 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lanthanide appended rotaxanes respond to changing chloride concentration†
Clémence Allaina, Paul D. Beer*a, Stephen Faulkner*a, Michael W. Jonesa, Alan M. Kenwrightb, Nathan L. Kilaha, Richard C. Knightona, Thomas Just Sørensena and Manuel Tropianoa
aChemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: stephen.faulkner@chem.ox.ac.uk
bDepartment of Chemistry, University of Durham, South Road, Durham DH1 3LE, UK
First published on 22nd October 2012
Abstract
Lanthanide appended rotaxanes have been prepared by the CuAAC ‘click’ reaction between an azide appended rotaxane and lanthanide complexes of propargyl DO3A. The resulting complexes are luminescent, and exhibit chloride responsive luminescence behavior consistent with the existence of two independent halide binding pockets, one in the rotaxane cavity and one on the ninth (axial) coordination site of the lanthanide. Strong halide binding to europium gives rise to changes in the relative intensity of the hypersensitive ΔJ = 2 transition compared to the rest of the europium emission spectrum, combined with quenching of the overall intensity of emission as a consequence of non-radiative quenching by the bound halide. The weaker interaction with the rotaxane pocket mediates a subsequent recovery of intensity of the europium centered luminescence despite the considerable separation between the lanthanide and the rotaxane binding pocket.
Luminescence from lanthanide ions has been widely studied for more than a quarter of a century, partly as a consequence of the usefulness of luminescent lanthanide complexes in imaging and assay.1,2 The long-lived luminescence from lanthanides can be exploited in combination with time-gating methodologies to give very low detection limits and eliminate background fluorescence from organic chromophores and biological molecules.3,4 The low extinction coefficients associated with lanthanide absorption spectra can be overcome by incorporating chromophores into the ligand structure; these act as antennae and mediate formation of the lanthanide emissive state,5 usually via the chromophore triplet state, though charge transfer mediated energy transfer is common for ytterbium complexes.6 Energy transfer can occur by both Dexter exchange and Förster energy transfer depending upon the structure of the complex.7
Studies of luminescence from lanthanide containing systems have, understandably, tended to focus upon the local environment at the metal centre, and upon the sensitizing chromophore. Considerable effort has focused in recent years upon the development of new organic and d-block transition metal chromophores as sensitizers of lanthanide emission.8 Furthermore, the energy transfer cascade in such systems has been exploited in the development of a range of responsive complexes for which the luminescence is modulated by perturbations to intermediate states (e.g. non-radiative quenching of the excited aryl singlet state by chloride, or quenching of the triplet state by molecular oxygen),9 by chemical or electrochemical modification of the chromophore,10 or by coordination to the lanthanide with concomitant displacement of solvent.11
While lanthanide containing assemblies, particularly helicates and coordination polymers, are now becoming commonplace,12 the incorporation of lanthanide complexes into mechanically interlocked structures has been largely neglected. In 2006 we reported the synthesis and properties of a d–f hybrid pseudorotaxane that was assembled through anion templation in non-competitive solvent media,13 but which dissociates in competitive solvents. Stable, interlocked, mechanically bonded rotaxanes appended with lanthanides have proved elusive until now. We have utilised anion templation to construct a range of mechanically interlocked host molecules which, upon removal of the templating anion, bind anions selectively in competitive water-containing solvent mixtures.14 We reasoned that coupling of the interlocked architecture to a lanthanide containing luminescent reporter would allow us to explore the possibility of time-gated luminescence detection of anions in the rotaxane cavity, while also permitting us to evaluate the anion coordination chemistry of both the rotaxane binding site and the ninth (axial) coordination site on the lanthanide. Here we report the synthesis of a lanthanide containing rotaxane and the rationalization of its luminescence response in the presence of chloride and other anions. The use of a kinetically and thermodynamically stable luminescent tag is shown to greatly lower the chloride detection limit thanks to the use of superior sensitivity of luminescence detection over NMR. Furthermore, the effect of changes remote from the lanthanide site is shown to influence the photophysical properties of the lanthanide, suggesting that it is always necessary to consider the system as a whole.
The synthetic approach used to prepare the target lanthanide containing rotaxanes involved the initial assembly of an appropriately functionalized rotaxane, which was then coupled to a kinetically stable lanthanide complex. A chloride anion templated condensation clipping reaction between a pyridinium chloride axle (2·Cl), diamine macrocycle precursor (3) and 5-azidoisophthaloyl chloride afforded the azide functionalized rotaxane 4·Cl (Scheme 1).
![Preparation of Ln·1 [Ln = Lu and Eu].](/image/article/2013/SC/c2sc21614k/c2sc21614k-s1.gif) | ||
| Scheme 1 Preparation of Ln·1 [Ln = Lu and Eu]. | ||
4·Cl formed crystals that were suitable for X-ray crystallography. The structure confirms the interlocked nature of the system with the templating chloride anion located in the rotaxane cavity encapsulated by the orthogonal hydrogen-bonding array, with a mean N–Cl distance of 3.37 Å. The complexes Ln·5 (Ln = Eu, Lu) were prepared by the literature procedure.10,15 We were fortunate that Lu·5 also gave crystals suitable for crystallographic study; these are the first crystals of such a system to be studied, and give a direct insight into the precursor structure in such systems. The structure of Lu·5 is unusual in that two complexes are bridged by a carbonate, while the structure is stabilized by two sodium ions in the asymmetric unit and multiple water molecules in an extensive, complex 3-dimensional hydrogen-bonding network. The crystal was found to be a non-merohedral twin by rotation about the c-axis with a twin ratio of ca. 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30. The structures are illustrated in Fig. 1, and further details are tabulated in the ESI.†
:30. The structures are illustrated in Fig. 1, and further details are tabulated in the ESI.†
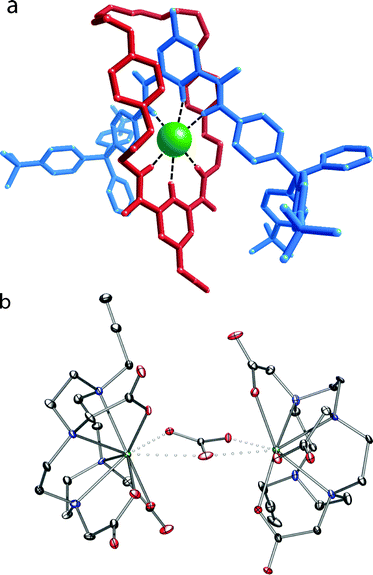 | ||
| Fig. 1 Representation of the solid state structure of (a) 4·Cl (Mercury capped stick depiction showing the components of the rotaxane) and (b) Lu·5 (ORTEP depiction) with thermal ellipsoids of Lu·5 displayed at 30% probability and the water, sodium, and hydrogen atoms omitted for clarity. | ||
CuAAC ‘click’ reaction between 4·Cl and Ln·5 yielded Ln·1 as its chloride complex, which upon halide anion removal from the rotaxane host cavity via washing with aqueous ammonium hexafluorophosphate was then converted to the non-coordinating hexafluorophosphate salt.
The rotaxane systems Ln·1 were characterized by mass spectrometry and NMR spectroscopy (further details are available in the ESI†). For Eu·1, the shifted regions of the spectra provide clear evidence for successful CuAAC ‘click’ reaction. Comparison of the 1H spectra for Ln·1 with published data for other related systems15a clearly indicates an octadentate ligand bound to the metal centre (as opposed to the heptadentate ligand in Ln·5), with a square antiprismatic geometry predominating at the lanthanide centre. This is in accordance with previous observations on the triazoloDO3A system.10,15 Though the protons on the rotaxane are too far from the paramagnetic lanthanide centre to be shifted significantly as a consequence of the presence of the lanthanide, the NMR spectrum of the Lu·1 complex is entirely consistent with the structure of the rotaxane domain being closely related to 4·Cl. Luminescence spectroscopy was used to probe the association of Ln·1 with chloride, dihydrogen phosphate and acetate in dichloromethane solution. There are two possible binding pockets for anions, since binding can occur on the ninth (axial) coordination site of the lanthanide metal as well as in the rotaxane binding pocket. Indeed, in dichloromethane solution binding to both is to be expected.
[Eu·1]·PF6 exhibits sensitized emission following excitation of the aryl chromophore, and the modulation of this sensitised emission upon titration with anions was used to probe the binding of these. Titration of a dichloromethane solution of [Eu·1]·PF6 with tetrabutylammonium chloride produced clear changes in the luminescence spectra. As the titration progressed, the luminescence intensity initially decreased sharply, before recovering as additional equivalents of chloride were added (Fig. 2), indicating that two binding events occur. Initially, increasing concentrations of chloride ions gave rise to a rapid decrease in lanthanide-centered emission accompanied by a change in the general band structure of the peaks arising from the ΔJ = 2 and the ΔJ = 4 transitions. Further increasing the concentration of chloride is followed by a more gradual increase in intensity without any change in appearance of the peaks in the spectrum. The initial binding process clearly involves a substantial change in the intensity of the hypersensitive ΔJ = 2 transition at 617 nm relative to the other europium centered transitions. This is clear evidence for binding of chloride to the ninth (axial) donor site on the lanthanide complex.16 For the first strong binding event, the association constant could be determined from the luminescence titration data. Fitting the observed data using Dynafit17 to a 1:1 model gave a binding constant for the association of chloride to [Eu·1]·PF6 in dichloromethane of 504000 M−1. The data did not allow for a determination of the association constant for the second binding event. Modelling the data shown in Fig. 2 allowed us to study the binding in depth and demonstrate that completely independent binding events18 have to be used to emulate the observed data; the representation was modelled using an association constant for the axial coordination site on the lanthanide (K1) of 750000 M−1 and an association constant (K2) for binding in the rotaxane pocket of 1360 M−1.
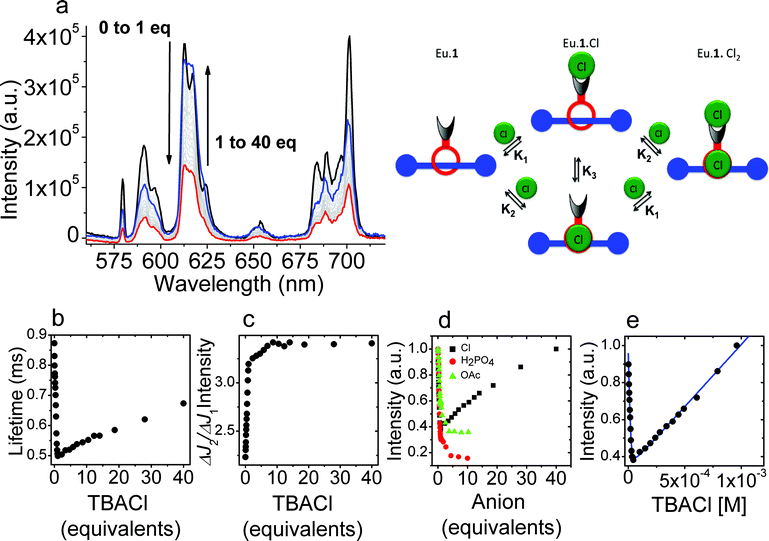 | ||
| Fig. 2 (a) Variation in the luminescence spectrum of Eu·1 upon titration with chloride (λex = 270 nm) in dichloromethane; (b) variation in luminescence lifetime of the europium centered emission arising from 5D0 as the titration progresses; (c) variation in the ratio of the intensities of the 5D0–7F2 and 5D0–7F1 transitions with increasing halide concentration; (d) variation in the luminescence intensity observed upon titration of Eu·1 with TBA·Cl, TBA·H2PO4 and TBA·OAc; (e) fitting the observed changes in luminescence intensity upon titration of Eu·1 with chloride in dichloromethane using an independent binding sites model as shown in the picture above. | ||
Time-resolved data casts further light upon the processes involved. The luminescence lifetimes of the europium centered emission initially falls off rapidly, as a consequence of intramolecular non-radiative quenching of the excited state of the lanthanide by a photo-electron transfer process involving the bound chloride. There is a subsequent recovery in lanthanide luminescence lifetime as a consequence of rigidification and changes in solvation following chloride binding in the rotaxane pocket. Importantly, the europium-centered emission acts as a sensor of the relatively remote structural changes that occur upon binding a chloride anion in the rotaxane cavity.
By contrast, when the binding of acetate and dihydrogenphosphate anions was studied in dichloromethane, we observed only one binding event, also accompanied by changes in the ΔJ = 2 transition, indicating that binding to the lanthanide center is completely predominant; no structural changes in the rotaxane was observed for these anions. In the case of acetate and dihydrogenphosphate titrants, the affinity of the axial donor site for the anion was reduced relative to the affinity for chloride (K1 = 58000 M−1 and 304000 M−1 for acetate and dihydrogenphosphate respectively). The absence of a second binding event in the rotaxane cavity is to be expected given the known selectivity of response to chloride for related rotaxane hosts.14 In more competitive solvent media comprising 1:1 dichloromethane–methanol mixtures, the affinity of anions for both lanthanide center and the rotaxane cavity was found to be diminished (see Table 1), and only binding to the former was observed in the luminescence titrations.
| Solvent | Added salt | K1a (M−1) | |
|---|---|---|---|
| a Obtained from ΔJ ratios binding plots.b Modelling the observed data to an independent binding model for the two binding sites gave two association constants (K1 = 750000 M−1 and 1360 M−1) that can be assigned to binding at the lanthanide and in the rotaxane cavity.c Confidence intervals determined as the values of K that are possible while keeping χ2 with 99% of the minimum value. | |||
| DCM | TBA·Cl | 504000b | [298900–1014000]c |
| TBA·H2PO4 | 304000 | [264200–354600]c | |
| TBA·OAc | 58000 | [52520–64300]c | |
| DCM–MeOH 1:1 | TBA·Cl | 95000 | [87554–102540]c |
| TBA·H2PO4 | 70000 | [54780–86250]c | |
| TBA·OAc | 23000 | [20290–26310]c | |
The assignment of binding sites was further supported by additional titrations of a model system containing only the lanthanide binding pocket in 1:1 DCM–MeOH (see ESI† for further details), which exhibited an association constant of 65000 M−1, similar to the association constant of the lanthanide site in Eu·1 with chloride in the same system. NMR studies on 4·PF6 were used to probe the affinity of the rotaxane for anions, revealing that the association constant with chloride (K1 = 1460 M−1) (CDCl3, MeOD, D2O, 45:45:10) was significantly greater than that for dihydrogenphosphate or acetate (K1 = 188 M−1 and K1 = 115 M−1 respectively). These observations lend further credence to the observation of two binding events in the titration of Eu·1 with chloride.
Conclusions
These results show that lanthanide containing rotaxanes can be constructed via a modular approach, where an appropriately functionalized rotaxane can be coupled to a lanthanide centre. Furthermore, the lanthanide ion is an effective component in anion responsive behaviour, both in the binding event directly and when acting as a sensor for remote changes of rotaxane host molecular structure due to specific anion binding. In the systems described in this manuscript, it is clear that two binding domains exist, and that binding of chloride ions can take place both through axial ligation to the lanthanide center and through binding in the rotaxane cavity. In this system, the two binding sites operate separately. However, the results reveal that lanthanides can act as efficient luminescent reporters and receptors in anion binding systems. Furthermore, the long-lived luminescence from lanthanide ions can be time-gated to remove background fluorescence and scatter, meaning that this work sets a clear precedent for the development of lanthanide-containing supramolecular sensing systems. The exploitation of changes to distant parts of the molecule in other fields is also possible, since the effects of rigidification and changes to energy transfer and non-radiative quenching pathways can clearly be exploited in a much greater range of systems than that described here.It is also clear from these results that axial anion coordination by lanthanides can be very effective in mediating a response, even in the case of simple mono-dentate anions such as halides. We are currently engaged in the preparation of a second generation of lanthanide containing mechanically bonded arrays, in which the lanthanide is incorporated into the rotaxane cavity.
Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's 7th Framework Programme (FP7/2007-2013)/ERC-Advanced Grant Agreement Number 267426) and a Marie-Curie Fellowship for CA. We also thank the University of Oxford, EPSRC and Helmore for support (studentships for MT and RK), The Danish Council for Independent Research, Technology and Production Sciences (grant 10-093546) for financial support for TJS, and the Royal Commission for the Exhibition of 1851 (research fellowship for NLK). We thank Diamond Light Source for an award of beam time on I19 (MT1858) and the University of Oxford Crystallography Service for instrument use and helpful discussions.Notes and references
- (a) S. Faulkner and J. L. Matthews, Fluorescent Complexes for Biomedical Applications in Comprehensive Coordination Chemistry II, ed. M. D. Ward, Pergamon, Oxford, 2004, vol. 9, pp. 913–944 Search PubMed; (b) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- (a) I. A. Hemmilä, Applications of Fluorescence in Immunoassays, Wiley, New York, Chichester, 1991 Search PubMed; (b) J.-C. G. Bünzli, S. Comby, A. S. Chauvin and C. D. B. Vandevyver, J. Rare Earths, 2007, 25, 257–274 CrossRef.
- P. Hanninen and H. Härmä, Lanthanide Luminescence, Springer Series on Fluorescence, Springer, Heidelberg, 2011, vol. 7 Search PubMed.
- A. Beeby, S. W. Botchway, I. M. Clarkson, S. Faulkner, A. W. Parker, D. Parker and J. Williams, J. Photochem. Photobiol., B, 2000, 57, 83–89 CrossRef CAS.
- For reviews, see (a) S. Faulkner, B. P. Burton-Pye and S. J. A. Pope, Appl. Spectrosc. Rev., 2005, 40, 1–31 CrossRef CAS; (b) S. Faulkner, L. S. Natrajan, W. S. Perry and D. Sykes, Dalton Trans., 2009, 3890–3899 RSC.
- A. Beeby, S. Faulkner and J. A. G. Williams, Dalton Trans., 2002, 1918–1922 RSC.
- T. Lazarides, D. Sykes, S. Faulkner, A. Barbieri and M. D. Ward, Chem.–Eur. J., 2008, 14, 9389–9399 CrossRef CAS.
- For a review see: M. D. Ward, Coord. Chem. Rev., 2007, 251, 1663–1677 CrossRef CAS.
- (a) D. Parker, R. S. Dickins, H. Puschmann, C. Crossland and J. A. K. Howard, Chem. Rev., 2002, 102, 1977–2010 CrossRef CAS; (b) T. Gunnlaugsson and J. P. Leonard, Chem. Commun., 2005, 3114–3131 RSC; (c) C. Allain and S. Faulkner, Future Med. Chem., 2010, 2, 339–350 CrossRef CAS.
- M. Tropiano, N. L. Kilah, M. Morten, H. Rahman, J. J. Davis, P. D. Beer and S. Faulkner, J. Am. Chem. Soc., 2011, 133, 11847–11849 CrossRef CAS.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS.
- (a) C. Piguet, M. Borkovec, J. Hamacek and K. Zeckert, Coord. Chem. Rev., 2005, 249, 705–726 CrossRef CAS; (b) K. Zeckert, J. Hamacek, J.-M. Senegas, N. Dalla-Favera, S. Floquet, G. Bernardinelli and C. Piguet, Angew. Chem., Int. Ed., 2005, 44, 7954–7958 CrossRef CAS; (c) L. Aboshyan-Sorgho, H. Nozary, A. Aebischer, J.-C. G. Bünzli, P.-Y. Morgantini, K. R. Kittilstved, A. Hauser, S. V. Eliseeva, S. Petoud and C. Piguet, J. Am. Chem. Soc., 2012, 134, 12675–12684 CrossRef CAS; (d) C. Lincheneau, F. Stomeo, S. Comby and T. Gunnlaugsson, Aust. J. Chem., 2011, 64, 1315–1326 CrossRef CAS.
- M. R. Sambrook, D. Curiel, E. J. Hayes, P. D. Beer, S. J. A. Pope and S. Faulkner, New J. Chem., 2006, 30, 1133–1136 RSC.
- (a) M. D. Lankshear and P. D. Beer, Acc. Chem. Res., 2007, 40, 657–668 CrossRef CAS; (b) L. M. Hancock and P. D. Beer, Chem.–Eur. J., 2009, 15, 42–44 CrossRef CAS; (c) L. M. Hancock, L. C. Gilday, S. Carvalho, P. J. Costa, V. Felix, C. J. Serpell, N. L. Kilah and P. D. Beer, Chem.–Eur. J., 2010, 16, 13082–13094 CrossRef CAS; (d) L. M. Hancock, L. C. Gilday, N. L. Kilah, C. J. Serpell and P. D. Beer, Chem. Commun., 2011, 47, 1725–1727 RSC; (e) N. H. Evans, C. J. Serpell and P. D. Beer, Chem. Commun., 2011, 47, 8775–8777 RSC.
- (a) M. Jauregui, W. S. Perry, C. Allain, L. R. Vidler, M. C. Willis, A. M. Kenwright, J. S. Snaith, G. J. Stasiuk, M. P. Lowe and S. Faulkner, Dalton Trans., 2009, 6283–6285 RSC; (b) G. J. Stasiuk and M. P. Lowe, Dalton Trans., 2009, 9725–9727 RSC.
- R. S. Dickins, J. I. Bruce, D. J. Tozer and D. Parker, Dalton Trans., 2003, 1264–1271 RSC.
- P. Kuzmic, Anal. Biochem., 1996, 237, 260–273 CrossRef CAS.
- M. Borkevec, G. J. M. Koper and C. Piguet, Curr. Opin. Colloid Interface Sci., 2006, 11, 280–289 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for synthetic and spectroscopic procedures, including the synthesis of 4·Cl: characterisation and structural data; binding studies and photophysical measurements, including detailed analysis of binding models. CCDC 903824 and 903825. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2sc21614k |
| This journal is © The Royal Society of Chemistry 2013 |