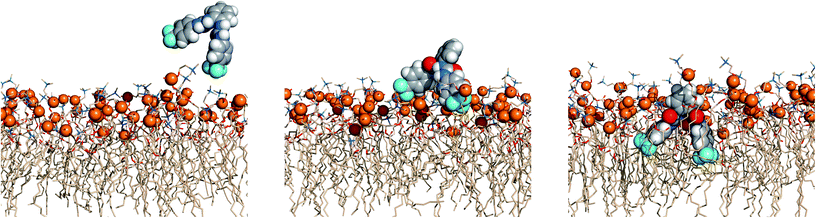 | ||
| Fig. 13 Passive diffusion of 5·Cl− along the POPC bilayer showing the interaction of 5 with the phospholipid heads with three sequential snapshots taken from a representative MD replicate (R1). The receptor and phosphorus atoms are drawn in space filling fashion with hydrogen atoms in white, oxygen atoms in red, nitrogen atoms in light blue and carbon atoms in grey (receptor) or wheat (phospholipids) colour. The van der Waals radius of P was arbitrarily set to 1.2 Å. The water slabs and the second monolayer of phospholipids were omitted for clarity. | ||
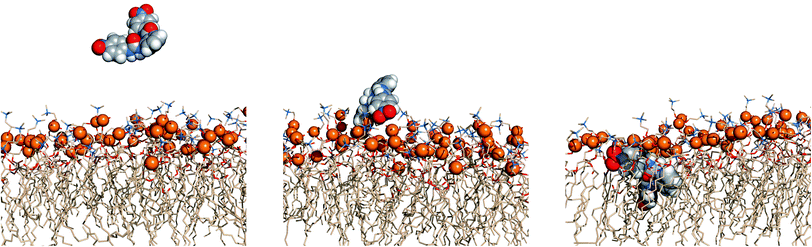 | ||
| Fig. 14 Passive diffusion of 6·Cl− along POPC bilayer illustrated with three sequential snapshots of a representative MD replicate (R2). Remaining details as given in Fig. 13. | ||
Fig. 13 and 14 show that the first interaction of 5 and 6 with membrane occurs through the p-CF3 or p-NO2 groups from an ortho-phenyldiamine substituent. Subsequently, regardless of the relative position of the receptors to the interface, both p-substituents of 5 are preferentially located between the lipids chains, pointing to the bilayer core, while the ortho-phenyldiamine head with the two syn urea binding units are closer to the water–lipid interface, thus able to form strong hydrogen bonding interactions with the phosphate head groups as shown in Fig. 15 (left). This is particularly evident in replicates R2 and R1 of complexes 5·Cl− and 5·HCO3−, respectively, where the receptor mainly remains at the water–lipid interface throughout the course of the MD simulation (see Fig. 12, top and middle plots, above and Fig. S96,† left and right top plots). In contrast, the slightly lighter receptor 6 (mol. wt 436.11 of 6vs. 482.12 of 5) permeates the phospholipid bilayer more deeply during the simulation time (see Fig. 12, bottom) with a non-specific spatial orientation consistent with the existence of sporadic N–H⋯OP hydrogen bonds. These interactions generally occur via a single urea binding unit, as depicted in Fig. 15 (right), and are observed during both MD simulations replicates of 6·Cl− (see Fig. S96,† bottom), in spite of this receptor higher propensity to establish hydrogen bonding interactions (see Table 3). In other words, these results seem to indicate that N–H binding sites of 6 are available to bind the anions and consequently to promote their transmembrane transport, in agreement with the chloride efflux experimental data.
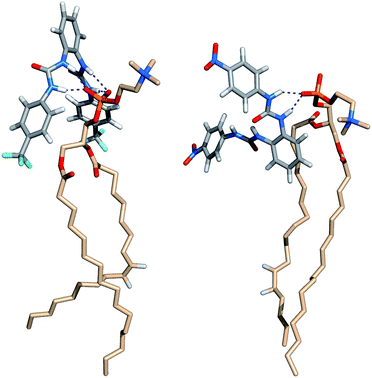 | ||
| Fig. 15 Insights into N–H⋯OP hydrogen bonding interactions between phospholipids and transporters 5 (left) and 6 (right). | ||
In order to ascertain the impact of 5 and 6 on the structural properties of the POPC bilayer, the aforementioned simulations were preceded by a simulation of the free membrane under the same computational conditions. The following structural membrane parameters, area per lipid, bilayer thickness, electronic density profile and order parameters were evaluated and compared with the corresponding experimental data and with those found for the last 10 ns of MD replicates of 5·Cl−, 6·Cl− and 5·HCO3− complexes. The first two parameters' values are listed in Table 5, whereas the corresponding electronic density profiles and order parameters are plotted in Fig. S100 to S105.† As would be expected, the average values of the area per lipid (64.38 Å2) and bilayer thickness (38.28 Å) calculated for the free membrane are similar to those recently obtained by neutron scattering (64.30 Å2 and 39.10 Å),71 and as reported when a POPC bilayer with 128 lipids is simulated using the aforementioned simulation conditions.69 The interaction of 5 and 6 with the membrane produces a slight decrease in the area per lipid (<4.8%) and, consequently, a slight increase in the bilayer thickness (<2.2%), indicating that the influence of both receptors on these two structural parameters is small. The order parameters for saturated (sn-1) and unsaturated (sn-2) POPC lipid chains of the free membrane (see Fig. S99†) follow the ones previously found with LIPID11.69 Similar profiles were obtained for the membrane simulations carried out with 5 and 6 (see Fig. S103 to S105†), suggesting that the order parameters are unaffected by the presence of the receptors. In fact, the single exception was found for the sn-2 tails of POPC in replicate R2 of 5·Cl− in which the receptor is preferentially located near of the water–lipid interface. Furthermore, the electronic density profiles for all simulated systems show that the bilayer structure is preserved without apparent interdigitation between the phospholipid tails as shown in Fig. S100 to S102, and further discussed in ESI.† In addition, the electronic density profiles for the receptors are consistent with their relative position to the water–lipid interface throughout the simulation time, as represented in Fig. 12.
| System | Area | Thickness | |
|---|---|---|---|
| a The sampling time for the pure membrane was 40 ns, while the remaining systems were analysed during the final 10 ns of the MD simulation time. | |||
| Pure membrane | |||
| Experimental71 | 64.30 | 39.10 | |
| Simulated | 64.38 ± 0.74 | 38.28 ± 0.37 | |
| Transporters in POPC | |||
| 5·Cl− | R1 | 62.16 ± 0.53 | 39.34 ± 0.24 |
| R2 | 61.25 ± 0.49 | 39.80 ± 0.31 | |
| 5·HCO3− | R1 | 62.67 ± 0.74 | 39.30 ± 0.44 |
| R2 | 61.50 ± 0.77 | 39.97 ± 0.30 | |
| 6·Cl− | R1 | 62.39 ± 0.40 | 39.14 ± 0.23 |
| R2 | 61.79 ± 0.91 | 39.67 ± 0.61 | |
The energy profiles associated with Cl− and HCO3− facilitated transport across a phospholipid bilayer by ortho-phenylenediamine-based bisureas (namely 4, 5 and 6), as well as the theoretical investigation of Cl−/HCO3− exchange mechanisms in more complex membrane systems (models of vesicles) and more computationally demanding, are currently in progress. However, the studies reported here have unequivocally shown that two of the most promising receptors are able to permeate the membrane, having a low impact in its structure, being capable of mediating anion transport as mobile carriers.
Conclusions
We have demonstrated that the ortho-phenylenebisurea motif is a new scaffold for the construction of transmembrane ion transporters that function by an anion antiport and in some cases a HCl co-transport mechanism. Addition of electron withdrawing groups to either the central core or the peripheral phenyl groups yielded potent anion transporters for the transmembrane transport of chloride, nitrate and bicarbonate with transport observed at receptor: lipid concentration ratios as low as 1:1000000. Compounds 2, 3, 5, 6 and 7 were found to be cytotoxic in a range of cancer cell lines. AO assays confirmed that these compounds can facilitate a reduction in pHi and the two most cytotoxic compounds (2 and 5) were found to induce apoptosis in A375 cells. We have also demonstrated that our most active transporters (4, 5 and 6) can facilitate the transmembrane transport of carboxylate species. We are continuing to study the transport of biologically relevant carboxylates. The results of these studies will be reported in due course. The MD results have demonstrated, at the atomic level, the ability of 5 and 6 to permeate the membrane while having a reduced influence on its properties.
Acknowledgements
PAG thanks the EPSRC for a PhD studentship (SJM) and for access to the crystallographic facilities at the University of Southampton.72 CJEH thanks the EPSRC for a Doctoral Prize. This work was supported by a research grant from the Spanish government and the European Union (FIS-PI10/00338). The modelling studies were supported by the FEDER, through the Operational Program Competitiveness Factors – COMPETE and National Funds through FCT (Fundação para a Ciência e a Tecnologia) under project PTDC/QUI-QUI/101022/2008. PJC thanks FCT for the postdoctoral grant SFRH/BPD/27082/2006.Notes and references
- J. I. Korenbrot, Annu. Rev. Physiol., 1977, 39, 19–49 CrossRef CAS.
- F. M. Ashcroft, Ion Channels and Disease, Academic Press, San Diego, 2000 Search PubMed.
- E. Cordat and J. R. Casey, Biochem. J., 2009, 417, 423–439 CrossRef CAS.
- (a) P. M. Quinton, Lancet, 2008, 372, 415–417 CrossRef CAS; (b) A. S. Garcia, N. Yang and P. M. Quinton, J. Clin. Invest., 2009, 119, 2613–2622 CrossRef.
- (a) N. Altan, Y. Chen, M. Schindler and S. M. Simon, J. Exp. Med., 1998, 187, 1583–1598 CrossRef CAS; (b) Y. Chen, M. Schindler and S. M. Simon, J. Biol. Chem., 1999, 274, 18364–18373 CrossRef CAS; (c) S. Matsuyama, J. Llopis, Q. L. Deveraux, R. Y. Tsien and J. C. Reed, Nat. Cell Biol., 2000, 2, 318–325 CrossRef CAS.
- For an excellent overview of the chemistry of the prodigiosins related to their structural, anion transport and anti-cancer properties see: (a) J. T. Davis, in Topics in Heterocyclic Chemistry, ed. P. A. Gale and W. Dehaen, Springer-Verlag, Berlin Heidelberg, 2010, vol. 24, pp. 145–176 Search PubMed. See also: (b) T. Sato, H. Konno, Y. Tanaka, T. Kataoka, K. Nagai, H. H. Wasserman and S. Ohkuma, J. Biol. Chem., 1998, 273, 21455–21462 CrossRef CAS; (c) J. L. Sessler, L. R. Eller, W.-S. Cho, S. Nicolaou, A. Aguilar, J. T. Lee, V. M. Lynch and D. J. Magda, Angew. Chem., Int. Ed., 2005, 44, 5989–5992 CrossRef CAS; (d) A. Fürstner, Angew. Chem., Int. Ed., 2003, 42, 3582–3603 CrossRef; (e) J. L. Seganish and J. T. Davis, Chem. Commun., 2005, 5781–5783 RSC; (f) H. Konno, H. Matsuya, M. Okamoto, T. Sato, Y. Tanaka, K. Yokoyama, T. Kataoka, K. Nagai, H. H. Wasserman and S. Ohkuma, J. Biochem., 1998, 124, 547–556 CrossRef CAS; (g) J. T. Davis, P. A. Gale, O. A. Okunola, P. Prados, J. C. Iglesias-Sánchez, T. Torroba and R. Quesada, Nat. Chem., 2009, 1, 138–144 CrossRef CAS; (h) M. Nguyen, R. C. Marcellus, A. Roulston, M. Watson, L. Serfass, S. R. M. Madiraju, D. Goulet, J. Viallet, L. Bélec, X. Billot, S. Acoca, E. Purisima, A. Wiegmans, L. Cluse, R. W. Johnstone, P. Beauparlant and G. C. Shore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19512–19517 CrossRef CAS.
- (a) B. A. McNally, A. V. Koulov, B. D. Smith, J.-B. Joos and A. P. Davis, Chem. Commun., 2005, 1087–1089 RSC; (b) A. P. Davis and J.-B. Joos, Coord. Chem. Rev., 2003, 240, 143–156 CrossRef CAS; (c) A. P. Davis, Coord. Chem. Rev., 2006, 250, 2939–2951 CrossRef CAS; (d) B. A. McNally, A. V. Koulov, T. N. Lambert, B. D. Smith, J.-B. Joos, A. L. Sisson, J. P. Clare, V. Sgarlata, L. W. Judd, G. Magro and A. P. Davis, Chem.–Eur. J., 2008, 14, 9599–9606 CrossRef CAS.
- (a) C. C. Tong, R. Quesada, J. L. Sessler and P. A. Gale, Chem. Commun., 2008, 6321–6323 RSC; (b) P. A. Gale, C. C. Tong, C. J. E. Haynes, O. Adeosun, D. E. Gross, E. Karnas, E. M. Sedenberg, R. Quesada and J. L. Sessler, J. Am. Chem. Soc., 2010, 132, 3240–3241 CrossRef CAS; (c) M. G. Fisher, P. A. Gale, J. R. Hiscock, M. B. Hursthouse, M. E. Light, F. P. Schmidtchen and C. C. Tong, Chem. Commun., 2009, 3017–3019 RSC.
- S. K. Berezin and J. T. Davis, J. Am. Chem. Soc., 2009, 131, 2458–2459 CrossRef CAS; S. Bahmanjah, N. Zhang and J. T. Davis, Chem. Commun., 2012, 48, 4432–4434 RSC.
- P. V. Santacroce, J. T. Davis, M. E. Light, P. A. Gale, J. C. Iglesias-Sánchez, P. Prados and R. Quesada, J. Am. Chem. Soc., 2007, 129, 1886–1887 CrossRef CAS; N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef.
- N. Busschaert, P. A. Gale, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis and W. A. Harrell, Jr, Chem. Commun., 2010, 46, 6252–6254 RSC.
- (a) H. Grasemann, F. Stehling, H. Brunar, R. Widmann, T. W. Laliberte, L. Molina, G. Döring and F. Ratjen, Chest, 2007, 131, 1461–1466 CrossRef CAS; (b) I. Izzo, S. Licen, N. Maulucci, G. Autore, S. Marzocco, P. Tecilla and F. De Riccardis, Chem. Commun., 2008, 2986–2988 RSC; (c) X. Li, B. Shen, X.-Q. Yao and D. Yang, J. Am. Chem. Soc., 2009, 131, 13676–13680 CrossRef CAS.
- (a) M. P. Gleeson, J. Med. Chem., 2008, 51, 817–834 CrossRef CAS; (b) E. H. Kerns and L. Di, Drug-like Properties: Concepts, Structure Design and Methods, Academic Press, Amsterdam, 2008 Search PubMed.
- (a) C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS; (b) C. A. Lipinski, J. Pharmacol. Toxicol. Methods, 2000, 44, 235–249 CrossRef CAS.
- S. J. Moore, M. Wenzel, M. E. Light, R. Morley, S. J. Bradberry, P. Gómez-Iglesias, V. Soto-Cerrato, R. Pérez-Tomás and P. A. Gale, Chem. Sci., 2012, 3, 2501–2508 RSC.
- C. J. E. Haynes, S. J. Moore, J. R. Hiscock, I. Marques, P. J. Costa, V. Félix and P. A. Gale, Chem. Sci., 2012, 3, 1436–1444 RSC.
- (a) S. J. Brooks, P. A. Gale and M. E. Light, Chem. Commun., 2005, 4696–4698 RSC; (b) S. J. Brooks, P. R. Edwards, P. A. Gale and M. E. Light, New J. Chem., 2006, 30, 65–70 RSC.
- Y.-J. Kim, H. Kwak, S. J. Lee, J. S. Lee, H. J. Kwon, S. H. Nam, K. Lee and C. Kim, Tetrahedron, 2006, 62, 9635–9640 CrossRef CAS.
- S. J. Brooks, P. A. Gale and M. E. Light, Supramol. Chem., 2007, 19, 9–15 CrossRef CAS.
- (a) C. Jia, B. Wu, S. Li, Z. Yang, Q. Zhao, J. Liang, Q.-S. Li and X.-J. Yang, Chem. Commun., 2010, 46, 5376–5378 RSC; (b) C. Jia, B. Wu, S. Li, X. Huang and X.-J. Yang, Org. Lett., 2010, 12, 5612–5615 CrossRef CAS.
- C. Jia, B. Wu, S. Li, X. Huang, Q. Zhao, Q.-S. Li and X.-J. Yang, Angew. Chem., Int. Ed., 2011, 50, 486–490 CrossRef CAS.
- (a) S. J. Brooks, P. A. Gale and M. E. Light, Chem. Commun., 2006, 4344–4346 RSC; (b) S. J. Brooks, S. E. García-Garrido, M. E. Light, P. A. Cole and P. A. Gale, Chem.–Eur. J., 2007, 13, 3320–3329 CrossRef CAS.
- (a) H. Yang, Z.-G. Zhou, J. Xu, F.-Y. Li, T. Yi and C.-H. Huang, Tetrahedron, 2007, 63, 6732–6736 CrossRef CAS; (b) D. A. Jose, D. K. Kumar, B. Ganguly and A. Das, Tetrahedron Lett., 2005, 46, 5343–5346 CrossRef CAS.
- V. Amendola, M. Boiocchi, D. Esteban-Gómez, L. Fabbrizzi and E. Monzani, Org. Biomol. Chem., 2005, 3, 2632–2639 CAS.
- M. O. Odago, D. M. Colabello and A. J. Lees, Tetrahedron, 2010, 66, 7465–7471 CrossRef CAS.
- (a) S. J. Brooks, P. A. Gale and M. E. Light, CrystEngComm, 2005, 7, 586–591 RSC; (b) S. Li, M. Wei, X. Huang, X.-J. Yang and B. Wu, Chem. Commun., 2012, 48, 3097–3099 RSC.
- J. van Esch, F. Schoonbeek, M. de Loos, H. Kooijman, A. L. Spek, R. M. Kellogg and B. Feringa, Chem.–Eur. J., 1999, 5, 937–950 CrossRef CAS.
- (a) C. Bied, J. J. E. Moreau and M. W. C. Man, Tetrahedron: Asymmetry, 2001, 12, 329–336 CrossRef CAS; (b) E. G. Klauber, C. K. De, T. K. Shah and D. Seidel, J. Am. Chem. Soc., 2010, 132, 13624–13626 CrossRef CAS.
- (a) M. T. Bogert and L. E. Wise, J. Am. Chem. Soc., 1912, 34, 693–702 CrossRef CAS; (b) L. Peyron and J. Peyron, Bull. Soc. Chim., 1953, 9, 846–852 Search PubMed; (c) S. N. Gavade, R. S. Balaskar, M. S. Mane, P. N. Pabrekar, M. S. Shingare and D. V. Mane, Chin. Chem. Lett., 2011, 22, 675–678 CrossRef CAS.
- (a) J. Scheele, P. Timmerman and D. N. Reinhoudt, Chem. Commun., 1998, 2613–2614 RSC; (b) Y. Ge, L. Miller, T. Ouimet and D. K. Smith, J. Org. Chem., 2000, 65, 8831–8838 CrossRef CAS.
- (a) P. Grammaticakis, Bull. Soc. Chim., 1959, 10, 1559–1570 Search PubMed; (b) H. Iwamura, T. Fujita, S. Koyama, K. Koshimizu and Z. Kumazawa, Phytochemistry, 1980, 19, 1309–1319 CAS.
- (a) M. Miyahara, Chem. Pharm. Bull., 1986, 34, 1950–1960 CrossRef CAS; (b) G. A. Artamkina, A. G. Sergeev and I. P. Beletskava, Russ. J. Org. Chem., 2002, 38, 538–545 CrossRef CAS; (c) G. A. Artamkina, A. G. Sergeev and I. P. Beletskaya, Tetrahedron Lett., 2001, 42, 4381–4384 CrossRef CAS; (d) B. J. Kotecki, D. P. Fernando, A. R. Haight and K. A. Lukin, Org. Lett., 2009, 11, 947–950 CrossRef CAS.
- (a) N. J. Andrews, C. J. E. Haynes, M. E. Light, S. J. Moore, C. C. Tong, J. T. Davis, W. A. Harrell Jr and P. A. Gale, Chem. Sci., 2011, 2, 256–260 RSC; (b) P. A. Gale, J. R. Hiscock, S. J. Moore, C. Caltagirone, M. B. Hursthouse and M. E. Light, Chem.–Asian J., 2010, 5, 555–561 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- A. Barnard, S. J. Dickson, M. J. Paterson, A. M. Todd and J. W. Steed, Org. Biomol. Chem., 2009, 7, 1554–1561 CAS.
- (a) M. Boiocchi, L. Del Boca, D. Esteban-Gómez, L. Fabbrizzi, M. Licchelli and E. Monzani, Chem.–Eur. J., 2005, 11, 3097–3104 CrossRef CAS; (b) D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, J. Org. Chem., 2005, 70, 5717–5720 CrossRef; (c) V. Amendola, D. Esteban-Gómez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343–353 CrossRef CAS; (d) M. Bonizzoni, L. Fabbrizzi, A. Taglietti and F. Tiengo, Eur. J. Org. Chem., 2006, 3567–3574 CrossRef CAS; (e) S. E. García-Garrido, C. Caltagirone, M. E. Light and P. A. Gale, Chem. Commun., 2007, 1450–1452 RSC.
- Y. Shao, B. Linton, A. D. Hamilton and S. G. Weber, J. Electroanal. Chem., 1998, 441, 33–37 CrossRef CAS.
- E. N. Kitova, A. El-Hawiet, P. D. Schnier and J. S. Klassen, J. Am. Soc. Mass Spectrom., 2012, 23, 431–441 CrossRef CAS.
- L. Jaquillard, F. Saab, F. Schoentgen and M. Cadene, J. Am. Soc. Mass Spectrom., 2012, 23, 908–922 CrossRef CAS.
- (a) B. D. Smith and T. N. Lambert, Chem. Commun., 2003, 2261–2268 RSC; (b) A. V. Koulov, T. N. Lambert, R. Shukla, M. Jain, J. M. Boon, B. D. Smith, H. Li, D. N. Sheppard, J.-B. Joos, J. P. Clare and A. P. Davis, Angew. Chem., Int. Ed., 2003, 42, 4931–4933 CrossRef CAS.
- M. Yano, C. C. Tong, M. E. Light, F. P. Schmidtchen and P. A. Gale, Org. Biomol. Chem., 2010, 8, 4356–4363 CAS.
- (a) W. W. Cleland, T. J. Andrews, S. Gutteridge, F. C. Hartman and G. H. Lorimer, Chem. Rev., 1998, 98, 549–561 CrossRef CAS; (b) A.-V. Rousselle and D. Heymann, Bone, 2002, 30, 533–540 CrossRef CAS; (c) D. Bok, G. Galbraith, I. Lopez, M. Woodruff, S. Nusinowitz, H. BeltrandelRio, W. Huang, S. Zhao, R. Geske, C. Montgomery, I. van Sligtenhorst, C. Friddle, K. Platt, M. J. Sparks, A. Pushkin, N. Abuladze, A. Ishiyama, R. Dukkipati, W. Liu and I. Kurtz, Nat. Genet., 2003, 34, 313–319 CrossRef CAS; (d) R. D. Vaughan-Jones, K. W. Spitzer and P. Swietach, J. Mol. Cell. Cardiol., 2009, 46, 318–331 CrossRef CAS.
- Y. Marcus, J. Chem. Soc., Faraday Trans., 1991, 87, 2995–2999 RSC.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS.
- P. A. Gale, J. Garric, M. E. Light, B. A. McNally and B. D. Smith, Chem. Commun., 2007, 1736–1738 RSC.
- N. R. Clement and J. M. Gould, Biochemistry, 1981, 20, 1534–1538 CrossRef CAS.
- (a) A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC; (b) A. L. Sisson, M. R. Shah, S. Bhosale and S. Matile, Chem. Soc. Rev., 2006, 35, 1269–1286 RSC; (c) B. A. McNally, E. J. O'Neil, A. Nguyen and B. D. Smith, J. Am. Chem. Soc., 2008, 130, 17274–17275 CrossRef CAS; (d) G. W. Gokel and N. Barkey, New J. Chem., 2009, 33, 947–963 RSC; (e) J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC; (f) P. A. Gale, Acc. Chem. Res., 2011, 44, 216–226 CrossRef CAS.
- C. Kirby, J. Clarke and G. Gregoriadis, Biochem. J., 1980, 186, 591–598 CAS.
- O. Murillo, I. Suzuki, E. Abel, C. L. Murray, E. S. Meadows, T. Jin and G. W. Gokel, J. Am. Chem. Soc., 1997, 119, 5540–5549 CrossRef CAS.
- (a) A. V. Hill, Biochem. J., 1913, 7, 471–480 CAS; (b) S. Bhosale and S. Matile, Chirality, 2006, 18, 849–856 CrossRef CAS.
- (a) P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43, 3714–3737 CrossRef CAS; (b) S. A. Wildman and G. M. Crippen, J. Chem. Inf. Comput. Sci., 1999, 39, 868–873 CrossRef CAS; (c) Fieldview version 2.0.2 for Macintosh, Cresset, 2011 Search PubMed.
- (a) D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420–427 CrossRef CAS; (b) S. R. Crouch, D. A. Skoog, D. M. West and F. J. Holler, Analytical Chemistry, An Introduction, 7th edn, Brooks/Cole, Belmont CA, USA, 1999, Appendix 2, p. A-3 Search PubMed.
- (a) F. Sancenón, R. Martínez-Máñez, M. A. Miranda, M.-J. Seguí and J. Soto, Angew. Chem., Int. Ed., 2003, 42, 647–650 CrossRef; (b) A. M. Costero, M. Colera, P. Gaviña and S. Gil, Chem. Commun., 2006, 761–763 RSC; (c) J. C. Kim, A. J. Lough, H. Park and Y. C. Kang, Inorg. Chem. Commun., 2006, 9, 514–517 CrossRef CAS; (d) Y.-P. Yen and K.-W. Ho, Tetrahedron Lett., 2006, 47, 7357–7361 CrossRef CAS; (e) Y.-P. Tseng, G.-M. Tu, C.-H. Lin, C.-T. Chang, C.-Y. Lin and Y.-P. Yen, Org. Biomol. Chem., 2007, 5, 3592–3598 RSC; (f) S. Goswami, N. K. Das, D. Sen, G. Hazra, J. H. Goh, Y. C. Sing and H.-K. Fun, New J. Chem., 2011, 35, 2811–2819 RSC.
- (a) E. Fan, S. A. Van, S. Kincaid and A. D. Hamilton, J. Am. Chem. Soc., 1993, 115, 369–370 CrossRef CAS; (b) T. R. Kelly and M. H. Kim, J. Am. Chem. Soc., 1994, 116, 7072–7080 CrossRef CAS; (c) K.-S. Jeong, J. W. Park and Y. L. Cho, Tetrahedron Lett., 1996, 37, 2795–2798 CrossRef CAS; (d) R. J. Fitzmaurice, G. M. Kyne, D. Douheret and J. D. Kilburn, J. Chem. Soc., Perkin Trans. 1, 2002, 841–864 RSC.
- (a) E. Foran and D. Trotti, Antioxid. Redox Signaling, 2009, 11, 1587–1602 CrossRef CAS; (b) J.-H. Yi and A. S. Hazell, Neurochem. Int., 2006, 48, 394–403 CrossRef CAS; (c) E. Masliah, L. Hansen, M. Alford, R. Deteresa and M. Mallory, Ann. Neurol., 1996, 40, 759–766 CrossRef CAS; (d) S. Li, M. Mallory, M. Alford, S. Tanaka and E. Masliah, J. Neuropathol. Exp. Neurol., 1997, 56, 901–911 CrossRef CAS; (e) P. F. Behrens, P. Franz, B. Woodman, K. S. Lindenberg and G. B. Landwehrmeyer, Brain, 2002, 125, 1908–1922 CrossRef CAS.
- J. S. Clark, D. H. Vandrope, M. N. Chernova, J. F. Heneghan, A. K. Stewart and S. L. Alper, J. Physiol., 2008, 586, 1291–1306 CAS.
- H. A. Krebs, The Citric Acid Cycle, Nobel Lecture, 1953 Search PubMed.
- (a) J. Kazius, R. McGuire and R. Bursi, J. Med. Chem., 2005, 48, 312–320 CrossRef CAS; (b) Q. Li, M. Minami, T. Hanaoka and Y. Yamamura, Toxicology, 1999, 137, 35–45 CrossRef CAS; (c) K. Shinoda, K. Mitsumori, K. Yasuhara, C. Uneyama, H. Onodera, K. Takegawa, M. Takahashi and T. Umemura, Arch. Toxicol., 1998, 72, 296–302 CrossRef CAS; (d) M. Sajan, G. Reddy and A. P. Kulkarni, Int. J. Toxicol., 2000, 19, 285–292 CrossRef CAS; (e) Y. Ohkuma and S. Kawanishi, Biochem. Biophys. Res. Commun., 1999, 257, 555–560 CrossRef CAS.
- M. F. McCarty and J. Whitaker, Alternative Med. Rev., 2010, 15, 264–272 Search PubMed.
- I. H. Madshus, Biochem. J., 1988, 250, 1–8 CAS.
- A. C. Allison and M. R. Young, Lysosomes in Biology and Pathology, North-Holland Publishing Co., Amsterdam, 1969, vol. 2 Search PubMed.
- M. Yamagate and I. F. Tannock, Br. J. Cancer, 1996, 73, 1328–1334 CrossRef.
- R. Pérez-Tomás, B. Montaner, E. Llagostera and V. Soto-Cerrato, Biochem. Pharmacol., 2003, 66, 1447–1452 CrossRef; P. I. Hernández, D. Moreno, A. A. Javier, T. Torroba, R. Pérez-Tomás and R. Quesada, Chem. Commun., 2012, 48, 1556–1558 RSC.
- S. J. Moore, M. G. Fisher, M. Yano, C. C. Tong and P. A. Gale, Dalton Trans., 2011, 40, 12017–12020 RSC; S. J. Moore, M. G. Fisher, M. Yano, C. C. Tong and P. A. Gale, Chem. Commun., 2011, 47, 689–691 RSC.
- A. De Milito, E. Lessi, M. Logozzi, F. Lozupone, M. Spada, M. L. Marino, C. Federici, M. Perdicchio, P. Matarrese, L. Lugini, A. Nilsson and S. Fais, Cancer Res., 2007, 67, 5408–5417 CrossRef CAS.
- D. A. Case, T. A. Darden, T. E. Cheatham, III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko, and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- (a) J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS; (b) J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2005, 26, 114 CrossRef CAS.
- (a) Å. A. Skjevik, B. D. Madej, R. C. Walker and K. Teigen, J. Phys. Chem. B, 2012 DOI:10.1021/jp3059992; (b) R. C. Walker and B. Madej, private communication, 2012.
- I. S. Joung and T. E. Cheatham, J. Phys. Chem. B, 2008, 112, 9020–9041 CrossRef CAS.
- N. Kučerka, M.-P. Nieh and J. Katsaras, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2761–2771 CrossRef.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic experimental details, stability constant determinations. Membrane transport experimental and further details of the modelling work in the paper. CCDC 892348. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21112b |
| ‡ Crystal data for carbonate complex of 7: C57H72N14O15, M = 1193.29, triclinic, a = 14.146(2) Å, b = 14.338(2) Å, c = 16.719(3) Å, α = 86.271(8)°, β = 81.687(8)°, γ = 61.358(5)°, V = 2944.5(8) Å3, T = 100(2) K, P − 1, Z = 2, 33949 reflections measured, 13317 unique (Rint = 0.0279), R1 = 0.0618 [F2 > 2σ(F2)], R1 = 0.0780 (all data). The counterions balancing the two negative charges of the carbonate ion comprise one whole and two half TEA molecules. The two half molecules lie on inversion centres and were refined using thermal parameter and geometrical restraints. |
| This journal is © The Royal Society of Chemistry 2013 |