Precursor-directed generation of amidine containing ammosamide analogs: ammosamides E–P†
Ende
Pan
,
Nathaniel W.
Oswald
,
Aaron G.
Legako
,
Janie M.
Life
,
Bruce A.
Posner
and
John B.
MacMillan
*
Department of Biochemistry, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd., Dallas, TX, USA. E-mail: john.macmillan@utsouthwestern.edu
First published on 8th October 2012
Abstract
Ammosamides E–F (1–2), are amidine analogs of the ammosamide family of alkaloids isolated from a marine-derived Streptomyces variabilis. Further studies with S. variabilis revealed a variety of aryl and alkyl amines added into the fermentation media could be efficiently incorporated into the ammosamide framework to generate a library of precursor-directed amidine analogs, ammosamides G–P (9–18). We demonstrate that the amines are introduced via non-enzymatic addition to the iminium ion of ammosamide C. Biological evaluation of the amidine analogs against quinone reductase 2 (QR2) showed low nM potency for a number of analogs. When tested for in vivo activity against a panel of non-small cell lung cancer (NSCLC) cell-lines there was a clear increase in potency by incorporation of lipophilic alkylamines, with the most potent compounds having sub μM IC50 values (0.4 to 0.8 μM).
Introduction
The ability to generate analog libraries of natural products can be a challenge, often times requiring complex total synthesis or selective modification of a specific functionality. It is this challenge that often hinders the optimization of the biological activity and pharmacokinetic properties of promising natural products. However, new strategies that combine chemical and biological approaches are emerging that offer great promise for generating analogs.1 In that context, we have been interested in the biological activity of ammosamides A–D (3–6), part of a growing family of biologically active pyrroloquinoline natural products isolated from actinomycetes and other sources over the past few years (Fig. 1).2–4 Other members of the pyrroloquinoline family of compounds include lymphostin (7) from a terrestrial actinomycete,5 damirone B from a caribbean sponge6 and mycenarubin (8) from a mushroom.7 Ammosamides A and B (3–4) were discovered based on their cytotoxicity to HCT-116 colon tumor cells and the mechanism of action attributed to the interaction of 3 with myosin.8 Further studies on 4 and synthetic analogs by Cushman and co-workers showed potent nM inhibition of quinone reductase-2 (QR2, NQO2), a cytosolic protein that has been implicated as a target for cancer chemoprevention, via inhibition of the conversion of quinone substrates into highly reactive and toxic species.9,10 Lymphostin and related analogs exhibit inhibition of mTOR and lymphocyte kinase.5,11 Based on the range of biological activity for pyrroloquinoline natural products, we were drawn to a marine-derived strain of Streptomyces variabilis that produced a variety of ammosamide analogs, including the oxidatively ring opened analog ammosamide D (6), previously reported from our lab.3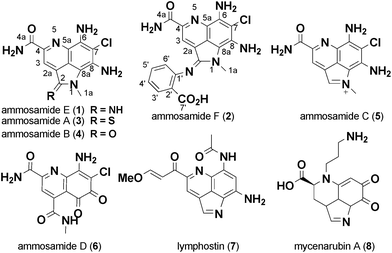 | ||
| Fig. 1 Structures of pyrroloquinoline family alkaloids. | ||
Analysis of cytotoxic fractions from S. variabilis by LC-UV-MS showed the presence of ammosamides A (3), B (4) and D (6) (based on their distinctive characteristic UV-Vis and MS profiles) as well as the presence of additional analogs, including ammosamide E (1), which contains an amidine functionality at C-2. In an attempt to increase production of 1 we fed tryptophan (1 g L−1), to the fermentation and found the predominant compound to be an amidine derivative that had incorporated 2-aminobenzoic acid to give ammosamide F (2).
Intrigued by the ability to induce new ammosamide analogs by providing biosynthetic precursors, we supplemented the media with a variety of aryl and alkylamines to generate a library of precursor-directed amidine containing analogs, ammosamides G–P (9–18). Herein we report the isolation and structural elucidation of 1 and 2, the precursor driven synthesis of 9–18 and the biological activity of all amidine analogs against QR2 and their cytotoxicity against a panel of non-small cell lung cancer (NSCLC) cell lines. Two of these analogs, ammosamide L (14) and ammosamide M (15) have significant improvement in cytotoxicity against NSCLC cell lines.
Results and discussion
Isolation of ammosamide E and F
Ammosamide E (1) was isolated from the fermentation of S. variabilis using a starch based media and extracted using XAD-7 resin.3 Purification by a combination of solvent–solvent partitioning, C18 flash chromatography and Sephadex LH20 size exclusion chromatography provided 1 as a red solid with UV absorption bands at 552, 423, 346 and 262 nm (MeOH). The positive ion HRESIMS revealed a pseudomolecular ion peak [M + H]+m/z 291.0720 corresponding to a molecular formula of C12H11ClN6O. The 1H NMR signals in DMSO-d6 exhibited six singlets: δH 9.46, 8.95, 8.90, 7.68, 7.23, 7.00 and one methyl singlet at δH 3.84 ppm, of which the signals at δH 8.95 and 3.84 were determined to be non-exchangeable protons upon addition of D2O to the NMR sample (Table 1).| No. | 1 | 2 | ||
|---|---|---|---|---|
| δ H, mult. (J) | δ C | δ H, mult. (J) | δ C | |
| 1a | 3.84 s | 32.9 | 3.81 s | 32.0 |
| 2 | — | 154.1 | — | 152.7 |
| 2a | — | 125.2 | — | 127.4 |
| 3 | 8.95 s | 118.3 | 7.93 s | 117.0 |
| 4 | — | 144.2 | — | 143.9 |
| 4a | — | 165.8 | — | 165.8 |
| 5a | — | 131.8 | — | 131.8 |
| 5b | — | 118.9 | — | 119.7 |
| 6 | — | 138.1 | — | 134.7 |
| 7 | — | 102.7 | — | 103.9 |
| 8 | — | 143.6 | — | 141.5 |
| 8a | — | 106.3 | — | 108.5 |
| CONH2 | 7.68, s | — | 7.58 s | — |
| 8.90, s | 8.82 s | |||
| NH2 (C6) | 7.23 br s | — | 6.77 br s | — |
| NH2 (C8a) | 7.00 s | — | 6.37 br s | — |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH NH |
9.46 br s | — | — | — |
| 1′ | — | — | — | 147.1 |
| 2′ | — | — | — | 122.5 |
| 3′ | — | — | 7.99 d (7.7) | 131.6 |
| 4′ | — | — | 7.21 t (7.7) | 124.0 |
| 5′ | — | — | 7.53 t (7.7) | 133.5 |
| 6′ | — | — | 7.42 d (7.7) | 122.5 |
| 7′ | — | — | — | 167.5 |
The 13C NMR data was similar to reported data for 3, suggesting the pyrroloquinoline ring system. Careful comparison of both sets of data (Table S1†) revealed one significant chemical shift change in 1 compared to 3 – an upfield 13C shift from δC 177.2 in 3 to δC 154.1 in 1. The distinctive chemical shift change and the HRMS data suggested the presence of an amidine functionality at C-2. The structure assignment of 1 was confirmed by analyzing the 2D NMR spectra and interpretation of the exchangeable protons (Fig. 2). In the HMBC, the non-exchangeable aromatic proton H-3 (δH 8.95) showed correlations with C-5b (δC 118.9), the amidine C-2 (δC 154.1) and the CONH2 (δC 165.8). The H-1a methyl singlet (δH 3.84 ppm) showed correlations to C-2 as well as C-8a (δC 106.3). HMBC correlations from the exchangeable amine protons at C-8 (δH 7.00) to C-7 (δC 102.7) and C-8a were also observed. On the basis of COSY and HMBC correlations, protons at δH 7.68 and 8.90 were assigned to be the primary amide protons. The broad singlet protons at δH 9.46 and 7.23 (integration to 2 protons) were thus ascribed to be the NH of the amidine and the exchangeable C-6 amine, respectively. The isolation of 1 represents the third functional group to be introduced to the C-2 position of the ammosamide core (thioamide, amide and amidine). The biosynthetic origin of 3 and 4 has been proposed to arise from nucleophilic oxidation or sulfuration of the iminium ion of ammosamide C (5).4 As addressed below, it is possible that 1 would arise via a similar process, via nucleophilic attack of an amine, followed by oxidation.
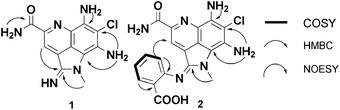 | ||
| Fig. 2 Key HMBC and COSY correlations for 1 and 2. | ||
We returned to the fermentation of S. variabilis strain SNA-020 in order to obtain additional material of 1, 3 and 4 for biological studies and based on tryptophan being the presumed biosynthetic precursor to the ammosamides, we supplemented the fermentation media with tryptophan (1 g L−1, 4 g L−1). The addition of tryptophan did not increase overall production of 1, 3 and 4, but there were obvious changes in the LC-UV-MS profile, suggesting formation of new analogs (Fig. 3). Supplementation with 1 g L−1 of tryptophan resulted in significant production of 3 and 4, but a series of more polar minor analogs appeared. Fermentation with 4 g L−1 of tryptophan resulted in the depletion of 3 and an increase in more polar metabolites, including one major product at 12.2 minutes by LC-UV-MS, a signal that had not been present in previous fermentations of S. variabilis.
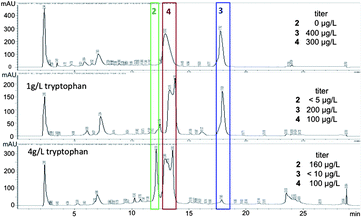 | ||
| Fig. 3 Change in metabolite profile upon addition of tryptophan. Titers of 2, 3 and 4 in the three conditions. | ||
Purification of the new metabolite by a combination of HP20, C18 flash chromatography and LH20 provided ammosamide F (2) as a dark green solid and displayed a similar UV spectrum to 1 (λmax (MeOH) 603, 424, 338 and 294 nm). HRESIMS revealed a pseudomolecular ion peak at m/z 411.0976 [M + H]+, significantly higher than any previous ammosamide analog. The 13C NMR for 2 showed 19 carbon atoms, providing a molecular formula of C19H16ClN6O3. Detailed analysis of the 13C NMR clearly showed the presence of the C-2 amidine carbon at δC 152.7 and the remaining carbons reminiscent of the pyrroloquinoline ring system (Table 1). The 1H NMR had two non-exchangeable doublets at δH 7.42 and 7.99 ppm as well as two non-exchangeable triplets at δH 7.21 and 7.53 ppm, suggesting a di-substituted aromatic ring. This was further confirmed by the presence of seven additional sp2 carbons, including a signal at δC 167.5, suggestive of a carboxylic acid. The absence of amidine NH proton at δH 9.46 allowed us to deduce 2 as an ammosamide amidine analog that incorporated 2-aminobenzoic acid (Fig. 2, Table 1). The regiochemistry of the amidine analog was determined to be trans, based on an NOE from H-6′ (δH 7.42) to H-3 (δH 7.93). As 2-aminobenzoic acid is an established product of tryptophan catabolism,12 we could rationalize 2 as resulting from nucleophilic addition of catabolically produced 2-aminobenzoic acid to the iminium ion of 5 and subsequent oxidation. Although, we have also considered, based on the disappearance of 3 under these conditions, that there could be an enzyme-mediated conversion of the thioamide of 3 to the amidine in 2. Both possibilities will be further discussed below. Irrespective of the mechanism of formation, we were intrigued by the potential utility of amidine formation.
Precursor-directed amidine analogs
Synthetic efforts by Cushman on the ammosamide family of compounds efficiently provided a diverse set of ammosamide analogs accessible through total synthesis.9b These results demonstrated methylation of the C-6 amine of 4 can greatly enhance the inhibition of quinone reductase (from 61 nM to 4.1 nM), whereas conversion of the pyrroloquinoline ring to a bicyclic quinoline ring greatly reduces activity. Due to the route to synthetic analogs, variation of the functionality at C-2 was not examined. With the ability to incorporate amines at C-2, we set out to generate a library of amidine containing ammosamide analogs by precursor-directed biosynthesis and determine the effect on biological activity (Fig. 4). A series of aryl and aliphatic amines were added to media (1 L) as a solution in DMSO, the media inoculated with S. variabilis and fermented under normal conditions for 7 days while monitoring for production of a given compound by LC-MS.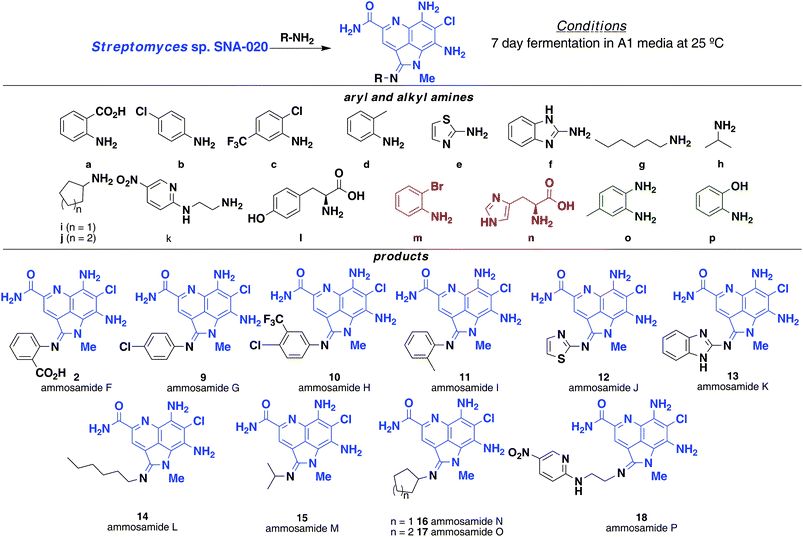 | ||
| Fig. 4 Precursor-directed biosynthesis of ammosamide F–P (2, 9–18) by addition of aryl and alkylamines. Amines m and n (red) were not incorporated while amines o and p (green) were toxic to the bacteria. | ||
Analysis of the fermentation broth extracts showed modest levels of production of amidine analogs for the majority of the arylamines used. As expected, direct addition of 1 g L−1 of 2-aminobenzoic acid yielded 2, the same product as obtained from feeding tryptophan. Additional arylamines that were successfully incorporated include entries b–f, yielding ammosamides G–K (9–13). The aryl amine precursors can contain a variety of substituents, including halogens, alkyl groups and phenols. Heteroaromatic amines such as e and f were readily incorporated with high efficiency. Not all arylamines worked efficiently; 2-bromoaniline (m) led to a large number of products that could not be purified, while 1,2-diaminobenzene (o) and 2-hydroxyaniline (p) were toxic to S. variabilis and therefore not incorporated.
With success incorporating arylamines, we turned to alkylamines, such as hexylamine (g), isopropylamine (h), cyclopentylamine (i), cyclohexylamine (j) and 2-(2-aminoethylamino)-5-nitropyridine (k), which were readily incorporated to give ammosamide L–P (14–18), respectively. Because feeding studies with tryptophan were key in discovering the potential of the precursor driven biosynthesis of the amidine analogs, we looked at a few additional amino acids including tyrosine and histidine. Based on LC-MS analysis, we could see tyrosine (i) incorporation, but attempts to isolate the analog resulted in decomposition. Derivatization with histidine (n) failed to produce an ammosamide analog, due to reduced growth of S. variabilis.1H and 13C NMR, HRMS and IR characterization of the new ammosamide analogs is included in the supporting information.†
Biological evaluation of ammosamide analogs
Biological evaluation of ammosamide analogs against NSCLC cell lines HCC44, HCC4017, Calu-3 and the non-cancerous human bronchial epithelial cell line HBEC30KT showed an interesting trend (Table 2). The majority of the molecules showed little to no cytotoxicity (>20 μM) against any of the cell lines tested. However, analogs 14–17, which incorporate aliphatic amines, exhibit the most potent IC50 values. The isopropylamine-containing analog 15 was the most potent, with IC50 values of 0.50 μM and 0.48 μM against HCC44 and HCC4017, respectively, followed by 14 with IC50 values of 1.1, 0.96 and 0.86 μM against HCC44, HCC4017, Calu-3, respectively. The overall cytotoxicity trend indicates introduction of lipophilic character to the polar ammosamide core structure enhances activity, possibly by increasing its ability to cross cell membranes. It is unclear whether the cytotoxicity of the ammosamide analogs is related to the inhibition of myosin as previously reported for 3 and 4.| # | QRa | HCC44a | HCC4017a | Calu-3a | HBEC 30KTa |
|---|---|---|---|---|---|
| a IC50 values in μM. Values are averages of three measurements. | |||||
| 1 | 0.040 | 86.0 | >100 | >100 | >100 |
| 2 | 0.140 | 78.0 | >100 | >100 | >100 |
| 3 | 0.023 | 20.2 | 24.5 | 19.4 | 16.7 |
| 4 | 0.061 | 15.3 | 15.9 | 17.6 | 21.4 |
| 6 | >10 | 3.1 | 2.4 | 1.4 | 2.6 |
| 9 | 0.052 | 23.2 | 22.9 | 10.4 | 21.8 |
| 10 | — | 23.9 | 24.2 | 14.5 | 20.2 |
| 11 | >10 | 38.1 | 26.1 | 17.6 | 26.9 |
| 12 | 0.250 | >100 | >100 | >100 | >100 |
| 13 | 0.500 | >100 | >100 | >100 | >100 |
| 14 | 0.017 | 1.1 | 2.87 | 0.86 | 1.1 |
| 15 | 0.020 | 0.50 | 0.48 | 0.77 | — |
| 16 | 0.050 | 18.5 | 12.4 | 22.4 | — |
| 17 | 0.020 | 15.5 | 11.1 | 20.2 | — |
| 18 | 0.170 | >100 | >100 | >100 | >100 |
With the cytotoxicity data in hand, we evaluated the inhibition of QR2 by 1–4, 6 and 9–18. In the work by Cushman and Mesecar et al.,9b X-ray crystal structures of QR2 in complex with 4 and the C-6 N-methyl analog of 4 demonstrate that there is a strong role for formation of a hydrogen bond network between the primary amide and Asn161 and between the C-8 NH2 and Thr71. The surprising aspect of the binding of 4 to QR2 is a H2O-mediated hydrogen bond from the primary amide NH2, the C-6 NH2 and the ring-nitrogen to the backbone carbonyl oxygen of Gly174. Interestingly, the structural data did not show any role for the interaction of the C-2 carbonyl in interactions with QR2. It has been demonstrated with amidine containing vancomycin analogs that the amidine functionality can act as both an H-bond donor and an H-bond acceptor, which allows the analog to retain activity against vancomycin resistant bacteria.13 We felt that incorporation of the amidine functionality would allow us to explore the role of additional hydrogen bonding interactions on the potency against QR2.
We evaluated the ammosamide analogs in the QR2 inhibition assay using human recombinant protein (Table 2). Ammosamide B (4), was previously found to have an IC50 value of 22 nM against QR2, while we found it to have a value of 61 nM in our assay system. In general, nearly all of the ammosamide analogs retain potent inhibition of QR2 in the range of 10–200 nM, with a few notable exceptions. Modification with hexylamine (14) gave slightly increased potency to 17 nM, while the other alkylamine containing analogs 15–17 show similar potency to 4.
The majority of the arylamine-containing analogs showed reduced potency to the 120–500 nM range. The simplest amidine analog, ammosamide E (1), was nearly equipotent to 4. Ammosamide H (10) and gave irreproducible results, potentially due to solubility issues. We are unable to make any conclusions on whether the amidine functionality is able to act as an H-bond donor or H-bond acceptor in the binding site of QR2. Further efforts combining derivatization at both C-2 and the C-6 NH2 could provide promising QR2 inhibitors.
Attempts at chemical conversion of thioamide to amidine
The precursor driven SAR study of the ammosamide amidine analogs provided access to a large variety of analogs that would have been difficult to access directly from 3 or 4. There are a number of methods for conversion of thioamides to amidines, such as use of the Mukaiyama reagent (2-chloro-1-methylpyridinium iodide)14 or the recently reported Ag(I)-promoted method of Boger.15When we attempted the direct conversion of 3 using the Mukaiyama reagent with 4-chloroaniline in methanol we detected trace amounts of the desired ammosamide analogs, but mostly unreacted starting material. Using AgBF4 with 4-chloroaniline gave rapid decomposition of 3, giving rise to multiple uncharacterizable products. Using 4 as the starting material, under the harsh conditions of neat POCl3, reaction with 4-chloroaniline resulted in a mixture of the desired amidine 9 (9%), the bis-amidine 19 (23%) and the major product 20 (39%), which has undergone conversion of the primary amide to a nitrile (Scheme 1).16 The above experiments suggested the difficulty of selectively introducing an amine to form ammosamide amidine analogs through a synthetic approach due to the highly reactive nature of 3 and 4. By tapping into the existing biosynthetic framework of the ammosamides, we can generate significant diversity in a mild, efficient manner.
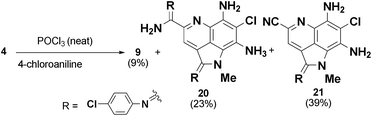 | ||
| Scheme 1 Attempts at chemical conversion of 4 to 9. | ||
There are a number of strategies for generating analogs of natural products for biological studies, with total synthesis and semi-synthesis being the most common. Total synthesis offers the flexibility to functionalize a variety of positions in a molecule, but typically requires multi-step processes, providing a material challenge to generate a large number of analogs. Semi-synthesis offers the advantage of starting with a complex natural product. However, for highly functionalized molecules, the challenge for semi-synthesis becomes chemoselectivity. The precursor-driven approach we have utilized overcomes the challenge of the chemoselectivity, while at the same time not requiring a multi-step total synthesis. Although it is important to point out that Cushman, as described previously, was able to efficiently exploit total synthesis to access ammosamide analogs that would not have been possible through precursor-directed synthesis.9b
Biogenesis of amidine analogs
The utilization of existing biosynthetic pathways to generate new natural products has been an area of interest for a long time, either through genetic manipulation of the biosynthetic gene cluster and/or incorporation of non-natural precursors.17 The simplicity of precursor-driven biosynthesis is attractive, as it does not necessarily require identification and manipulation of the biosynthetic gene cluster.18–20 For example, generation of analogs of the uridyl peptide antibiotic pacidamycin was accomplished by feeding studies with halogenated and methyl substituted tryptophan analogs in the wild type producer Streptomyces coeruleorubidus.19 Another example is the incorporation of substituted 5- and 6-monofluorinated tryptophan into diazepinomicin, a farnesylated trihydroxy-dibenzodiazepinone isolated from Micromonospora sp.20In the case of the ammosamide amidine analogs, we considered two possible routes to amidine formation (Fig. 5). The first was an enzyme-mediated conversion of 3 (or 4) to the amidine derivatives via nucleophilic attack at C-2 and subsequent loss of sulfur (or oxygen). To probe this possibility, we began by removing bacterial cells from a 4-day fermentation of S. variabilis via centrifugation. Incubation of the supernatant with 4-chloroaniline for 24 h at 30 °C resulted in production of 9. Based on these results, for the conversion to be enzyme-mediated, it would require the enzyme to be excreted by the bacteria into the media, which is highly unlikely. However, to definitively rule out this possibility, cells were again removed from a 4-day fermentation of S. variabilis and the resulting supernatant filtered through a 5 kDa filter. Under these conditions an excreted enzyme should be removed from the supernatant, resulting in the inability to generate an amidine analog when incubated with an amine. However, after incubation of the filtered supernatant with 4-chloroaniline for 24 h at 30 °C we could detect significant production of 9, thus ruling out the enzymatic pathway.
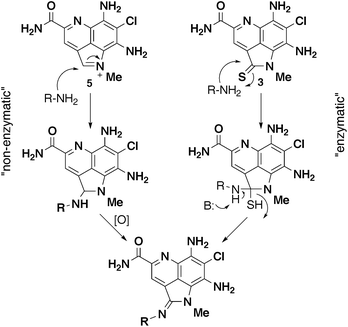 | ||
| Fig. 5 Two possible biogenic routes to amidine analogs. | ||
The second possibility we considered was a non-enzyme catalyzed nucleophilic addition of an amine to the activated iminium ion of 5 (Fig. 5). This would be followed by subsequent oxidation of the resulting benzylic position to give an amidine. This hypothesis is consistent with the observations from the cell-free supernatant experiments with 4-chloroaniline. Moreover, we are able to detect 5 in the fermentation supernatant. To validate this hypothesis, we utilized synthetically prepared 5 along with an amine in the fermentation media.
Semi-synthetically prepared 54 was incubated for 24 hours in A1 media (pH 6.5) with 4-chloroaniline at 30 °C to give 20% yield of 9, with the remainder of the material being 5 (unreacted starting material) and <5% of 4 (Scheme 2). The clean conversion demonstrated that the amidine analogs are most likely derived via a non-enzymatic addition of amine to the iminium ion of 5.
Surprisingly, when this reaction was attempted in H2O or CH3CN–H2O the reaction only shows trace conversion (<5% by LC-MS). A more exhaustive exploration of the role of pH and metal ions could shed light on promotion of the reaction in organic solvents or biological buffers.
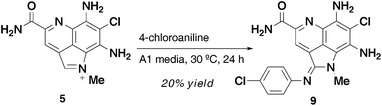 | ||
| Scheme 2 Conversion of 5 to 9. | ||
Conclusion
In conclusion, the ammosamide class of natural products have been shown to possess a variety of biological activities, which can be modified through the formation of amidine-containing analogs. In particular by varying the nature of the amine incorporated into the amidine there is a dramatic effect on the in vivo activity. From a more general perspective, the precursor-directed generation of analogs in this study is a deviation from previous studies that employ the bacterial biosynthetic machinery to generate new analogs. Rather, we are harnessing a highly reactive, yet stable intermediate that can be intercepted even in the milieu of the fermentation. Although infrequent, there are previous reports of non-enzymatic steps in natural product biosynthesis that take advantage of reactive intermediates.21 One well studied example is the non-enzymatic formation of an oxazolone between the polyketide core of jadomycin and L-isoleucine.22 Precursor-directed biosynthesis was used to incorporate a number of amino acids (both proteinogenic and non-proteinogenic) to form a variety of oxazolone analogs of jadomycin.23 Biological evaluation of the jadomycin analogs generated in this study showed the DNA-damaging activity of the jadomycins is tunable depending upon the nature of the amino acid side-chain.23a Another recent example is the generation of derivatives of the antibiotic elansolid via a conjugate addition of arylamines to a p-quinone methide generated under mildly basic conditions.24 Each of these examples; the ammosamides, jadomycin and elansolid take advantage of a different reactive functional group and a different chemical transformation for analog generation, but represent a powerful approach to obtain analogs of natural products with suitable reactive intermediates.Acknowledgements
Funding provided by the NIH (R01 CA149833, P01 CA095471), the Cancer Center Support Grant (5P30 CA142543) and the Welch Foundation (I-1689). JBM is a Chilton/Bell Foundation Endowed Scholar. We thank Ko Uyeda for assistance with the QR2 assay.Notes and references
- R. J. M. Goss, S. Shankar and A. A. Fayad, Nat. Prod. Rep., 2012, 29, 870–889 RSC.
- C. C. Hughes, J. B. MacMillan, S. P. Gaudencio, P. R. Jensen and W. Fenical, Angew. Chem., Int. Ed., 2009, 48, 725–727 CrossRef CAS.
- E. Pan, M. Jamison, M. Youssufuddin and J. B. MacMillan, Org. Lett., 2012, 14, 2390–2393 CrossRef CAS.
- C. C. Hughes and W. Fenical, J. Am. Chem. Soc., 2010, 132, 2528–2529 CrossRef CAS.
- (a) H. Nagata, K. Ochiai, Y. Aotani, K. Ando, M. Yoshida, I. Takahashi and T. J. Tamaoki, J. Antibiot., 1997, 50, 537–542 CrossRef CAS; (b) Y. Aotani, H. Nagata and M. Yoshida, J. Antibiot., 1997, 50, 543–545 CrossRef CAS.
- B. Stierle and D. J. Faulkner, J. Nat. Prod., 1991, 54, 1131–1133 CrossRef.
- S. Peters and P. Spiteller, Eur. J. Org. Chem., 2007, 1571–1576 CrossRef CAS.
- C. C. Hughes, J. B. MacMillan, S. P. Gaudencio, W. Fenical and J. J. LaClair, Angew. Chem., Int. Ed., 2009, 48, 728–732 CrossRef CAS.
- (a) P. V. N. Reddy, B. Banerjee and M. Cushman, Org. Lett., 2010, 12, 3112–3114 CrossRef CAS; (b) P. V. N. Reddy, K. C. Jensen, A. D. Mesecar, P. E. Fanwick and M. Cushman, J. Med. Chem., 2012, 55, 367–377 CrossRef CAS.
- (a) L. Buryanovskyy, Y. Fu, M. Boyd, Y. Ma, T. Hsieh, J. M. Wu and Z. Zhang, Biochemistry, 2004, 43, 11417–11426 CrossRef CAS; (b) B. Calamini, B. D. Santarsiero, J. A. Boutin and A. D. Mesecar, Biochem. J., 2008, 413, 81–91 CrossRef CAS.
- A. Miyanaga, J. E. Janso, L. McDonald, M. He, H. Liu, L. Barbieri, A. S. Eustáquio, E. N. Fielding, G. T. Carter, P. R. Jensen, X. Feng, M. Leighton, F. E. Koehn and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 13311–13313 CrossRef CAS.
- C. E. Dalgliesh, W. E. Knox and E. Neuberger, Nature, 1951, 168, 20–22 CrossRef CAS.
- J. Xie, J. G. Pierce, R. C. James, A. Okano and D. L. Boger, J. Am. Chem. Soc., 2011, 133, 13946–13949 CrossRef CAS.
- S. Flemer and J. S. Madalengoitia, Synthesis, 2011, 1638–1648 CAS.
- A. Okano, R. C. James, J. G. Pierce, J. Xie and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 8790–8793 CrossRef CAS.
- C. T. Brain and S. A. Brunton, Tetrahedron Lett., 2002, 43, 1893–1895 CrossRef CAS.
- (a) K. J. Weissman, Trends Biotechnol., 2007, 25, 139–142 CrossRef CAS; (b) S. Weist and R. D. Süssmuth, Appl. Microbiol. Biotechnol., 2005, 68, 141–150 CrossRef CAS; (c) J. R. Jacobsen, C. R. Hutchinson, D. E. Cane and C. Khosla, Science, 1997, 277, 367–369 CrossRef CAS.
- J. Gerard, R. Lloyd, T. Barsby, P. Haden, M. T. Kelly and R. J. Andersen, J. Nat. Prod., 1997, 60, 223–229 CrossRef CAS.
- S. Greschow, E. J. Rackham, B. Elkins, P. L. A. Newill, L. M. Hill and R. J. M. Goss, ChemBioChem, 2009, 10, 355–360 CrossRef.
- A. S. Ratnayake, J. E. Janso, X. Feng, G. Schlingmann, I. Goljer and G. T. Carter, J. Nat. Prod., 2009, 3, 496–499 CrossRef.
- (a) A. R. Howard-Jones and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 11016–11017 CrossRef CAS; (b) S. Hanessian and J. P. Maianti, Chem. Commun., 2010, 46, 2013–2015 RSC; (c) E. McCoy, M. C. Galan and S. E. O'Connor, Bioorg. Med. Chem. Lett., 2006, 16, 2475–2478 CrossRef CAS; (d) J. Rohr, J. Chem. Soc., Chem. Commun., 1990, 16, 2475–2479 Search PubMed.
- U. Rix, J. Zheng, L. Remsing-Rix, L. Greenwell, K. Yang and J. J. Rohr, J. Am. Chem. Soc., 2004, 126, 4496–4497 CrossRef CAS.
- (a) K. M. Cottreau, C. Spencer, J. R. Wentzell, C. L. Graham, C. N. Borissow, D. L. Jakeman and S. A. McFarland, Org. Lett., 2010, 12, 1172–1175 CrossRef CAS; (b) S. N. Dupuis, T. Veinot, S. M. A. Monro, S. E. Douglas, R. T. Syvitski, K. B. Goralski, S. A. McFarland and D. L. Jakeman, J. Nat. Prod., 2011, 74, 2420–2424 CrossRef CAS; (c) S. N. Dupuis, A. W. Robertson, T. Veinot, S. M. A. Monro, S. E. Douglas, R. T. Syvitski, K. B. Goralski, S. A. McFarland and D. L. Jakeman, Chem. Sci., 2012, 3, 1640–1644 RSC.
- H. Steinmetz, W. Zander, M. A. M. Shushni, R. Jansen, K. Gerth, R. Dehn, A. Kirschning and R. Müller, ChemBioChem, 2012, 13, 1813–1817 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, NMR data tables, NMR spectra. See DOI: 10.1039/c2sc21442c |
| This journal is © The Royal Society of Chemistry 2013 |