DOI:
10.1039/C3AN01569F
(Paper)
Analyst, 2014,
139, 251-258
Paper-based colorimetric immunosensor for visual detection of carcinoembryonic antigen based on the high peroxidase-like catalytic performance of ZnFe2O4–multiwalled carbon nanotubes
Received
18th August 2013
, Accepted 4th October 2013
First published on 8th October 2013
Abstract
A new paper-based colorimetric immunosensor for the detection of carcinoembryonic antigen (CEA) was developed based on the intrinsic peroxidase activity of ZnFe2O4–multiwalled carbon nanotubes (ZnFe2O4@MWNTs). The immunosensor platform was prepared by depositing chitosan and porous gold onto filter paper and entrapping the primary antibodies (Ab1) onto the layers. Secondary antibodies (Ab2) were assembled on the surface of the functionalized ZnFe2O4@MWNTs. The immunosensor response was quantified as a color change resulting from ZnFe2O4@MWNTs catalyzing the oxidation of 3,3′,5,5′-tetramethylbenzidine in the presence of H2O2. The catalytic performance of ZnFe2O4@MWNTs was higher than ZnFe2O4 due to the high electrical conductance of MWNTs, moreover, the electron communications between ZnFe2O4@MWNTs and substrates are electrically “wired”. Detection was achieved by measuring the color change when the concentrations of CEA were different. The color change can be quantified with the naked eye but a digitalized picture can also be used to provide more sensitive comparison to a calibrated color scheme. This method was simple for CEA detection with a linear range from 0.005 to 30 ng mL−1 and a detection limit of 2.6 pg mL−1. Such an equipment-free immunoassay has great potential in resource-limited environments.
1. Introduction
Detection by means of colorimetric sensors is based on the change in color as a result of signal–analyte interaction. So the key challenge for colorimetric sensors is to transform the detection events into color changes. Recently, with the widespread development of nanoscience and technology, a number of smart materials including gold nanoparticles (AuNPs), magnetic nanoparticles (MNPs), cerium oxide nanoparticles and multiwalled carbon nanotubes (MWNTs) have shown much potential for colorimetric biosensing on account of their physical or chemical properties. Lou1 reported an AuNPs-based colorimetric method for the determination of mercury ions (Hg2+) and silver ions (Ag+) in which the color change from red to blue seems more eye-sensitive. But the color change based on AuNPs is highly dependent upon their size, shape, capping agents, and the medium refractive index2 which is difficult to control, they are also very expensive. These shortcomings limit their applications.
Peroxidase can catalyze the oxidation of substrates to produce a color change, thus it has great potential for practical application in many fields. The catalytic color reaction provides another means for colorimetric sensing. This type of assay has been reported initially by the Whitesides group for the simultaneous detection of glucose and bovine serum albumin (BSA) in urine based on the reaction of H2O2 with horseradish peroxidase (HRP) and potassium iodide, resulting in a color change from clear to brown.3 Unfortunately, natural enzymes have some intrinsic drawbacks such as low stability, sensitivity of catalytic activity to environmental conditions, difficulty in preparation and purification. To overcome these drawbacks, much effort has been devoted to developing peroxidase mimetics. There has been an explosive growth in the use of nanomaterials as catalysts over the past few decades because of their unique size, shape, composition and structure-dependent properties.4 Some metal oxide such as Co3O4 nanoparticles,5 V2O5 nanowires6 and CeO2 nanoparticles7 can catalyze the reaction of the peroxidase substrate to produce a color change. However, the application of these nanomaterials is restricted because of difficult separation and instability.
Recently, some MNPs like Fe3O48 and CoFe2O49 have been shown to exhibit significant peroxidase-like activity and they are more stable with exposure to air and can be separated by means of a magnet. However, the dispersibility is poor. Therefore it is crucial to look for stable, easily separated artificial enzyme mimetics. Subsequently, Su et al. have reported that ZnFe2O4,10 one of the spinel ferrite compounds, exhibits intrinsic peroxidase-like activity and is a promising peroxidase mimetic with better stability, dispersibility and ease of separation. However, due to the poor separation efficiency of electrons and holes, the catalytic activity of pure ZnFe2O4 is relatively weak. Pure ZnFe2O4 MNPs need to be modified in order to promote the charge-transfer process. MWNTs play an important role in catalysis because they are excellent electron acceptors and electronic conductors and can be used to prepare composite materials with some nanoparticles. These composites not only solve the problem of the smaller specific surface area of conventional catalysts, but also eliminate the problems associated with powder agglomerates of pure catalysts. Moreover, MWNTs alone have catalytic activity, as previously reported11 which may contribute to the catalytic performance of the composites.
Since color changes can be observed by the naked eye, colorimetric sensors do not require expensive or sophisticated instrumentation. The need for simple, economical, portable and integrated diagnostic methods with minimal-infrastructure is required in resource-limited settings such as those encountered by emergency first responders, primary-care physicians, patients at home and forensic investigators. Further in response to the need for such detection techniques, microfluidic paper-based devices (μPAD) are considered to be the ideal biosensor platforms to develop point-of-care diagnostic devices, because the paper is an inexpensive sensor platform.
Apart from the low cost, there are also some advantages such as small size, and reduced sample and reagent consumption.
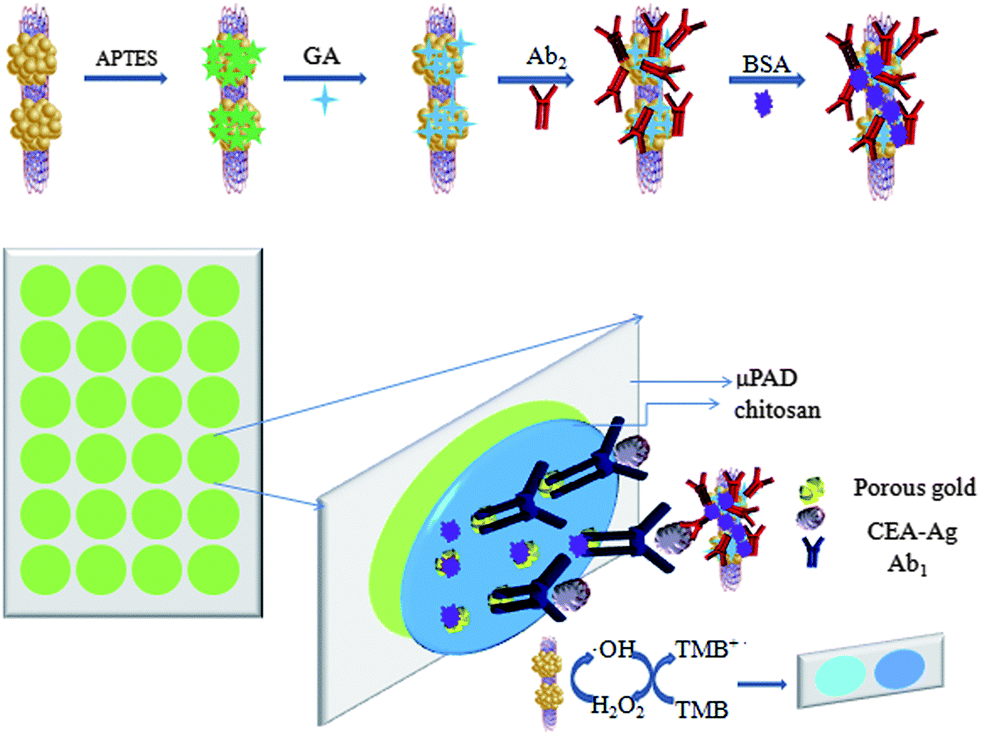
In this work, we report a one-step facile hydrothermal route to directly fabricate ZnFe2O4–multiwalled carbon nanotubes (ZnFe2O4@MWNTs) nanocomposites which exhibited good catalytic properties, stability, dispersibility, and rapid separation compared to HRP. To utilize the high peroxidase-like catalytic performance of ZnFe2O4@MWNTs, we used this composite to fabricate a novel paper-based colorimetric immunosensor for the determination of CEA successfully. Secondary antibodies (Ab2) were immobilized onto the surface of ZnFe2O4@MWNTs by (3-aminopropyl)triethoxysilane (APTES), glutaraldehyde (GA) as coupling agents. Primary antibodies (Ab1) were conducted in the paper microzones which were functionalized with chitosan and porous gold. The presence of CEA was indicated by a color change, the results may be easily interpreted by the naked eye without the need for complex instrumentation.
2. Materials and methods
2.1. Reagents
All reagents were of analytical-reagent grade or the highest purity available and directly used for the following experiments without further purification and the aqueous solutions unless indicated were prepared with ultrapure water. The standard CEA, Ab1 to CEA antigen and Ab2were purchased from Shanghai Linc-Bio Science Co. Ltd (Shanghai, China), and the standard CEA, Ab1, Ab2 stock solutions were prepared with phosphate-buffered saline (PBS, pH 7.4, 10 mM) and stored at 4 °C. BSA was bought from Shanghai Boao Bio-Technology Co., Ltd (Shanghai, China). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Alfa Aesar China Ltd. Whatman chromatography paper 1#(200 mm × 200 mm) was obtained from GE Healthcare World-wide (Pudong Shanghai, China) and used with further adjustment of size. The MWNTs were purchased from Nanoport Co. Ltd. (Shenzhen, China). Disodium hydrogen phosphate (Na2HPO4), potassium dihydrogen phosphate (KH2PO4), H2O2 (30%, w/v), polyethylene glycol-4000, glutaraldehyde, APTES, chitosan and zinc chloride (ZnCl2) were obtained from Chemical Reagent Co. (Tianjin, China). 3,3′,5,5′-Tetramethylbenzidine (TMB) was purchased from Sigma-Aldrich. The MWCNTs were purchased from Nanoport Co. Ltd. (Shenzhen, China). Sulphuric acid (H2SO4, 98%), nitric acid (HNO3), sodium acetate (NaAc), acetic acid (HAc), FeCl3·6H2O and ethylene glycol were obtained from Sinopfarm Chemical Reagent Co., Ltd (Shanghai, China). The washing buffer was PBS (10 mM, pH 7.4), PBS of various pH was prepared by adjusting the ratio of Na2HPO4 to KH2PO4. The clinical serum samples were provided by Shandong Tumor Hospital.
2.2. Apparatus
A Fourier transform infrared (FT-IR) Spectrum RX (PerkinElmer Spectoment) instrument was used for infrared spectroscopy (IR) measurements. Scanning electron microscope (SEM) images was obtained using a JSM-6700F microscope (Japan). Transmission electron microscope (TEM) images were obtained using a JEOL JEM-1400 microscope (Japan). The microzone plate patterns were predesigned on a personal computer using CorelDraw X6. The screen matches with the designed microzone plate pattern were fabricated by assembling 200 mesh of nylon on an aluminium frame at a local printing shop. UV-vis absorption spectra were recorded on a Shimadzu UV-2550 UV-vis spectrophotometer (Shimadzu, Japan). The photoluminescence (PL) spectra were obtained on a LS-55 spectrofluorometer (P.E. USA).
2.3. Fabrication of paper microzone plate
The paper microzone plate was fabricated using a method developed recently in our laboratory. The model paper microzone plate in this work had a diameter of 6 mm with a center-to-center distance of 8 mm. The screen with a specific pattern was placed close to a piece of chromatography paper. Then a solid wax which was used as a squeegee was rubbed through the screen stencil to form wax patterns on the paper surface. The wax-screen-printed paper was placed in an oven set at 130 °C for 150 s, and then the wax melted and penetrated the thickness of the paper to create completely hydrophobic barriers in the paper to form the microzones. There was no need for any other complicated instruments such as a wax-printer.
2.4. Synthesis of ZnFe2O4@MWNTs
MWNTs were initially sonicated in a mixture solution containing H2SO4 and HNO3 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) for 4 h, and then the mixture was filtered and washed repeatedly with ultrapure water until the pH was about 7.0. During this process, carboxyl groups were generated on the MWNT surface.
:1, v/v) for 4 h, and then the mixture was filtered and washed repeatedly with ultrapure water until the pH was about 7.0. During this process, carboxyl groups were generated on the MWNT surface.
The synthesis of ZnFe2O4@MWNTs nanocomposite catalyst with 10% MWNTs, was as follows:12 0.34 g of ZnCl2, 1.35 g of FeCl3·6H2O and 62 mg of acidified MWNTs were added to 40 mL ethylene glycol with sonication for 30 min at room temperature to form a clear solution. Then 3.6 g of NaAc and 1.0 g of polyethylene glycol-4000 were added. The mixture was stirred vigorously for 30 min, yielding a stable bottle-yellow homogeneous emulsion. The resulting mixture was sealed in a Teflon-lined stainless-steel autoclave and heated to 200 °C for 8 h, followed by cooling to room temperature. The black products were washed several times with ethanol and dried at 60 °C for 3 h. For comparison, the same method was used to synthesize pure ZnFe2O4.
2.5. Bioconjugation of catalyst with secondary antibodies
10 mg ZnFe2O4@MWNTs were dispersed in a mixture of 20 mL of ethanol and 5 mL of water by sonication. APTES (5 μL) was then added, and the mixture was mechanically stirred for 7 h. Next, it was washed with ethanol to remove surplus APTES with the aid of magnet. 1 mg of APTES modified ZnFe2O4@MWNTs hybrid was dispersed in PBS containing 3 mL of 2.5% GA solution. The mixture was mechanically stirred for 2 h, and the GA modified ZnFe2O4@MWNTs (ZnFe2O4@MWNTs/APTES/GA) hybrids were magnetically washed with ultrapure water. 10 μL EDC (20 mg mL−1) and NHS (20 mg mL−1) were added to the solution and stirred for 1 h to activate the carboxyl groups. This was followed by washing twice with PBS then 30 μg mL−1 of Ab2 was added to the solution to react for 45 min at room temperature under stirring. Following that, 10 μL 1% BSA solution was added to block the possible remaining active sites followed by washing with PBS. The resulting compounds were washed with PBS and dispersed in PBS then stored at 4 °C before use.
2.6. Fabrication of proposed immunosensor
To adapt the colorimetric immunosensor to a paper format, the corresponding captured antibodies were immobilized onto different paper zones on one paper microzone plate through chitosan coating and porous gold cross-linking. Typically, as shown in Scheme 1, 5.0 μL of 1% chitosan was dropped onto each paper zone and dried at room temperature. To increase the effective surface area of the working zones, 5.0 μL of porous gold (that was fabricated using a method developed in our laboratory13) was added and dried in the air. Porous gold was entrapped within the chitosan layer through complexation. Each working zone was washed twice with PBS to the top of each paper zone, and then the excess PBS was brought in contact with a piece of blotting paper from the bottom of the paper zones. This effective washing procedure was used in this work consistently. Subsequently, 5 μL of Ab1 (30 μg mL−1) was added to each functional paper zone and incubated at room temperature for 1 h, followed by washing three times with PBS to remove redundant Ab1. Then, 10 μL of 1% BSA was applied to each paper zone and incubated for 30 min at room temperature to block nonspecific binding sites. Finally, the antibodies modified paper zones were washed and stored at 4 °C before use.
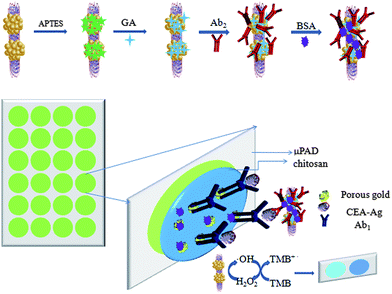 |
| | Scheme 1 Schematic representation of the fabricated paper-based colorimetric immunosensor. | |
2.7. CEA detection with the colorimetric immunosensor
The colorimetric detection of CEA could be achieved using functionalized ZnFe2O4@MWNTs instead of the traditional HRP. 4 μL of different concentrations of CEA were added to each corresponding paper zone and allowed to incubate for 40 min. Each paper zone was then washed using the procedure mentioned above. 5 μL of ZnFe2O4@MWNTs/APTES/GA/Ab2/BSA compounds were added to the corresponding paper zones and incubated for 40 min. After rinsing and drying, 10 μL of 20 mM TMB, 5 μL of 2% H2O2 and 10 μL of 0.2 mM HAc–NaAc buffer (pH 4.0) were added to each paper zone and incubated for 20 min at 30 °C then kept in an ice-water bath for 10 min to terminate the reaction. A fast color reaction occurs, which can be easily judged by the naked eye. The grey intensity was measured and used to construct the calibration curve which was used to determine the analytical performance of the immunosensor.
3. Results and discussion
3.1. Characterization of ZnFe2O4 and ZnFe2O4@MWNTs
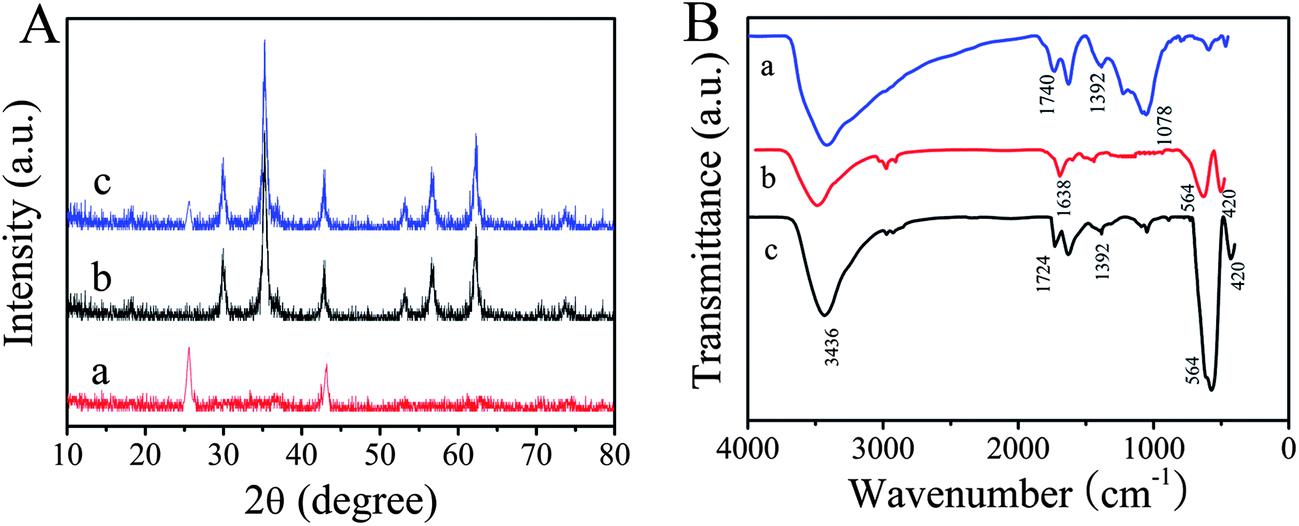
X-ray diffraction (XRD) patterns of the as-prepared MWNTs, ZnFe2O4 and ZnFe2O4@MWNTs are shown in Fig. 1A. In curve a, the peaks at 2θ values of 25.7° and 43.2° were the (002) and (100) characteristic graphitization wall crystal planes of MWNTs. In curve b, the peaks at 2θ values of 29.9, 35.2, 42.8, 53.2, 56.8, and 62.4° can be indexed to (220), (311), (400), (422), (511), and (440) crystal planes of spinel ZnFe2O4, respectively. All the diffraction peaks can be directly indexed to spinel-type ZnFe2O4 (JCPDS 22-1012). According to Sherrer's formula, the average crystallite size, which was calculated based on the XRD pattern (311), was approximately 21 nm. But in curve c, it can be seen that the typical diffraction peaks of MWNTs (002)/(100) were weak/disappeared and the diffraction peaks of spinel-type ZnFe2O4 were observable. The disappearance of diffraction peak (100) of MWNTs was attributed to the crystal growth of ZnFe2O4 on MWNTs. These results indicated that the MWNTs were decorated with ZnFe2O4 MNPs during the hydrothermal reaction.
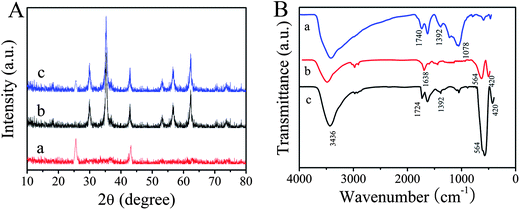 |
| | Fig. 1 (A): XRD patterns of the as-prepared MWNTs (a), ZnFe2O4 (b) and ZnFe2O4@MWNTs (c). (B): FT-IR spectra of MWNTs (a), ZnFe2O4 (b) and ZnFe2O4@MWNTs (c). | |
FT-IR was used to identify the functional groups of the compounds. Fig. 1B shows the FT-IR spectra of the MWNTs (curve a), pure ZnFe2O4 (curve b) and ZnFe2O4@MWNTs (curve c). In curve a, the peak located at 1740 cm−1 could be attributed to characteristic bands of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of carboxylic acid and carbonyl moieties, the characteristic band at 1392 cm−1 corresponded to O–H group and the peak appearing at around 1078 cm−1 is due to C–O. The results indicated that carboxylic groups were generated on the surface of the MWNTs. For ZnFe2O4, the two strong absorption peaks at around 564 and 420 cm−1 were attributed to the stretching vibrations of the Zn–O bonds in tetrahedral positions and the Fe–O bonds in octahedral positions respectively.14 The band at 3436 cm−1 and 1638 cm−1 can be attributed to the stretching vibration and bending vibration of O–H which indicated that a large amount of O–H groups were generated on the surface of ZnFe2O4. Meanwhile, the spectrum of ZnFe2O4@MWNTs, wasalmost completely exist made up of Zn–O Fe–O stretching vibrations, O–H stretching vibration and bending vibration, indicating that ZnFe2O4 was loaded on the surface of the MWNTs. In addition, CO stretching vibrations of carboxyl groups were blue-shifted to 1724 cm−1 which further demonstrated that ZnFe2O4 was loaded on the surface of the MWNTs.
O of carboxylic acid and carbonyl moieties, the characteristic band at 1392 cm−1 corresponded to O–H group and the peak appearing at around 1078 cm−1 is due to C–O. The results indicated that carboxylic groups were generated on the surface of the MWNTs. For ZnFe2O4, the two strong absorption peaks at around 564 and 420 cm−1 were attributed to the stretching vibrations of the Zn–O bonds in tetrahedral positions and the Fe–O bonds in octahedral positions respectively.14 The band at 3436 cm−1 and 1638 cm−1 can be attributed to the stretching vibration and bending vibration of O–H which indicated that a large amount of O–H groups were generated on the surface of ZnFe2O4. Meanwhile, the spectrum of ZnFe2O4@MWNTs, wasalmost completely exist made up of Zn–O Fe–O stretching vibrations, O–H stretching vibration and bending vibration, indicating that ZnFe2O4 was loaded on the surface of the MWNTs. In addition, CO stretching vibrations of carboxyl groups were blue-shifted to 1724 cm−1 which further demonstrated that ZnFe2O4 was loaded on the surface of the MWNTs.
The morphology of the as-synthesized MWNTs, ZnFe2O4 and ZnFe2O4@MWNTs were observed using TEM and are shown in Fig. 2. It can be observed that the MWNTs had a uniform tubulose nanostructure and good dispersion after treatment with mixed acid as shown in Fig. 2A. The obtained ZnFe2O4 were spherical and dispersive with an average size of about 80–100 nm and the surface seems to be rough. These results confirm the fact that the porous ZnFe2O4 were made up of small building blocks of MNPs with a size of about 21 nm, which was consistent with the XRD data analysis as shown in Fig. 2B. Fig. 2C displays the TEM image of ZnFe2O4@MWNTs in which the MWNTs were decorated with ZnFe2O4 and no obvious aggregation, the porous structure of the shell of ZnFe2O4 also can be observed clearly. Accordingly, the formation route to anchor ZnFe2O4 onto the MWNTs may be proposed as being the intercalation and adsorption of zinc and iron ions into the MWNTs, followed by the nucleation and growth of ZnFe2O4. With the porous structure, high electronic conductivity and larger surface area, ZnFe2O4@MWNTs can offer more active adsorption sites and catalytic reaction centers.
 |
| | Fig. 2 TEM image of the as-synthesized MWNTs (A), ZnFe2O4 (B) and ZnFe2O4@MWNTs (C). | |
3.2. Characterization of the modified papers
The morphologies of the modified paper were characterized by SEM. The smooth microfibers of pure cellulose paper are shown in Fig. 3A. Fig. 3B shows that the porous structures of the paper disappeared and that layers of chitosan film were then formed because chitosan were attached onto the cellulose paper through electrostatic adsorption and hydrogen bonding due to their surface charges and structural similarities. We can clearly see that a layer of porous gold film was bound on the chitosan layer by means of amino groups as shown in Fig. 3C.
 |
| | Fig. 3 (A): SEM images of pure paper fibers, (B): chitosan assembled paper fibers, and (C): porous gold/chitosan modified paper fibers, the inset is an enlargement of (C). | |
3.3. Mechanism of peroxidase-like activity of ZnFe2O4@MWNTs
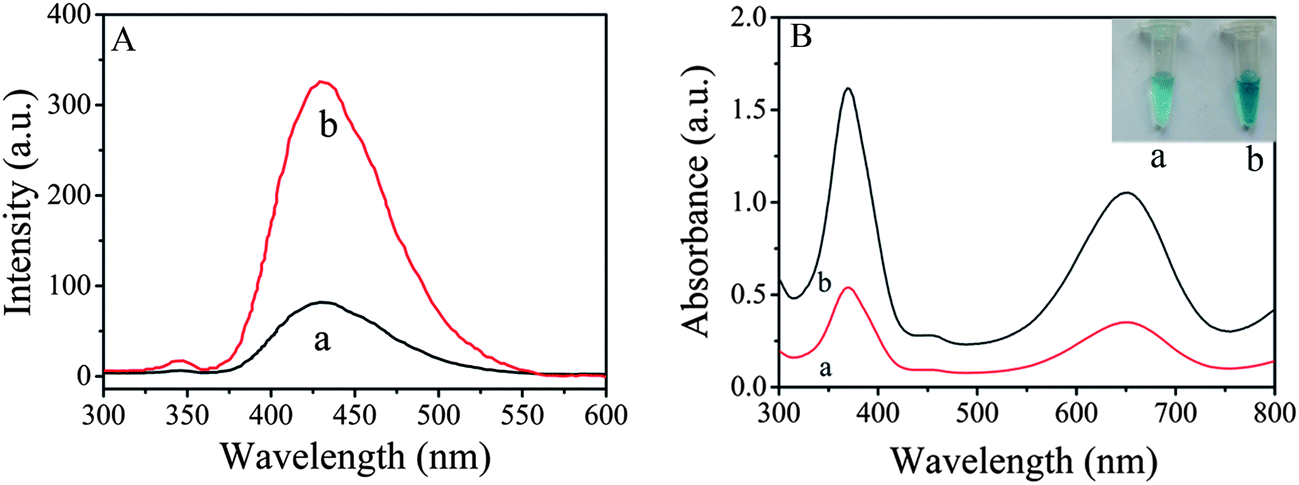
Fig. 4A shows fluorescence spectra of the supernatant solutions of the same amount of ZnFe2O4 (curve a) and ZnFe2O4@MWNTs (curve b) containing 3 mM terephthalic acid (TA) and 2% H2O2 in 0.2 mM HAc–NaAc buffer (pH 4.0). The spectral feature was 429 nm, where TA easily reacted with ˙OH to form 2-hydroxy terephthalic acid (TAOH). We may draw the conclusion that TAOH was generated during the reaction process of TA with ˙OH. It also can be seen that the fluorescence intensity of ZnFe2O4@MWNTs was higher than that of pure ZnFe2O4. The significant enhancement in activity can be attributed to the fact that not only can the MWNTs and ZnFe2O4 both generate strong oxidant ˙OH via decomposition of H2O2, but also promote the effective separation of photogene rated charge carriers through the remarkable synergistic effect of the combination of ZnFe2O4 and MWNTs. In addition, the generated ˙OH was stabilized by ZnFe2O4via partial electron exchange interaction which may enhance the catalytic ability.9
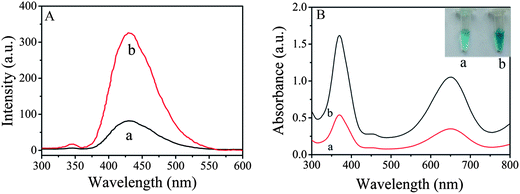 |
| | Fig. 4 (A): The effect of ZnFe2O4 (a) and ZnFe2O4@MWNTs (b) on the formation of hydroxyl radical with TA as a fluorescence probe. Reaction conditions: 3 mM TA and 2% H2O2 in 0.2 mM HAc–NaAc buffer (pH 4.0). (B): UV-vis absorption spectra of TMB–H2O2 mixed solution in the presence of BSA, Ab2, GA and APTES modified ZnFe2O4 (a) and ZnFe2O4@MWNTs (b). Experimental conditions: 20 mM TMB, 2% H2O2, 0.2 mM acetate buffer (pH 4.0). The inset of (B) is the images of (a) and (b). | |
A possible mechanism for the catalytic activity of ZnFe2O4@MWNTs is as follows: First, electron hole pairs are generated upon visible-light excitation on the ZnFe2O4 surface, followed by instant transfer of electrons to MWNTs via a percolation mechanism and the negatively charged MWNTs activate the hydrogen peroxide to produce OH radical. Then the electrons are directly trapped by the Fe3+ to form Fe2+, which can react with H2O2 to produce OH radical. TMB is oxidized by ˙OH to form a blue colored product. The blue signal comes from the charge-transfer complex, which consists of a cation free radical and TMB.15
| | | ZnFe2O4 + hν → ZnFe2O4 + (h + e) | (1) |
| | | ZnFe2O4 (e) + MWNTs → MWNTs (e) + ZnFe2O4 | (2) |
| | | MWNTs (e) + H2O2 → ˙OH + OH− | (3) |
| | | Fe2+ + H2O2 → Fe3+ + ˙OH + OH− | (5) |
| | | ˙OH + TMB + H+ → TMB˙+ + H2O | (6) |
3.4. Peroxidase-like activity of ZnFe2O4@MWNTs/APTES/GA/Ab2/BSA
Previous studies have shown that ZnFe2O4 possesses an intrinsic peroxidase-like activity10 and the enzyme activity decreases after surface modification. A number of factors such as the size and density of packing of the modifying groups may influence the extent to which the surface-modifying groups shield the surface from the substrate and affect activity.8 To investigate the peroxidase-like activity of immunosensors (BSA, Ab2, GA and APTES modified ZnFe2O4 and ZnFe2O4@MWNTs), the catalytic oxidation of peroxidase substrate TMB was tested in the presence of H2O2 using the same amount of immunosensors. As shown in Fig. 4B, we observed that each centrifuge tube showed a blue color indicating that these modifying groups did not affect the catalytic property of the immunosensors. However, the absorption intensity of centrifuge tube a was lower than that of centrifuge tube b, this result indicated that MWNTs play an important role in the catalysis progress due to the high electrical conductance and their inherent catalytic properties. Because of the high affinity between TMB and MWNTs, large numbers of TMB molecules were absorbed.16 In addition, electron communications between ZnFe2O4 and substrates are electrically “wired” which is attributed to the high electrical conductance.17
3.5. Optimization of experimental conditions
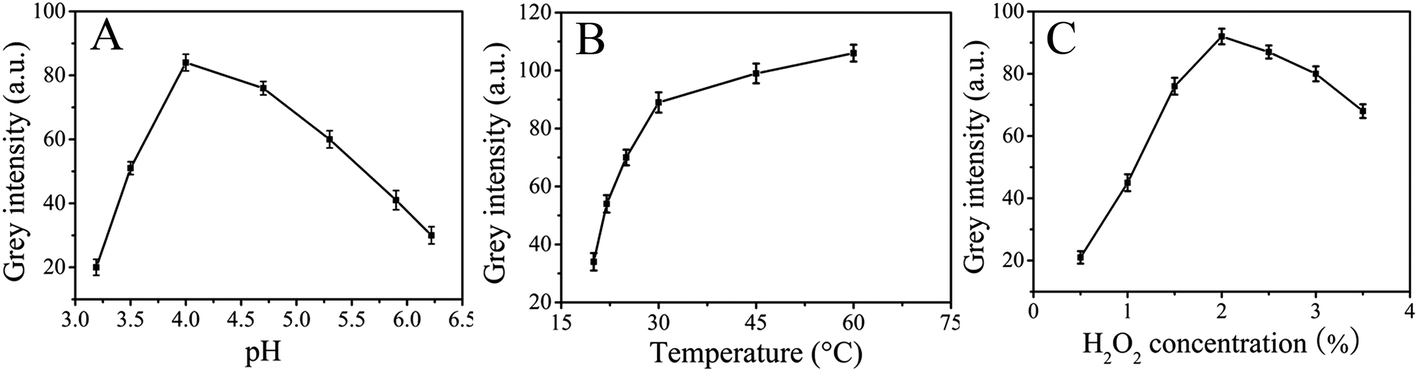
The grey intensity of paper-based colorimetric immunosensors is strongly dependent on pH, temperature and H2O2 concentration. We examined the influence of pH in the range 3.19–6.22. Fig. 5A shows the pH-dependent response curve which indicated that the catalytic oxidation of TMB with the immunosensor in the presence of H2O2 was much faster when pH 4.0 buffer solution was used. When pH < 4.0, the iron ions could be leached from the ZnFe2O4 of the immunosensor, however, when pH > 4.0, nearly no catalytic activity could be detected. In view of the high grey intensity of the catalytic reaction and to ensure that the catalytic activity originated from modified ZnFe2O4@MWNTs themselves, 0.2 mM pH 4.0 acetate buffer solution was taken as the optimal buffer solution. To examine the influence of the reaction temperature, catalytic reactions were investigated at different temperatures from 20 to 60 °C as shown in Fig. 5B. The grey intensity of the immunosensor in the presence of TMB–H2O2 increased with increasing temperature. Thus, we chose 30 °C as the reaction temperature. We also investigated the influence of H2O2 concentration in the range of 0.5% to 3.5% (v/v) as shown in Fig. 5C. For a certain amount of immunosensor, the number of active sites on its surface was limited under certain conditions. When the H2O2 concentration was relatively low, the surface active sites of the immunosensor were not fully occupied by H2O2 and few ˙OH radicals were generated. As a consequence the grey intensity was lower. When the H2O2 concentration increased, abundant H2O2 was adsorbed on the immunosensor surface which could generate more ˙OH radicals. When the H2O2 concentration reached a certain value, the amount of H2O2 achieved dynamic saturation and the grey intensity would reach saturate. Thus, 2% was chosen as the optimum concentration.
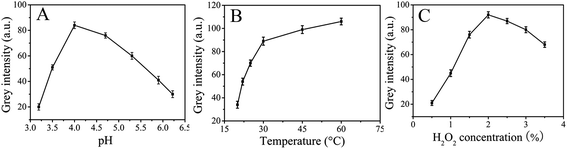 |
| | Fig. 5 (A): Effect of the pH, (B): effect of the temperature, and (C): effect of the H2O2 concentration. | |
3.6. Sensitivity of the immunosensor
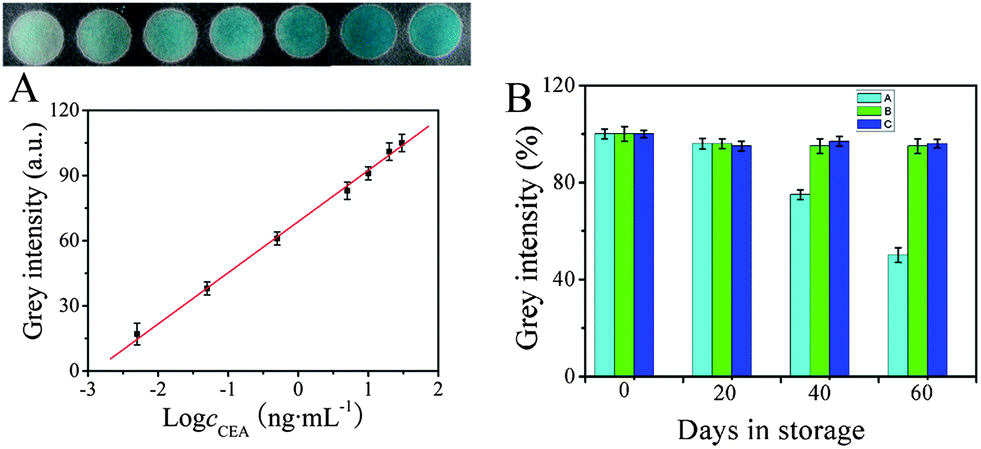
Under the optimized conditions, a series of immunosensors were prepared for the detection of different concentrations of CEA. 5 μL of ZnFe2O4@MWNTs/APTES/GA/Ab2/BSA compounds were added to the corresponding paper zones. Following that, H2O2, TMB and buffer were added to each paper zone and incubated for 20 min at 30 °C then kept in an ice-water bath for 10 min to terminate the reaction. The digital images were acquired on a scanner after the zones had dried and the maximum grey intensity of each zone was measured using ImageJ software. It was clearly observed that the grey intensity of each immunosensor was proportional to the logarithm of the CEA concentration in the range of 0.005–30 ng mL−1. As seen in Fig. 6A, when the CEA concentration was low, a feeble result (with a color similar to that observed on the zone) was obtained on the corresponding test zone, while increasing concentrations of CEA resulted in an increase in the grey intensity. Moreover, as shown in the calibration curve, the limit of detection of CEA was determined to be 2.6 pg mL−1 (determined using the 3σs/S criteria, where S is the slope of the linear calibration curve, and σs is the standard deviation of intensity in a blank solution, n = 11). The low detection limit, large linear range and sensitivity of the immunosensor may be attributed to such factors. (I): more chitosan molecules were strongly attached onto the cellulose paper by electrostatic adsorption and hydrogen bonding.18,19 As a result, more porous gold was bound on the chitosan layer through amino groups. Consequently, a great deal of Ab1 was stabilized effectively. (II): more importantly, the high sensitivity could also be attributed to the high peroxidase-like activity of the modified ZnFe2O4@MWNT which has a greater surface-to-volume ratio as well as a high affinity for molecules to interact with substrates.16 In addition, in comparison with other previously reported methods for nanomaterials-based immunosensing of CEA as shown in Table 1, the proposed immunosensor had a relative large linear range and low detection limit, and is a promising sensitive method for the quantification of CEA.
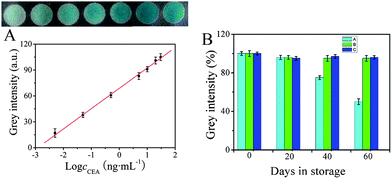 |
| | Fig. 6 (A): Colorimetric response of the immunosensors to various concentrations of CEA (ng mL−1): (a): 0.005, (b):0.05, (c):0.5, (d): 5, (e): 10, (f): 20, (g): 30 and CEA calibration curve plotted on a logarithmic scale. (B): The stability of the immunosensor at room temperature (A), in a refrigerator (B) and in a freezer (C). | |
3.7. Reproducibility and stability of the immunosensor
As reproducibility is a very important feature for an immunosensor, we evaluated the reproducibility with intra- and inter-assay precision. To evaluate the inter-assay precision, a series of five immunosensors prepared independently but following the same experimental protocol were used for the detection of 5 ng mL−1 CEA. The average grey intensity after the addition of TMB–H2O2 was 83 with a calculated relative standard deviation (RSD) of 6.5%. A series of five immunosensors prepared from the same batch were used to evaluate the intra-assay precision at a concentration level of 5 ng mL−1, the grey intensity was 89 with a calculated RSD of 7.4%. These results demonstrated the acceptable reproducibility and precision of the proposed immunosensor.
The storage stability of the immunosensors was evaluated by measuring the colorimetric response of the modified papers and the ZnFe2O4@MWNTs/APTES/GA/Ab2/BSA stored at room temperature (25 °C), in a refrigerator (4 °C) and in a freezer (−20 °C). Fig. 6B shows the average storage stability under three conditions over a period of 60 days. 5 ng mL−1 CEA was used for each test and each experiment was performed in triplicate. The stability of the immunosensors stored at room temperature was 96% of the initial response in the first 20 days and then decreased to 50% in the next 40 days. A remarkable average stability of up to 96% and 95% of the initial response was retained after 60 days of storage in the refrigerator and freezer. The results demonstrate the excellent storage stability of the immunosensor.
3.8. Application to sample analysis
The performance of the immunosensor was tested in the analysis of human serum. The serum was provided by Shandong Tumor Hospital and was analysed using the immunosensor. The results were compared with reference to a commercialized enzyme-linked immunosorbent assay (ELISA) method and are shown in Table 2. The relative difference between the two methods indicated that there was no significant difference between the results obtained by the two methods. The standard addition method was also used to evaluate the practical application of this immunosensor. Serum samples were prepared by adding 5.0 ng mL−1 of CEA to human serum samples. The recoveries indicated that this immunosensor may hold great promise as a viable alternative method for the determination of CEA in real clinical serum samples. As a simple, easy-to-perform, naked eye and rapid detection immunosensor, this method could be used for further assessment of the health status of the individual at home by themselves and may be useful for rural populations where clinical laboratory facilities are limited.
Table 1 Analytical performances of various nanomaterials-based CEA immunosensors
| Nanomaterial |
Linear range/ng mL−1 |
LOD/ng mL−1 |
Ref. |
| Graphene nanocomposites |
0.5–60 |
0.1 |
20
|
| Fe3O4@Ag coated carbon paste |
1.5–200 |
0.5 |
21
|
| SiO2@C-dots |
0.01–50 |
0.006 |
22
|
| Gold–silver hollow microspheres |
0.005–50 |
0.001 |
23
|
| ZnFe2O4–multiwalled carbon nanotubes |
0.005–30 |
0.0026 |
This work |
Table 2 CEA determination in serum by the paper-based colorimetric immunosensor
|
|
1 |
2 |
3 |
4 |
5 |
|
The average value of seven successive determinations.
|
| Founda/ng mL−1 |
0.032 |
3.64 |
10.33 |
17.56 |
24.32 |
| RSD |
4.12 |
3.24 |
2.61 |
2.85 |
3.36 |
| Spike/ng mL−1 |
5.0 |
5.0 |
5.0 |
5.0 |
5.0 |
| Proposed immunosensora/ng mL−1 |
5.059 |
8.81 |
15.21 |
22.68 |
29.03 |
| Recovery (%) |
100.54 |
103.4 |
97.6 |
102.4 |
94.2 |
| ELISAa |
0.034 |
3.51 |
10.18 |
17.92 |
25.11 |
| Relative difference (%) |
−5.88 |
3.70 |
1.47 |
−2.01 |
−3.15 |
4. Conclusions
ZnFe2O4@MWNTs were prepared through a one-step hydrothermal approach. The significant enhancement in catalytic activity can be ascribed to the efficient separation of electron carriers in the ZnFe2O4 and MWNTs coupling system, and the concerted effects of individual components on their integrated properties. In addition, ZnFe2O4 itself has magnetic properties, which can make the ZnFe2O4@MWNTs composite magnetically separable in a suspension system. Compared with the traditionally used label HRP, ZnFe2O4@MWNTs are much cheaper and more stable. On the basis of this, we demonstrated a novel paper-based immunosensor for the colorimetric determination of CEA. We have demonstrated the ability of the paper-based colorimetric immunosensor to detect CEA in a real serum sample within a total analysis time substantially less than that of currently available rapid screening methods. At present, we believe that this is a cost-effective and simple method without the need for complicated instrumentation. Also this work facilitates the study of MNPs as peroxidase mimics and their utilization in biotechnology and clinical diagnosis.
Acknowledgements
This work was financially supported by the Natural Science Research Foundation of China (21207048, 21277058, 51273084) and the Natural Science Foundation of Shandong Province, China (ZR2012BZ002).
References
- T. T. Lou, Z. P. Chen, Y. Q. Wang and L. X. Chen, ACS Appl. Mater. Interfaces, 2011, 3, 1568 CAS.
- R. Klajn, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2010, 39, 2203 RSC.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318 CrossRef CAS PubMed.
- Y. Khalavka, J. Becker and C. Sonnichsen, J. Am. Chem. Soc., 2009, 131, 1871 CrossRef CAS PubMed.
- J. S. Mu, Y. Wang, M. Zhao and L. Zhang, Chem. Commun., 2012, 48, 2540 RSC.
- R. Andre, F. Natalio, M. Humanes, J. Leppin, K. Heinze, R. Wever, H. C. Schroeder, W. E. G. Mueller and W. Tremel, Adv. Funct. Mater., 2011, 21, 501 CrossRef CAS.
- M. Ornatska, E. Sharpe, D. Andreescu and S. Andreescu, Anal. Chem., 2011, 83, 4273 CrossRef CAS PubMed.
- L. Z. Gao, J. Zhuang, L. Nie, J. B. Zhang, Y. Zhang, N. Gu, T. H. Wang, J. Feng, D. L. Yang, S. Perrett and X. Y. Yan, Nat. Nanotechnol., 2007, 2, 577 CrossRef CAS PubMed.
- W. B. Shi, X. D. Zhang, S. H. He and Y. M. Huang, Chem. Commun., 2011, 47, 10785 RSC.
- L. Su, J. Feng, X. M. Zhou, C. L. Ren, H. H. Li and X. G. Chen, Anal. Chem., 2012, 84, 5753 CrossRef CAS PubMed.
- Y. J. Song, X. H. Wang, C. Zhao, K. G. Qu, J. S. Ren and X. G. Qu, Chem.–Eur. J., 2010, 16, 3617 CrossRef CAS PubMed.
- H. Deng, X. L. Li, Q. Peng, X. Wang, J. P. Chen and Y. D. Li, Angew. Chem., 2005, 117, 2842 CrossRef.
- J. J. Lu, S. G. Ge, L. Ge, M. Yan and J. H. Yu, Electrochim. Acta, 2012, 80, 334 CrossRef CAS PubMed.
- G. L. Fan, Z. J. Gu, L. Yang and F. Li, Chem. Eng. J., 2009, 155, 534 CrossRef CAS PubMed.
- L. Z. Gao, J. M. Wu and D. Gao, ACS Nano, 2011, 5, 6736 CrossRef CAS PubMed.
- X. L. Zuo, C. Peng, Q. Huang, S. P. Song, L. H. Wang, D. Li and C. H. Fan, Nano Res., 2009, 2, 617 CrossRef CAS.
- T. J. McDonald, D. Svedruzic, K. H. Kim, J. L. Blackburn, S. B. Zhang and P. W. King, Nano Lett., 2007, 7, 3528 CrossRef CAS PubMed.
- P. Myllytie, J. Salmi and J. Laine, BioResources, 2009, 4, 1647 CAS.
- H. Orelma, I. Filpponen, L. S. Johansson, J. Laine and O. J. Roja, Biomacromolecules, 2011, 12, 4311 CrossRef CAS PubMed.
- X. Chen, X. L. Jia, J. N. Han, J. Ma and Z. F. Ma, Biosens. Bioelectron., 2013, 50, 356 CrossRef CAS PubMed.
- D. P. Tang, R. Yuan and Y. Q. Chai, J. Phys. Chem. B, 2006, 110, 11640 CrossRef CAS PubMed.
- Y. Zhang, W. Y. Liu, S. G. Ge, M. Yan, S. W. Wang, J. H. Yu, N. Q. Li and X. R. Song, Biosens. Bioelectron., 2013, 41, 684 CrossRef CAS PubMed.
- Z. F. Fu, H. Liu and H. X. Ju, Anal. Chem., 2006, 78, 6999 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.