The design and investigation of porphyrins with liquid crystal properties at room temperature†
Christopher J.
Wilson
,
Daniela A.
Wilson‡
,
Ross W.
Boyle
* and
Georg H.
Mehl
*
Department of Chemistry, University of Hull, Kingston-upon-Hull, East Yorkshire HU6 7RX, UK. E-mail: g.h.mehl@hull.ac.uk; r.w.boyle@hull.ac.uk
First published on 9th November 2012
Abstract
The design, synthesis and investigation of a series of porphyrins displaying smectic liquid crystal phase behaviour at room temperature are described. The molecules are constructed in a modular approach, consisting of porphyrin units linked via hydrocarbon and siloxane spacers to calamitic cyanobiphenyl units. The investigation of the phase behaviour, based on optical polarizing microscopy, differential scanning calorimetry and XRD studies, is discussed in detail.
Introduction
Porphyrins play a vital role in photosynthesis, cellular respiration, oxygen transport, and as components of metalloenzymes.1 The functions performed by porphyrins in natural systems have driven research into the use of synthetic porphyrins in many areas including artificial photosynthetic materials,2 as oxidation catalysts3 and in photodynamic therapy.4 Incorporation of porphyrins into functional materials is an area which has received considerable attention, due to the possibility of using these materials to fabricate devices, which retain some of the photochemical and redox properties associated with porphyrins and their metal complexes. Porphyrins have previously been incorporated into bulk materials either by electropolymerisation onto electrodes, where they have shown applications in the electrochemical activation of thiols,5 or by incorporation into polymers for use as antibacterial materials.6 Octa-substituted porphyrins with liquid crystalline (LC) behaviour were first reported more than thirty years ago with the materials having only a very narrow phase range, however, over the years the stability of the mesophase has been expanded considerably.7 Tetraphenyl porphyrins with LC behaviour have also been reported and for such materials LC behaviour close to room temperature has been described.8 The predominant LC behaviour found with both octa-substituted and tetraphenyl porphyrins is columnar, due to the planar discotic structure of the porphyrin macrocycle.9In order to obtain the liquid crystalline phase behaviour with a layer like register and to break the dominating effect of the macrocycle on mesophase formation, a number of zinc(II) complexes of 5,15-diphenyl porphyrins have been explored previously. It was demonstrated that by attachment of suitable substituents on the two phenyl rings a calamitic type behaviour could be induced.10 Melting points of calamitic type LC porphyrins were found to be high, however, incorporation of secondary substituents on the 2-positions of the phenyl ring has reduced transition temperatures considerably, pointing thus towards a methodology for modifying such materials, resulting in lower liquid crystal phase transition temperatures.11
To date there are no single molecule based examples of calamitic porphyrins exhibiting liquid crystalline phases at or close to room temperature. The aim of the work reported here was to obtain porphyrins which show smectic phase behaviour and to devise a systematic route towards this goal. To achieve this objective the porphyrin macrocycle was extended in one direction, destabilizing potential columnar phase formation completely. An additional target was to control the transition temperatures with application accessible phase transitions being the focus. Moreover we demonstrate that the concept is architecturally robust and that LC behaviour can be obtained with or without metals in the central cavity.
Results and discussion
As a core structure 5,15-di(4-hydroxyphenyl)porphyrin was selected. The porphyrin core is essentially planar and the two phenyl rings positioned in the 5 and 15 positions whilst essentially being orthogonal to the porphyrin have some rotational flexibility. In order to induce calamitic phase behaviour when functionalizing an essentially discotic centre, the attachment of calamitic mesogens using a flexible spacer was employed. This has been explored for a number of systems12 and has been reviewed recently.13In order to promote low melting points, or the formation of glass transitions a tetramethyl disiloxane group was introduced into the spacer unit separating the porphyrin from the calamitic mesogenic unit. The spacer units connecting porphyrin to siloxane, and siloxane to mesogen groups were selected to be of different lengths to ensure a liquid like character of the spacer over a very wide temperature range. One of the spacers was selected to consist of a pentyl chain, the other of an undecyl chain to induce a lowering of melting points and glass transition temperatures, by disfavouring antiparallel assembly of sublayers of the final materials. Such an approach has been found to be very versatile, particularly when using the cyanobiphenyl unit to promote the calamitic phase behaviour.14
Synthesis
In order to explore the role of mesogenic groups on the liquid crystalline properties of 5,15-diphenyl porphyrins, we sought to devise a versatile method by which these groups could be attached easily to the common porphyrin core. Hydrosilylation of both the porphyrin and mesogens could be easily achieved via alkyl chains possessing terminal double bonds distal to the porphyrin core; this encouraged us to explore the route further.15 The 5,15-di(4-hydroxyphenyl)porphyrin core was synthesised according to the procedure of Boyle et al.16 to yield compound 1 and functionalised by Williamson etherification with 11-bromoundec-10-ene to yield 5,15-di(11-endec-1-enylphenyl)porphyrin, compound 2 (Scheme 1). The first mesogenic unit bearing a terminal alkene was prepared according to literature procedures from 4-cyano-4′-hydroxy biphenyl and 1-bromopent-4-ene using similar etherification conditions to those used for the porphyrins to yield 4′-(pent-4-enyloxy)biphenyl-4-carbonitrile 3.17 With the two coupling partners in hand, the first hydrosilylation was attempted. Alkene 3 was functionalised with tetramethyldisiloxane using platinum(0)-1,1,3,3-tetramethyl-1,3-divinyldisiloxane (Karstedt's catalyst) to yield the silylated compound 4 in a yield of 90% according to literature procedures.18 Siloxane 4 was further reacted with porphyrin 2, again in the presence of Karstedt's catalyst. Thin layer chromatographic investigation of the reaction product revealed two major products, which were separated by column chromatography. Interestingly, it was found that the two reaction products, which were obtained in approximately equal amounts, were the desired di-hydrosilylated product 5 and a mono-hydrosilylated product 6 with the remaining terminal double bond internalised to a less reactive form observed in the 1H NMR as a multiplet at 5.4 ppm. Although the yield of the di-hydrosilylated porphyrin (50%) was somewhat disappointing, the overall yield of both di- and mono-hydrosilated products was excellent (90%). The hydrosilylation reaction of porphyrin 2 thus generated two potentially interesting compounds for study, both in acceptable yields.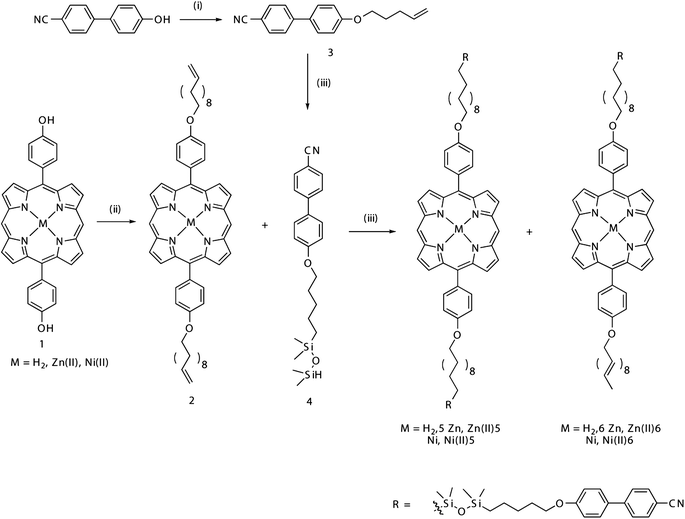 | ||
| Scheme 1 Reagents and conditions: (i) K2CO3, butanone, KI, C5H9Br, (ii) K2CO3, butanone, KI, C11H21Br and (iii) Karstedt's cat., toluene, 1,1,3,3-tetramethyldisiloxane. | ||
DSC and optical polarising microscopy
Porphyrins 5 and 6 were investigated first by differential scanning calorimetry (DSC) (Fig. 1 and ESI Fig. 2†) and their thermal properties are summarized in Table 1. The mono hydrosilylated porphyrin 6 showed a first transition at 105 °C (3.0 kJ mol−1) and a second transition at 163 °C (9.5 kJ mol−1), while the dihydrosilylated porphyrin 5 showed a single transition at 178 °C (18 kJ mol−1). Further investigation by optical polarising microscopy (OPM) (Fig. 2 and ESI Fig. 3–5†) indicated that, in the case of 6, the two phase changes observed are the crystalline to SmA transition at 105 °C and the SmA to isotropic liquid transition at 163 °C.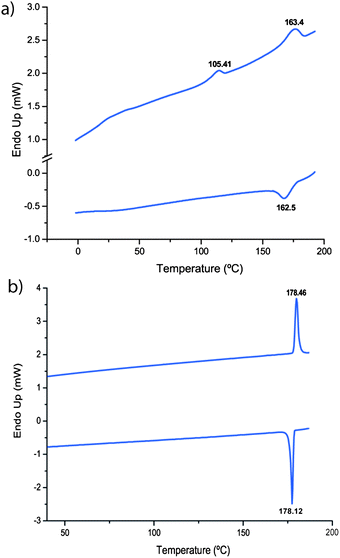 | ||
| Fig. 1 Differential scanning calorimetry plots for (a) compound 6 and (b) compound 5. | ||
| Compound | Transition temperatures (°C), enthalpies (ΔH/(kJ mol−1)) |
|---|---|
| 6 | Cr 105 (3.0) SmA 163 (9.5) Iso |
| Iso 162 (−9.5) SmA glass | |
| 6-Zn(IIiiii) | Cr 117 (9.2) SmA 150 (10.2) Iso |
| Iso 149 (−8.8) SmA 93 (−3.5) SmX | |
| 6-Ni(IIiiii) | Cr 93 (1) SmA 163 (10.4) Iso |
| Iso 162 (−10.1) SmA glass | |
| 5 | SmA 178 (18) Iso |
| Iso 178 (−19) SmA | |
| 5-Zn(IIii) | SmA 145 (9.5) Iso |
| Iso 145 (−9.5) SmA | |
| 5-Ni(IIii) | Cr 56 (10.0) SmA 157 (16.0) Iso |
| Iso 157 (−16) SmA glass |
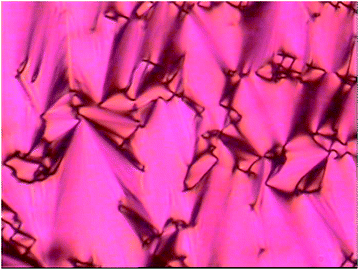 | ||
| Fig. 2 Optical polarising microscopy image of 5 at 20 °C (magnification 100×). | ||
Porphyrin 5, bearing two mesogenic cyanobiphenyl groups linked by alkyl chains and siloxy groups, exhibited SmA behaviour at room temperature (Fig. 2), only transforming to an isotropic liquid at 178 °C, arguably a very wide liquid crystal phase range.
The most striking feature of increasing the number of calamitic mesogenic groups from one in 6 to two in 5 is not the relatively small increase in the stability of the mesophase by 15 °C, but the strong destabilisation of the crystalline phase present in 6 but absent in 5 and the formation of a very wide range SmA phase. Based on the positive results obtained for the free base porphyrins this method was extended to explore the effect of complexation of the porphyrin with metal ions on the liquid crystal phases.
To achieve this, porphyrin 2 was metalated with zinc(II) acetate and nickel(II) acetate respectively using previously published conditions.19 Formation of the metal complex was evidenced by the collapse of the four Q-bands in the UV-spectrum along with the loss of the aminal protons of the macrocycle at −3 ppm in the 1H NMR.19 Hydrosilylation of the two resulting metallo-porphyrins, Zn(II)2 and Ni(II)2, proceeded smoothly and in a similar manner to that observed for the metal free porphyrin 2. Approximately equal amounts of the di-hydrosilylated products, 5-Zn(IIii) and 5-Ni(IIii), and mono-hydrosilylated products, 6-Zn(IIiiii) and 6-Ni(IIiiii), were obtained. DSC analysis of the metal complexes indicated the formation of liquid crystalline behaviour for all of the hydrosilylated porphyrin products, however the di-substitution products 5-Zn(IIii) and 5-Ni(IIii) both showed liquid crystalline behaviour at lower temperatures, when compared with their mono-substituted analogues (Table 1). The zinc dimer 5-Zn(IIii) exhibits a transition at 145 °C turning from a SmA phase to an isotropic liquid, the corresponding nickel dimer undergoes this transition at 157 °C. The nickel complex 5-Ni(IIii) shows a low temperature crystalline or partially crystalline phase with a melting point of 56 °C. It is noted that the transition enthalpy with a value of 10 kJ mol−1 is relatively low for a crystal to liquid crystal transition, suggesting the possible formation of a higher ordered lamellar phase. The lower transition temperatures for the metalated porphyrins 5-Zn(IIii) and 5-Ni(IIii) when compared to the bare porphyrin 5 indicates that the electron density associated with the incorporation of metal ions does not enhance LC properties, indeed the metal centre reduces the LC phase stability. For the metallo-porphyrins containing only one calamitic mesogenic group the difference in the isotropisation temperatures was around 13 °C. The melting points vary from 93 °C for 6-Ni(IIiiii) to 117 °C for 6-Zn(IIiiii); 6-Ni(IIiiii) can be supercooled to a room temperature glassy state, whereas 6-Zn(IIiiii) at 93 °C forms a monotropic higher ordered LC phase, which will be discussed in more detail below.
Notable is that the metal containing dimers exhibit lower isotropisation temperatures than the respective mono-functionalized materials. At present there is no conclusive explanation for this behaviour, though it can be surmised that in the dimeric materials a layer register is induced at both ends of the molecules, and, given the detrimental effect of the metal centres on the LC phase behaviour, this induces a stability penalty, when compared to the mono-substituted compounds where the porphyrin ends have more freedom to pack efficiently.
Optical polarising microscopy
A striking observation for all of the investigated materials was the ease of LC texture formation, a feature more typical for low molar mass liquid crystals, with the rapid formation of typical defect textures when cooled from the isotropic state, an encouraging feature of the molecular design approach adopted here. An example is shown in Fig. 2, where the focal texture of 5 in the SmA phase is depicted, the red colouring is due to the absorption properties of the porphyrin core in the visible region of the electromagnetic spectrum. The low temperature liquid crystal phase of 6-Zn(IIiiii) is characterized by the rapid formation of a schlieren texture, when cooled, evolving as a front in the homeotropic areas of the preceding SmA phase (Fig. 3 and ESI Fig. 5†).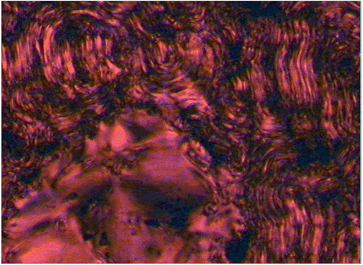 | ||
| Fig. 3 Optical polarising microscopy image showing the transition from the SmA to SmX phase in compound 6-Zn(IIiiii) on cooling from 130 °C to 92 °C (magnification 100×). | ||
Additionally a fingerprint like texture developed from the focal conic and fan areas of 6-Zn(IIiiii). The formation of schlieren and fingerprint texture suggests the formation of a higher ordered tilted smectic phase. Though these fingerprint regions were not investigated in great detail, it is noted that after formation no significant change in the apparent spacings of the fingerprints with changing the temperature was observed.
X-ray diffraction results
In order to elucidate the mechanism of self-assembly operating in these porphyrin based LC systems using X-ray diffraction patterns datasets were collected spanning both the small and wide angle regions. The data gathered from the small angle region allowed for the phase structure to be elucidated whereas the wide angle region delivers crucial information on the nature of the ordering in these systems. The symmetric and non-symmetric structures, bearing two and one cyanobiphenyl groups respectively, were analysed by X-ray diffraction (Fig. 4 and ESI Fig. 6 and 7†). A representative example is shown for compound 5 and the corresponding d-spacings are presented in Table 2. The temperature dependent layer spacing for the 01 reflection is shown in Fig. 5. In all cases for the highest temperature phase three sharp reflections were observed, corresponding to the 01, 02 and 03 reflections of a layered structure together with a characteristically broad halo in the wide angle region. The q-values of the collected intensity maxima and the associated d-spacings for the small angle region of the symmetric compound 5 are listed in Table 2. The layer spacings corresponding to the 01 reflection for all compounds are shown in Table 3.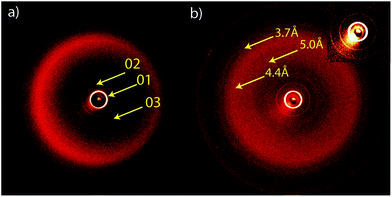 | ||
| Fig. 4 (a) 2D powder diffraction of the non-symmetric compound 6 and (b) 2D powder diffraction of symmetric compound 5; inset: expansion of the small angle region. | ||
| Peak | q nm−1 | d-Spacing Å |
|---|---|---|
| 001 | 0.199 | 31.4 |
| 002 | 0.399 | 15.7 |
| 003 | 0.599 | 10.5 |
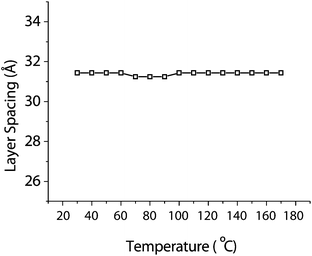 | ||
| Fig. 5 Temperature dependent layer spacings for compound 5. | ||
| Compound | q nm−1 | d-Spacing Å |
|---|---|---|
| 5 | 0.199 | 31.4 |
| 6 | 0.196 | 32.1 |
| Zn(IIiiiiii)5 | 0.191 | 32.9 |
| Zn(IIiiii)6 | 0.191 | 32.9 |
| Ni(IIii)5 | 0.200 | 31.4 |
| Ni(IIii)6 | 0.200 | 31.4 |
Although the experiments were carried out in the presence of a ∼0.5 T magnetic field perpendicular to the capillary axis, complete preferential axis alignment was not obtained for any of the samples, though some preferential alignment could be observed with the meridional small angle and the equatorial wide angle intensities being stronger.
This would suggest a minimal preferential orientation of the molecules parallel to the magnetic field. Temperature dependent XRD studies showed that a single liquid crystal phase structure for almost all of the materials (exception high ordered low temperature phase for Zn(IIiiii)6) was observed throughout the mesophase range. All observations were in good agreement with the DSC data and no discernible change in the layer spacing was seen, an example for temperature dependent behaviour is shown in Fig. 5. Radial integration of the wide angle region of the 2D diffractograms for the high temperature phase showed a characteristic main broad peak centred around q = 1.4 Å−1 = 4.4 Å. Closer inspection revealed that the peak was not symmetric in nature. The presence of multiple peaks in the wide-angle region of the XRD diffractograms of polyphilic liquid crystals displaying smectic phase morphology has been reported previously and attributed to in plane correlations in the SmA phase.20 It was possible to fit the broad peak to three Lorentzian type peaks by least squares fitting. The peaks are centred around q: 1.25 Å−1 = 5.0 Å, 1.44 Å−1 = 4.4 Å and 1.68 Å−1 = 3.7 Å respectively (Fig. 6). In line with earlier work this asymmetric peak appearance has been attributed as follows: the first of the three peaks at q = 1.25 = 5.0 Å is associated with the liquid correlations of the linear calamitic aromatic groups in the SmA phase. The remaining peaks at q = 1.44 Å−1 = 4.4 Å, the dominant peak in the wide angle region to the lateral distance of the molten aliphatic chains, and q = 1.68 Å−1 = 3.7 Å were attributed to intra-layer correlations of the large porphyrin groups. It is noted that the value of the peak at q = 1.68 Å−1 = 3.7 Å is at the value typically observed for the separation of extended aromatic ring systems in columnar phases.13 It can clearly be seen that the 4.4 Å peak dominates over the much less intense peak at 3.7 Å. For the low temperature smectic phase of 6-Zn(IIiiii) observed by both optical polarising microscopy and DSC, as well as X-ray diffraction experiments, it was not possible to assign a LC phase structure unambiguously. The XRD results indicate clearly the absence of a hexatic ordering and a partial crystallisation of sublayers as has been observed earlier (see ESI†).12a,c
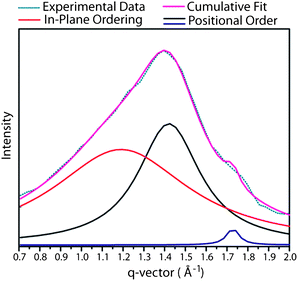 | ||
| Fig. 6 Example of the least squares fit to the broad scattering of compound 5. | ||
Molecular organisation
A model for the molecular organisation has to take into account the observation that the observed layer spacings are much smaller than the estimated molecular lengths of 91 Å and 68 Å respectively (see ESI†). Moreover the enhanced in-plane ordering imparted by the large planar aromatic core of the porphyrin macrocycle needs to be considered.20 Using the collected XRD data and well established methods the reconstructed electron density profiles were calculated.21 The models are described in detail in the ESI† (Fig. 13 and 14). A number of likely arrangements are possible and they are outlined in Fig. 7.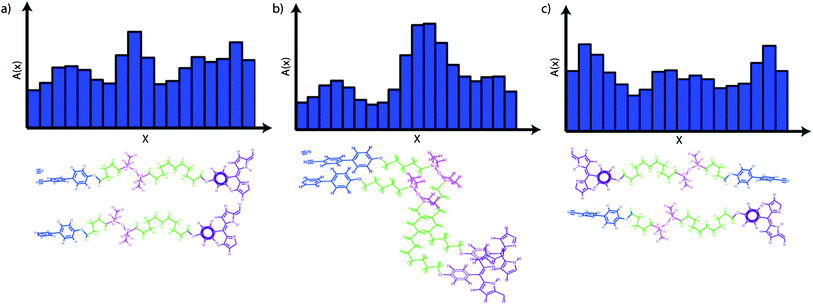 | ||
| Fig. 7 Composite models showing three potential molecular packing models and the corresponding cross-section profiles A(x). | ||
The three principal assembly modes are: sub-layer formation with regions of concentration of cyanobiphenyl-, siloxane- and porphyrin groups with some disorder in the hydrocarbon groups as shown in Fig. 7a (only one half of a molecule is shown), a sublayer formation as shown in Fig. 7a but with strongly tilted and highly ordered aliphatic chains (Fig. 7b) and the formation of antiparallel correlations as shown in Fig. 7c. Some related schematic assembly models are shown in Fig. 8. Though principally possible, antiparallel correlations shown in Fig. 7c resulting in a self-assembly behaviour shown in Fig. 8b seem to be the most unlikely model of the series. This is due to the smallest modulation in the estimated electron densities and mainly as there is very little shape amphiphilic microsegregation and a co-facial assembly of the disks which has been observed for multimesogenic systems, consisting of LC groups of different types before.12a,c A model of microsegregated rod shaped and disk shaped groups, as shown in Fig. 8a, is in our view the most likely arrangement and the building up of this assembly via some sub-layers with rather disordered hydrocarbon chains as shown in Fig. 7a seems to be the most likely case. This model also fits with the observation that the materials containing only one calamitic mesogen (materials 6, 6-Zn(IIiiii) and 6Ni-(IIii) have only slightly smaller layer spacings than the materials 5, 5-Zn(IIii) and 5-Ni(IIii), they show very similar OPM textures to the dimer mesogen materials and full miscibility in the liquid crystal state. As previously discussed by Bruce et al.10,11 co-facial interactions of the porphyrin macrocycles drive self-assembly in liquid crystalline 5,15-diphenyl porphyrin systems. Where this mechanism predominates, defects arising from the asymmetric substitution pattern in the molecule are much better tolerated than in a head to tail motif where inter-layer interactions should be much less favoured. It could even be expected that in this configuration a nematic phase would arise. Hence the model which is based on a co-facial arrangement of porphyrins with distinct cyanobiphenyl sublayering prohibiting columnar phase formation is favoured, as it can explain quite easily the observed properties both for the symmetric and non-symmetric compounds.
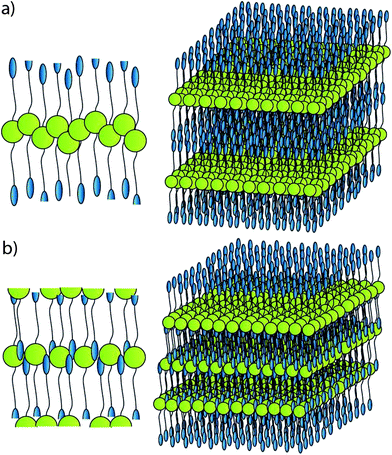 | ||
| Fig. 8 (a) Proposed co-facial packing model for a single sub-layer unit cell and the associated proposed bulk packing model. (b) Proposed head to tail packing model showing a single sub-layer unit cell and the associated proposed bulk packing model. | ||
Conclusions
In this report we present the first examples of liquid crystalline porphyrin based materials displaying lamellar phases at room temperature and being stable over large temperature ranges. Furthermore we provide a model for the mechanisms driving assembly in porphyrin based liquid crystalline systems. The porphyrin macrocycle dominates liquid crystal phase formation and strong interactions govern the nature of the phase formed. Variation of the macrocycle structure and the connection to calamitic mesogens via hydrocarbon siloxane spacers allow for a suppression of the crystalline phase behaviour and the formation of a SmA phase up to 178 °C. The selected architecture is robust, and the inclusion of metals does not change the transitions significantly. This design, maximizing metalated porphyrin interactions within layers whilst controlling the organisation of these moieties in smectic layers, might provide a useful tool to enhance the utilisation of porphyrin based materials.Notes and references
- (a) The Porphyrin Handook, ed. K. M. Kadish, K. M. Smith and R. Guillard, Academic Press, San Diego, 2000 Search PubMed; (b) L. R. Milgrom, Colours of Life, Oxford University Press, Oxford, 1997 Search PubMed.
- (a) S. D. Straight, G. Kodis, Y. Terazono, M. Hambourger, T. A. Moore, L. A. Moore and D. Gust, Nat. Nanotechnol., 2008, 3, 280–283 CrossRef CAS; (b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS.
- Metalloporphyrins in Catalytic Oxidations ed. R. A. Sheldon, Marcel Dekker Inc., New York, 1994 Search PubMed.
- (a) L. B. Josefsen and R. W. Boyle, Met.-Based Drugs, 2008, 1, 276109 Search PubMed; (b) I. J. MacDonald and T. J. J. Dougherty, J. Porphyrins Phthalocyanines, 2001, 5, 105–129 CrossRef CAS; (c) E. D. Sternberg, D. Dolphin and C. Bruckner, Tetrahedron, 1998, 54, 4151–4202 CrossRef CAS.
- S. Griveau, V. Albin, T. Pauporte, J. H. Fagal and F. Bedioui, J. Mater. Sci., 2002, 12, 225–232 CAS.
- J. Mosinger, O. Jirsak, P. Kubat, K. Lang and B. Mosinger, J. Mater. Chem., 2007, 17, 164–166 RSC.
- (a) J. W. Goodby, P. S. Robinson, B. K. Teo and P. E. Cladis, Mol. Cryst. Liq. Cryst., 1980, 56, 303–309 CrossRef CAS; (b) M. Castella, F. Lopez-Calahora and D. Velasco, Liq. Cryst., 2002, 29, 559–565 CrossRef CAS; (c) M. Castella, F. Lopez-Calahora, D. Velasco and H. Finkelmann, Chem. Commun., 2002, 2348 RSC; (d) A. Segade, M. Castella, F. Lopez-Calahora and D. Velasco, Chem. Mater., 2005, 17, 5366–5374 CrossRef CAS; (e) C. M. Drain, A. Varotto and I. Radivojevic, Chem. Rev., 2009, 109, 1630–1658 CrossRef CAS.
- (a) B. A. Gregg, M. A. Fox and A. J. Bard, J. Am. Chem. Soc., 1989, 111, 3024–3029 CrossRef CAS; A. Nowak-Krol, D. Gryko and D. T. Gryko, Chem.–Asian J., 2010, 5, 904–909 Search PubMed.
- (a) Y. Shimizu, A. Ishikawa and S. Kusabayashi, Chem. Lett., 1986, 7, 1041–1044 CrossRef; (b) A. S. Ahsen, A. Segade, D. Velasco and Z. Z. Ozturk, J. Porphyrins Phthalocyanines, 2009, 13, 1188–1195 CrossRef CAS; J. Z. Li, T. Tang, F. Li and M. Li, Dyes Pigm., 2007, 77, 395–401 Search PubMed; (c) I. N. Fedulova, N. A. Bragina, N. V. Novikov, A. F. Mironov, V. V. Bykova, N. V. Usul'tseva and G. A. Ananeva, Mendeleev Commun., 2008, 18, 324–326 CrossRef CAS.
- (a) D. W. Bruce, D. A. Dunmur, L. S. Santa and M. A. Wali, J. Mater. Chem., 1992, 2, 363–364 RSC; (b) D. W. Bruce, M. A. Wali and Q. M. Wang, J. Chem. Soc., Chem. Commun., 1994, 2089–2090 RSC.
- (a) Q. M. Wang and D. W. Bruce, Chem. Commun., 1996, 2505–2506 RSC; (b) Q. M. Wang and D. W. Bruce, Tetrahedron Lett., 1996, 45, 7641–7644 CrossRef.
- (a) P. H. J. Kouwer and G. H. Mehl, J. Mater. Chem., 2009, 19, 1564–1575 RSC; (b) P. H. J. Kouwer, J. Pourzand and G. H. Mehl, Chem. Commun., 2004, 66 RSC; (c) P. H. J. Kouwer and G. H. Mehl, Angew. Chem., Int. Ed., 2003, 42, 6015 CrossRef CAS; (d) H. K. Bisoyi, V. A. Raghuathan and S. Kumar, Chem. Commun., 2009, 7003–7006 RSC; (e) S. K. Pal, S. Kumar and J. Seth, Liq. Cryst., 2008, 35, 521–525 CrossRef CAS; (f) K. U. Jeong, A. J. Jing, B. Mansdorf, M. J. Graham, D. K. Yang, F. W. Harris and S. Z. D. Cheng, Chem. Mater., 2005, 19, 2921 CrossRef; (g) M. L. Rahman, C. Tschierske, M. Yusoff and S. Silong, Tetrahedron Lett., 2005, 46, 2303–2306 CrossRef CAS; (h) C. H. Yu, C. Schubert, B. J. Tang, C. Welch, M. G. Tamba and G. H. Mehl, J. Am. Chem. Soc., 2012, 134, 5076–5079 CrossRef CAS; (i) C. Schubert, M. G. Tamba and G. H. Mehl, Chem. Commun., 2012, 48, 6851–6853 RSC.
- (a) S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hagele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem., Int. Ed., 2007, 46, 4832–4887 CrossRef CAS; (b) C. T. Imrie and P. A. Henderson, Chem. Soc. Rev., 2007, 36, 2096–2124 RSC; (c) Chemistry of Discotic Liquid Crystals, ed. S. Kumar, CRC Press, Boca Raton, 2011 Search PubMed.
- (a) G. H. Mehl and J. W. Goodby, Chem. Commun., 1999, 13 RSC; (b) K. J. Shepperson, P. H. J. Kouwer, W. F. Jager, W. J. Mijs, S. J. Picken and G. H. Mehl, Mol. Cryst. Liq. Cryst., 2004, 411, 387 CrossRef.
- (a) B. D. Karstedt, US Pat., 3775452, Dow Corning 1973; (b) D. Apreutesei and G. H. Mehl, J. Mater. Chem., 2007, 44, 4711–4715 RSC.
- R. W. Boyle, C. Bruckner, J. Posakony, B. R. James and D. Dolphin, Org. Synth., 1999, 287–293 CAS.
- K. J. Shepperson, T. Meyer and G. M. Mehl, Mol. Cryst. Liq. Cryst., 2004, 411, 1227–1233 CAS.
- R. Alvarez and G. H. Mehl, Mol. Cryst. Liq. Cryst., 2005, 439, 2125–2133 CAS.
- T. P. Wijesekera and D. Dolphin in Metalloporphyrins in Catalytic Oxidations, ed. R. A. Sheldon, Marcel Dekker Inc., New York, 1994. p218 Search PubMed.
- L. M. Blinov, T. A. Lobko, B. I. Ostrovskii, S. N. Sulianov and F. G. Tournilhac, J. Phys. II, 1993, 3(7), 1121–1139 CrossRef CAS.
- (a) G. Ungar, K. Nobel and V. Percec, J. Mater. Sci., 2000, 35, 5241–5246 CrossRef CAS; (b) B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Percec, Chem. Rev., 2009, 109(11), 6275–6540 CrossRef CAS; (c) B. M. Rosen, D. A. Wilson, C. J. Wilson, M. Peterca, B. C. Won, C. Huang, L. R. Lipski, X. Zeng, G. Ungar, P. A. Heingy and V. Percec, J. Am. Chem. Soc., 2009, 131(47), 17500–17521 CrossRef CAS; (d) M. Peterca, M. R. Immam, P. Leowanawat, B. M. Rosen, D. A. Wilson, C. J. Wilson, X. Zeng, G. Ungar, P. A. Heiney and V. Percec, J. Am. Chem. Soc., 2010, 132(32), 11288–11305 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2tc00286h |
| ‡ Current address: Institute for Molecules and Materials, Radboud University Nijmegen, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2013 |