A protected annealing process for the production of high quality colloidal oxide nanoparticles with optimized physical properties
Isabelle
Maurin
,
Géraldine
Dantelle
,
Jean-Pierre
Boilot
and
Thierry
Gacoin
*
Laboratoire de Physique de la Matière Condensée, Groupe de Chimie du Solide, Ecole Polytechnique – CNRS, Route de Saclay, 91128 Palaiseau, France. E-mail: thierry.gacoin@polytechnique.edu; Tel: +33 331-693-34656
First published on 17th September 2012
Abstract
Colloidal synthesis of inorganic materials is usually achieved in solvents under milder conditions of temperature than those usually used for the preparation of bulk compounds. As commonly shown on oxide nanoparticles with sizes of more than typically 20 nm, this leads to particles exhibiting an altered crystallinity with the presence of more or less extended defects that may impact the physical properties of the particles. Considering that post-annealing treatments on powders of nanoparticles inevitably lead to their sintering (i.e. growth and irreversible aggregation), the influence of these defects has rarely been discussed. For a few years, we have been investigating a post-synthesis process of “protected annealing”. The basic principle relies on thermal treatment of preformed particles that have been previously dispersed into a sol–gel silica matrix. After annealing at temperatures up to 1000 °C, the dissolution of the host matrix allows recovery of a suspension of particles with the same size as the pristine particles and an almost perfect crystallinity. Investigation of the physical properties of these particles permitted us to show their optimized properties, thus discriminating the influence of altered crystallinity compared to small size or surface effects and high surface area. Results are presented in the case of oxide phosphors (YVO4:Eu, YAG:Ce), photocatalytic TiO2 and magnetic γ-Fe2O3 particles. Finally, an extension of the process is shown in the case of a reactive protected annealing, when the thermal treatment is associated with a change of chemical composition of the particles, thus allowing the investigation of systems that are difficult to obtain through conventional colloid chemistry. This latter strategy is presented in the cases of Zn2SiO4:Mn nanophosphors, Co-doped γ-Fe2O3 and N-doped TiO2.
Introduction
Nanoparticles research represents a very active field of investigations motivated by specific properties associated with their small size. This indeed leads to a high surface to volume ratio, leading to processing issues, for example, of transparent coatings and nano-objects that may be manipulated and coupled to other systems such as biological species. It is also well-known that inorganic materials considered as nano-objects may present new physical properties due to their small sizes which are close to the characteristic lengths of some physical properties. Well-known examples are quantum confinement in semiconductors, plasmonic properties of noble metal particles, coulomb blockade in metals, superparamagnetism in magnetic materials, etc. These original properties are interesting from a fundamental point of view of the solid state physics of nanomaterials, and also because they could be used in many innovative applications.Chemical synthesis of well-controlled nanoparticles often puts a limit on investigations. Although many works report on the elaboration of nanoparticles as agglomerated nanopowders, colloidal suspensions of individualized particles allow further processing of materials with a controlled microstructure or investigation of the properties of single nano-objects. Top-down approaches such as grinding or dispersion of nanopowders may be considered for some applications but they do not allow a good control of size and dispersion state unless a low yield size selection step is achieved. Thus, colloid chemistry still appears as the most powerful approach and this field of chemistry has made huge progress in the last twenty years. Main improvements were obtained through increasing the control of nucleation/growth steps while preserving particle dispersion. Starting from preliminary works on precipitation in water from salts, the extension toward the use of organometallic precursors in high boiling point coordinating solvents has paved the way toward an always increasing number of new systems.1,2
Concerning their size, nanoparticles are usually considered to be less than 100 nm. It is worth noticing that one could separate nanoparticles into two classes. The first one corresponds to the 1 to about 10 to 20 nm range, for which the influence of size and surface on physical properties is significant. The second class corresponds to the 20 nm to 100 nm range, for which properties are not so drastically affected by size and surface effects and, in this case one can expect to benefit from both small size and bulk material properties. Nevertheless, this is true only if a good crystallinity of particles is obtained through the elaboration process.
This question is usually irrelevant to very small particles which are obtained with a good crystallinity through nucleation/growth from individual seeds. In the case of particles with a larger size, the growth rate is much higher and may proceed from the aggregation of several primary particles leading to porosity or defects in the structure of particles. Moreover, these particles may undergo a transition due to a competition between the surface and volume energy so that two different crystalline structures may coexist. Fig. 1 presents typical TEM images of different oxide nanoparticles of the second class made by conventional colloid chemistry, either through direct precipitation from metal salts (YVO4,3 Fe2O3 (ref. 6 and 7)), thermohydrolysis (ZrO2,8 TiO2 (ref. 5 and 9)) or solvolysis (ref. 4 and 10). The inhomogeneous contrast in most of these particles is clear evidence of their poor overall crystallinity. The improvement of crystallinity may be obtained by playing on the experimental parameters of elaboration, mainly the temperature similarly to what is known for common solid state reactions for bulk materials synthesis. This has led to processes such as hydrothermal treatments or solvothermal routes that indeed improve the crystallinity of particles from dissolution/reprecipitation processes which can lead to the crystal reconstruction. It remains that in many cases, the elaboration of particles in the 20 to 80 nm size range is still a challenge as post-treatments in solution are not completely efficient. Poor crystallinity thus results in the alteration of physical properties that does not allow us to take full advantage of the intrinsic properties of the bulk materials. Moreover, it prevents us from answering the question of knowing exactly for which size a particle exhibits properties that are not affected by its surface.
 | ||
| Fig. 1 TEM images of various oxide nanoparticles obtained from aqueous precipitation. (a) YVO4:Eu (ref. 3), (b) YAG:Ce (ref. 4), (c) TiO2 (ref. 5), and (d) Fe2O3 (ref. 6). | ||
In this context, further improvement of crystallinity may only be obtained through increasing the temperature of a post-annealing treatment to reach temperatures that correspond to those used for bulk material synthesis. These temperatures, which are commonly of more than 700 °C, are not compatible with solvents, and such treatments achieved on powders of nanoparticles lead to their sintering (irreversible aggregation and grain growth).
For that reason, our work over the last few years has been to develop a so-called protected annealing process, which consists of achieving a thermal treatment at very high temperatures on particles that are dispersed in an inorganic matrix, inert at the considered temperatures but that can be further dissolved to recover the annealed particles as colloidal suspensions.11
We mainly focused our work on oxide materials with either luminescence or magnetic properties, to investigate the influence of defects on these properties. An extension called the ‘‘reactive protected annealing process’’ involves chemical modification of the particles through solid state reactions that may be involved in addition to the annealing process. We showed that this approach also allows the elaboration of nanosized systems that cannot be synthesized through conventional colloid chemistry mainly because reaction between precursors is ineffective for the precipitation of the expected phase either in terms of chemical composition, crystallinity or crystalline phase, size control or redox state of the constituents.
Basic principle of the protected annealing process
The basic principle of the process is schematically described in Fig. 2. Pristine particles are obtained through conventional colloid chemistry. In the case of oxides, it is generally achieved through a precipitation reaction starting from precursor salts dissolved in water, but particles resulting from glycothermal reaction and further dispersed in ethanol are also considered. Particle dispersion may be achieved using their electrostatic charge at the appropriate pH, but it may also be improved through the addition of a surface complexing agent, usually a polymer such as polyacrylic acid or polyethylene glycol.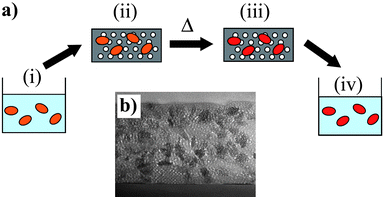 | ||
| Fig. 2 (a) Schematic description of the protected annealing process. The initial point (i) is an aqueous or alcoholic solution of nanoparticles, which are then dispersed in a surfactant templated silica matrix. After gelation, this composite is calcinated at 450 °C in air to obtain a mesoporous silica matrix embedding the nanoparticles (ii). A high temperature treatment is applied, leading to annealed nanoparticles dispersed in the silica matrix (iii). The last step of the process is to remove the silica matrix by a fluorhydric acid treatment, leading to an aqueous solution containing the annealed nanoparticles (iv). (b) SEM image of a typical mesoporous silica matrix containing TiO2 nanoparticles (adapted from ref. 12). | ||
The first step is the elaboration of a composite material consisting of a host matrix in which the nanoparticles are homogeneously dispersed. This is achieved by simple mixing of the colloidal suspension with a solution of the precursors of the matrix which will lead to the composite system after aging and drying. The choice for the host matrix has to meet mainly three requirements. First, the matrix should be processable from precursors in solution in order to allow the appropriate mixing and dispersion of the particles. Second, it must be inorganic and inert to undergo high temperature treatments without degradation or reaction with the particles. Finally, it must be dissolvable under mild conditions in order to preserve the annealed particles.
In our case, we focused our attention on silica that was found to satisfy all the above listed requirements, but other experiments were also performed using zinc oxide. Silica appears as a good candidate first because its sol–gel chemistry is well-known. It can be achieved under conditions that are compatible with the stability of colloidal particles (low ionic strength, in water or water–ethanol mixtures). Surface complexation by hydrosiloxane groups may also provide some stabilization of the particles. The sol-to-gel transition allows the entrapment of the particles that thus remain well dispersed during solvent removal. Secondly, silica can be dissolved through reaction with KOH or with HF. The kinetics of dissolution of the annealed sol–gel silica was found to be greatly improved when considering the surfactant-templated mesoporous silica. This approach only requires the adjunction of a copolymer that will act as a template for the pore formation, but may also improve the particle dispersion through adsorption on their surface. The porosity may also limit secondary solid state reaction between the nanoparticles and the silica phase through reducing their common interface.
The thermal treatment is achieved in two steps. The first one is typically at 450 °C in order to remove the organic template. The second step corresponds to the annealing treatment of the particles. The maximum possible temperature is determined either by reaction between silica and the particles or by the temperature at which crystallization of silica into cristobalite is observed. In the latter case, the main limitation is due to the very difficult dissolution of this phase. For the systems we investigated, maximum annealing temperatures were typically of 1000 °C, mostly related to silica crystallization. In some cases, the annealing treatments may be achieved under controlled atmosphere, for example H2–N2 mixture, in order to control the oxidation state of some elements present in the particles. This is the case, for example, of luminescent YAG:Ce nanoparticles in order to preserve the active 3+ state of the cerium ions.4
Structural evolution through annealing
After calcination and matrix dissolution, the particles are recovered as a colloidal suspension, usually in water with controlled pH or by adding surface modifiers such as polyacrylic acid (PAA) to ensure good dispersion. IR spectroscopy and chemical analysis allow us to check that appropriate conditions (dissolution time, HF concentration) and further washing lead to a complete elimination of all silica species. Dynamic light scattering analysis confirms that the particle size and dispersion state are globally maintained, with only small aggregates that can be easily removed by centrifugation. This is the first evidence of the ability of the silica matrix to prevent particle sintering during the thermal treatment. | ||
| Fig. 3 TEM images of pristine YVO4:Eu and TiO2 particles respectively (a and c) and corresponding images of annealed particles (b and d). | ||
TEM observations of YVO4 and YAG:Ce samples after their annealing revealed a surprising microstructure with the presence of cavities (Fig. 4). In some cases, the cavities exhibit a shape and an orientation that correspond to some specific crystallographic planes. In the case of YAG:Ce particles, cavities are observed in almost all the particles with a very spherical shape.
 | ||
| Fig. 4 TEM images of annealed particles exhibiting cavities resulting from pore coalescence: (a) YVO4:Eu nanoparticles and (b) annealed YAG:Ce nanoparticles. | ||
The origin of these cavities is explained as resulting from the coalescence of pores that were present within the pristine particles. Such a porosity may explain the poor homogeneity in their TEM images. It can be characterized by specific area measurements and BET analysis on a powder of pristine particles. For example, YVO4 particles exhibit a specific surface of 206 m2 g−1.13 This corresponds to the value expected when considering that pristine particles are formed through the agglomeration of about 7 nm sized primary particles, thus revealing a possible mechanism for the particle formation. A similar microstructure seems to be commonly reported on oxide particles of sizes above 10 nm, such as YAG,4 ZrO2,8 and TiO2,9 suggesting a similar mechanism of particle formation. It is interesting to note that some differences are observed concerning the efficiency of the annealing process to remove all the porosity.
Indeed, annealing of TiO2 particles at 1000 °C leads to a complete densification of the particles. In the case of YVO4 (at the same temperature), densification is either complete or coalescence of the porosity produces the formation of one or two cavities as shown above. In the case of YAG, we observe a partial coalescence of the pores into several cavities. This difference between the investigated systems may be understood as a consequence of their refractory nature and their ability to sinter.
Using a protected annealing process, a similar behaviour was observed in TiO2 and Fe2O3 particles for which the anatase and maghemite structures were respectively preserved even for annealing temperatures up to 900 °C. As shown in Fig. 5, maghemite (γ-Fe2O3) nanoparticles dispersed in a silica matrix retain their spinel structure up to 900 °C, while a transition into the stable hematite (α-Fe2O3) form is observed at 450 °C for powder of particles. This result, similar to what has been previously reported by other authors,21,22 proves the efficiency of the confinement in the silica matrix to prevent the growth of particles through coalescence and sintering. It can also be noted that the absence of transition is a clear indication of the good dispersion of the particles instead of the formation of aggregates that would be able to grow upon heating, thus leading to a phase transition.
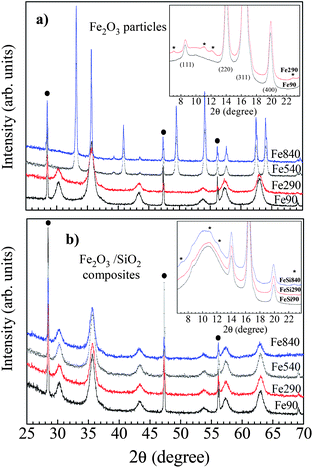 | ||
| Fig. 5 X-ray diffraction profiles (λ = 1.5418 Å) of 7 nm Fe2O3 nanoparticles either annealed as a powder or dispersed in a silica matrix (annealing temperatures: 90 °C, 290 °C, 540 °C and 840 °C). The peaks related to a Si calibrant are indicated by filled circles. Inset: an enlarged view of synchrotron X-ray data (λ = 0.7211 Å), showing the P4132 extra reflections (asterisks) related to Fe vacancies ordering. Adapted from ref. 6. | ||
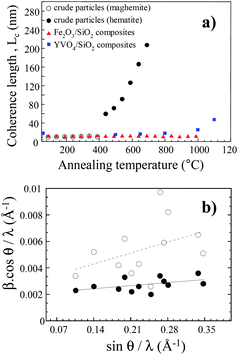 | ||
| Fig. 6 (a) Coherence length value, Lc, as a function of the annealing temperature for crude Fe2O3 particles (open and filled circle), for Fe2O3 particles embedded within silica (filled triangles) and for YVO4:Eu nanoparticles also embedded into silica (filled squares). (b) Williamson–Hall plot for YVO4:Eu nanoparticles before (open circles) and after (filled circles) application of the annealing process at 1000 °C. | ||
In the case of particles dispersed in a silica matrix, one would have expected first an increase of the coherence length up to the point at which particles are completely annealed and monocrystalline, and then a saturation up to a temperature at which there is either a parasitic reaction with silica, or a significant diffusion within the matrix leading to some kind of Ostwald ripening. Such a behaviour is actually not observed in the case of YVO4 (and similarly YAG), in which the coherence length values increase continuously starting from about 16 nm at 400 °C and reaching 47 nm at 1100 °C (Fig. 6a). At this point, this length almost corresponds to the geometrical size of the particle as determined by TEM suggesting that the saturation might have taken place above the maximum temperature of the annealing process. In the case of protected γ-Fe2O3 particles, the coherence length remains almost constant up to 1000 °C. This is explained by the small size of the initial particles (7 nm) that are already monocrystalline after synthesis.
Assuming a Cauchy line shape for the Bragg reflections, the full width at half maximum H of each peak can be written as:
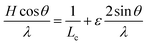 | (1) |
Structural disorder (strains and defects) can also be assessed through a more precise analysis of diffraction data, for example by using synchrotron radiation. Analysis performed on data from YAG nanoparticles showed a lattice parameter of 12.0785 (1) Å in the pristine particles that decreased down to 12.01410 (3) Å for annealed particles, much closer to the bulk value (12.01300 (1) Å). This effect is consistent with the observations of Hosokawa and colleagues on unprotected nanopowders annealed at 1000 °C.25 Similar to results on other systems,26 the authors suggest that the presence of hydroxyl groups in the porosity of the pristine particles causes the observed lattice expansion.
However, only large densities of extended defects such as stacking faults or dislocations induce a significant line broadening or a change in the lattice parameter value, while a low density of uncorrelated point defects will rather impact the repartition of the scattered intensity in the reciprocal space. For instance, for γ-Fe2O3 particles below 15 nm synthesized by room-temperature co-precipitation of ferric and ferrous salts in water, the main source of disorder is a randomized distribution of iron vacancies within the octahedral sites of the spinel structure (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group). Partial vacancy ordering occurs by subsequent annealing above 300 °C (Fig. 5), as evidenced by the appearance of additional peaks corresponding to the P4132 space group.6
m space group). Partial vacancy ordering occurs by subsequent annealing above 300 °C (Fig. 5), as evidenced by the appearance of additional peaks corresponding to the P4132 space group.6
Influence of crystallinity and annealing on physical properties
The initial idea of this work was to investigate the influence of bulk defects on the physical properties of nanoparticles, and provide a way for optimization in cases where it would have been strongly limiting. This would also allow more precise investigation of the size for which surface properties begin to alter the physical properties significantly as compared to those of the bulk compounds. The answer to this question can indeed only be provided in the case of well-crystallized particles in order to discriminate between bulk and surface defects. Finally, it must be noted that since elaboration processes were shown to lead to porous particles in some cases, the difference between surface atoms and bulk defects is not always so clear. Concerning the physical properties that may be affected by bulk defects, surface and size, and since we are concerned mainly with oxides, we focused our attention on rare-earth doped oxides (YVO4 and YAG:Ce) and magnetic iron oxide nanoparticles.Many optimized compositions have been developed, usually for industrial purposes. Our research activities on rare-earth doped nanoparticles are mainly concentrated on two systems: YVO4:Eu and YAG:Ce. The reason for this choice was first given by the performance of the bulk materials, but also because of the possible synthesis of nanoparticles through conventional colloid chemistry routes.3,10
The emission properties of Eu3+-doped YVO4 pristine nanoparticles have been investigated in detail.28 The spectroscopic features were nearly similar to those of the bulk materials with a maximum excitation wavelength at 280 nm (vanadate absorption) and the main emission at 617 nm from the 5D0–7F2 transition of Eu3+. A maximum quantum yield of 10 to 20% was found corresponding to a maximum Eu doping concentration of 20%. Although it is not so low when compared to other luminescent particles dispersed in water without surface modification, the difference in the bulk properties (68% of quantum yield for 5% Eu doping) was initially attributed to alteration of energy transfers and quenching of the emission through surface defects and hydroxyl groups.28
Nevertheless, this result was not satisfactory since the large size of the particles (30 to 50 nm) led us to expect a rather limited contribution of the surface. The protected annealing process was achieved and properties of the resulting annealed particles were compared to those of pristine particles and bulk reference materials.
Fig. 7a displays the evolution of emission quantum yield of the different samples (pristine particles, annealed particles and bulk references) as a function of their europium content. These plots give a good indication of the emission yield and of the efficiency of the energy transfers leading to emission from the europium. It appears clearly that, contrary to pristine particles, annealed samples display an overall evolution that is now very similar to the one of the bulk references, with an optimum doping at 5%, attesting that energy transfers have been largely improved by the annealing treatment due to a better crystallinity and a large decrease of porosity.
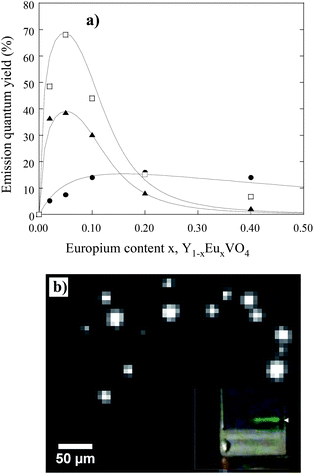 | ||
| Fig. 7 (a) Evolution of the luminescence quantum yield of Y1−xEuxVO4 as-made nanoparticles (●), annealed nanoparticles (□) and bulk material (□), as a function of the Eu doping concentration x (adapted from ref. 13). (b) Wide-field fluorescence microscopy image of annealed Y0.78Yb0.2Er0.02VO4 nanoparticles deposited onto a glass slide. The nanoparticle emission is centered at 550 nm under a 980 nm excitation (8 kW cm−2). This image was taken with an acquisition time of 500 ms.27 | ||
Nevertheless, the optimum quantum yield of the annealed particles (40%) remained lower than for the bulk reference (68%) which seemed surprising considering the low porosity and the specific surface area of the particles, their excellent crystallinity and similar energy transfer processes attested by the same shape of the curves shown in Fig. 7a. The reason was found when considering a dielectric effect intrinsic to the small size of the particles.13 Indeed, light emission measurements were achieved on annealed nanoparticles dispersed in water, whose refractive index (n = 1.33) is much lower than that of the bulk YVO4 (n ∼ 2). In a rough approximation,29,30 the radiative lifetime τrad of a transition from an ion embedded in a media of refractive index n can be written as:
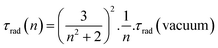 | (2) |
A decrease of the effective refractive index results in an increase of radiative lifetime and thus a decreased quantum yield when assuming a constant non-radiative decay time. To verify this hypothesis, YVO4 nanoparticles were embedded into two media (SiO2 and TiO2) with two different refractive indexes (nSiO2 = 1.5 and nTiO2 = 2) and annealed for 12 hours at 600 °C. The 5D0 level lifetime of Eu3+ was then measured for these two samples. Experimentally, we found τSiO2/τTiO2 = 2.6 which corresponds exactly to the value (2.6) calculated from eqn (2).
Considering that the radiative lifetime of the emission from the nanoparticles is increased by a lower effective refractive index than in the bulk compound, this explains their lower quantum yield if non-radiative processes are not affected by such dielectric effects. This appears as an intrinsic limitation of the optical properties of the nanoparticles when compared to bulk materials, annealed particles thus being the best that can be obtained in terms of emission properties.
A straightforward application of the protected annealing process concerns the investigation of up-conversion properties of Yb–Er doped samples. This process is based on the excitation of two Yb ions at about 975 nm that transfer their energy to a neighboring Er ion which further emits photons in the visible range (mainly around 520–550 nm and 660 nm). This process is well-known to be very sensitive to non-radiative relaxations through phonons from the matrix, and is all the more sensitive to defects and surface groups such as hydroxyls with high energy phonons. Unsurprisingly, the first experiments have shown that no up-conversion could be detected on pristine YVO4:Yb:Er particles. The same experiments made on annealed particles showed a drastic enhancement of the emission yield, to a point that detection at the single particle level could be achieved as shown in Fig. 7b.27 This confirms the high efficiency of the annealing in reducing non-radiative processes. Moreover, the estimation of the quantum yield provided a value of about 1.3% for an excitation intensity of 8 kW cm−2, which is not so low in comparison with those reported on halide nanoparticles whose phonon energy is much more favorable for the UC process. Our interpretation is that the excellent quality of the annealed particles compensates the less favorable phonon energy range assuming that the reported halide particles may not be so well crystallized.
We have recently investigated the influence of post-synthesis heat treatments carried out up to 900 °C on 7 and 14 nm sized γ-Fe2O3 nanoparticles prepared by room temperature coprecipitation in water.6 Thermal annealings up to 900 °C were shown to afford a control of the crystalline state of the particles, preventing both grain growth and transformation into hematite (see Fig. 5 and 6a). Heat treatments above 290 °C led to a partial ordering of the Fe vacancies, evidenced by synchrotron X-ray powder diffraction and FT-IR spectroscopy measurements. An increased magnetic anisotropy was also reported, presumably representative of a surface contribution. This latter effect could be ascribed to a modification of the surface state upon heating, involving the removal of surface hydroxyl groups. For the 7 nm particles, a slight decrease in saturation magnetization was observed after annealing. Analysis of the thermal dependence of the saturation magnetization indicated that this result was related to an increased number of weakly coupled and misaligned spins at the surface of the particles.6 All experimental findings were actually consistent with a propagation of the shell of non-collinear spins toward the center of the particles. Comparison with Monte Carlo simulations suggested that this expansion was mainly driven by the increased surface anisotropy.43 In contrast, for the 14 nm particles, an increased saturation magnetization was reported after thermal treatment, confirming that the previous evolution was mostly ascribed to surface effects. For large particles, the volume contribution dominates and the saturation magnetization value increases due to Fe vacancy ordering, as reported by other authors.44
Extension to a reactive protected annealing process
As previously shown, the protected annealing process makes possible the thermal treatment of particles preserving their size and agglomeration state. The permissible temperatures are compatible with those commonly used for solid state reactions to form ceramics. Initially developed to improve the crystallinity of preformed particles, the extension of the process to the elaboration of new compositions that cannot be reached by colloid chemistry is straightforward. In this case, the idea is to target compositions in which one component can be prepared by colloid chemistry in order to control the final size of the product.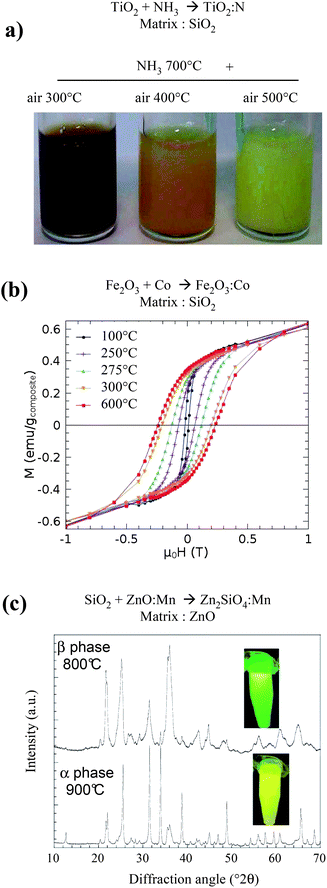 | ||
| Fig. 8 Examples of application of a reactive protected annealing process. (a) Pictures of N-doped TiO2 colloids obtained from TiO2 particles dispersed in a porous silica matrix and annealed under NH3 at 700 °C, then in air at different temperature to eliminate residual Ti3+.46 (b) Magnetization versus magnetic field loops measured at 10 K for Co doped γ-Fe2O3 nanoparticles dispersed in silica, after annealing at temperatures ranging between 100 and 600 °C.57 (c) X-ray diffraction of Zn2SiO4:Mn particles obtained through reaction of SiO2 nanoparticles in a Mn-doped ZnO matrix, evidencing a size induced phase transition and the corresponding change of luminescence from green to yellow.48 | ||
Conclusions and perspectives
Physical properties of nanoparticles usually differ significantly from those of the bulk counterparts. It is commonly accepted that it results from both effects intrinsic to small size (quantum confinement, superparamagnetism, dielectric confinement, etc.) and more extrinsic effects associated to surface states, which are largely enhanced in this size range. The influence of defects in the volume of the particles, resulting from an altered crystallinity, has rarely been discussed. Their influence could be non-negligible considering the low temperature of synthesis required by colloid chemistry and the growth mechanism of particles, especially when considering sizes of several tens of nanometers. The main difficulty in the investigation of such effects comes from the possibility to produce particles with optimized crystallinity while preserving their size, surface and dispersion state. In this context, we have developed for a few years a so-called “protected annealing process”. It consists of (i) dispersing preformed colloidal nanoparticles into a refractory matrix, (ii) achieving thermal treatments of annealing up to high temperatures (typically 1000 °C) and (iii) dissolving the matrix to recover the annealed particles as a colloidal suspension. Applications of this process were investigated in our group on various systems, especially oxides such as luminescent rare-earth doped oxides (YVO4:Eu, YAG:Ce), photocatalytic TiO2 and magnetic γ-Fe2O3. This allowed us to provide new insights into the physical properties of these systems and to discriminate between size, surface and altered crystallinity effects. As a further development of this strategy, we also studied the application of the process for the elaboration of systems obtained following a solid state reaction at high temperature, commonly used in ceramic synthesis, but still preserving the size and dispersion state of the particles. We show that, in the case of Zn2SiO4:Mn nanophosphors, Co doped γ-Fe2O3 and N doped TiO2, such a “reactive protected annealing process” gives access to new systems by increasing the temperatures of synthesis when compared to those used in conventional colloid chemistry. This strategy is a way to join colloid chemistry and conventional solid state chemistry and should lead to numerous investigations for the elaboration of complex systems with excellent crystallinity and thus optimized physical properties.The authors gratefully acknowledge the contribution of Caroline Bertail, Geneviève Mialon, Morgan Gohin, Amélie Revaux, Charlotte Vichery and Lucie Devys to this work.
Notes and references
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS.
- A. Huignard, T. Gacoin and J.-P. Boilot, Chem. Mater., 2000, 12, 1090 CrossRef CAS.
- A. Revaux, G. Dantelle, N. George, R. Seshadri, T. Gacoin and J.-P. Boilot, Nanoscale, 2011, 3, 2015 RSC.
- Commercial TiO2 suspensions CristalACTiV™ S5-300A from CristalGlobal.
- C. Vichery, I. Maurin, P. Bonville, J.-P. Boilot and T. Gacoin, J. Phys. Chem. C, 2012, 116, 16311 Search PubMed.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247 CrossRef.
- D. Carriere, M. Moreau, P. Barboux and J.-P. Boilot, Langmuir, 2004, 20, 3449 CAS.
- E. Puzenat and P. Pichat, J. Photochem. Photobiol., A, 2003, 160, 127 CrossRef CAS.
- R. Kasuya, T. Isobe and H. Kuma, J. Alloys Compd., 2006, 820, 408.
- G. Mialon, M. Gohin, T. Gacoin and J.-P. Boilot, ACS Nano, 2008, 2, 2505 CrossRef CAS.
- E. Allain, S. Besson, C. Durand, M. Moreau, T. Gacoin and J.-P. Boilot, Adv. Funct. Mater., 2007, 17, 549 CrossRef CAS.
- G. Mialon, S. Turkcan, A. Alexandrou, T. Gacoin and J.-P. Boilot, J. Phys. Chem. C, 2009, 113, 18699 CrossRef CAS.
- A. Navrotsky, J. Chem. Thermodyn., 2007, 39, 2 CAS.
- R. C. Garvie, J. Phys. Chem., 1965, 69, 1238 CrossRef CAS.
- G. Caboche, F. Chaput, J.-P. Boilot and J.-C. Niepce, Silic. Ind., 1993, 58, 103 Search PubMed.
- S. Sakurai, A. Namai, K. Hashimoto and S.-I. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 18299 CrossRef CAS.
- T. Belin, N. Millot, N. Bovet and M. Gailhanou, J. Solid State Chem., 2007, 180, 2377 CrossRef CAS.
- G. Schimanke and M. Martin, Solid State Ionics, 2000, 136–137, 1235 Search PubMed.
- A. Navrotsky, L. Mazeina and J. Majzlan, Science, 2008, 319, 1635 CrossRef CAS.
- C. Chanéac, E. Tronc and J.-P. Jolivet, Nanostruct. Mater., 1995, 6, 715 CrossRef.
- C. Chanéac, E. Tronc and J.-P. Jolivet, J. Mater. Chem., 1996, 6, 1905 RSC.
- P. Scherrer, Gött. Nachr., 1918, 2, 98 Search PubMed.
- G. K. Williamson and W. H. Hall, Acta Metall., 1953, 4, 22 Search PubMed.
- S. Hosokawa, Y. Tanaka, S. Iwamoto and M. Inoue, J. Alloys Compd., 2008, 451, 309 CrossRef CAS.
- I. F. Guilliat and N. H. Brett, J. Mater. Sci., 1970, 5, 615 Search PubMed.
- G. Mialon, S. Turckan, G. Dantelle, D. Collins, M. Hadjipanayi, R. Taylor, T. Gacoin, A. Alexandrou and J.-P. Boilot, J. Phys. Chem. C, 2010, 114, 22449 CrossRef CAS.
- A. Huignard, V. Buissette, A.-C. Franville, T. Gacoin and J.-P. Boilot, J. Phys. Chem. B, 2003, 107, 6754 CrossRef CAS.
- R. S. Meltzer, S. P. Feofilov, B. Tissue and H. B. Yuan, Phys. Rev. B: Condens. Matter, 1999, 60, R14012 CrossRef CAS.
- V. LeBihan, A. Pillonnet, D. Amans, G. Ledoux, O. Marty and C. Dujardin, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 113405 CrossRef.
- X. Batlle and A. Labarta, J. Phys. D: Appl. Phys., 2002, 35, R15 CrossRef.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS.
- Q. A. Pankhurst, N. K. T. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- E. Tronc, A. Ezzir, R. Cherkaoui, C. Chanéac, M. Noguès, H. Kachkachi, D. Fiorani, A.-M. Testa, J.-M. Grenèche and J.-P. Jolivet, J. Magn. Magn. Mater., 2000, 221, 63 CrossRef CAS.
- S. Morup, J. Magn. Magn. Mater., 2003, 266, 110 Search PubMed.
- A. Millan, A. Urtizberea, N. J. O. Silva, F. Palacio, V. S. Amaral, E. Snoeck and V. Serin, J. Magn. Magn. Mater., 2007, 312, L5 CrossRef CAS.
- A. G. Roca, R. Costo, A. F. Rebolledo, S. Veintemillas-Verdaguer, P. Tartaj, T. Gonzalez-Carreño, M. P. Morales and S. J. Serna, J. Phys. D: Appl. Phys., 2009, 42, 224002 CrossRef.
- M. P. Morales, S. Veintemillas-Verdaguer, M. I. Montero and C. J. Serna, Chem. Mater., 1999, 11, 3058 CrossRef CAS.
- P. Dutta, A. Manivannan, M. S. Seehra, N. Shah and G. P. Huffman, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 174428 CrossRef.
- X. Batlle, N. Pérez, P. Guardia, O. Iglesias, A. Labarta, F. Bartolomé, L. M. García, J. Bartolomé, A. G. Roca, M. P. Morales and C. J. Serna, J. Appl. Phys., 2011, 109, 07B524 CrossRef.
- B. Luigjes, S. M. C. Woudenberg, R. De Groot, J. D. Meeldijk, H. M. Torres Galvis, K. P. de Jong, A. P. Philipse and B. Erné, J. Phys. Chem. C, 2011, 115, 14598 CrossRef CAS.
- M. Levy, A. Quarta, A. Espinosa, A. Figuerola, C. Wilhelm, M. Garcia-Hernandez, A. Genovese, A. Falqui, D. Alloyeau, R. Buonsanti, P. Cozzoli, M. Garcia, F. Gazeau and T. Pellegrino, Chem. Mater., 2011, 23, 4170 CAS.
- O. Iglesias and A. Labarta, J. Magn. Magn. Mater., 2005, 290–291, 738 Search PubMed.
- M. P. Morales, C. J. Serna, F. Bodker and S. Morup, J. Phys.: Condens. Matter, 1997, 9, 5461 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- M. Gohin, I. Maurin, T. Gacoin and J.-P. Boilot, J. Mater. Chem., 2010, 20, 8070 RSC.
- M. Gohin, E. Allain, N. Chemin, I. Maurin, T. Gacoin and J.-P. Boilot, J. Photochem. Photobiol., A, 2010, 216, 142 Search PubMed.
- C. Bertail, S. Maron, V. Buissette, T. Le Mercier, T. Gacoin and J.-P. Boilot, Chem. Mater., 2011, 23, 2961–2967 CrossRef CAS.
- Q. A. Pankhurst, J. Connely, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS.
- R. Hergdt, S. Dutz and M. Röder, J. Phys.: Condens. Matter, 2008, 20, 385214 CrossRef.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204 CrossRef CAS.
- S. G. Kwon, Y. Piao, J. Park, S. Angappane, Y. Jo, N. M. Hwang, J. G. Park and T. Hyeon, J. Am. Chem. Soc., 2007, 129, 12571 CrossRef CAS.
- E. Blums and A. Yu. Chukhrov, J. Magn. Magn. Mater., 1993, 122, 110 Search PubMed.
- R. Massart, E. Dubois, V. Cabuil and E. Hasmonay, J. Magn. Magn. Mater., 1995, 149, 1 CrossRef CAS.
- J. C. Slonczewski, Phys. Rev., 1958, 110, 1341 CrossRef CAS.
- G. Salazar-Alvarez, J. Sort, A. Uheida, M. Muhammed, S. Suriñach, M. D. Baró and J. Nogués, J. Mater. Chem., 2007, 17, 322 RSC.
- C. Vichery, I. Maurin, J.-P. Boilot and T. Gacoin, J. Appl. Phys., 2012, 111, 07B541 Search PubMed.
- T. S. Ahmadi, M. Haase and H. Weller, Mater. Res. Bull., 2000, 35, 1869 Search PubMed.
- J. S. An, J. H. Noh, I. S. Cho, H. S. Roh, J. Y. Kim, S. H. Han and K. S. Hong, J. Phys. Chem. C, 2010, 114, 10330 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |