Temperature directed-assembly of coated-laponite nanoparticles in pluronic micellar solutions†
Imane
Boucenna
*a,
Marie-Alice
Guedeau-Boudeville
a,
Alain
Lapp
b,
Pierre
Colinart
a,
Amsha
Proag
a,
Laurent
Royon
a and
Ahmed
Mourchid
*a
aMatière et Systèmes Complexes, UMR 7057 CNRS and Université Paris Diderot, cc 7057, 75205 Paris Cedex 13, France
bLaboratoire Léon Brillouin, UMR 12 CNRS and CEA, F-91191 Gif-sur-Yvette, France
First published on 16th October 2012
Abstract
We investigate the microstructure of copolymer micellar solutions with embedded laponite colloidal particles. The solutions exhibit microscopic confinement of the coated nanoparticles triggered by increasing the temperature above the gelation temperature. The copolymer is pluronic which self-assembles into well-defined nanostructured arrays. These self-assembly properties are governed by copolymer–solvent interaction through temperature. In the presence of laponite nanoparticles, a large amount of copolymer unimers adsorb onto laponite. Above the gelation temperature of the pure copolymer micelles in solution, the study demonstrates that the remaining micelles form three-dimensional polycrystallites and trigger the confinement of the nanoparticles in the interstices between them. This conclusion is supported by phase contrast optical and fluorescence microscopy techniques which show the appearance of well-contrasted grains that form when the temperature increases, with excluded fluorescent nanoparticles from the grains. Finally, the contrast matching small-angle neutron scattering displays the development of an interparticle correlation peak revealing their strong confinement in the sample.
Introduction
Assembly refers to the process by which molecules, nanoparticles and other discrete building blocks aggregate to form supramolecular assemblies, either spontaneously due to specific forces acting between them or indirectly through functionalization or by application of an external stimulus.1 At the nanoscale level, the process of organization yields a variety of species including spherical, rod-like, plate-like, plain or hollow morphologies. At the subsequent scale level, both self and induced assemblies can be exploited to create three-dimensional crystals or more complex but well-defined three-dimensional superstructures. In this case, the induced or directed assembly is realized by using a template or by applying an external stimulus.2–4 The achieved structures are governed by the balance of attractive forces (such as covalent or hydrogen bonding, electrostatic interactions like attraction between oppositely charged ligands or dipole–dipole interactions, depletion forces), and repulsive forces (such as steric forces and electrostatic repulsion between ligands of like charge).4 An ideal induced assembly method of nanoparticles should permit the construction of desired topologically complex assemblies and allow for the manipulation of their features in order to improve the structural and mechanical properties of the composite materials.5 All the progress made in this burgeoning field is motivated by its technological promise in applications.2 Still, the realization of three-dimensional superstructures at the nanometric and micrometric scale remains a challenging task as compared to naturally occurring composites such as nacre.6,7 Indeed, this hierarchically structured material made of layered mineral aragonite particles held by biopolymers show excellent high performance mechanical properties which are believed to be a consequence of its unique multiscale structure. The remarkable features of alike natural materials are inspiring new strategies to control assembly and phase separation in composite materials such as in mixtures of organic and inorganic components.It is thus obvious that the supramolecular assembly is now a subject of intense interest. Among the most studied materials are the colloidal nanoparticles assembled in various complex media such as soft templates like DNA, surfactants or block copolymers.2,5,8–12 The combination of the relative ease of fabrication, processing and flexibility in property-tailoring has turned the superstructures of nanoparticles into promising materials. The multitude applications range from optoelectronics, photonics, spintronics, catalysis, solar energy conversion, thermoelectrics to biomedical applications.2 The morphology of either the nanoparticles or the template is an important factor in the geometrical packing in organized structures. The realization of assembly of non-spherical nanoparticles still remains challenging because of their anisotropic shape and mutual interaction which control the topology of the supra-assemblies. On the other hand, the existence of interaction between a template and the nanoparticles might lead to the arrangement of the particles into more complex architectures which are predefined by the shape of the template.13
In this context, Shen et al. reported more recently a novel method for inducing the assembly of gold nanoparticles in aqueous solution.14 The strategy takes advantage of the formation of polycrystalline ice upon freezing which triggers the segregation between the resulting ice polycrystals and the nanoparticles, thus leading to particle confinement. Therefore the polycrystals are used as templates. Upon decreasing the temperature, three-dimensional ice grains form and squeeze the gold nanoparticles. The freezing process, as it proceeds, provokes the confinement of the nanoparticles in the interstices between the ice polycrystals. They were able to obtain one-dimensional superstructures of nanoparticles of uniform cross-section by freezing aqueous solution of spherical gold nanoparticles. They reported that below the freezing point water-filled veins form between the microstructures of polycrystalline ice and interconnect. The gold nanoparticles always closely pack together in chains in the formed veins. They can take a triangular prism shape at the junction of ice grains. One major finding of this study is that, while the contour length of the veins can lie in the macroscopic range, their width can go down to the nanometric level. The assembly is forced by the confining capability of the veins and the observed superstructures are dictated by the shape of the veins. These authors also used anisotropic gold nanorods and showed that they should be able to move in the tiny veins along with the liquid phase during the final stage of freezing to form multiple-line chains or triangular prisms. The rod axes align parallel to the longitudinal direction of the chains. It was concluded that the steric influence from the neighboring gold nanorods and the movement of the liquid phase may also contribute to the alignment. This successful assembly demonstrated the robust nature of their proposed method.
In a previous study, we used small-angle neutron scattering, SANS, differential scanning calorimetry, DSC, and rheology techniques to investigate in detail the structure and properties of aqueous solutions of pluronic F127 and laponite particles in the concentrated regime.15,16 Pluronic block copolymers, PEO-PPO-PEO, in aqueous solution self-assemble into well-defined nanostructured arrays. These self-assembly properties are governed by copolymer–solvent interaction. At a relatively low temperature both PPO and PEO are hydrophilic and the copolymer chains are fully soluble in the aqueous solution. As the temperature increases, the PPO block becomes hydrophobic and individual micelles begin to form when the temperature reaches the critical micelle temperature, cmt. We showed that a large amount of pluronic unimers is adsorbed onto the colloidal particles due to their high specific area. However, the effect of added laponite on the structure of the micelles in solution is not visible below the fluid–crystal transition temperature of the pure copolymer solution, which coincides with its gelation temperature Tgel0. Above Tgel0, where the pure micellar solutions exhibit a well-ordered face-centered cubic (fcc) lattice, addition of laponite is found to promote structural disorder. The adsorption of the copolymer unimers onto laponite is the driving mechanism observed in this investigation. The large number of copolymer unimers removed from the micellar population and adsorbed onto laponite gives more free volume to the remaining micelles in solution and tends to promote the fluid phase. While the whole system remains homogenous and does not show phase separation, investigation by optical microscopy techniques reveals that above the nanometric length scale the copolymer–laponite aqueous solutions display patterned microstructures. Therefore, the aim of this study is to shed light on these observed textures. We will show that self-assembly of polymeric micelles, which is governed by polymer–solvent interactions through temperature variation,17 constitutes an ideal process for directing the assembly of colloidal nanoparticles. This phenomenon is reminiscent of micellar polycrystalline grains formation whose growth is limited by the presence of laponite nanoparticles. The micelles form a three-dimensional polycrystalline structure, instead of a single crystal, thus confining the nanoparticles in the interstices between them. The process constitutes an assembly strategy which produces confinement of colloidal nanoparticles between the polycrystalline grains induced by the increase of temperature.
Materials and methods
Materials
The copolymer pluronic F127 used in these experiments was purchased from Sigma-Aldrich and used without further purification. The reported chemical structure for copolymer chains is (EO)100-(PO)65-(EO)100 and its nominal weight average molar mass is 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 g mol−1. De-ionized water (Millipore) was used to prepare the samples for the rheological and microscopic measurements, while D2O (99.97% deuterated, from Eurisotop France) and solvent-matched particle solutions were used for the SANS studies.
600 g mol−1. De-ionized water (Millipore) was used to prepare the samples for the rheological and microscopic measurements, while D2O (99.97% deuterated, from Eurisotop France) and solvent-matched particle solutions were used for the SANS studies.
The copolymer solutions were prepared by slowly adding the powder to water adjusted to pH 10. They were left for 1 week at low temperature (T = 0 °C) to ensure complete dissolution of the copolymer. The nominal weight fraction WP of the copolymer is defined as WP = mF127/(mF127 + mwater), where mF127 and mwater are the weights of copolymer and water.
The synthetic clay used was laponite RDS from Laporte Industries (Warrington, UK). The chemical formula of laponite is Si8Mg5.45Li0.4H4O24Na0.7.18 Laponite has a density of 2.65 g cm−3 and the individual particles have an average diameter of 250 Å and a thickness of 9.1 Å, which yield a specific area of 900 m2 g−1. Laponite dispersions were always prepared at pH = 10 to avoid particle dissolution.19–21
The mixtures of copolymer and laponite in aqueous dispersions were prepared by slowly adding laponite powder to the previously prepared copolymer solution. The copolymer–laponite dispersions were stirred for one week using a magnetic stirrer to fully dissolve the laponite. The solutions were stored at 0 °C for at least 1 week to ensure complete dissolution and homogeneity of the samples, and then they were kept in the refrigerator for long-time storage (∼4 °C). The nominal weight fraction of laponite in the mixtures is defined as WL = mRDS/(mRDS + mF127 + mwater) where mRDS is the weight of laponite. We will present results on mixtures with WP = 16 wt% and WL ranging between 0 and 3 wt%.
Preparation of fluorescent laponite
We labeled the laponite with a cationic fluorescent dye, 9-amino acridine, in order to localize the laponite particles inside the sample mixtures by two- and three-dimensional optical microscopy.22 The fluorescent molecule was added to laponite solutions, at a weight fraction of 1 wt%, under magnetic stirring. The molar concentration of acridine used in this study is 150 μM which corresponds to 2.913 mg of the dye for 1 g of laponite. After two days, we dialyzed the laponite–fluorescent dye solutions in regenerated cellulose membranes with a molecular weight cutoff of 12000–14000 (SpectraPor) against a reservoir of de-ionized water at pH = 10 for one week. The solution in the reservoir was changed several times. Finally the dialyzed labeled laponite solution was freeze-dried and adequately stored prior to use.
Small-angle neutron scattering
SANS measurements were carried out on the PAXY beamline at the CNRS-CEA Léon Brillouin joint Laboratory, LLB, located in Saclay, France. Two beamline configurations were chosen to cover scattering wave vectors q between 0.006 and 0.3 Å−1. Typical configurations were chosen by setting the incident neutron wavelength to 4 and 6 Å and the distance between the two-dimensional detector and sample to 2 and 6.7 m, respectively. The SANS intensities were collected on samples with varying laponite weight fractions. A quartz cell with solvent was used to quantify the background scattering and was subtracted from the data. The spectra were further normalized by incoherent scattering of H2O in a 1 mm quartz cell, with the empty cell scattering subtracted off. The resulting data were integrated over the azimuthal angle. Both beamline configurations yielded overlapping data without scale adjustment. The scattering intensities, which were also corrected for incoherent scattering estimated from the signal at high q, were subsequently obtained on the absolute scale (cm−1).Rheological measurements
The temperature-induced changes in the rheological properties were recorded by using a stress-controlled rheometer (Carrimed CSL100) with a cone and plate geometry (40 mm diameter with 1° cone angle) equipped with a solvent trap to prevent solvent evaporation. Oscillatory experiments were typically performed at a stress of 0.1 Pa, well within the linear viscoelastic regime for our systems which was established via strain sweep experiments. The measurement of G′ and G′′, the elastic and viscous moduli respectively, was performed as a function of temperature from 10 to 50 °C at a heating rate of 1 °C min−1 and a constant frequency of 1 Hz.Microscopic observations
Optical microscopy was carried out on a Nikon microscope (Diaphot). The samples were placed in a sealed cell of fixed thickness: 100 μm and 1 mm, and the temperature was varied between the cmt and 50 °C at a heating rate of 0.066, 0.2 and 1 °C min−1. A comparable experimental protocol was used for the fluorescence observation on samples (50 and 100 μm thickness) with labeled laponite particles. The excitation signal was collected at λ = 380 nm and the emission signal was collected at λ ≥ 420 nm.We also used an inverted microscope (Olympus IX81) with a spinning disk device in order to investigate the fluorescence imaging inside the bulk samples with labeled laponite. The sample, placed in a 270 μm thickness optical cell at fixed temperature, was illuminated with a 405 nm laser. The emission signal, at λ ≥ 465 nm, was collected using a 512 × 512 iXon camera during an exposure period of 60 ms. The optical cross-sections ranged between the sample–cell interface up to 80 μm inside the sample.
Results and discussion
Rheology data
The inset in Fig. 1 shows a typical result of the dynamic rheology measurement on a sample at WP = 16 wt% and WL = 3 wt%. The data represent the temperature variation of G′ and G′′, the elastic and loss moduli respectively recorded at a fixed frequency of 1 Hz and applied shear stress of 0.1 Pa. We recall that G′ and G′′ show two distinct domains as a function of temperature. Below a temperature defined as Tgel, G′ and G′′ are very weak with G′′ > G′. In this region G′′ exponentially decreases, reaches a minimal value when T reaches the cmt and increases again as the temperature continues to increase. At and above the Tgel temperature, G′ and G′′ abruptly increase towards a high value. Tgel is called the temperature of gelation and corresponds to the transition point from a viscoelastic liquid (G′ < G′′) to a viscoelastic solid (G′ > G′′).15 Besides Tgel, we identify the threshold temperature of transition which corresponds to the onset of gelation, Tonset, i.e., the temperature point at which the elastic modulus starts to increase sharply.23 The onset of gelation denotes the percolation processes in this type of copolymer micellar solutions. This phenomenon has been observed in a similar pluronic system.24 The addition of laponite particles to the copolymer solution induces a shift in both temperatures to higher temperatures as is presented in Fig. 1. Tgel and Tonset increase from 30 to 48 °C and from 30 to 40 °C respectively when WL increases from 0 to 3 wt% in a copolymer solution of WP = 16 wt%. We notice that sol–gel transition is abrupt in pure copolymer solutions while it is more gradual in the solutions with increasing amount of colloidal particles.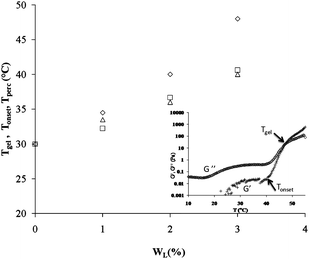 | ||
| Fig. 1 Evolution of the gelation temperature (◊), Tgel, onset gelation temperature (□), Tonset, and temperature of percolation (Δ), Tperc, as a function of weight fraction of laponite in a micellar pluronic solution at Wp = 16 wt%. Tgel and Tonset correspond to G′ = G′′ and onset of gelation respectively. Tperc corresponds to the crowding of the imaged samples with polycrystalline grains. Inset: typical elastic (G′, +) and viscous (G′′, ◊) moduli at a frequency of 1 Hz and a stress of 0.1 Pa as a function of temperature for the mixture of pluronic at a concentration of 16 wt% and laponite at a concentration of 3 wt%. | ||
The texture of the solutions with added laponite particles (WL between 0 and 3 wt%) as a function of temperature was observed by optical phase contrast microscopy. We investigated the three regions with distinct rheological comportments as shown in the inset of Fig. 1. For the sake of simplicity, we only show the microscopy images of the sample at WL = 3 wt%, which are presented in Fig. 2, recorded at T = 37, 38, 39, 40 (close to Tonset = 40.6 °C), 45 and 50 °C, and at an average heating rate of 0.2 °C min−1. However, additional data are presented in the ESI (Fig. S1†).
 | ||
| Fig. 2 Observation by optical microscopy of a sample at a polymer weight fraction of 16 wt% and laponite weight fraction of 3 wt% with increasing measurement temperature. As the temperature increases, polycrystalline grains form and occupy the probed surface at the temperature Tperc. The scale bar is 100 μm. | ||
The optical microscopy reveals that when the temperature is below the temperature of gelation of the pure copolymer solution, T ≤ Tgel0 = Tonset = 30 °C, the samples do not show noticeable textures. The images are uniform in intensity. We conclude that the microstructure of the samples is homogenous. When the temperature of observation increases above the Tgel0 for the pure copolymer solution, the samples with increasing WL show the appearance of well-contrasted grains whose number density sharply increases above a temperature close to Tonset as is shown in Fig. 2. Indeed, the difference in the number density of grains observed at T = 37 and 38 °C fully illustrate their rapid establishment over 1 °C.
It is obvious from the data gained with optical microscopy that the appearance of the grains in heated samples coincides with the transition from the micellar fluid phase to the crystal phase. In fact, we have shown that above 30 °C, the copolymer solutions exhibit a fcc crystal phase,25 with increasingly high structural disorder as a function of laponite concentration.15 The ratio of the peak intensities, measured by SANS, of the first to the second correlation peak, in laponite contrast-matched samples shows a continuous decrease of this ratio as a function of laponite concentration. This decrease associated with addition of laponite is consistent with a Debye–Waller factor26 rapidly decreasing for the copolymer–laponite solutions with the highest WL. It suggests that the addition of laponite to the copolymer perturbs the crystalline order of the micellar solution. Moreover, we noticed that when T ≥ Tgel0 the two-dimensional scattering patterns of the copolymer solutions without laponite particles reveal the presence of anisotropic scattering spikes in the first correlation peaks, in agreement with the results reported previously by Prud'homme et al.27 These features are a clear indication that the samples are both crystallized and exhibit a preferential orientation. All the two-dimensional scattering patterns on the samples with added laponite particles show uniform correlation rings, for measurement temperature between cmt and 60 °C, which suggest that they are isotropic (Fig. S2†). We concluded that the copolymer chains preferentially adsorb onto laponite nanoparticles which results in an increase in free volume of the remaining micelles and yield the observed enhancement of the structural disorder of the micellar solutions.15 Investigation of the system in the temperature range above Tgel by optical microscopy techniques reveals that above the nanometric length scale the copolymer–laponite aqueous solutions display patterned microstructures. We thus hypothesize that the observed texture are polycrystals constituted of copolymer micelles arranged in a fcc structure and that laponite nanoparticles are expelled from these polycrystalline grains. This situation should be analogous to the one observed during the polycrystallization of ice in the presence of gold nanoparticles described by Shen and co-workers.14 In order to test this hypothesis, we performed simultaneous phase contrast optical microscopy and fluorescence microscopy on copolymer–laponite solutions with labeled laponite. First, we checked that the copolymer-labeled laponite solutions are homogenously fluorescent below Tgel0 which corresponds to the temperature of crystallization of the samples. Above Tgel0, fluorescent zones become visible in the images. We carried out the following protocol for the observation of fluorescence. First the samples were loaded into a thin cell in order to restrict the thickness of the observation layer which enhances the contrast between fluorescent and non-fluorescent zones. Second, the sample cells were placed at a temperature slightly below Tgel temperature of the solution in order to have fewer number of grains. This procedure requires a long waiting time for the grains to form. Fig. 3 displays the microcrystals obtained, at 35 °C, for the copolymer and labeled laponite solution with WP = 16 wt% and WL = 3 wt%. In order to carry out a rigorous comparison, the same observation field was imaged by phase contrast microscopy and the resulting micrograph is also shown in Fig. 3. The fluorescence image clearly shows dark geometrical grains dispersed in a continuous fluorescent medium. Because the fluorescent dye is only present on the nanoparticles, we conclude that the fluorescent nanoparticles are excluded from the grains that form when the temperature increases above Tgel0.
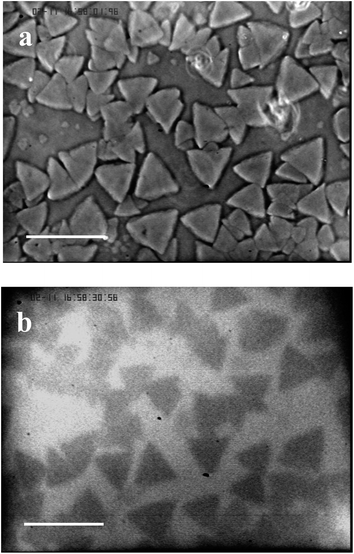 | ||
| Fig. 3 Comparison of optical (a) and fluorescence (b) imaging of the sample at a polymer weight fraction of 16 wt% and labeled laponite weight fraction of 3 wt%. The measurement temperature is 35 °C, the waiting time is 18 hours and the scale bar is 100 μm. | ||
It is thus reasonable to assume that the micelles, when they undergo the fluid–crystal transition induced by temperature, form microcrystallites when laponite is present in the solutions, instead of a macroscopic crystal. The dark microcrystallites represent the grains free from labeled laponite, thus they appear dark on the fluorescence images. This conclusion is further supported by the observed crystallite morphologies which show geometrical forms when the waiting time is long enough to allow slow crystallization of the micelles free of defects.
In order to gain quantitative insights into the optical microscopy images and the formation of polycrystalline grains at varying temperature, we chose to quantitatively analyze the phase contrast optical microscopy images by locating the temperature value which corresponds to the onset of transition from the liquid phase to the viscoelastic phase. From a mechanical point of view, this transition point corresponds to the percolation threshold for an assembly of particles which occurs when the volume fraction reaches a critical value of transition.28 Thus we approximate the polycrystalline grains as spherically shaped morphologies. In this case, we expect that percolation transition would yield a viscoelastic solid when the volume fraction of grains reaches the theoretical value for spheres of 29%.29 On the other hand, we need to determine the total area of the section of grains visible on the two-dimensional optical microscopy images or their corresponding surface fraction for each sample. Indeed, there is a relationship between the volume fraction of three-dimensional morphologies in a bulk material and the equivalent surface fraction seen on a two-dimensional random section of the sample.30 The surface fraction of particles on a section of the sample is defined as the sum of the sections of all particles identified on a two-dimensional image per unit area of the image. For morphologies of the same shape with moderate or weak size polydispersity, it was shown that the number per unit area, NA, of objects that can be seen on a section of the sample is related to the number per unit volume, NV, of particles homogenously distributed in the sample by the following expression: NA = NV × 2R where R is the average tangent radius of the morphologies under observation.31 For spherically shaped particles, the mean tangent radius reduces to their mean radius. Moreover, on the one hand, the surface fraction of imaged particles is equal to NA × πR2 = NV × 2πR3 = (3/2) × NV × 4πR3/3, and, on the other hand, the volume fraction is given by ϕ = NV × 4πR3/3. In this case, if the volume fraction of a bulk sample is equal to 0.29 then we estimate the corresponding surface fraction of visualized particles on a random section to be equal to 3 × 0.29/2 = 0.435. Thus we determined the temperature at which the surface fraction of the grains formed on the upper surface of the observation cell is equal to 43% which we denote as Tperc. Tperc is expected to correspond to the temperature point where the growing grains start to form a percolating network. The quantitative analysis of the microscopy images of copolymer micellar solutions with laponite particles shows a rapid increase of the surface occupied by the grains which occurs over a small range of temperature. Moreover, the corresponding Tperc temperature increases as the concentration of laponite increases as is shown in Fig. 1. Remarkably the evolution of Tperc closely follows the variation of Tonset as a function of addition of laponite particles to the copolymer solutions. The coincidence of both temperature values and their sharp increase with addition of laponite strongly suggests that the elasticity of the copolymer solutions with added laponite particles is triggered by the formation of networked solution of polycrystalline micelles forming grains of finite-size. This observation further reinforces our assertion that the observed textures are copolymer micellar polycrystals and that laponite nanoparticles are expelled towards the interstice between them. Their localization in this constrained space is equivalent to a confinement and should lead to inhomogeneous particle dispersion inside the sample with the coexistence of zones with clustered nanoparticles and zones poor or exempt from them. This scenario of density fluctuation of the nanoparticles would have a clear signature in the interparticle structure factor, especially in the most scattering concentrated samples of laponite. Thus, we expect to measure a correlation peak in the SANS intensity of copolymer-matched solutions when only the particle signal is present.32 For this reason, we performed SANS experiments on copolymer samples with WP = 16 wt% and WL = 3 and 5 wt%. We chose to investigate these concentrated samples of copolymer and laponite solutions because the weak contrast of laponite requires a long measurement time at a temperature above Tgel and compares the scattering curve with the data measured on the same sample at room temperature. In this experiment, the scattering length density of the pluronic copolymer chains is taken equal to the average of the PEO and PPO values. This approximation is valid because the scattering length densities of the two blocks are nearly close as compared to the laponite value.33
The scattering curve of the copolymer and laponite solution at room temperature, with the solvent scattering length density adjusted to that of the copolymer, follows the variation of the form factor of the laponite particles. This feature demonstrates that when the temperature is lower than Tgel but higher than the cmt, the nanoparticles are randomly dispersed in the copolymer micellar solution. Above Tgel all the features of an interacting nanoparticle solution with the appearance of a correlation peak are observed. The correlation peak is due to interparticle interactions because the solvent exactly matches the scattering length density of the copolymer. In Fig. 4 we show the measured signal of the copolymer–laponite solution with WP = 16 wt% and WL = 3 wt% normalized by the scattering intensity of a laponite sample with WP = 0 wt% and WL = 3 wt%, i.e., the form factor of the nanoparticles. The solvent composition is 15% D2O and 85% H2O and both scattering intensities were recorded at T = 40 °C, at the transition temperature Tonset of this copolymer–laponite solution. The evolution of the normalized scattering intensity as a function of the wave vector q clearly exhibits the correlation peak which is located at an intermediate q = 0.039 Å−1. Thus the measured peak strongly points to the effect of confinement of laponite particles in the interstices of the copolymer polycrystalline grains triggered by the increase of temperature above the fluid–crystal transition temperature. The position of the correlation peak, qo, is associated with the distance between neighboring laponite particles. The knowledge of this distance allows a straightforward determination of the local concentration of nanoparticles in the confinement area if we assume that they all are aligned because of the excluded volume effect.34 In this case, their effective volume fraction, ϕeff, is simply equal to the ratio of the particle thickness, e = 9.1 Å, to the average distance between them: ϕeff = e × qo/2π = 9.1 × 0.039/2π = 0.057. This effective volume fraction corresponds to a local concentration close to 15 wt%. This value is far above the average laponite concentration in the sample: WL = 3 wt%. The difference between the actual nanoparticle concentration in the sample and the value estimated from the scattering data strongly supports our conclusion that the nanoparticles are non-uniformly dispersed in the sample upon formation of the polycrystalline micellar grains. It demonstrates that the sample is composed of regions where the local concentration is much higher than the average concentration, which in turn triggers the appearance of a correlation peak.
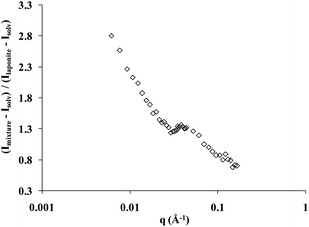 | ||
| Fig. 4 Scattering intensity of the copolymer–laponite solution with WP = 16 wt% and WL = 3 wt% normalized by the scattering intensity of a laponite sample with WP = 0 wt% and WL = 3 wt%. The contrast of the copolymer in this solvent is zero. The measurement temperature is T = 40 °C. | ||
An additional confirmation of the segregation induced by micellar crystallization in this copolymer–laponite nanoparticle solutions was carried out through the investigation of the microstructure by confocal microscopy by using fluorescent nanoparticles. The microscopic images revealed that upon increasing the temperature, non-fluorescent microscopic and disconnected grains begin to grow in a surrounding continuous and fluorescent medium. Optical observation through the sample of different cross-sections reveals that these grains are three-dimensional micron-sized morphologies of nearly spherical shape. They primarily appear on the sample cell interfaces and continue their growth deeply inside the sample as time proceeds. A sequence of scans of the sample at different depths, with WP = 16 wt% and WL = 2 wt%, which are displayed in the ESI clearly supports our conclusion (Fig. S3†).
Conclusions
Using rheology, neutron scattering and optical microscopy, we studied the mechanical properties and the structure of mixtures of pluronic copolymers and laponite colloidal nanoparticles in aqueous solutions. The copolymer micellar solutions undergo a phase transition from fluid-like to fcc crystal when the temperature increases from the critical micellar temperature to above the gelation temperature. Upon addition of laponite nanoparticles to the copolymer solution, the fcc crystal phase is disturbed by the nanoparticles which act as doping defects inside the solution and have a propensity to favor the disordered phase. The consequences are noticeable in both rheological measurements and microscopic observations.We found that the appearance of microscopic grains, composed of polycrystalline micelles, provokes the segregation of the nanoparticles from the polycrystalline grains, thus they are primarily located in the interstices between them. The results shown here are further demonstrations of a general behavior of colloidal particles dispersed in a simple or complex template, which exhibit a tendency for the confinement when a phase transition occurs. Such behavior demands special attention and care when using colloidal nanoparticles in a suspending medium inclined to undergo a phase transition, especially towards a crystalline phase. Our measurements are in agreement with the observations recently reported on the behavior of different kinds of nanoparticles in polycrystalline ice and in micellar copolymer solution similar to the one we used.14,35 In the previous study, it was found that the volume of the grains decreases when the heating rate increases, while the effective volume fraction of nanoparticles in the interstices remains almost constant. These observations suggest that the thickness of the interstices should increase with the heating rate. Unfortunately such evolution is not easily detectable with microscopic techniques.
All these observations point to the fact that the surface chemistry of the nanoparticles plays a minor role in the comportment. Indeed, the processes found in this study are believed to be generic and seem to hold for a variety of nanoparticles of different size and morphology and in different suspending media, either a simple liquid like water or a more complex fluid such as a micellar copolymer solution. Thus, such observed mechanism in composite materials provides a powerful and facile strategy to trigger the assembly of nanoparticles especially when using a templating copolymer solution whose properties can be easily tuned like the case presented in this study.
Acknowledgements
We acknowledge the support of the CNRS/CEA joint Léon Brillouin laboratory in providing the neutron facilities used in this work.References
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- Z. H. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzan, ACS Nano, 2010, 7, 3591–3601 CrossRef.
- K. Liu, N. Zhao and E. Kumacheva, Chem. Soc. Rev., 2011, 40, 656–671 RSC.
- C. L. Chen and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 6902–6903 CrossRef CAS.
- J. Wang, Q. Cheng and Z. Tang, Chem. Soc. Rev., 2012, 41, 1111–1129 RSC.
- M. Fritz and D. E. Morse, Curr. Opin. Colloid Interface Sci., 1998, 3, 55–62 CrossRef CAS.
- Z. H. Nie, D. Fava, M. Rubinstein and E. Kumacheva, J. Am. Chem. Soc., 2008, 130, 3683–3689 CrossRef CAS.
- C. de Mello Donega, Chem. Soc. Rev., 2011, 40, 1512–1546 RSC.
- S. B. Darling, Energy Environ. Sci., 2009, 2, 1266–1273 RSC.
- A. Haryono and W. H. Binder, Small, 2006, 2, 600–611 CrossRef CAS.
- R. J. Spontak, R. Shankar, M. K. Bowman, A. S. Krishnan, M. W. Hamersky, J. Samseth, M. R. Bockstaller and K. Rasmussen, Nano Lett., 2006, 6, 2115–2120 CrossRef CAS.
- T. Huang, Q. Zhao, J. Xiao and L. Qi, ACS Nano, 2010, 4, 4707–4716 CrossRef CAS.
- X. Shen, L. Chen, D. Li, L. Zhu, H. Wang, C. Liu, Y. Wang, Q. Xiong and H. Chen, ACS Nano, 2011, 5, 8426–8433 CrossRef CAS.
- I. Boucenna, L. Royon, P. Colinart, M. A. Guedeau-Boudeville and A. Mourchid, Langmuir, 2010, 26, 14430–14436 CrossRef CAS.
- I. Boucenna, L. Royon and P. Colinart, J. Therm. Anal. Calorim., 2009, 98, 119–123 CrossRef CAS.
- K. J. Mortensen, J. Phys.: Condens. Matter, 1996, 8, 103–124 CrossRef CAS.
- A. Mourchid, A. Delville, J. Lambard, E. Lecolier and P. Levitz, Langmuir, 1995, 11, 1942–1950 CrossRef CAS.
- A. Mourchid and P. Levitz, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 57, 4887–4890 Search PubMed.
- A. Mourchid, E. Lecolier, H. Van Damme and P. Levitz, Langmuir, 1998, 14, 4718–4723 CrossRef CAS.
- S. R. Bhatia, J. Barker and A. Mourchid, Langmuir, 2003, 19, 532–535 CrossRef CAS.
- R. A. DellaGuardla and J. K. Thomas, J. Phys. Chem., 1983, 87, 3550–3557 CrossRef CAS.
- M. A. V. Axelos and M. Kolb, Phys. Rev. Lett., 1990, 64, 1457–1460 CrossRef.
- L. Lobry, N. Micali, F. Mallamace, C. Liao and S. H. Chen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1999, 60, 7076–7087 CrossRef CAS.
- I. W. Hamley, S. M. Mai, A. J. Ryan, J. P. A. Fairclough and C. Booth, Phys. Chem. Chem. Phys., 2001, 3, 2972–2980 RSC.
- A. C. Lawson, B. Martinez, J. A. Roberts, J. W. Richardson and B. I. Bennett, Los Alamos Sci., 2000, 26, 190–201 Search PubMed.
- R. K. Prud'homme, G. Wu and D. K. Schneider, Langmuir, 1996, 12, 4651–4659 CrossRef CAS.
- S. R. Bhatia, A. Mourchid and M. Joanicot, Curr. Opin. Colloid Interface Sci., 2001, 6, 471–478 CrossRef CAS.
- R. Consiglio, D. R. Baker, G. Paul and H. E. Stanley, Physica A, 2003, 319, 49–55 CrossRef.
- E. R. Weibel and D. Paumgartner, J. Cell Biol., 1978, 77, 584–597 Search PubMed.
- E. R. Weibel, G. S. Kistler and W. F. Scherle, J. Cell Biol., 1966, 30, 23–38 Search PubMed.
- D. C. Pozzo and L. M. Walker, Colloids Surf., A, 2004, 240, 187–198 CrossRef CAS.
- A. Nelson and T. Cosgrove, Langmuir, 2005, 21, 9176–9182 CrossRef CAS.
- S. T. Hyde, Colloids Surf., A, 1995, 103, 227–247 CrossRef CAS.
- N. Ghofraniha, E. Tamborini, J. Oberdisse, L. Cipelletti and L. Ramos, Soft Matter, 2012, 8, 6214–6219 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2sm26851e |
| This journal is © The Royal Society of Chemistry 2013 |