A toolbox of differently sized and labeled PMMA nanoparticles for cellular uptake investigations†
Antje
Vollrath
ab,
Anja
Schallon
ab,
Christian
Pietsch
ab,
Stephanie
Schubert
bc,
Takahiro
Nomoto
d,
Yu
Matsumoto
e,
Kazunori
Kataoka
*defg and
Ulrich S.
Schubert
*ab
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstrasse 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
cInstitute of Pharmacy, Department of Pharmaceutical Technology, Friedrich Schiller University Jena, Otto-Schott-Str. 41, 07745 Jena, Germany
dDepartment of Bioengineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
eCenter for Disease Biology and Integrative Medicine, Graduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
fDepartment of Materials Engineering, Graduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
gCenter for NanoBio Integration (CNBI), The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: kataoka@bmw.t.u-tokyo.ac.jp
First published on 16th October 2012
Abstract
The cellular internalization of defined PMMA nanoparticles was investigated. For this purpose, the biocompatible copolymer p(MMA-stat-MAA)0.91:0.09 was synthesized by RAFT polymerization and labeled with three different fluorescent dyes (λEx = 493, 557, and 653 nm). Nanoparticles were formulated from the differently labeled copolymers into samples with relatively narrow size distribution (diameter d < 100 nm, 100 to 200 nm, >300 nm) under appropriate conditions of nanoprecipitation and were subsequently characterized by DLS and SEM. Mixtures of the differently sized nanoparticle samples were applied for internalization studies using monolayer cultured HeLa cells. The localization of the nanoparticles was detected after certain time points up to 24 h by CLSM, using LysoTracker as a marker for late endosomes and lysosomes. In investigations by flow cytometry, a fast uptake of medium sized nanoparticles was found, whereas the large and small nanoparticles exhibited a slower internalization. However, small and medium sized nanoparticles were detected in the late endosomes/lysosomes, whereas the large nanoparticles exhibit little co-localization with LysoTracker. Moreover, it could be shown by using different inhibitors for clathrin-dependent (chlorpromazine), caveolin-dependent (filipin III) endocytosis and macropinocytosis (EIPA) that nanoparticles with d < 200 nm were internalized via clathrin-dependent endocytosis, whereas those with d > 300 nm were internalized via macropinocytosis.
Introduction
Polymeric nanoparticles (NPs) can provide manifold opportunities for drug and gene delivery,1,2 because molecules and particles at the nanometer scale offer important benefits like an enhanced permeability and retention effect (EPR), resulting in an improved bioavailability and fewer side effects. In addition, NPs offer the possibility of delivering nucleic acids (siRNA, DNA), proteins or other active substances into targeted organs or cells.1–6 However, many points in the field of nanotechnology still need to be considered very carefully with regard to unknown long-time consequences such as the accumulation of the particles in the vessels/liver, the uptake through skin and lungs, and the removal by the reticuloendothelial system.7 Hence, a detailed understanding of the interaction of NPs with their environment is essential for the development of defined drug delivery systems as well as for specific diagnostic applications.Since the last decade, a number of studies have been published investigating the cellular uptake mechanisms of NPs with respect to various physical, chemical, and biological parameters.8–13 The size of the NPs was found to play the key role in the final particle–cell interaction.14–16 In detail, depending on their size, particles can enter cells either by phagocytosis or pinocytosis; the latter mechanism can be further subdivided into clathrin- and caveolin-dependent endocytosis as well as clathrin/caveolin-independent endocytosis such as macropinocytosis.17–20 Phagocytosis and macropinocytosis are processes to engulf large particles up to the range of 10 μm, whereas the clathrin- and caveolin-dependent endocytosis are the main processes for the internalization of smaller particles below 500 nm.18 In addition, the shape of the NPs significantly influences uptake of NPs by phagocytosis. Rod-like and oblate ellipsoidal NPs with a high aspect ratio were taken up more efficiently in comparison to their spherical counterparts, due to their higher surface area and, therefore, a better attachment to the cell membrane.13,16,21,22 In addition, the internalization of particles into the cells is influenced by their surface charge: an increased positive charge leads to a higher cellular uptake due to electrostatic interactions with the negatively charged cell membrane.23 As the surface of NPs can be further functionalized with different reactive groups (COOH and NH2), amino acids, sugar units, antibodies, or peptides, cellular uptake of NPs can be inhibited or advanced.24–26 Nevertheless, no final conclusion can be drawn about the size, shape and charge dependency of cellular uptake of NPs, as the studies published so far are all based on different materials. Inorganic (gold,27,28 silica particles,29,30 and quantum dots31) and organic (poly(lactic-co-glycolic acid) (PLGA),15 polystyrene (PS),32,33 and chitosan34,35) materials were studied and many contradictory results were reported by different investigators.36 Furthermore, cell internalization studies frequently used NP beads with non-defined surfaces because conventional preparation techniques (e.g. the emulsion technique) require the use of surfactants, which also influence cellular uptake.37 Hence, further studies on NP–cell-interactions with well-defined NPs, in the absence of any surfactants, would be helpful to gain a better insight into the involved parameters and structure–property relationships.
The present study aims at investigating the cell interaction of polymeric NPs with well-defined characteristics based on poly(methyl methacrylate) (PMMA) derivatives, poly(methyl methacrylate)-stat-poly(methacrylic acid) (p(MMA-stat-MAA)) copolymer. The p(MMA-stat-MAA) copolymer is based on the same monomers as the pharmaceutically important coating material EUDRAGIT® S100, which is applied for pH-dependent drug release.31 Recently, it has been demonstrated that PMMA systems are suitable for diagnostic applications in cell imaging and as a gene carrier due to their non-toxicity and good biocompatibility.38–41 Here, the poly(methacrylic acid) segment of p(MMA-stat-MAA) was labeled with various dyes (DY-495 (green), DY-547 (orange), and DY-647 (red)) for tracking of the NPs.42 NPs of the labeled p(MMA-stat-MAA) copolymers (p(MMA-stat-MAdye (green, orange, or red))) were prepared by the solvent-evaporation (nanoprecipitation) method43 and characterized comprehensively by dynamic light scattering (DLS) and scanning electron microscopy (SEM).44 The stability of the NPs was demonstrated by DLS measurements after autoclave treatment, after incubation in cell culture media, and during pH titration studies, respectively. Cytotoxicity assays were performed to prove the biocompatibility and non-toxicity of the NPs. The cellular uptake studies used HeLa cells and three differently sized and individually labeled NPs. Flow cytometry was applied for studies on time- and concentration-dependent cellular uptake and confocal laser scanning microscopy (CLSM) for cellular distribution and co-localization studies. In addition, cellular internalization was investigated by inhibitors of clathrin- and caveolin-dependent endocytosis as well as of macropinocytosis.
Experimental
Materials
All reagents were purchased from commercial sources (Fluka and Sigma Aldrich). MMA and MAA were purchased from Sigma Aldrich and purified with an inhibitor-remover before use. 2,2′-Azobis(iso-butyronitrile) (AIBN) was recrystallized from methanol prior to use. 2-Cyano-2-propyl dithiobenzoate (CPDB) was purchased from Sigma Aldrich. Purified N,N-dimethylacetamide (DMA) and dimethylformamide (DMF) were obtained from VWR. The fluorescent dyes λ = 495 nm (DY-495), λ = 557 nm (DY-547), and λ = 653 nm (DY-647) were purchased from DYOMICS GmbH. AlamarBlue, LysoTracker Green, and OptiMEM were obtained from Life Technologies. Hoechst 33342, 5-(N-ethyl-N-isopropyl)amiloride (EIPA), filipin III, and chlorpromazine were purchased from Sigma Aldrich. Cell culture materials were received from Greiner Bio One, cell culture media and solutions from Biochrome, Greiner, and PAA. Unless otherwise stated, the chemicals were used without further purification.Synthesis of p(MMA-stat-MAA)
P(MMA-stat-MAA) was prepared by copolymerization of MMA with MAA using the reversible addition–fragmentation chain transfer (RAFT) polymerization method.37,38 In a typical RAFT copolymerization experiment, 4.325 g of MMA monomer (43.2 × 10−3 mol), 0.413 g of MAA monomer (4.8 × 10−3 mol), 19.7 mg of AIBN initiator (0.12 × 10−3 mol), 106.3 mg of CPDB RAFT agent (0.48 × 10−3 mol) and 5.8 mL of ethanol were mixed together with anisole as the internal standard (1.2 mL) in a 25 mL reaction vial. The monomer concentration was kept at 4 mol L−1. Subsequently, the reaction solution was placed in a preheated oil bath at 70 °C for 10 hours. The copolymer was purified by precipitation into a large volume of cold diethyl ether and dried under reduced pressure. Conversion was measured by 1H NMR spectroscopy using anisole as the internal standard. 1H NMR (DMSO-d6, 300 MHz): δ = 12.5 (–OH), 7.83, 7.64, and 6.46 (Ar–H, CPDB), 3.55 (OCH3), 2.25–0.3 (backbone) ppm. SEC (DMA, LiCl, and PMMA standard): Mn = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and PDI = 1.19. Elemental analysis: p(MMA-stat-MAA) C: 58.18 and H: 7.91%.
000 g mol−1 and PDI = 1.19. Elemental analysis: p(MMA-stat-MAA) C: 58.18 and H: 7.91%.
Labeling of p(MMA-stat-MAA)
500 mg of the p(MMA-stat-MAA) polymer (3.1 × 10−4 mol) were dissolved in 2 mL of dried DMF, and 50 μL (3.0 × 10−4 mol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was added as well as 500 μL of a stock solution (c = 0.1 mg mL−1 in DMF) of the desired dye DY-495 (green, 8.5 × 10−8 mol), DY-547 (orange, 7.5 × 10−8 mol), or DY-647 (red, 7.1 × 10−8 mol). The labeled polymers were purified by repeated precipitation in water and extensive dialysis afterwards. The products were obtained by freeze drying in 70% overall yield. SEC (DMA, LiCl, and PMMA standard): p(MMA-stat-MAgreen): Mn = 23500 g mol−1 and PDI = 1.21; p(MMA-stat-MAorange): Mn = 21800 g mol−1 and PDI = 1.22; p(MMA-stat-MAred): Mn = 23800 g mol−1 and PDI = 1.22; elemental analysis: p(MMA-stat-MAgreen) C: 56.85, H: 8.18, and N: 2.43; p(MMA-stat-MAorange) C: 58.51, H: 8.12, and N: 2.40; p(MMA-stat-MAred) C: 58.17, H: 8.08, and N: 2.46.
Preparation of the NP suspension
NPs were prepared by nanoprecipitation with subsequent solvent evaporation. For this purpose, the polymers were dissolved in acetone and filtered through a 2 μm filter prior to use. NP suspensions with different sizes were prepared by variation of the initial conditions of the formulation. For the small (diameter d < 100 nm) and medium (100 to 200 nm) sized NPs, the acetone solution was dropped into deionized water (AW) with a concentration of 1 mg mL−1 and 10 mg mL−1, respectively. By dropping water to the polymer solution (WA) with a concentration of 4 mg mL−1, large NPs (d > 300 nm) were prepared. For all suspensions, the acetone–water ratio was chosen to be 0.25. Furthermore, the dropping speed was approximately 50 μL per second, and the stirring speed was set to 1000 rpm (Magnetic Stirrer MR Hei-Standard). Afterwards, the acetone was evaporated from the solution by stirring overnight at room temperature, the suspensions were filtered using a filter paper and diluted to a final concentration of 0.5 mg mL−1. The suspensions were stored in a fridge at 4 °C and before further usage they were vortexed to ensure a homogenous particle suspension.Dynamic light scattering (DLS) and zeta potential measurements
For DLS investigations, a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) operating with a laser beam at 633 nm and a scattering angle of 173° was used. Each sample was analyzed in triplicate at 25 °C in a polycarbonate zeta cell. For size measurements, three runs were applied for 30 s; for the zeta potential measurements, three runs were applied for 10 s. The intensity, volume and number distribution of the NPs were calculated applying the NNLS mode.Scanning electron microscopy (SEM)
SEM images were obtained using a LEO-1450 VP, Leo Elektronenmikroskopie GmbH, Oberkochen, Germany. The sputter coating device BAL-TEC SCD005 (Balzers, Liechtenstein; 60 mA, 80 s) was used. The system was operated from 8 to 10 kV.Cell lines and culture conditions
The HeLa (CCL-2, ATCC) and L929 (CCL-1, ATCC) cell lines used in the uptake and cytotoxicity experiments were maintained in suitable cell culture media supplemented with 10% fetal calf serum (FCS), 100 μg mL−1 streptomycin, 100 IU mL−1 penicillin, and 2 mM L-glutamine (4 mM for L929). The cells were cultured at 37 °C in a humidified 5% CO2 atmosphere.Flow cytometry analysis
For the determination of cellular uptake of NPs via flow cytometry, 105 cells per well were seeded in 12-well plates and incubated for 24 h. Thirty minutes prior to the incubation with the NPs, the cells were rinsed with PBS and supplemented with OptiMEM. The NPs were added to the cells and the plates were incubated for the indicated time. Afterwards, the cells were harvested by trypsinization and resuspended in PBS supplemented with 10% FCS. To determine the relative uptake of NPs, 10000 cells were quantified by flow cytometry using a Cytomics FC 500 (Beckman Coulter).
Microscopy studies
HeLa cells were cultured on 35 mm glass dishes (Iwaki, Japan) at 2 × 105 cells per dish. After 24 h, the medium was exchanged with OptiMEM, and the cells were incubated for 30 min before the addition of the NP suspensions. The nuclei and the late endosomes/lysosomes were stained with Hoechst 33342 and LysoTracker Green, respectively, before CLSM imaging. CLSM images were acquired 24 h after the administration of NPs, using a Zeiss LSM 780 (Carl Zeiss). Excitation wavelengths were 405 nm, 488 nm, 561 nm and 633 nm for Hoechst 33342, LysoTracker Green or DY-495 stained NPs, DY-547 stained NPs, and DY-647 stained NPs, respectively. Co-localization was quantified, using Imaris software (Bitplane AG, Zurich, Switzerland).Inhibition of endosomal pathways
The cells were seeded as described before. The growth media were changed to OptiMEM and incubated for 30 min before 10 μg mL−1 chlorpromazine, 1 μg mL−1 filipin III, or 100 μM EIPA were added and incubated for a further 30 min. Afterwards, the NPs were added, and the cells were incubated for the indicated time.Cell viability
For L929 cells, the cytotoxicity assay was performed according to ISO10993-5. In detail, the cells were seeded at 10000 cells per well in a 96-well plate and incubated for 24 h. No cells were seeded in the outer wells. The growth media were replaced by OptiMEM. Afterwards, NP dilutions in the concentration range from 78 to 254 μg mL−1 were added, and the cells were incubated at 37 °C for further 24 h. Subsequently, the medium was replaced by DPBS and AlamarBlue, as recommended by the supplier. After incubation for 4 h, the fluorescence was measured at Ex 570/Em 610 nm in a microplate reader (Genios Pro, Tecan GmbH), with untreated cells serving as controls.
Results and discussions
Polymer preparation and characterization
To provide the possibility of functionalization after the polymerization, carboxylic acid groups were introduced into the PMMA chain. Therefore, MMA was copolymerized with MAA using the RAFT polymerization method.37,38 This technique allows the synthesis of tailored polymers with control over molar mass and the composition of the copolymer, i.e., the ratio between MAA and MMA.In the copolymerization reaction, a final conversion of 87% (both monomers) was reached. MMA and MAA are statistically distributed in the polymer backbone due to the same reactivity ratio of the MMA and MAA units.45 The ratio between both MMA and MAA in p(MMA-stat-MAA) was determined by 1H NMR spectroscopy. The observed ratio of 91 to 9% agrees well with the theoretical value of 90 to 10%. The low content of MAA ensures the stability at pH > 7 of the NPs and seems to be beneficial for a well-defined NP formation in aqueous systems. A molar mass of Mn = 16000 g mol−1 with a polydispersity index of 1.15 (DMA, LiCl) was determined by size exclusion chromatography (SEC) for the final copolymer p(MMA-stat-MAA)0.91:0.09 (Table 1).
| Sample | M n a [g mol−1] | PDISEC (RI)a | C [%] | H [%] | N [%] |
|---|---|---|---|---|---|
| a Calculated from SEC (DMA and LiCl) and PMMA calibration. | |||||
| P(MMA-stat-MAA)0.91:0.09 | 16000 |
1.15 | 58.18 | 7.91 | — |
| P(MMA-stat-MAgreen) | 23500 |
1.21 | 56.85 | 8.18 | 2.43 |
| P(MMA-stat-MAorange) | 21800 |
1.22 | 58.51 | 8.12 | 2.40 |
| P(MMA-stat-MAred) | 23800 |
1.22 | 58.17 | 8.08 | 2.46 |
In order to obtain differently labeled polymers, three different dyes (DY-495, DY-547 and DY-647) were chosen, because the dyes provide high quantum yields, good photostability, and are available with different functional groups (Scheme 1).42 The labeling procedure was performed by the reaction of the COOH groups with the NH2-functionalized dyes using EDC as the coupling reagent. The absence of free dye was proven by fluorescence measurements of the washing water after purification. The fluorescence emission spectra of the labeled copolymers p(MMA-stat-MAgreen), p(MMA-stat-MAorange), and p(MMA-stat-MAred) are displayed in the ESI, Fig. S1.† All the copolymers show distinct peaks at 525 nm, 568 nm, and 668 nm, respectively. In comparison to the initial emission of the pure dyes, no significant change in the fluorescence behavior was obtained even after conjugation into the copolymer segments. A comparison of the SEC graphs of the unmodified p(MMA-stat-MAA) and the labeled copolymers revealed a slight change in the elution volume, indicating that the molar mass slightly increased due to dye conjugation and that the polymer was not degraded or cross-linked during the labeling procedure. The overlay of the diode array detector (DAD) and refractive index (RI) traces of the labeled samples confirm the covalent attachment of the dyes (ESI, Fig. S2†). Furthermore, the elemental analysis revealed an increase in the nitrogen content, which also indicates the attachment of the dyes in the polymer backbone.
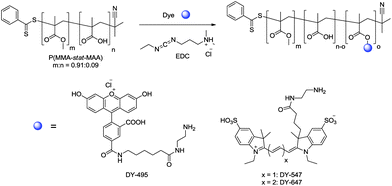 | ||
| Scheme 1 Schematic representation of the reaction of p(MMA-stat-MAA)0.91:0.09 with the DY-495, DY-547, and DY-647 using EDC as a coupling reagent. | ||
Preparation and characterization of NPs
Differently sized NPs were prepared by nanoprecipitation with subsequent solvent evaporation, a technique, which is not only simple and cost-effective but also fast and easy.43 By variation of the initial conditions of the formulation, such as the solvent/non-solvent ratio and the concentration of the polymer solution, well-defined NPs with different sizes can be prepared. In comparison to other procedures commonly used, e.g., the emulsification technique, no surfactants are necessary for the NP preparation. This represents an important benefit of the nanoprecipitation technique. It was proven that surfactants significantly affect the interaction of NPs with cells as well as their cellular uptake.37 Using nanoprecipitation, it is known that the final NP sizes can be tuned from 50 nm up to 1 μm by varying the initial polymer concentration in the organic phase and/or by changing the dropping method (polymer solution into water or water into polymer solution). In order to yield small (S; d < 100 nm), medium (M; d between 100 and 200 nm), and large (L, d > 300 nm) NPs, different nanoprecipitation conditions were applied. By dropping the acetone–polymer solution into water, smaller NPs were obtained in comparison to the reverse technique. For the preparation of the small NPs, polymer acetone solutions with a concentration of 1 mg mL−1 were dropped into water. For the medium sized NPs the same procedure with an increased concentration of 10 mg mL−1 was used. The large NPs were generated by dropping water into polymer solutions with a concentration of 4 mg mL−1. For all suspensions, a solvent/non-solvent ratio of 0.25 and no surfactants were used. After subsequent removal of the acetone by overnight evaporation, the NPs were filtered, diluted to a concentration of 0.5 mg mL−1, and characterized by DLS and SEM. These complementary techniques give sufficient information about the size, shape, and surface characteristics of the particle systems.44 For the small NPs, a size of 80 (±10) nm was measured by DLS in water. The medium NPs revealed a size of 150 (±10) nm, whereas for the large NPs a size of 400 (±50) nm was obtained. Low PDIP values confirm a narrow size distribution of the NPs, which was further verified by the SEM investigations. A detailed analysis of a representative small, medium and large NP batch is displayed in Fig. 1. As the major focus of this research was set on the influence of the size on the cell internalization, a charge effect needs to be excluded. Thus, only a low degree of labeling was performed in order to keep the charge density similar. The zeta potential of the NP was measured in water (pH = 6), resulting in comparable values with 30 ± 10 mV for all nanoparticle suspensions. The zeta potential values larger than 20 mV further indicate high repulsion forces and colloidal stability of the NPs in suspension. However, the stability of the NPs should also be confirmed under different conditions, and, therefore, a NP suspension with medium sized NPs was investigated in more detail. For this purpose, all NPs were centrifuged at 15000 rpm for 20 min, autoclaved, or incubated in PBS or cell culture media as well as titrated in the pH range of 4 to 10. The resulting suspensions were analyzed again by DLS and SEM. Neither the size distribution nor the zeta potential value changed, which proves the high stability of the p(MMA-stat-MAdye) NPs (ESI, Fig. S3†). In combination with the easily tunable NP size, the absence of surfactants, and the variety on possible modifications/labels of the original polymers, the NPs represent excellent and well-defined materials for further cell internalization experiments.
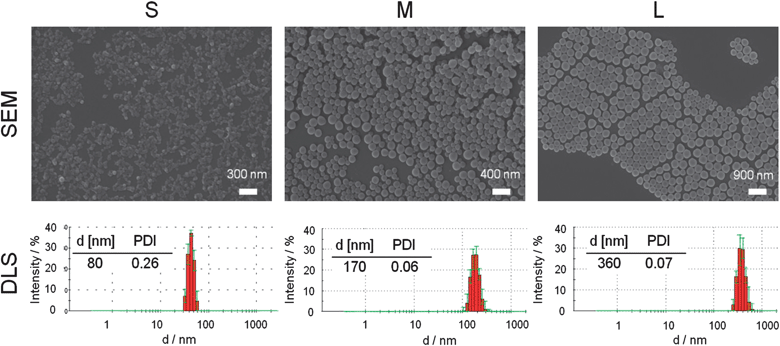 | ||
| Fig. 1 Representative SEM images and DLS intensity size distribution with corresponding Z average value and polydispersity PDIP of the small, medium and large particles of the p(MMA-stat-MAdye) copolymer. | ||
Cytotoxicity of the polymers in L929 cells
To evaluate the cytotoxicity of the NPs, we used L929 cells, because they were commonly used for the investigation of cytotoxicity, as they are sensitive and recommended by ISO10993-5. For the cell experiments, the NPs (which originally were in distilled water) were buffered with DPBS before being added to the cells. In preliminary experiments, cytotoxicity tests with AlamarBlue were performed for 24 h, to evaluate the metabolic activity of L929 cells exposed to the NPs. Small (<100 nm) and large (>300 nm) labeled NPs were investigated. As shown in Fig. S4 (ESI†), all types of NPs did not cause a significant cytotoxicity after 24 h of incubation at the investigated concentrations up to 260 μg NPs per mL (p > 0.01; ANOVA). Moreover, neither size nor labeling of the NPs showed an influence on the cytotoxicity. This is in agreement with studies of other groups using EUDRAGIT S100.38–40 Thus, the NPs used here are not toxic even at the maximum concentration of 150 μg mL−1.Validation of cellular uptake measurement by flow cytometry via fluorescence and side scatter
To investigate cellular uptake of the NPs into HeLa cells by flow cytometry, we first confirmed the validation of cellular uptake measurement by flow cytometry. The fluorescence channel as well as the side scatter (SSC) and forward scatter (FSC) channels were used. The SSC is directly related to the cell granularity and was used as an indicator of cellular uptake.46 On the other hand, the FSC is correlated with the cell size and was used as an additional factor. After overnight incubation with the NPs, the cells showed a high cellular uptake that was detected by the fluorescence intensity measurements. This result was also confirmed by an increase in the SSC channel (ESI, Fig. S5†): the SSC signal increased with the increase of the incubation time, whereas the FSC signal did not change. Moreover, a strong correlation between fluorescence and SSC was detected when HeLa cells were incubated with increasing sizes of DY-547 labeled NPs. To exclude a high influence of NPs on the cell surface, the outer fluorescence was quenched with trypan. No differences in the non-quenched ones could be observed. Therefore, the uptake of NPs can be detected by fluorescence and SSC measurements.Dependency of cellular uptake on the NP concentration
A concentration-dependent cellular uptake was investigated after 24 h using four different NP concentrations (25, 50, 100, and 150 μg mL−1, respectively). The cellular uptake was quantified using flow cytometry. It should be noted that larger NPs contain a higher content of labeled polymers leading to an increased fluorescence signal compared to smaller NPs. To suppress the influence of the cell size on granularity, the SSC was measured relative to the FSC as the cofactor. For analysis, the fluorescence (mean fluorescence intensity = MFI) and SSC/FSC of the treated cells were plotted relative to untreated cells (Fig. 2). It was observed that higher NP concentrations lead to an increase in MFI and SSC/FSC (Fig. 2A). In particular, for the medium and large sized NPs, a clear concentration-dependent cellular uptake was observed. In detail, for incubation with 150 μg mL−1, a three times higher relative SSC/FSC value was obtained for the medium and large sized NPs (3.4 ± 0.2 and 3 ± 0.4, respectively) in comparison to the control cells. For the small NPs only a relative SSC/FSC value of 2.6 ± 0.4 was detected. The fluorescence measurement also confirmed these results, but is influenced by the amount of fluorescent polymer per particle. The number of “positive cells” (cells that internalize NPs) was increased with higher NP concentration: 95% positive cells were found for c = 150 μg mL−1 and 75% for 50 μg mL−1, respectively (data not shown). Furthermore, the relative SSC/FSC as well as the relative MFI showed no significant differences between small, medium and large NPs at 50 μg mL−1 (p > 0.05). It could be shown that all NPs were taken up by HeLa cells at the used concentrations.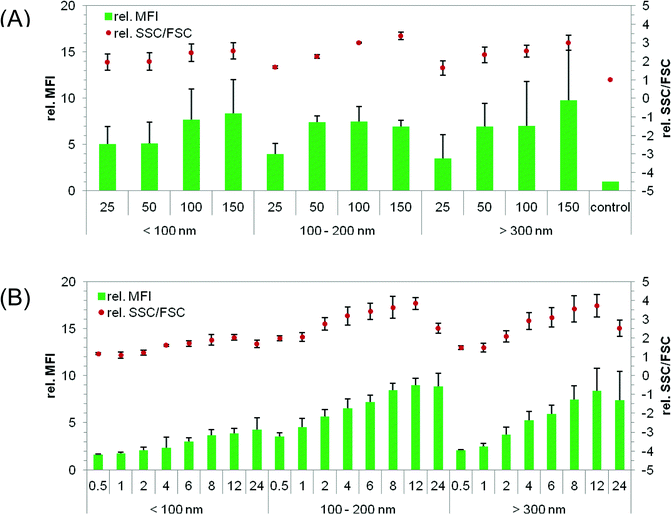 | ||
| Fig. 2 Cellular uptake of NPs into HeLa cells measured by flow cytometry: the cells were seeded at a concentration of 105 cells per mL 24 h before particle incubation with NPs. The uptake of NPs was investigated in serum-reduced media and measured by two methods: the relative change of fluorescence (MFI) and granularity (SSC/FSC) compared to non-treated cells. (A) Different concentrations of differently sized particles were analyzed after 24 h of incubation. (B) Time-dependent uptake of 50 μg NPs per mL. Data represent mean ± SD, n ≥ 3. | ||
Cultivation conditions – influence on the NP uptake by HeLa cells
The majority of experiments were performed with serum-reduced media (OptiMEM), but additionally the cellular uptake of NPs was further investigated in growth media to exclude an influence of serum proteins on the uptake. No significant differences could be obtained between serum-reduced and serum-containing (growth media) conditions for 50 μg mL−1 small and large NPs. In all cases, 60% to 70% of positive cells and an increased SSC/FSC signal could be detected after 24 h. The results indicate that the used media have no influence on the cellular uptake.Dependency of cellular uptake on the incubation time
The size of the NPs showed no influence on the cytotoxicity and cellular uptake after 24 h incubation at a NP concentration of 50 μg mL−1 as described above. To gain a deeper understanding of the cellular uptake, a time-dependent cellular uptake was evaluated. In these experiments, HeLa cells were incubated with p(MMA-stat-MAorange) NPs for 30 min up to 24 h, and MFI as well as SSC/FSC were analyzed by flow cytometry. In Fig. 2B, the cellular uptake is presented as relative MFI and SSC/FSC of treated cells to non-treated cells. It could be observed that all the NPs were taken up by the cells in a time-dependent manner. In detail, small NPs < 100 nm reached a relative MFI of around 4 and an increase of SSC/FSC of around 2 after 24 h at 50 μg mL−1. This is in agreement with the results observed by the concentration-dependent uptake in Fig. 2A. A significant increase (p < 0.01) in granularity (relative SSC/FSC) was detectable after 4 h incubation. The highest relative SSC/FSC was found after 12 h incubation. This was also the case for medium and large sized NPs (medium NP: MFI 8.5, SSC/FSC 3.5; large NP: MFI 7.5–8, SSC/FSC 3.5). Furthermore, no significant differences were detected between medium and large NPs after 4 to 24 h incubation (p > 0.05). The internalization of all NPs reached a plateau after 8 h, as indicated by the lack of further significant increase in fluorescence and granularity (p > 0.05). A difference in the cellular uptake of the differently sized NPs was, however, observed at early time points. In detail, small NPs < 100 nm and large NPs > 300 nm showed no increase in granularity for 2 h and 1 h, respectively (p > 0.05). In contrast to this, an increase in granularity was detectable with medium sized NPs (p < 0.01), which can be due to different reasons. First of all, the effect may be caused by different endosomal pathways. The delayed cellular uptake of small NPs can also be explained by a slower sedimentation of the NPs compared to the larger ones. This could lead to a later contact between the membrane of cells and NPs, and an eventual delayed cellular uptake. Besides, a decrease in relative SSC/FSC was observed at 24 h compared to that at 12 h in particular with medium and large sized NPs. This could not be observed by fluorescence. The reason for this decrease in granularity is not understood by now and requires further investigations.In conclusion, medium NPs of 100 to 200 nm were found to have the fastest cellular uptake in HeLa cells compared to smaller and larger ones. A fast cellular uptake of medium sized NPs could be due to fast sedimentation and fast internalization into cells, indicating an endosomal pathway. Large NPs were taken up very slowly despite fast sedimentation, indicating a macropinocytotic uptake, because endosomal pathways like clathrin- or caveolin-dependent endocytosis are faster compared to macropinocytosis.47
Intracellular localization study of differently sized NPs
The cellular uptake mechanisms of HeLa cells were investigated by CLSM. For this purpose, the intracellular distribution of NPs after 24 h incubation was studied in living cells. Small NPgreen, medium NPorange and large NPred suspensions were used at 50 μg mL−1 each. The nuclei of the cells were stained with Hoechst 33342. A representative NP distribution in HeLa cells is presented in Fig. 3. Cells containing NPs of different sizes are plotted separately (Fig. 3A–C) as well as the corresponding cell nuclei (Fig. 3D). In addition, two overlays are presented. The overlay of all channels is presented in Fig. 3F. No NPs could be observed in the cell nuclei. The intracellular distribution of small and medium sized NPs is comparable. In detail, a perinuclear localization was observed (compare Fig. 3A and B) in the cytoplasm. Hence, a strong co-localization was found with NPs below 200 nm. In contrast, the intracellular localization of small and large NPs is different, as presented in Fig. 3A and C, and merged in Fig. 3E. Co-localizations between small and large sized NPs would be indicated by yellow signals. Here, only a few yellow signals were detectable indicating only some co-localization. This could also be observed in Fig. 3F, where the observed cyan staining indicates a co-localization of small and medium sized NPs, whereas only a few purple (co-localization of medium and large NPs) and yellow signals could be found. Furthermore the co-localization was quantified by Pearson's correlation coefficient (PCC) (Fig. 3G). The PCC between the small and medium sized NPs showed the highest value, suggesting that the small and medium sized NPs might have a similar cellular uptake pathway. To exclude the possibility that this cellular distribution is caused by the dye, other NPs with comparable sizes but different dyes were investigated (e.g. small NPorange and large NPgreen). This combination of NPs was also incubated with HeLa cells and treated as described before. Thereby, the intracellular distribution shows no dependency on the dye and, thus, indicates no influence of the chemical nature of dyes used on the uptake behavior of HeLa cells. Additionally, HeLa cells incubated with small, medium, and large NPs at 4 °C revealed no fluorescence inside the cells (ESI, Fig. S6†). This indicates an active cellular uptake mechanism of all sized NPs via endocytosis. Hence, a different intracellular distribution of large NPs (>300 nm) compared to small (<100 nm) and medium (100 to 200 nm) sized NPs was clearly proven. This indicates a different internalization of NPs below 200 nm compared to larger ones. Furthermore, the microscopy data support the results observed by flow cytometry, where differences in time-dependent uptake could be found with regard to the NP size. Although, medium and large sized NPs sediment faster onto the cells, the large NPs were not internalized within the first hour, whereas medium sized NPs were detected in cells after 30 min.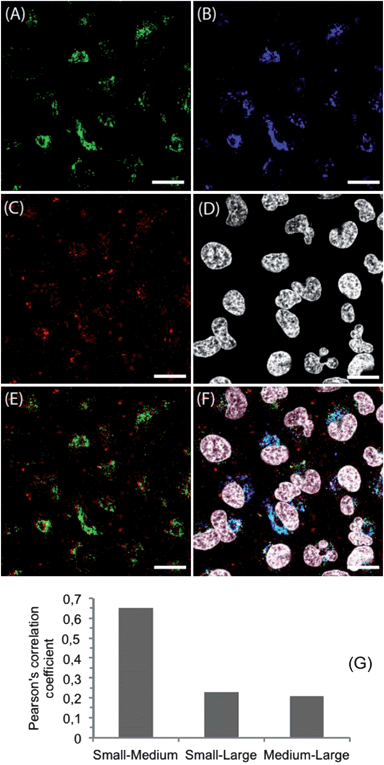 | ||
| Fig. 3 Confocal microscope images of HeLa cells incubated with NPs. Cells were seeded at a density of 105 cells per mL for 24 h in medium before particle incubation. NPs with different sizes and labels (A: small NPgreen; B: medium NPorange; C: large NPred) were added at 50 μg mL−1 simultaneously and incubated for 24 h. (D) Cell nuclei were stained with Hoechst 33342. (E) Overlay of small NPgreen and large NPred. (F) Overlay image of labeled NPs and cell nuclei. (G) Pearson's correlation coefficient of labeled NPs. The scale bars indicate 20 μm. | ||
Investigation of cellular pathways
The intracellular localization and the cellular uptake kinetics of NPs showed strong dependency on size (Fig. 2B and 3). To further elucidate this observation, the intracellular localization was investigated by using LysoTracker, a well known substance for staining acidic late endosomes and lysosomes. As NPs <200 nm showed the same cellular localization in all experiments, only medium (100 to 200 nm) NPs are presented here. They show a stronger fluorescence signal and could be detected more easily compared to small NPs, due to more labeled polymers inside the NPs. In Fig. 4, the stained lysosomes (A), medium NPs (B), large NPs (C), and cell nuclei (D) are presented. Moreover, the overlay of lysosomes and large NPs (E) and the overlay of all dyes used (F) are shown. Again, no NPs were found in the nuclei. It should be noted that almost all medium sized NPs were detected in the late endosomes or lysosomes indicated by a high PCC of 0.61 and a cyan staining in Fig. 4F due to the merging of medium sized NPorange (plotted in blue) with LysoTracker (A). An adverse intracellular localization was found by using large NPs showing little co-localization with medium sized NPs and a PCC of 0.19, as described before. Furthermore, little co-localization was observed with late endosomes or lysosomes, indicated by slight yellow signals in Fig. 4E and a PCC of only 0.15.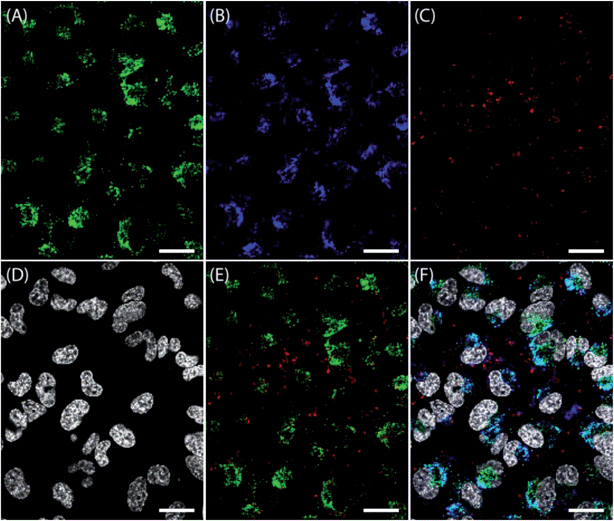 | ||
| Fig. 4 Confocal microscope images of HeLa cells incubated with particles. Cells were seeded at a density of 105 cells per mL for 24 h. The medium was changed and incubated for 30 min. NPs at different sizes and labels (B: medium NPorange; C: large NPred) were added at 50 μg mL−1 simultaneously and incubated for 24 h. (A) LysoTracker Green was added 10 min before microscopic analysis and incubated at 37 °C to stain acidic late endosomes and lysosomes. (D) Cell nuclei were stained with Hoechst 33342. (E) Overlay of stained lysosomes and large NPred. (F) Overlay image of labeled NPs, lysosomes and cell nuclei. The scale bars indicate 20 μm. | ||
The large NPs were taken up by the cells, but the internalization or the cellular pathway seems to be different to the medium ones. Whereas smaller NPs revealed a fate in late endosomes or lysosomes, the cellular uptake mechanism of larger NPs is not fully understood. It can be postulated that NPs smaller than 200 nm were internalized via endocytosis and ended up in the late endosomes or lysosomes. The missing co-localization of larger NPs > 300 nm with late endosomes or lysosomes indicates an uptake mechanism that does not end in the lysosomes at the investigated time points, as a release of NPs into the cytoplasm is not supported.
Inhibition of endocytic pathways
To investigate the cellular uptake mechanism of differently sized NPs, internalization routes were inhibited. Chlorpromazine was used for inhibition of clathrin-dependent and filipin III for inhibition of caveolin-dependent endocytosis. It was described in the literature that NPs < 200 nm are predominantly taken up by these pathways.18,48 In contrast to smaller NPs, larger NPs can be taken up by macropinocytosis or phagocytosis and were inhibited by EIPA.18,48 The NP internalization was analyzed by flow cytometry (ESI, Fig. S7†). Inhibition of cellular uptake was investigated with small and large sized NPs, as it was shown that the intracellular localization of small and medium sized NPs is comparable. As mentioned above, the MFI and the SSC/FSC showed no strong increase after 2 h (Fig. 2), because the NP concentration in cells has an influence on the signal intensity of SSC/FSC and MFI. As the cells internalize only a few NPs after 2 hours, the SSC/FSC and MFI could not be used in this case. Therefore, the percentage of cells taking up NPs was used (ESI, Fig. S7†). In the case of using small NPs and the three different inhibitors, it was found that NPs < 200 nm were excluded not by filipin III and EIPA but by chlorpromazine, indicating an uptake via clathrin-dependent endocytosis. To prove this result, CLSM studies were also performed. In Fig. S8 (ESI†), medium sized NPs (green) were not detectable in cells incubated with chlorpromazine (A) but detectable in cells incubated with filipin III. As controls, polystyrene (PS) beads of 150 nm were used (blue emission). These PS beads could be excluded by filipin III, indicating an uptake via caveolin-dependent endocytosis, as also confirmed in the literature.18 This suggests that surfactants, which are usually necessary to prepare commercial NPs, have an influence on the uptake in cells. By using inhibitors, it can be assumed that NPs < 200 nm internalize via clathrin-dependent endocytosis, in particular the 100 nm NPs used in this study. In the case of internalization of large NPs, an exclusion could be observed by using EIPA, but not when chlorpromazine and filipin III are used (ESI, Fig. S7†), indicating an uptake of larger NPs via macropinocytosis. As already reported in the literature,49 EIPA caused also a weak inhibition of small sized NPs. These blocking study of differently sized NPs showed that defined small p(MMA-stat-MAA) NPs were predominantly taken up by clathrin-dependent endocytosis, whereas larger ones were predominantly taken up by macropinocytosis.Conclusion
The preparation of a differently labeled p(MMA-stat-MAA) copolymer and its nanoprecipitation into defined NPs with various sizes (<100 nm, 100 to 200 nm, and >300 nm) without the usage of surfactants were demonstrated. These tailor-made NPs are promising for studying the influence of the surface, charge or size of the NPs on their internalization into cells, as no stabilizers were used, which have an influence on cellular uptake. We showed that the size itself of the NPs has a strong influence on the uptake in HeLa cells. This is further influenced by the concentration used and incubation time. The medium sized NPs were taken up faster compared to small and large ones. All NPs were found inside the cells, whereas small and medium sized NPs showed the same cellular distribution and were detectable in lysosomes. In contrast, large NPs showed less co-localization with smaller NPs and were not detectable in the lysosomes. By using inhibitors, we have shown that clathrin-dependent endocytosis is the predominant pathway for smaller NPs < 200 nm, whereas macropinocytosis is responsible for larger NPs. In further studies, the influence of the zeta potential and of the additional functional groups on the particle surface will be studied, again in the absence of surfactants.Acknowledgements
The financial support from the Thuringian Ministry for Education, Science and Culture (grant #B514-09051, NanoConSens), the Deutsche Forschungsgemeinschaft (Japan-Germany exchange program, DFG and JSPS), the Carl-Zeiss Foundation (JCSM Strukturantrag), the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST Program, JSPS), and the Core Research Program for Evolutional Science and Technology (CREST) from the Japan Science and Technology Corporation (JST) is gratefully acknowledged. We express our gratitude to Steffi Stumpf, EMZ Jena, for assistance in the SEM investigations, Melanie Nikolajski, University of Jena, for help with the pH dependent zeta measurements, Tomoya Suma, the University of Tokyo, for help with FC studies, and Dr Xueying Liu, the University of Tokyo, for the CLSM measurements.References
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872 CrossRef CAS.
- N. Sanvicens and M. P. Marco, Trends Biotechnol., 2008, 26, 425 CrossRef CAS.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136 CrossRef CAS.
- K. Miyata, T. Nomoto, H. Takemoto, H. J. Kim, Y. Matsumoto, M. Oba, N. Nishiyama and K. Kataoka, Abstr. Pap. Am. Chem. Soc., 2010, 110, 178 Search PubMed.
- Y. Liu, H. Miyoshi and M. Nakamura, Int. J. Cancer, 2007, 120, 2527 CrossRef CAS.
- X. A. Wu and H. M. Mansour, Int. J. Nanotechnol., 2011, 8, 115 Search PubMed.
- G. Oberdorster, V. Stone and K. Donaldson, Nanotoxicology, 2007, 1, 2 CrossRef CAS.
- J. A. Kim, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 62 CAS.
- V. Mailander and K. Landfester, Biomacromolecules, 2009, 10, 2379 CrossRef.
- L. E. Euliss, J. A. DuPont, S. Gratton and J. M. DeSimone, Chem. Soc. Rev., 2006, 35, 1095 RSC.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmacol., 2008, 5, 505 CrossRef CAS.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610 CrossRef CAS.
- J. Wang, J. D. Byrne, M. E. Napier and J. M. Desimone, Small, 2011, 7, 1919 CrossRef CAS.
- G. M. Whitesides, Nat. Biotechnol., 2003, 21, 1161 CrossRef CAS.
- M. Gaumet, R. Gurny and F. Delie, Eur. J. Pharm. Sci., 2009, 36, 465 Search PubMed.
- J. P. Best, Y. Yan and F. Caruso, Adv. Healthcare Mater., 2012, 1, 35 Search PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182 CrossRef CAS.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159 CrossRef CAS.
- H. J. Gao, W. D. Shi and L. B. Freund, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9469 CrossRef CAS.
- S. Zhang, J. Li, G. Lykotrafitis, G. Bao and S. Suresh, Adv. Mater., 2009, 21, 419 CrossRef CAS.
- S. E. A. Gratton, P. A. Ropp, P. D. Pohlhaus, J. C. Luft, V. J. Madden, M. E. Napier and J. M. DeSimone, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11613 CrossRef CAS.
- N. Doshi and S. Mitragotri, PLoS One, 2010, 5, 1.
- J. Dausend, A. Musyanovych, M. Dass, P. Walther, H. Schrezenmeier, K. Landfester and V. Mailander, Macromol. Biosci., 2008, 8, 1135 CrossRef CAS.
- A. Musyanovych, J. Dausend, M. Dass, P. Walther, V. Mailaender and K. Landfester, Acta Biomater., 2011, 7, 4160 Search PubMed.
- S. Lorenz, C. P. Hauser, B. Autenrieth, C. K. Weiss, K. Landfester and V. Mailander, Macromol. Biosci., 2010, 10, 1034 CrossRef CAS.
- M. Gaumet, R. Gurny and F. Delie, Int. J. Pharm., 2010, 390, 45 Search PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145 CrossRef CAS.
- Arnida, M. M. Janat-Amsbury, A. Ray, C. M. Peterson and H. Ghandehari, Eur. J. Pharm. Biopharm., 2011, 77, 417 CrossRef.
- W.-K. Oh, S. Kim, M. Choi, C. Kim, Y. S. Jeong, B.-R. Cho, J.-S. Hahn and J. Jang, ACS Nano, 2010, 4, 5301 CrossRef CAS.
- P. Decuzzi, B. Godin, T. Tanaka, S. Y. Lee, C. Chiappini, X. Liu and M. Ferrari, J. Controlled Release, 2010, 141, 320 CrossRef CAS.
- Z. Popovic, W. Liu, V. P. Chauhan, J. Lee, C. Wong, A. B. Greytak, N. Insin, D. G. Nocera, D. Fukumura, R. K. Jain and M. G. Bawendi, Angew. Chem., Int. Ed., 2010, 49, 8649 CrossRef CAS.
- W. Zauner, N. A. Farrow and A. M. R. Haines, J. Controlled Release, 2001, 71, 39 CrossRef CAS.
- O. Lunov, T. Syrovets, C. Loos, J. Beil, M. Delecher, K. Tron, G. U. Nienhaus, A. Musyanovych, V. Mailander, K. Landfester and T. Simmet, ACS Nano, 2011, 5, 1657 CrossRef CAS.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657 CrossRef CAS.
- Y.-L. Chiu, Y.-C. Ho, Y.-M. Chen, S.-F. Peng, C.-J. Ke, K.-J. Chen, F.-L. Mi and H.-W. Sung, J. Controlled Release, 2010, 146, 152 CrossRef CAS.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176 CrossRef.
- S. K. Sahoo, J. Panyam, S. Prabha and V. Labhasetwar, J. Controlled Release, 2002, 82, 105 CrossRef CAS.
- M. Feng and P. Li, Acta Pharm. Sinica, 2005, 40, 893 Search PubMed.
- K. M. Ho, W. Y. Li, C. H. Wong and P. Li, Colloid Polym. Sci., 2010, 288, 1503 CrossRef CAS.
- P.-J. Lou, W.-F. Cheng, Y.-C. Chung, C.-Y. Cheng, L.-H. Chiu and T.-H. Young, J. Biomed. Mater. Res., Part A, 2009, 88, 849 Search PubMed.
- A. Vollrath, D. Pretzel, C. Pietsch, I. Perevyazko, S. Schubert, G. M. Pavlov and U. S. Schubert, Macromol. Rapid Commun., 2012 DOI:10.1002/marc.201200329.
- J. Pauli, T. Vag, R. Haag, M. Spieles, M. Wenzel, W. A. Kaiser, U. Resch-Genger and I. Hilger, Eur. J. Med. Chem., 2009, 44, 3496 CrossRef CAS.
- S. Schubert, J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581 RSC.
- I. Y. Perevyazko, A. Vollrath, S. Hornig, G. M. Pavlov and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3924 CrossRef CAS.
- J. Brandrup, E. H. Immergut and E. A. Grulke, in Polymer Handbook, Wiley-Interscience, 4th edn, 1999 Search PubMed.
- A. Palecanda and L. Kobzik, Methods, 2000, 21, 241 Search PubMed.
- C. S. S. R. Kumar, “Nanocomposites, Nanomaterials for Life Sciences”, in Nanocomposites, Wiley-VCH, 1st edn, 2010, p. 466 Search PubMed.
- S. Grosse, Y. Aron, G. Thevenot, M. Monsigny and I. Fajac, J. Controlled Release, 2007, 122, 111 CrossRef CAS.
- M. Fretz, J. Jin, R. Conibere, N. A. Penning, S. Al-Taei, G. Storm, S. Futaki, T. Takeuchi, I. Nakase and A. T. Jones, J. Controlled Release, 2006, 116, 247 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c2sm26928g |
| This journal is © The Royal Society of Chemistry 2013 |