Biomimetic intracellular matrix (ICM) materials, properties and functions. Full integration of actuators and sensors
Toribio F.
Otero
* and
Jose G.
Martinez
Universidad Politécnica de Cartagena. ETSII, Center for Electrochemistry and Intelligent Materials, Paseo Alfonso XIII. Aulario II, 30203, Cartagena, Spain. E-mail: toribio.fotero@upct.es; Fax: +34 968 325915; Tel: +34 968 325519
First published on 22nd October 2012
Abstract
The electrochemistry of conducting polymers, and other organic compounds, originates biomimetic (intracellular matrix, ICM, reactive gels) materials, properties and devices here reviewed. One reaction changes several properties (multifunctionality): one device can integrate different actuators (artificial muscles, batteries, smart windows) and sensors (temperature, concentration, mechanical). Actuating (current and charge) and sensing (potential) magnitudes are present in the two connecting wires, and can be read by the computer, at any working time mimicking brain–organs dialog. The theoretical description of any multi-functional device envisages intelligent gel robots. The kinetic magnitudes of the reaction become a function of the conformational structure: predictive structural, chemical and biochemical kinetics are emerging.
 Toribio F. Otero | Toribio F. Otero received his PhD in Chemistry in 1978 from the Complutense University of Madrid. The thesis was supervised at the Institute Rocasolano from the CSIC. In 1989 he became full Professor of Physical Chemistry and Macromolecules at the University of the Basque Country. In 2002 he became full Professor of Physical Chemistry at the Technical University of Cartagena. He has published over 250 papers, 20 book chapters and 7 patents, and supervised 23 doctoral theses. His work mainly concerns electrogeneration, electrochemical properties, electrochemical applications and theoretical modeling of conducting polymers and carbon based molecular materials. |
 Jose G. Martinez | Jose G. Martinez received a degree in electronic technical engineering in 2007 and a degree in automatics and electronics engineering in 2009 both from the Technical University of Cartagena. Currently he is pursuing his PhD in biomimetic devices from conducting polymers supported by Spanish Government. |
1. Introduction
Life as-we-know-it originated by chemical reactions of dense gels in the internal structure of the cell components. Most of these reactions link conformational movements in reactive biopolymers, change of intramolecular and intermolecular interactions, and ionic and water exchanges with the surroundings. The description of life as simultaneous physical and chemical processes is outside current theoretical models. Life and biological materials constitute a permanent inspiration for scientists and engineers to develop new biomimetic devices. Opening new ways to advance on the modelling of living processes is a permanent goal for scientists.Bio-mimetic materials include the surface and bulk modification of materials with bioactive molecules to produce scaffolds acting as extracellular matrix (ECM) allowing the modulation of cellular functions such as adhesion, proliferation and migration.1 In a similar way to ECM they act as sol–gel materials (silicon based materials) for the bioencapsulation in mineral hosts, of cells or biomolecules, for the development of new biosensors and bioreactors.2 Biomimetic mineralization is a different approach for the synthesis of advanced materials with complex shape, hierarchical organization, controlled size, and shape polymorphism under ambient conditions in aqueous environments.3 Stimuli responsive materials (SRMs) try to mimic biological functions undergoing relatively large, reversible and abrupt physical or structural changes in response to small external changes in the local environmental conditions.4a–c All these materials act from outside the cells. Smart healable materials constitute a very active area mimicking self-repair tissues.4d
Nowadays new reactive materials (most as gels) are emerging that can be considered, during reactions, as biomimetic material models of the intracellular matrix (ICM).5a,b They are available from different carbon based materials as conducting polymers, porphyrines, carbon nanotubes or graphenes, to mention just a few. They present high biocompatibility and, in the absence of their chemical reactions, they can be used as extracellular matrix (ECM) materials for biomedical applications.5c–e In aqueous solutions conducting polymers behave as soft and wet biomimetic (polymer–ions–solvent) reactors under flow of electric currents. These are the most studied (if still superficially) of the above-mentioned reactive biomimetic materials6 and they constitute the focus of this feature article. Concepts, properties, devices and conclusions can be extended to the different reactive materials for future studies. The basic breaking point for this feature article is the shift of the material composition (polymer–ions–solvent) by several orders of magnitude under reaction control, mimicking intracellular matrix (ICM) chemical changes in cells of working organs from mammals. Here we highlight and review the state of the art of this emerging world of biomimetic soft and wet reactive gels: their basic electrochemical reactions and the properties shifting their magnitude with the composition giving biomimetic functions altogether to reactive biomimetic devices and tools.
A second breaking concept linked to these materials, the reactive multifunctionality, requires some attention here. Several properties, functions and tools are present and work simultaneously in one device (full integration of sensors and actuators) driven by the same reaction. Individual devices and tools have attracted the interest of engineers, medical doctors, biologists, physicists or chemists providing different physical,7 mathematical,8 image treatment9 and chemical emerging theoretical models trying to describe experimental results and opening the way to design new devices, applications and tools.
Here we will focus on reviewing the model that combines electrochemistry and polymer science to describe and quantify their unique dual and simultaneous sensing–actuating properties that mimic brain–organs dialog. The study of the chemical reaction kinetics provides unexpected results for the activation energy, the kinetic constant and the reaction order: they change with the conformational structure of the initial state of the reaction including information about the conformational structure of the reactive polymeric chains.10 The state of the art of this emerging structural chemical kinetics and the perspectives open for a better understanding of biological reactions and processes like brain memory will be reviewed.
2. Electrochemical basic reactions in conducting polymers (CPs)
Each of the constitutive polymer chains of a film can be oxidized (if they have low first, second, … nth, ionization potentials) or reduced (for low first, second, … nth, electron affinities) involving the extraction or the injection, respectively, of n consecutive electrons. Positive charges (p-doping), or negative charges (n-doping), are generated along the chains. Counterions and solvent are exchanged with the electrolyte for charge and osmotic balance. The materials become reactive polyelectrolyte gels having a variable polymer–ion composition. The n consecutive redox steps can be summarized, according to the different materials, by three basic reactions and a general reaction:5b,d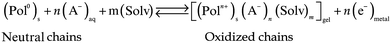 | (1) |
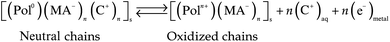 | (2) |
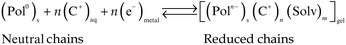 | (3) |
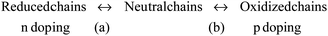 | (4) |
Reactions (1) and (2) represent oxidation (p-doping) processes for CPs or CP–macroion blends, respectively. Reaction (3) represents reduction (n-doping) processes. Reaction (1) requires for charge balance a prevalent exchange of anions between the film and the electrolyte, while reactions (2) and (3) require the exchange of cations.
During these reactions the distribution of the double bonds in the polymer chains changes (Fig. 1a) from σ bonds and free rotation among consecutive monomeric units to conjugated bonds with a flat quinoid structure along several monomeric units: the reversible electrochemical reaction controls reverse conformational movements of the chains.
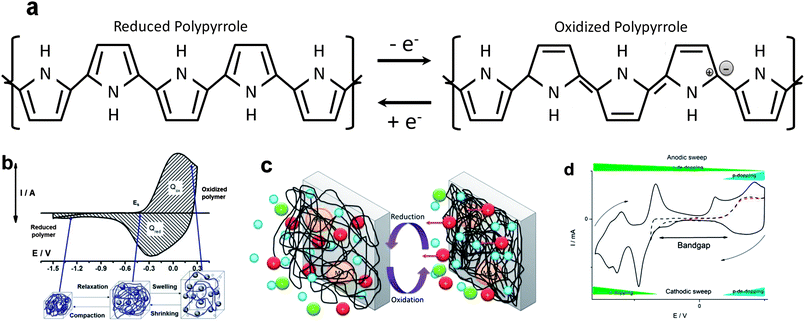 | ||
| Fig. 1 (a) Change of the double bond distribution during electrochemical reactions in polypyrrole; (b) cyclic voltammogram from a polypyrrole film and volume changes of a cubic sample inside a film with prevalent exchange of anions (reaction (1)) during reactions; (c) reversible changes of volume followed by a conducting polymer film with prevailing exchange of cations (reactions (2) or (3)); (d) evolution of p-doping and n-doping processes along the anodic and cathodic potential sweeps for poly(2-ethylheptyl-DTCPD-co-3-methylthiophene). Dotted red voltammogram shows the p-doping/de-doping domain. Dotted black voltammogram indicates the beginning of the n-doping process, allowing the determination of the bandgap. The full line voltammogram shows either p or n doping/de-doping processes (reproduced from ref. 12d, with permission of the American Chemical Society). | ||
The material swells by oxidation during reaction (1) (free volume is generated by conformational movements during the reaction to lodge charge balance ions and solvent) and shrinks by reduction (Fig. 1b).7b,c,g,m,11
During reactions (2) and (3) the material shrinks by oxidation and swells by reduction (Fig. 1c).
2.1. Double doping and bandgap
Some CPs such as PEDOT,12a polythiophenes12b or polyfluorenes12c can be either oxidized or reduced (Fig. 1d) from the neutral state (reaction (4)). The energy difference between the beginnings of the two processes (first ionization potential and first electronic affinity) is the electrochemical bandgap.12d2.2. Chemical composition shift
Reactions (1)–(3) occur through n consecutive steps involving one electron per step: for an ideal monodisperse (all the chains have the same length) polymer film the polymer composition moves through n consecutive stoichiometric polymer–counterion ratios. A large distribution of chain lengths exists in actual films: each of the consecutive ionization potentials takes a different value for the different chain lengths (Hückel principle)13a giving infinite average non-stoichiometric polymer–counterion composition ratios. For intermediate oxidation charges electrochromic thin films give (see below) infinite intermediate absorbance values (concentration of charged chromophores and balancing counterions in the film).13b The final conclusion is that the counterion content of the polymer (w/w percentage) can be changed under reverse electrochemical control for several orders of magnitude, from 0.01 to 40%, the higher value depending on the ion weight.3. Biomimetic consecutive events, properties and functions
Electrochemical reactions ((1), (2) or (3)) in an aqueous solution involve (Fig. 1): flow of electrons across the polymer–metal interface, flow of ions and water molecules across the polymer–solution interface, re-organization (breaking and formation) by reaction of double bonds along the chains giving conformational movements and shrinking or swelling macroscopic processes. Strong changes in the molecular interactions (polymer–polymer, polymer–solvent, polymer–ions, ions–solvent) take place inside the dense reactive material along the reaction time. Described steps in a reactive gel mimic intracellular matrix (ICM) materials and consecutive events taking place in cells of working biological organs.These properties of the CPs, the characteristic magnitudes of which are a function of the polymer–ion composition ratio, are reactive (electrochemical) properties that mimic biological functions performed by specialized cells in different organs. Table 1 summarizes the reactive properties, the driving actions and the inspired biological organs.6d,14
| Property | Action | Inspired organ |
|---|---|---|
| Electrochemomechanical | Change of volume | Muscles |
| Electrochromic | Change of colour | Mimetic skins |
| Charge storage | Current generation | Electric organs |
| Chemical or pharmacological storage | Chemical modulation or chemical dosage | Glands |
| Electron/neurotransmitter | Channel V action | Nervous interface |
| Electroporosity (permselectivity) | Transversal ionic flow | Membrane |
| Electron/ion transduction | ΔV (chemical/physical properties) | Bio-sensors |
The progressive and reversible variation of the value of every property under faradic control of the gel composition, driven by reactions (1), (2) or (3), envisages the development of devices and products mimicking biological functions. The simultaneous variation of different materials' properties driven by the same reaction envisages the full integration of different functions in one device: multifunctionality.
4. Artificial muscles and actuators
Natural muscles are biological organs developed by evolution to transform chemical energy into mechanical energy and heat. Their actuation involves: (a) aqueous media, (b) an electric pulse arriving from the brain (the pulse generator) to the muscle through the nervous system, (c) liberation of calcium ions inside the sarcomere, (d) chemical reactions (ATP hydrolysis), (e) conformational changes along natural polymeric chains (actin and myosin) with change of the sarcomere volume and (f) water exchange. This natural reactive motor works at constant temperature (outside the Carnot's servitude) and involves simultaneous sensing processes providing the living beings with a perfect consciousness of position (related to any other body organ), required energy, capability or not to produce this energy, obstacles and difficulties present during displacement, among others. Muscles, nerves and brain constitute the permanent inspiration for scientific and technological development of artificial intelligent motor systems.4.1. Chemistry versus physics
Electrochemical reactions (1)–(3) induce reversible changes in the polymer film volume. These changes of volume induced by the ionic exchange have been translated into linear or bending (Fig. 2A–C) macroscopic movements allowing the construction of macroscopic or microscopic tools and devices as reported by different reviews.4 The consecutive events mimic those above described for natural muscles: artificial muscles based on reactions driven by current flow through CPs (also from other carbon based reactive molecules and materials) in electrolytes (liquid or solid) constitute the forefront of the biomimetic actuators. In parallel, from the last decade of the past century a plethora of actuators, or artificial muscles, based on physical interactions (electrostrictive, piezoelectric, ferroelectric, coulombic, electrophoretic or electroosmotic, among others) between electric fields and polymers, some of them including solvents and ions, or organic molecules are being developed.4c,5b,15 They mimic some of the consecutive events taking place in natural muscles except chemical reactions. Whether the markets will accept better devices not including or those including chemical reactions is a deeply controversial subject in scientific meetings, founding agencies and foundations.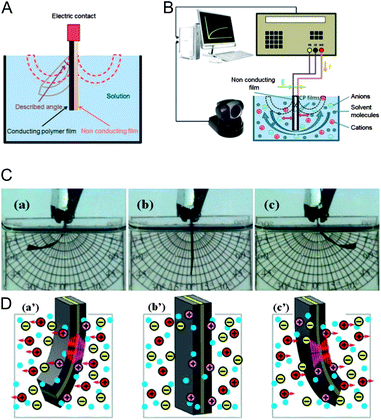 | ||
| Fig. 2 (A) Bi-layer structure of an artificial muscle. (B) Experimental setup for three-layer artificial muscles: an elastic polymer tape is adhered to two films (one by side) of a conducting polymer acting as reactive electrodes (oxidation and reduction). (C) Pictures showing the bending movement during flow of a direct current: the sense of the movement is reversed by changing the sense of the current flow. (D) Starting from the vertical position (b′) movements of the ions when the current flow reduces the right film and oxidizes the left film (a′) or vice versa (c′). Reproduced from ref. 11c, with permission of the Royal Society of Chemistry. | ||
When comparing the elasticity, strain and stress production, controllability, and molecular interactions, natural muscles can be surpassed by polymeric actuators. In relation to energy conversion efficiency, hierarchical structure (from molecular to macroscopic three-dimensional) and durability, natural muscles are the champions.5a,c,15b,16
5. Electrochemical and polymeric model
Artificial muscles are electrochemical motors. Faraday's laws when applied to reactions (1)–(3) determine the number of ions exchanged between the polymer film and the solution, the film volume increment, the described movement (angle, α) and the angular rate (ω) of bending muscles, as corroborated by experimental results (Fig. 3a–c for artificial muscles exchanging anions and Fig. 3a′–c′ for those exchanging cations).17a,b In this way, the change of the same specific ionic composition in the CP (mol g−1) per unit time (s−1) will produce the same angular rate whatever be the muscle geometry or the involved mass of CP (Fig. 3d and d′).| α (rad) = kQ | (5) |
| ω (rad s−1 g−1) = k′i | (6) |
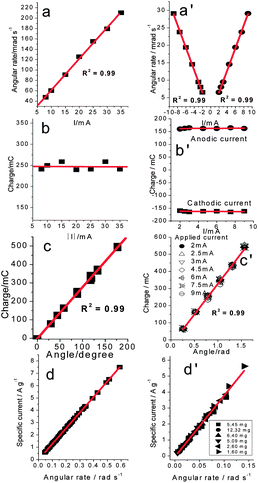 | ||
| Fig. 3 Artificial muscles are electrochemical devices with linear relationships between electrically driven magnitudes and angular movements: (a–d) for muscles exchanging anions (pPy/ClO4−) (reproduced from ref. 17a, with permission from the Royal Society of Chemistry); (a′–d′) for muscles exchanging cations (pPy DBS/tape) (reproduced from ref. 17b with permission from Elsevier). The same driving charge per unit of reduced polymer mass produces the same angular movement whatever is the mass of the conducting polymer in the device (d and d′). | ||
It could be concluded that artificial muscles are electrical motors working under faradic control of the driving current5b,d also acting as positioning devices (electro-chemo-positioning):17ceqn (5) states the required charge to attain a new angle (position). The faradic control must persist whatever be the geometry, or the actuating CP mass (film thickness) of the device.
Many different applications, both macroscopic and microscopic, have been developed: propulsion or locomotion systems for swimming devices7d,18a,b or cilia actuators for mini-robots;18c Braille displays;18d steerable microcatheters;18e,f integrated cell sensors;18g,h micro-pumps,18i,j micro-actuators18k–m and microtools constituted by bilayers18n–s or trilayers.18t
6. Electrochromic windows, mirrors or screens19a–c
Reactions (1)–(3) generate/destroy new molecular orbitals inside the LOMO–LUMO gap. New electronic transitions in the visible and IR19d range allowed absorption of photons and promotion of colour change (Fig. 4a) of thin polymer films. The concentration variation (c, mol L−1) of the new chromophores inside the volume (V) of the film is under faradic control of the charge consumed (Q) for the reactions (c = Q/FV). Fig. 4b shows this variation stimulated by a potential step (red line) and the evolution of the consumed current (blue dotted line) with time (there, Q = i∫tdt). The light absorption is defined by this concentration through the Lambert–Beer equation. The absorption variation can be tuned through infinitesimal steps by flow of infinitesimal anodic or cathodic charges. Fig. 4c shows the absorption variation during a voltammetric oxidation. A reverse evolution occurs during the voltammetric reduction.19e Different chromophores (polarons, polaron pairs, bipolarons, etc.) can be simultaneously present in the film for high oxidation states. The progressive transformation between them with deep oxidation gives absorption bands whose intensity passes through a maximum (Fig. 4c) showing wavelength shift (Fig. 4d) that is attributed to the induced conformational energetic changes. Materials and devices mind the behaviour of mimetic skins. Smart windows, mirrors and colour filters are being developed both in visible and IR regions.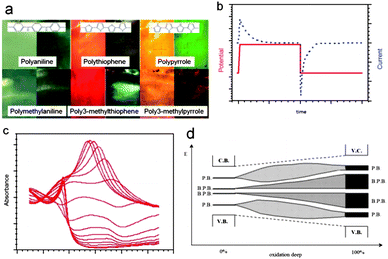 | ||
| Fig. 4 (a) Colour change between oxidized (left side) and reduced (right side) films of different conducting polymers. (b) The colour change is studied by consecutive potential steps (red line); the films respond with a current flow (black line) required to induce the reactions; (c) evolution of the polypyrrole absorbance during oxidation, reproduced from ref. 19e, with permission from the ACS; and (d) scheme of the polaronic and bipolaronic band evolution with deep oxidation. | ||
7. Polymeric batteries20a and/or supercapacitors20b,c
Independent of the chromophore's nature positive (reactions (1) and (2)) or negative (reaction (3)) charges are stored in polymers. In the absence of any parallel reaction the charge spent during the reaction is stored by the polymeric chains. Any subsequent discharge, or charge, promotes a variation of the number of charges stored in each polymer chain: chemical potential is kept constant across the material. A change of paradigm is produced: in classical inorganic batteries the reactive front during charge/discharge, including nanoparticles, is bi-dimensional (Fig. 5a). Now the electroactive materials can behave as a three-dimensional electrode at the polymer chain level (Fig. 1b–c and 5b–c). By using a polymeric membrane (only ionic conductor) and two films of conducting polymers (electronic and ionic), one storing positive charges and the second storing negative charges, a full polymeric battery having a large potential cell (>1.5 V) can be constructed. The three-dimensional nature of the two polymeric electrodes gives very fast charge–discharge processes. The devices, despite their faradic nature, used to be named supercapacitors in the literature. This confusing basic concept could induce design engineering mistakes and the non-optimum design of devices as batteries for ultrafast charge processes.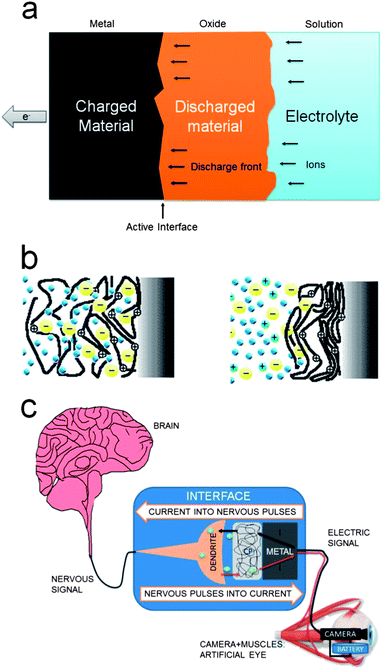 | ||
| Fig. 5 (a) Scheme of the advancing two-dimensional front during discharge of a battery for macroscopic materials or inside microscopic grains; (b) reactive organic materials behave as three-dimensional electrodes at the molecular level during charge/discharge; (c) schematic representation of the transduction, through an artificial synaptic space, between a video camera and the nervous system, reproduced from ref. 5d, with permission from Elsevier. | ||
The availability of CPs exchanging anions or cations during oxidation processes (reactions (1) or (2), respectively) and the fact that the oxidation, or the reduction, reaction occurs along several hundreds of millivolts provide active electrodes for multiple designs of polymeric or mixed (organic/inorganic) batteries. Due to the non-stoichiometric nature of the material the potential of the battery decreases during discharge: hundreds of millivolts when p-doped and n-doped materials are used, continuously when the same materials in oxidized and neutral initial states are used as battery electrodes.
Materials and devices mimic composition and behaviour of electric organs from animals (fishes, electric heels).
8. Smart drug delivery or chemical capture21a,b
Reactions (1)–(3) represent a reversible faradic storage/delivery (or stripping) of the concomitant counter-ions or co-ions (Fig. 5b–c). Those reactions are being used to store and concentrate ions, from very dilute solutions, in films of conducting polymers by film oxidation under constant potential. These ions are recovered by stripping (reverse reaction) in a small solvent volume for analytical purposes.When the counterion or the co-ion has a chemical, medical, pharmacological or biological interest a required number of moles (n) can be liberated in a short period of time, under faradic control of the charge (n = Q/F). The concentration variation per unit of time (c′, mol L−1 s−1) in a volume V around the material is under control of the applied current (I, A).5c,21c–f
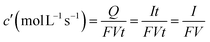 | (7) |
Large ions (of pharmacological interest, fertilizers or neurotransmitters) remain trapped inside the polymer. Low swelling volume (free volume in the polymer) or strong van der Waals polymer–ion forces can be the origin of the trapping effect. Until now we do not know how to manage either, cross-linking degrees during electropolymerization to produce large swelling ratios or intermolecular polymer–ion forces to avoid the trapping effects during redox reactions. Anyway, thousands of new materials based on conducting polymers are still waiting for their synthesis or are being explored for those aims.
These materials and devices mimic natural glands from living beings.
9. Computer/neuron dialog: artificial synapses22a,b
Nervous pulses include electrical (potential and charges, positive or negative) and chemical (ions and neurotransmitter molecules) information. The flow of the same charge carries many orders of magnitude more information in nerves than in electronic devices (only one carrier species bearing only one negative charge without any differentiated chemical information between two carriers). Reactions (1)–(3) can be used to store one of the ions participating in nervous pulses (Cl−, Na+, K+, Ca2+, glutamate, aspartate, D-serine, γ-aminobutyric acid—GABA). Its faradic liberation under control of the opposite reaction translates quantitatively (Faraday's laws) electronic signals (not understood by neurons) from the electrical generator into ionic signals in a narrow aqueous volume around the material of the ions required for synapses. The fast increase of the ion's concentration very close to a neuron will activate the ion channels or the entrance of neurotransmitters through the neuron membrane: the electronic pulse from the connecting metal becomes translated to a nervous pulse through neurons (Fig. 5d). The dialog between electronic devices, neurons and brain can be envisaged.5d,22c–f In the literature artificial synapses are mainly referred to as the (only) simulations of the potential component of natural nervous pulses through circuits of dry semiconductors using mathematical models. Here we refer to the physical and chemical mimicking of the real wet synaptic space between neurons by substituting one of the neurons. The driving reaction occurs (Fig. 1b) at low and biocompatible overpotentials (a few mV) inside the potential window without water discharge, hence avoiding H+ or OH− and radical's generation, not compatible with a neuron's life, by current flow. In addition, most of the oxidized conducting polymers present a great biocompatibility with neurons22g–l and cells in general.The opposite process attempting the translation of nervous pulses arriving from the brain into electronic signals that can be amplified and manipulated to move artificial arms or legs constitutes the broad field of bio-sensors, immaterial whether reactive or not, using conducting polymers.5c,21a,f,22m–r
10. Smart membranes23a,b
Reactions (1)–(3) control (Fig. 5b–c) the available free volume between polymer chains through the film. Used as a membrane (Fig. 6a) between two aqueous media (Fig. 6b) the oxidation state, under faradic control of the oxidation charge, of the film allows tuning the average distance between chains in the polymeric membrane to the diameters of the different ions23b–d present in solution for migration, diffusion or permeation across the membrane. Only those ions having smaller diameters than the controlled average distance between chains can migrate. They also can act as switches between the oxidized state and the reduced state allowing, or not, the transversal ionic flow.23c–f | ||
| Fig. 6 (a) Schematic representation of the transversal sequence of components in the final free-standing pPy membrane (14 mm of diameter). (b) Schematic picture of the diffusion cell showing the membrane (m) in between half-cells I and II. The membrane oxidation state is controlled by a potentiostat (P1). The potential gradient across the membrane is controlled by a bipotentiostat (P2) which imposes the required current. (c) Evolution of the current density when the potential gradient across the membrane (ΔΦm) is varied from −0.5 to +0.5 V. Pre-polarization potentials (Ep), indicated in the figure, were applied for 600 s to attain different oxidation degrees of the pPy film. Electrolyte: a 0.1 M KCl aqueous solution at both sides of the membrane. (d) Ionic conductivity through the pPy membrane (σionic) as a function of the pre-polarization potential (Ep) for different studied electrolytes and concentrations at 296 K (reproduced from ref. 23a with permission from Elsevier). | ||
For the transversal migration of ions the slope Δi/ΔΦm, where Δi is the transversal ionic flow under a transversal applied potential of ΔΦm, increases with the oxidation state of the film and decreases with its reduction state (Fig. 6c).23a,g–i The oxidation state of the membrane tunes the free volume between chains in the film and the transversal conductivity of different ions (Fig. 6d). Ionic transport numbers close to one for anions through membranes following reaction (1) during oxidation and different ionic diffusivities for ions having similar crystallographic radius indicate that CP membranes are ion selective membranes.
These materials mimic opening and closing of ionic channels across biological membranes under control of the transversal electrochemical (ionic) potential.
11. Smart surfaces24a–c
They are being investigated by tuning properties like wettability. The reduced material is mainly constituted by reduced polymer chains; meanwhile in the oxidised state those chains bear positive charges: the reaction induces great changes in the polymer–solvent interactions at the polymer–solution interface.12. Sensors and sensing devices
Conducting polymers can be considered as: (a) biosensors by including biological compounds that connect with its electron–ion equilibrium22m,p,25a,b and (b) new reactive sensors of any surrounding variable acting on the reaction ((1)–(3)) rates during the device action.5a,b,25c–e12.1. Reactive devices sense working variables: mimicking brain–organs dialog
Being driven by reactions ((1)–(3)) artificial muscles, and any of the reactive electrochemical devices based on conducting polymers, will sense the change of any physical or chemical variable acting on the reaction rate while working. By studying the muscle displacement always across the same angle (α) under flow of a constant current the evolution of the muscle potential during the movement changes when any of the variables acting on the reaction rates (electrolyte concentration, temperature, applied current or mass trailed by the muscle) changes, Fig. 7a, full black lines for [A−].25c,f–h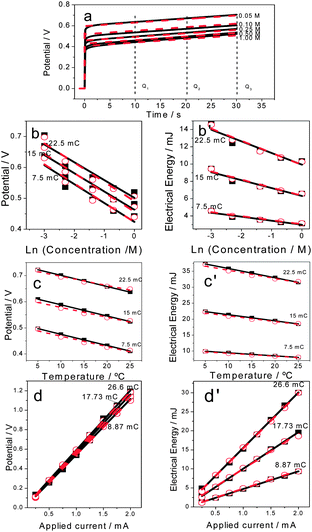 | ||
| Fig. 7 (a) Anodic experimental (full lines) and theoretical (dotted lines) chronopotentiograms obtained by a flow of ±0.75 mA through polypyrrole/tape bilayer muscles with 1.6 mg of active pPy describing every time movement of π/2 radians in different concentrations of LiClO4 aqueous solutions. (b) Experimental and theoretical (eqn (8) and (9)) potentials in different electrolyte concentrations after flow of three (always the same) intermediate charges, which means when the muscles go through the same intermediate angles: (c) at different temperatures and (d) under different driving currents; (b′) evolution of the experimental and theoretical consumed electrical energies after the same times of current flow as a function of the electrolyte concentration: (c′) at different temperatures and (d′) under different applied currents (reproduced from ref. 25i and j with permission from the ACS). | ||
The oxidation of a polymer chain occurs through n consecutive steps, each involving the extraction of one electron at increasing oxidation potentials. The potential evolution E(t) of a bilayer muscle during flow of a constant anodic current, ia, is described by a stair function of n consecutive steps.25i,j
| E(t) = ∑En(t)pn(t) = E1(t)p1(t) + E2(t)p2(t) + … + En(t)pn(t) | (8) |
Being En(t):
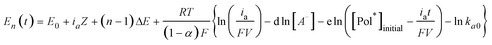 | (9) |
Eqn (9) is the basic multifunctional actuating–sensing equation. The evolution of the muscle potential includes the description of the muscle movement: the angular rate (eqn (6)) controlled by the current (ia) and the position (eqn (5)) controlled by the charge (Q = iat). It also includes the working variables: temperature, electrolyte concentration, current, and film volume. This equation was checked by using experimental results from either self-supported films of CPs or bilayer muscles and by changing one of the variables, keeping a constant value for each of the rest.5b,d A good fit is attained between theoretical and experimental results, E(t), for different temperatures, electrolyte concentrations (Fig. 7a, red dotted lines) or imposed currents.25i,j After the same duration of current flow the muscle potential results in a semilogarithmic function of the electrolyte concentration (Fig. 7b), a linear function of the working temperature (Fig. 7c), or a linear function of the applied current (Fig. 7d), as described by eqn (9), overlapping with experimental results. The muscle senses those variables while working.
Being electrical machines, by integration of eqn (9), the evolution of the electrical energy (Ua) consumed by the artificial muscle during the time (t) of anodic current (ia) flow is attained:
| Ua = ia∫E(t)dt | (10) |
The consumed energy results in a sensing function of the same surrounding and working variables also including actuating characteristics: current (ia) and charge (iat). Theoretical description fits experimental results (Fig. 7b′–d′).25i,j The slope (sensitivity) increases for deeper oxidation states of the device: this is a characteristic of the reactive sensing materials because the reaction becomes more difficult when the concentration of one of the reactants (the active centres of the chains) becomes lower.
Eqn (8)–(10) describe the dual actuating–sensing behaviour of any electrochemical device. The same device is a muscle and several sensors working simultaneously. All the information about the actuator position, movement rate, or consumed energy (current and charge) and the sensors (potential evolution) is present, at any time, in the two connecting wires. This information is instantaneously read by the computer at any working moment: the system mimics brain–muscle permanent communication (Fig. 8).
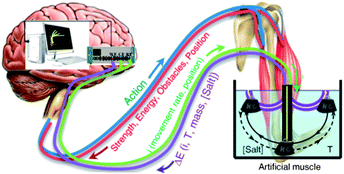 | ||
| Fig. 8 Brain–muscle feedback communication and scheme of the mimetic computer-generator/sensing artificial muscle system. Reproduced from ref. 25i with permission from the ACS. | ||
These equations explain the empirical results for different artificial muscles in different electrolytes sensing electrolyte concentrations, working temperatures or driving currents.25i,j In the near future the theoretical description from eqn (9) of artificial muscles sensing simultaneously: the mechanical ambient, the trailed weights,7e,26a–d or the presence of obstacles in its way (tactile muscles)26e,f is expected.
Eqn (9) describes the full actuating and sensing characteristics of the device. Up to now only dual devices (muscle sensing one variable) were checked. Taking into account the different nature of the coefficients for each variable in eqn (9), the simultaneous variation of two or more variables is expected that can be deconvoluted in the near future from the experimental results to get each variation. Moreover any three layer muscle is, simultaneously, a battery that will allow recovering a fraction of the consumed energy.
13. Other multidevices
By using three layer interpenetrated polymer network artificial muscles, when the two external films of CP are thin enough the films change colour (visible and IR domains).27a,b The muscle is, at the same time, an electrochromic device that can tune IR radiation reflection or transmission.Irrespective of the above-mentioned electrochemical devices, an exponential increase of the number of papers and patents per device and per year exists from 1990. Different products are arriving in the market and some of the start-up companies initiated 15 years ago are being incorporated by different multinationals indicating a progressive consolidation of the market.5d
14. Structural chemical kinetics
Reactions (1)–(3) induce structural changes: conformational movements in chains and relaxation, swelling or shrinking, closing and compaction processes in the reactive dense gel.10 The reaction kinetics until the same final state changes dramatically when initiated from different states of packed conformations (Fig. 9a, for the current evolution, Fig. 9b, for the concentration evolution; both are correlated by eqn (7)).28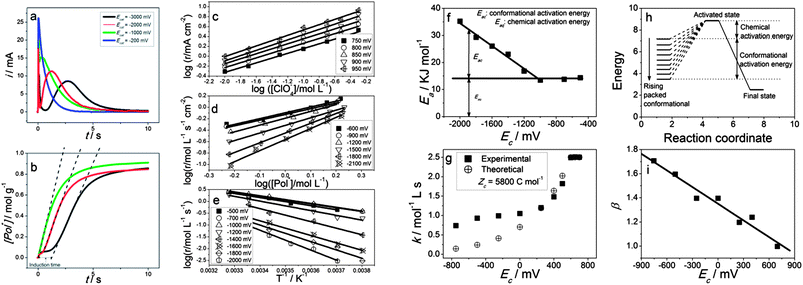 | ||
| Fig. 9 (a) Chronoamperometric responses of a PEDOT-coated platinum electrode in a 0.1 mol L−1 LiClO4 acetonitrile solution, submitted to potential steps from different cathodic potentials kept for 30 s, to 500 mV, to get different reduced states of packed conformations. (b) Evolution of the concentration of active centres in the film obtained from (a) through eqn (7). Dotted lines represent the initial reaction rate. Reproduced from ref. 28, with permission from Wiley; (c) evolution of the initial oxidation rate as a function of the electrolyte concentration (every line) for different final oxidation states (parallel lines); (d) initial oxidation rates for different concentrations of active centres in the film (line) and influence of the initial packed conformational state (different lines) obtained by reduction at different cathodic potentials for the same time; (e) initial oxidation rate evolution for different temperatures (line) and influence (different lines) of the initial packed conformational state. Evolution of: (f) the activation energy, (g) the reaction coefficient, or (i) the reaction order related to the concentration of active centres as a function of the packed conformations attained by reduction at rising cathodic potentials; (h) schema of the chemical and conformational activation energies following (f). | ||
Conformational and structural magnitudes induced by reactions are not included by today's chemical kinetic models. The study of the reaction ((1)–(3)) kinetics has been performed for different conducting polymers and electrolytes by submitting the polymer films to potential steps and changing one of the variables (concentrations of the electrolyte in the solution, concentration of active centres in the film or temperatures) every time.28 The initial oxidation rates follow the linear or semilogarithmic variations expected for a chemical reaction (Fig. 9c–e): the rate coefficient, the activation energy and reaction orders were obtained. The methodology was repeated for every system of polymer/electrolyte by using a different initial state of packed conformations every time (Fig. 9c–e, different lines). A new set of values was obtained for the rate coefficient, the activation energy and the reaction orders from each initial state of packed conformations (Fig. 9 f, g and i). Decreasing rate coefficients or increasing activation energies and reaction orders related to the concentration of active centres in the film were attained when the reactions started from rising packed conformations. This means that the kinetic magnitudes include and quantify conformational and structural energetic states. So, the reaction activation energy includes the constant reaction activation energy and the conformational energy of the initial packed state (Fig. 9h). The electrochemically stimulated conformational relaxation model describes the empirical variation of the kinetic magnitudes.10 The kinetics of those biochemical reactions involving conformational movements of the biopolymers could be now reconsidered and quantified.
14.1. Packed conformations, kinetics and memory
It is well known that the genetic code stores and transfers most of the biological information to the subsequent generations. The chemical sequence of the aminoacids, the double helix and its bending structure determine the correctness or not of the transfer memory. Summarizing, information and genetic memory are linked in biology to the chemical composition and structure of macromolecules.Now, in films of conducting polymers, different open (swollen), or rising packed, conformational states are attained by electrochemical reduction (reaction (1)) for the same time at increasing cathodic potentials, starting every time from the same oxidized state. The stored conformational energy becomes a linear function (Fig. 9f) of the packed conformational states: every chain can store thousands of different reproducible energetic states (packed conformations) constituting an energetic memory. This memory, linked to rising reduced chemical states, is read and erased by oxidation: the structure relaxes and ions and solvent penetrate from the solution. The consecutive events mimic those taking place in trans-membrane ion channels from neurons and the response (Fig. 1a) changes for different packed conformations. Low overoxidations promote different cross-linking degrees and permanent changes (permanent memory) on the chronoamperometric responses.29 The hypothesis that our memory, or a fraction of it, can be linked to conformational energetic states of channel proteins in neurons could be envisaged for a future exploration.
15. Scientific and technological challenges
There does not exist a general accepted kinetic and mechanistic model for the polymerization or electropolymerization processes: crosslinking degree, average length of the linear ideal chain or concentration of degradation points inside a film cannot be controlled. This fact and the lack of an uncontroversial chemical model for the description of the reactive properties retard the design of efficient devices. The model must include structural electrochemical kinetics, polymer science, mechanics, and thermodynamics of membranes, batteries and polymers.The structural chemical kinetic model must state the bases for the development of predictive structural chemical models for biology or medicine. New structural ways to improve health or to advance the knowledge of the brain molecular mechanisms like memory can be explored.
The control of the electropolymerization mechanism will allow the production of tailored materials able to store and release, under quantitative faradic control, large ions: neurotransmitters, fertilizers, pharmacological, and so on. With the improvements of both models in hand the production of most efficient devices, as those mentioned above, can be envisaged. A deeper knowledge of degradation kinetics and mechanism will allow longer life times for the devices.
From the technological point of view new engineering efforts are required to produce three-dimensional tools, from molecular to macroscopic scale, giving any geometrical shape. The integration of the multi sensing–actuating devices will require a different formation for scientists and engineers, allowing the development of different generations of intelligent tools and gel robots.
A great effort is required to overcome, in parallel, a strong social resistance and engineering difficulties to use reactive electrochemical devices. Its origin seems related to the liquid leakage from old batteries. Apart from elaborated foods only a few reactive products can be found in the market: batteries, fuel cells, smart windows or mirrors and some sensors. In order to produce devices closer to current dry technologies, engineers push the new reactive polymeric products (actuators, batteries, supercapacitors, smart mirrors,…) to work in the solid state, using solid electrolytes losing plasticity and actuation rates.
16. Conclusions
Carbon based molecular materials, from conducting polymers to graphenes, produce soft, wet, ionic and electrochemically reactive dense gels mimicking intracellular matrix (ICM) in living cells. The material composition (reactive polymer, ions and water) mimics, in its simplest expression, nuclear and protoplasmic composition in living cells.The concentration of the balancing counterions changes, under reversible electrochemical control, by several orders of magnitude. The magnitude of the concentration dependent properties of the material such as conductivity, volume, colour, stored charge, porosity, stored chemicals, delivered chemicals, wettability, and so on, can be tuned in a large range under electrochemical control. These properties mimic biological functions; hence, they are biomimetic properties.
Based on these reactive properties new biomimetic (reactive) devices are being developed: artificial muscles, smart windows and mirrors, all polymeric batteries, smart membranes, smart drug delivery systems, nervous interfaces, sensors and biological sensors. All these devices are electrochemical devices working under control of Faraday's laws.
Their chemical nature provides those devices with a unique property unparallel in present day technologies: the working device senses immediately the change of any actuating or surrounding physical or chemical variable acting on the driving reaction. A new technological forefront and breaking technological border is being open: any of those devices include several actuators and sensors working simultaneously (several devices in one). Driving and sensing signals are present, and are read by the computer at any time, in the same two connecting wires. The old engineering dream of the full integration of actuators and sensors in one device has become a reality. The system mimics brain–organs permanent dialog in animals.
The multi-device is described by actuating–sensing equations founded by the integration of electrochemical and polymeric principles. Activation energy, reaction coefficient and reaction order for the driving reaction are no longer constants, as described in chemical and biological books; they change with, and quantify, the packed conformations of the initial state of the reaction. The chemical kinetics becomes structural chemical kinetics opening the possibility to develop predictive biochemical models.
Acknowledgements
Authors acknowledge financial support from Spanish Government (MCINN) Projects MAT2008-06702 and MAT2011-24973, Seneca Foundation Project 08684/PI/08 and to the European network for Artificial Muscles ESNAM. Jose G. Martinez acknowledges Spanish Education Ministry for a FPU grant (AP2010-3460).Notes and references
- (a) F. Brandl, F. Sommer and A. Goepferich, Biomaterials, 2007, 28, 134–146 CrossRef CAS; (b) P. X. Ma, Adv. Drug Delivery Rev., 2008, 60, 184–198 CrossRef CAS; (c) H. Shin, S. Jo and A. G. Mikos, Biomaterials, 2003, 24, 4353–4364 CrossRef CAS; (d) M. Uchida, M. T. Klem, M. Allen, P. Suci, M. Flenniken, E. Gillitzer, Z. Varpness, L. O. Liepold, M. Young and T. Douglas, Adv. Mater., 2007, 19, 1025–1042 CrossRef CAS; (e) L. Zhang and T. J. Webster, Nano Today, 2009, 4, 66–80 CrossRef CAS.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013–1030 RSC.
- (a) L. A. Estroff and A. D. Hamilton, Chem. Mater., 2001, 13, 3227–3235 CrossRef CAS; (b) P. Fratzl, H. S. Gupta, E. P. Paschalis and P. Roschger, J. Mater. Chem., 2004, 14, 2115–2123 RSC; (c) A. W. Xu, Y. Ma and H. Coelfen, J. Mater. Chem., 2007, 17, 415–449 RSC.
- (a) E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS; (b) X. Hou, W. Guo and L. Jiang, Chem. Soc. Rev., 2011, 40, 2385–2401 RSC; (c) S. Singamaneni, M. C. LeMieux, H. P. Lang, C. Gerber, Y. Lam, S. Zauscher, P. G. Datskos, N. V. Lavrik, H. Jiang, R. R. Naik, T. J. Bunning and V. V. Tsukruk, Adv. Mater., 2008, 20, 653–680 CrossRef CAS; (d) E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS.
- (a) T. F. Otero, J. Mater. Chem., 2009, 19, 681–689 RSC; (b) L. Valero Conzuelo, J. Arias-Pardilla, J. V. Cauich-Rodriguez, M. Afra Smit and T. Fernandez Otero, Sensors, 2010, 10, 2638–2674 CrossRef; (c) N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS; (d) T. F. Otero, J. G. Martinez and J. Arias-Pardilla, Electrochim. Acta DOI:10.1016/j.electacta.2012.03.097; (e) E. Smela, Adv. Mater., 2003, 15, 481–494 CrossRef CAS.
- (a) Handbook of Conducting Polymers, ed. T. A. Skotheim and J. R. Reynolds, CRC Press, New York, 2006 Search PubMed; (b) From Non-Covalent Assemblies to Molecular Machines, ed.J. P. Sauvage and P. Gaspard, Wiley-VCH, Weinheim, 2011 Search PubMed; (c) J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS; (d) T. F. Otero, in Structural Biological Materials. Design and Structure Properties Relationships, ed. M. Elices and R. W. Cahn, Pergamon, Amsterdam, 2000, p. 187 Search PubMed; (e) G. G. Wallace, G. M. Spinks, L. A. P. Kane-Maguire and P. R. Teasdale, Conductive Electroactive Polymers: Intelligent Polymer Systems, CRC Press, London, 2008 Search PubMed.
- (a) W. J. Albery and A. R. Mount, J. Chem. Soc., Faraday Trans., 1993, 89, 327–331 RSC; (b) G. Alici, B. Mui and C. Cook, Sens. Actuators, A, 2006, 126, 396–404 CrossRef; (c) G. Alici and N. N. Huynh, Sens. Actuators, A, 2006, 132, 616–625 CrossRef; (d) G. Alici, G. Spinks, N. N. Huynh, L. Sarmadi and R. Minato, Bioinspiration Biomimetics, 2007, 2, S18–S30 CrossRef CAS; (e) G. Alici, Sens. Actuators, B, 2009, 141, 284–292 CrossRef; (f) J. Bisquert, G. G. Belmonte, F. F. Santiago, N. S. Ferriols, M. Yamashita and E. C. Pereira, Electrochem. Commun., 2000, 2, 601–605 CrossRef CAS; (g) M. Christophersen, B. Shapiro and E. Smela, Sens. Actuators, B, 2006, 115, 596–609 CrossRef; (h) P. Du, X. Lin and X. Zhang, Sens. Actuators, A, 2010, 163, 240–246 CrossRef; (i) Y. Fang, X. Tan, Y. Shen, N. Xi and G. Alici, Mater. Sci. Eng., C, 2008, 28, 421–428 CrossRef CAS; (j) Y. Fang, X. Tan and G. Alici, IEEE Trans. Contr. Syst. Tech., 2008, 16, 600–612 CrossRef; (k) J. M. Gere and B. J. Goodno, Mechanics of Materials, Cengage Learning, Toronto, 2009 Search PubMed; (l) G. Paasch, Electrochem. Commun., 2000, 2, 371–375 CrossRef CAS; (m) Q. B. Pei and O. Inganas, J. Phys. Chem., 1992, 96, 10507–10514 CrossRef CAS; (n) Q. B. Pei and O. Inganas, J. Phys. Chem., 1993, 97, 6034–6041 CrossRef CAS; (o) X. M. Ren and P. G. Pickup, J. Electroanal. Chem., 1997, 420, 251–257 CrossRef CAS; (p) T. Shoa, J. D. Madden, N. R. Munce and V. Yang, Polym. Int., 2010, 59, 343–351 CrossRef CAS; (q) T. Shoa, D. S. Yoo, K. Walus and J. D. Madden, IEEE ASME Trans. Mechatron., 2011, 16, 42–49 CrossRef; (r) S. Timoshenko, J. Opt. Soc. Am. Rev. Sci. Instrum., 1925, 11, 233–255 CAS.
- (a) G. Alici, P. Metz and G. M. Spinks, Smart Mater. Struct., 2006, 15, 243–252 CrossRef CAS; (b) P. Metz, G. Alici and G. M. Spinks, Sens. Actuators, A, 2006, 130, 1–11 CrossRef; (c) B. Shapiro and E. Smela, J. Intell. Mater. Syst. Struct., 2007, 18, 181–186 CrossRef CAS.
- (a) R. Verdu, R. Berenguer, J. Morales, G. Vazquez, T. F. Otero and L. Weruaga, IEEE International Conference on Image Processing, 2003, pp. 397–400 Search PubMed; (b) R. Verdu, J. Morales, A. J. Fernandez-Romero, M. T. Cortes, T. F. Otero and L. Weruaga, Smart Structures and Materials: Electroactive Polymer Actuators and Devices (EAPAD), 2002, pp. 253–261 Search PubMed.
- T. F. Otero and J. G. Martinez, Structural and bio-mimetic chemical kinetics – a review (kinetic magnitudes include structural information), Adv. Funct. Mater., 2012 DOI:10.1002/adfm.201200719.
- (a) L. Bay, K. West, P. Sommer-Larsen, S. Skaarup and M. Benslimane, Adv. Mater., 2003, 15, 310–313 CrossRef CAS; (b) M. Cho, J. Choi, T. Kim and Y. Lee, Sens. Actuators, B, 2011, 156, 218–221 CrossRef; (c) F. Garcia-Cordova, L. Valero, Y. A. Ismail and T. Fernandez Otero, J. Mater. Chem., 2011, 21, 17265–17272 RSC; (d) K. Kaneto, H. Fujisue, M. Kunifusa and W. Takashima, Smart Mater. Struct., 2007, 16, S250–S255 CrossRef CAS; (e) R. Kiefer, S. Y. Chu, P. A. Kilmartin, G. A. Bowmaker, R. P. Cooney and J. Travas-Sejdic, Electrochim. Acta, 2007, 52, 2386–2391 CrossRef CAS; (f) A. Mazzoldi, C. Degl'Innocenti, M. Michelucci and D. De Rossi, Mater. Sci. Eng., C, 1998, 6, 65–72 CrossRef; (g) T. Otero, G. Vazquez Arenas and J. Lopez Cascales, Macromolecules, 2006, 39, 9551–9556 CrossRef CAS; (h) T. Otero, J. Lopez Cascales and G. Vazquez Arenas, Mater. Sci. Eng., C, 2007, 27, 18–22 CrossRef CAS; (i) B. H. Qi, W. Lu and B. R. Mattes, J. Phys. Chem. B, 2004, 108, 6222–6227 CrossRef CAS; (j) J. M. Sansinena, J. B. Gao and H. L. Wang, Adv. Funct. Mater., 2003, 13, 703–709 CrossRef CAS; (k) E. Smela and N. Gadegaard, Adv. Mater., 1999, 11, 953–957 CrossRef CAS; (l) G. M. Spinks, L. Liu, G. G. Wallace and D. Z. Zhou, Adv. Funct. Mater., 2002, 12, 437–440 CrossRef CAS.
- (a) H. J. Ahonen, J. Lukkari and J. Kankare, Macromolecules, 2000, 33, 6787–6793 CrossRef CAS; (b) C. Arbizzani, M. Catellani, M. Mastragostino and C. Mingazzini, Electrochim. Acta, 1995, 40, 1871–1876 CrossRef CAS; (c) M. Ranger and M. Leclerc, Can. J. Chem., 1998, 76, 1571–1577 CAS; (d) J. Arias-Pardilla, W. Walker, F. Wudl and T. Otero, J. Phys. Chem. B, 2010, 114, 12777–12784 CrossRef CAS.
- (a) J. M. André, J. Delhalle and J. L. Brédas, Quantum Chemistry Aided Design of Organic Polymers – An Introduction to the Quantum Chemistry of Polymers and its Applications, World Scientific Publishing, Singapore, 1991 Search PubMed; (b) T. F. Otero and I. Boyano, ChemPhysChem, 2003, 4, 868–872 CrossRef CAS.
- T. F. Otero, in Modern Aspects of Electrochemistry, ed.J. O. M. Bockris, R. E. White and B. E. Conway, Kluwer Academic/Plenum Publisher, New York, 1999, p. 307 Search PubMed.
- (a) P. Brochu and Q. Pei, Macromol. Rapid Commun., 2010, 31, 10–36 CrossRef CAS; (b) T. Mirfakhrai, J. D. Madden and R. H. Baughman, Mater. Today, 2007, 10, 30–38 CrossRef CAS; (c) D. Pugal, K. Jung, A. Aabloo and K. J. Kim, Polym. Int., 2010, 59, 279–289 CrossRef CAS; (d) R. Tiwari and E. Garcia, Smart Mater. Struct., 2011, 20, 083001 CrossRef.
- (a) ActuatorWeb, http://actuatorweb.org, accessed in 2012; (b) I. W. Hunter, S. Lafontaine, T. D. Doukoglou, L. A. Jones, M. J. Korenberg, P. M. F. Nielsen, R. F. Kirsch and R. E. Kearney, Microrobotics and the Study of Muscle – Special Problems in Control, System-Identification and Modeling, Identification and System Parameter Estimation Symposia, 1992 Search PubMed; (c) C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397–2409 RSC; (d) Y. Z. Long, M. M. Li, C. Gu, M. Wan, J. L. Duvail, Z. Liu and Z. Fan, Prog. Polym. Sci., 2011, 36, 1415–1442 CrossRef CAS; (e) J. D. W. Madden, N. A. Vandesteeg, P. A. Anquetil, P. G. A. Madden, A. Takshi, R. Z. Pytel, S. R. Lafontaine, P. A. Wieringa and I. W. Hunter, IEEE J. Oceanic Eng., 2004, 29, 706–728 CrossRef.
- (a) T. F. Otero and M. T. Cortes, Chem. Commun., 2004, 284–285 RSC; (b) L. Valero, J. Arias-Pardilla, J. Cauich-Rodriguez, M. Smit and T. Otero, Electrochim. Acta, 2011, 56, 3721–3726 CrossRef CAS; (c) T. F. Otero and J. Rodriguez, in Intrinsically Conducting Polymers: An Emerging Technology, ed. M. Aldissi, Kluwer, Dordrecht, 1993, p. 179 Search PubMed.
- (a) S. McGovern, G. Alici, V. T. Truong and G. Spinks, Smart Mater. Struct., 2009, 18, 095009 CrossRef; (b) S. T. McGovern, M. Abbot, R. Emery, G. Alici, V. T. Truong, G. M. Spinks and G. G. Wallace, Polym. Int., 2010, 59, 357–364 CrossRef CAS; (c) G. Alici and D. Gunderson, A Bio-Inspired Robotic Locomotion System Based on Conducting Polymer Actuators, IEEE ASME International Conference on Advance Intelligent Mechatronics, 2009 Search PubMed; (d) G. M. Spinks, G. G. Wallace, J. Ding, D. Z. Zhou, B. B. Xi, T. R. Scott and V. T. Truong, Electroactive polymer actuator devices (EAPAD), 2003 Search PubMed; (e) A. DellaSanta, A. Mazzoldi and D. De Rossi, J. Intell. Mater. Syst. Struct., 1996, 7, 292–300 CrossRef; (f) K. K. Lee, N. R. Munce, T. Shoa, L. G. Charron, G. A. Wright, J. D. Madden and V. X. Yang, Sens. Actuators, A, 2009, 153, 230–236 CrossRef; (g) E. Smela, M. Christophersen, S. B. Prakash, M. Urdaneta, M. Dandin and P. Abshire, Integrated cell-based sensors and “cell clinics” utilizing conjugated polymer actuators, Electroactive Polymer Actuators and Devices (EAPAD), SPIE Proceedings, 2007, 6524, G5240 Search PubMed; (h) E. W. H. Jager, C. Immerstrand, K. H. Peterson, K. E. Magnusson, I. Lundstrom and O. Inganas, Biomed. Microdevices, 2002, 4, 177–187 CrossRef; (i) S. Ramirez-Garcia and D. Diamond, Sens. Actuators, A, 2007, 135, 229–235 CrossRef; (j) Y. Wu, D. Zhou, G. M. Spinks, P. C. Innis, W. M. Megill and G. G. Wallace, Smart Mater. Struct., 2005, 14, 1511–1516 CrossRef CAS; (k) X. He, C. Li, F. Chen and G. Shi, Adv. Funct. Mater., 2007, 17, 2911–2917 CrossRef CAS; (l) D. Pede, E. Smela, T. Johansson, M. Johansson and O. Inganas, Adv. Mater., 1998, 10, 233–237 CrossRef CAS; (m) S. A. Wilson, R. P. Jourdain, Q. Zhang, R. A. Dorey, C. R. Bowen, M. Willander, Q. U. Wahab, M. Willander, M. Safaa, O. Nur, E. Quandt, C. Johansson, E. Pagounis, M. Kohl, J. Matovic, B. Samel, W. van der Wijngaart, E. W. Jager, D. Carlsson, Z. Djinovic, M. Wegener, C. Moldovan, R. Iosub, E. Abad, M. Wendlandt, C. Rusu and K. Persson, Mater. Sci. Eng., R, 2007, 56, 1–129 CrossRef; (n) E. W. H. Jager, E. Smela, O. Inganas and I. Lundstrom, Synth. Met., 1999, 102, 1309–1310 CrossRef CAS; (o) E. W. H. Jager, E. Smela and O. Inganas, Science, 2000, 290, 1540–1545 CrossRef CAS; (p) E. W. H. Jager, O. Inganas and I. Lundstrom, Science, 2000, 288, 2335–2338 CrossRef CAS; (q) E. W. H. Jager, O. Inganas and I. Lundstrom, Adv. Mater., 2001, 13, 76–80 CrossRef CAS; (r) E. Smela, O. Inganas, Q. B. Pei and I. Lundstrom, Adv. Mater., 1993, 5, 630–632 CrossRef CAS; (s) E. Smela, Adv. Mater., 1999, 11, 1343–1345 CrossRef CAS; (t) R. Kiefer, X. Mandviwalla, R. Archer, S. S. Tjahyono, H. Wang, B. MacDonald, G. A. Bowmaker, P. A. Kilmartin and J. Travas-Sejdic, The Application of Polypyrrole Trilayer Actuators in Microfluidics and Robotics, Electroactive Polymer Actuators and Devices (EAPAD), 2008 Search PubMed.
- (a) C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397–415 CrossRef CAS; (b) R. Baetens, B. P. Jelle and A. Gustavsen, Sol. Energy Mater. Sol. Cells, 2010, 94, 87–105 CrossRef CAS; (c) A. L. Dyer and J. R. Reynolds, in Handbook of Conducting Polymers, ed. T. A. Skotheim, J. R. Elsenbaumer and J. R. Reynolds, CRC Press, Boca Raton, 2007, vol. 1 Search PubMed; (d) P. Verge, P. H. Aubert, F. Vidal, L. Sauques, F. Tran-Van, S. Peralta, D. Teyssie and C. Chevrot, Chem. Mater., 2010, 22, 4539–4547 CrossRef CAS; (e) T. F. Otero and M. Bengoechea, Langmuir, 1999, 15, 1323–1327 CrossRef CAS.
- (a) J. A. Irvin, D. J. Irvin and J. D. Stenger-Smith, in Handbook of Conducting Polymers, ed. T. A. Skotheim, J. R. Elsenbaumer and J. R. Reynolds, CRC Press, Boca Raton, 2007, pp. 9.1–9.29 Search PubMed; (b) P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS; (c) G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- (a) D. Svirskis, B. E. Wright, J. Travas-Sejdic, A. Rodgers and S. Garg, Sens. Actuators, B, 2010, 151, 97–102 CrossRef; (b) M. R. Abidian, D. H. Kim and D. C. Martin, Adv. Mater., 2006, 18, 405–409 CrossRef CAS; (c) D. Svirskis, J. Travas-Sejdic, A. Rodgers and S. Garg, J. Controlled Release, 2010, 146, 6–15 CrossRef CAS; (d) R. T. Richardson, A. K. Wise, B. C. Thompson, B. O. Flynn, P. J. Atkinson, N. J. Fretwell, J. B. Fallon, G. G. Wallace, R. K. Shepherd, G. M. Clark and S. J. O'Leary, Biomaterials, 2009, 30, 2614–2624 CrossRef CAS; (e) A. Guiseppi-Elie, Biomaterials, 2010, 31, 2701–2716 CrossRef CAS; (f) S. Geetha, C. R. K. Rao, M. Vijayan and D. C. Trivedi, Anal. Chim. Acta, 2006, 568, 119–125 CrossRef CAS.
- (a) C. E. Schmidt, V. R. Shastri, J. P. Vacanti and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8948–8953 CrossRef CAS; (b) K. A. Ludwig, J. D. Uram, J. Yang, D. C. Martin and D. R. Kipke, J. Neural Eng., 2006, 3, 59–70 CrossRef; (c) D. T. Simon, S. Kurup, K. C. Larsson, R. Hori, K. Tybrandt, M. Goiny, E. Jager, M. Berggren, B. Canlon and A. Richter-Dahlfors, Nat. Mater., 2009, 8, 742–746 CrossRef CAS; (d) R. Ravichandran, S. Sundarrajan, J. R. Venugopal, S. Mukherjee and S. Ramakrishna, J. R. Soc., Interface, 2010, 7, S559–S579 CrossRef CAS; (e) R. A. Green, N. H. Lovell and L. A. Poole-Warren, Acta Biomater., 2010, 6, 63–71 CrossRef CAS; (f) M. Asplund, T. Nyberg and O. Inganas, Polym. Chem., 2010, 1, 1374–1391 RSC; (g) M. R. Abidian, J. M. Corey, D. R. Kipke and D. C. Martin, Small, 2010, 6, 421–429 CrossRef CAS; (h) S. J. Chen, C. W. Yuan, X. D. Wang, P. Y. Zhang and X. S. Gu, Prog. Biochem. Biophys., 2000, 27, 212–214 CAS; (i) J. E. Collazos-Castro, J. L. Polo, G. R. Hernandez-Labrado, V. Padial-Canete and C. Garcia-Rama, Biomaterials, 2010, 31, 9244–9255 CrossRef CAS; (j) X. Y. T. Cui and D. C. Martin, Electroactive Polymers and Rapid Prototyping, 2002, pp. 101–106 Search PubMed; (k) P. M. George, A. W. Lyckman, D. A. Lavan, A. Hegde, Y. Leung, R. Avasare, C. Testa, P. M. Alexander, R. Langer and M. Sur, Biomaterials, 2005, 26, 3511–3519 CrossRef CAS; (l) H. Gerding, J. Neural Eng., 2007, 4, S30–S37 CrossRef CAS; (m) T. Ahuja, I. A. Mir, D. Kumar and Rajesh, Biomaterials, 2007, 28, 791–805 CrossRef CAS; (n) M. Ates and A. Sarac, Prog. Org. Coat., 2009, 66, 337–358 CrossRef CAS; (o) S. Cosnier and M. Holzinger, Chem. Soc. Rev., 2011, 40, 2146–2156 RSC; (p) Deepshikha and T. Basu, Anal. Lett., 2011, 44, 1126–1171 CrossRef; (q) H. Le, H. Sauriat-Dorizon and H. Korri-Youssoufi, Anal. Chim. Acta, 2010, 674, 1–8 CrossRef CAS; (r) B. Liu and G. C. Bazan, Chem. Mater., 2004, 16, 4467–4476 CrossRef CAS.
- (a) M. J. Ariza and T. F. Otero, J. Membr. Sci., 2007, 290, 241–249 CrossRef CAS; (b) J. Pellegrino, Adv. Membr. Technol. Appl., 2003, 25, 289–305 Search PubMed; (c) E. Smela and N. Gadegaard, J. Phys. Chem. B, 2001, 105, 9395–9405 CrossRef CAS; (d) P. Burgmayer and R. W. Murray, J. Electroanal. Chem., 1983, 147, 339–344 CrossRef CAS; (e) M. N. Akieh, S. F. Ralph, J. Bobacka and A. Ivaska, J. Membr. Sci., 2010, 354, 162–170 CrossRef CAS; (f) P. Burgmayer and R. W. Murray, J. Phys. Chem., 1984, 88, 2515–2521 CrossRef CAS; (g) M. J. Ariza and T. F. Otero, Colloids Surf., A, 2005, 270, 226–231 CrossRef; (h) W. E. Price, C. O. Too, G. G. Wallace and D. Zhou, Synth. Met., 1999, 102, 1338–1341 CrossRef CAS; (i) C. Weidlich and K. M. Mangold, Electrochim. Acta, 2011, 56, 3481–3484 CrossRef CAS.
- (a) J. H. Chang and I. W. Hunter, Macromol. Rapid Commun., 2011, 32, 718–723 CrossRef CAS; (b) J. Isaksson, C. Tengstedt, M. Fahlman, N. Robinson and M. Berggren, Adv. Mater., 2004, 16, 316–320 CrossRef CAS; (c) J. Isaksson, N. D. Robinson and M. Berggren, Thin Solid Films, 2006, 515, 2003–2008 CrossRef CAS.
- (a) M. Singh, P. K. Kathuroju and N. Jampana, Sens. Actuators, B, 2009, 143, 430–443 CrossRef; (b) L. Xia, Z. Wei and M. Wan, J. Colloid Interface Sci., 2010, 341, 1–11 CrossRef CAS; (c) J. Arias-Pardilla, C. Plesse, A. Khaldi, F. Vidal, C. Chevrot and T. Otero, J. Electroanal. Chem., 2011, 652, 37–43 CrossRef CAS; (d) Y. A. Ismail, J. G. Martinez, A. S. Al Harrasi, S. J. Kim and T. F. Otero, Sens. Actuators, B, 2011, 160, 1180–1190 CrossRef CAS; (e) L. Valero, J. Arias-Pardilla, M. Smit, J. Cauich-Rodriguez and T. F. Otero, Polym. Int., 2010, 59, 337–342 CrossRef CAS; (f) T. F. Otero and J. M. Sansinena, Bioelectrochem. Bioenerg., 1995, 38, 411–414 CrossRef CAS; (g) T. F. Otero and M. T. Cortes, Electrochemical Characterization and Control Triple-Layer Muscles, 2000 Search PubMed; (h) T. F. Otero, in Intelligent Materials, ed. M. Shahinpoor and H. J. Schenider, Royal Society of Chemistry, Cambridge, 2008, p. 142 Search PubMed; (i) T. F. Otero, J. J. Sanchez and J. G. Martinez, J. Phys. Chem. B, 2012, 116, 5279–5290 CrossRef CAS; (j) J. G. Martinez and T. F. Otero, J. Phys. Chem. B, 2012, 116, 9223–9230 CrossRef CAS.
- (a) T. F. Otero and J. M. Sansinena, Bioelectrochem. Bioenerg., 1997, 42, 117–122 CrossRef CAS; (b) T. F. Otero and J. M. Sansinena, Adv. Mater., 1998, 10, 491–494 CrossRef CAS; (c) T. F. Otero and M. T. Cortes, Sens. Actuators, B, 2003, 96, 152–156 CrossRef; (d) T. F. Otero and M. Broschart, J. Appl. Electrochem., 2006, 36, 205–214 CrossRef CAS; (e) T. F. Otero and M. T. Cortes, Adv. Mater., 2003, 15, 279–282 CrossRef CAS; (f) T. F. Otero, I. Boyano, M. T. Cortes and G. Vazquez, Electrochim. Acta, 2004, 49, 3719–3726 CrossRef CAS.
- (a) C. Plesse, A. Khaldi, Q. Wang, E. Cattan, D. Teyssie, C. Chevrot and F. Vidal, Smart Mater. Struct., 2011, 20, 124002 CrossRef; (b) I. Fabre-Francke, P. H. Aubert, S. Alfonsi, F. Vidal, L. Sauques and C. Chevrot, Sol. Energy Mater. Sol. Cells, 2012, 99, 109–115 CrossRef CAS.
- T. F. Otero and M. Caballero Romero, Polym. Int., 2010, 59, 329–336 CrossRef CAS.
- T. F. Otero and I. Boyano, Electrochim. Acta, 2006, 51, 6238–6242 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |