Isolating single stranded DNA using a microfluidic dialysis device
Yixiao
Sheng
a and
Michael T.
Bowser
*b
aDepartment of Biomedical Engineering, University of Minnesota, Minneapolis, MN 55455, USA
bDepartment of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA
First published on 30th October 2013
Abstract
Isolating a particular strand of DNA from a double stranded DNA duplex is an important step in aptamer generation as well as many other biotechnology applications. Here we describe a microfluidic, flow-through, dialysis device for isolating single-stranded DNA (ssDNA) from double-stranded DNA (dsDNA). The device consists of two channels fabricated in polydimethylsiloxane (PDMS) separated by a track etched polycarbonate membrane (800 nm pore size). To isolate ssDNA, dual-biotin labelled dsDNA was immobilized onto streptavidin-coated polystyrene beads. Alkaline treatment was used to denature dsDNA, releasing the non-biotinylated ssDNA. In the flow-through dialysis device the liberated ssDNA was able to cross the membrane and was collected in an outlet channel. The complementary sequence bound to the bead was unable to cross the membrane and was directed to a waste channel. The effect of NaOH concentration and flow rate on purity and yield were compared. >95% ssDNA purity was achieved at 25 mM NaOH. However, lower flow rates were necessary to achieve ssDNA yields approaching the 50% theoretical maximum of the concurrent-flow device. Under optimized conditions the microfluidic isolation achieved even higher purity ssDNA than analogous manual procedures.
Introduction
ssDNA has broad applications in therapeutics,1–5 diagnostics,6–10 biosensing11–17 and sequencing technologies.18–21 Unlike dsDNA, the unpaired bases of ssDNA allow it to participate in complex three dimensional folding giving rise to unique structural conformations including hairpins,8,22 pseudoknots23,24 and quadruplexes.25,26 Formation of conformations such as these are key to the success of technologies such as Systematic Evolution of Ligands by Exponential enrichment (SELEX)27–29 and single-stranded conformation polymorphism (SSCP).30,31 ssDNA templates are also critical components of hybridization-based detection schemes to detect single-nucleotide polymorphisms (SNP),32–34 pyrosequencing technology,19,35,36 solid phase DNA sequencing,37,38 and sensing in DNA chips and microarrays.17,39–41Aptamers are 40–120 nucleotide sequences of ssDNA or RNA that have been selected to fold into conformations with affinity for specific targets.27–29,42–44 Aptamers have been selected for a variety of targets ranging from small inorganic molecules to whole cells.23,45–51 Applications of aptamers are broad ranging from purification52 and detection53 to diagnostic54 and therapeutic agents.55 The SELEX process used to isolate aptamers begins with a ssDNA or RNA library of sequences containing a 20–60 bp random region flanked by forward and reverse primers. Binding sequences are enriched using an affinity separation and then amplified using PCR. A key step of the process is recovering the forward strand ssDNA from the dsDNA generated by PCR allowing the aptamers to refold into their binding conformations prior to further rounds of enrichment. There are several methods for recovering ssDNA from dsDNA including denaturing urea–polyacrylamide gel (PAGE),56–59 asymmetrical PCR,60–64 lambda exonuclease digestion,65–68 and separation with streptavidin-coated beads.20,40,51,69–72 In denaturing PAGE, a functional group is conjugated to one of the PCR primers. The resulting forward and reverse ssDNA have different sizes allowing them to be separated by denaturing gel electrophoresis.57 While effective, denaturing PAGE is a labour intensive and expensive approach.73 Asymmetrical PCR preferentially amplifies one strand over the other by adding different concentrations of the two PCR primers.60 While this provides a simple and economic solution, the enriched ssDNA pool also contains dsDNA, necessitating further purification using gel electrophoresis. The recovery process is slow, generates low yields of ssDNA and is not amenable to integration into a microfluidic format.74,75 Lambda exonuclease digestion selectively digests the 5′-phosphorylated strands of dsDNA. To recover ssDNA the complementary sequence is preferentially digested by introducing a 5′-phosphate group during PCR amplification.65,66 Phenol–chloroform extraction followed by ethanol precipitation is necessary to remove the lambda exonuclease enzyme at the conclusion of the digestion. dsDNA may also accumulate in the reaction mixture due to incomplete digestion of the phosphorylated double-stranded PCR product.76
The most common method for isolating ssDNA in aptamer selections is immobilization of biotinylated dsDNA onto streptavidin-coated beads followed by alkaline denaturation.51 Many aptamers have been isolated using this method.20,40,51,69 In the current paper, we have incorporated this approach into a flow through, microfluidic dialysis device, providing a simple procedure for isolating ssDNA from dsDNA.
A Lab on a Chip or Micro Total Analysis System (μTAS), is a miniaturized analysis system integrated onto a single chip, often conducting multiple functions such as sample preparation, separation, detection, etc. Microfluidic devices offer a number of important advantages including short analysis time, high sensitivity, low consumption of sample and reagents, and low cost.77,78 Several groups have reported microfluidic devices that are useful for aptamer selections. Park and Ahn developed a microfluidic device for binding nucleic acids to multiple proteins immobilized in sol–gel arrays followed by aptamer elution and recovery.79 The Soh group demonstrated an M-SELEX system which manipulated small numbers of beads to isolate high-affinity aptamers rapidly.80,81 Jing and Bowser isolated aptamers after a single round of selection in a μFFE device.82 Huang et al. have used a magnetic bead-based microfluidic system which integrated a ssDNA incubation module with an on-chip nucleic acid amplification module.83 Ferguson et al. developed a microfluidic electrochemical DNA sensor which integrated symmetric PCR, ssDNA generation and sequence-specific electrochemical detection onto a single chip.84 Lambda exonuclease digestion was used to generate ssDNA. While impressive, each of these microfluidic SELEX systems only addressed isolated steps of aptamer selection, not the entire SELEX process. Identifying strategies for incorporating ssDNA isolation into a more complex device is a key step in developing a fully integrated microfluidic SELEX system. For example, integration of μFFE selection82 and continuous PCR85 with the online ssDNA purification described here suggests that a simple, continuous flow strategy for microscale aptamer generation may be feasible.
Experimental
Reagents and chemicals
PDMS was made using a Sylgard 184 silicone elastomer kit (Dow Corning, Midland, MI). Piranha solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2SO4:H2O2, Ashland Chemical, Dublin, OH) was used to clean silicon wafers. Diluted HCl was used to reactivate the PDMS surface. SU8-2050 (Microchem Corp., Newton, MA) was used to fabricate the master. All DNA samples were purchased from Integrated DNA technologies (Coralville, IA). Tris EDTA (TE) buffers consisted of 10 mM Tris(hydroxymethyl)aminomethane (Tris), 1 mM ethylenediaminetetraacetic acid disodium salt (EDTA), 150 mM NaCl (Spectrum Chemical, New Brunswick, NJ) and 0.002% Triton X-100, adjusted to pH 7.5 using 1 M HCl. TE buffer was degassed before use. 12.5 mM, 18.75 mM, 25 mM, 50 mM, 100 mM and 300 mM NaOH were prepared to denature dsDNA. 1.26 μm streptavidin-coated polystyrene beads (Spherotech, Lake Forest, IL) were used to capture biotinylated dsDNA. Wash buffer comprised of 5 mM Tris, 0.5 mM EDTA, 1 mM NaCl and 0.05% Tween 20, adjusted to pH 7.5 using 1 M HCl. Binding buffer consisted of 10 mM Tris, 1 mM EDTA and 2 M NaCl, adjusted to pH = 7.5 using 1 M HCl. TBE buffer (Tris 89 mM, boric acid 89 mM, EDTA 2 mM) with 10% glycerol (Invitrogen, Grand Island, NY, purity ≥ 99.5%) was used as CE running buffer. 8% linear polyacrylamide (LPA) (MW = 600000–1000000, Polysciences, Inc, Warrington, PA) in TBE buffer with 10% glycerol was prepared and filtered through a 0.45 μm membrane filter. Poly(ethylene oxide) (PEO) was dissolved in deionized water to 0.2% by mass with 0.1 M HCl in a water bath at 95 °C with stirring. All buffer solutions were prepared using nuclease free water (Integrated DNA technologies, Coralville, IA) and filtered through a 0.2 μm membrane before use. All other reagents and chemicals were from Sigma-Aldrich (St. Louis, MO).
:1 H2SO4:H2O2, Ashland Chemical, Dublin, OH) was used to clean silicon wafers. Diluted HCl was used to reactivate the PDMS surface. SU8-2050 (Microchem Corp., Newton, MA) was used to fabricate the master. All DNA samples were purchased from Integrated DNA technologies (Coralville, IA). Tris EDTA (TE) buffers consisted of 10 mM Tris(hydroxymethyl)aminomethane (Tris), 1 mM ethylenediaminetetraacetic acid disodium salt (EDTA), 150 mM NaCl (Spectrum Chemical, New Brunswick, NJ) and 0.002% Triton X-100, adjusted to pH 7.5 using 1 M HCl. TE buffer was degassed before use. 12.5 mM, 18.75 mM, 25 mM, 50 mM, 100 mM and 300 mM NaOH were prepared to denature dsDNA. 1.26 μm streptavidin-coated polystyrene beads (Spherotech, Lake Forest, IL) were used to capture biotinylated dsDNA. Wash buffer comprised of 5 mM Tris, 0.5 mM EDTA, 1 mM NaCl and 0.05% Tween 20, adjusted to pH 7.5 using 1 M HCl. Binding buffer consisted of 10 mM Tris, 1 mM EDTA and 2 M NaCl, adjusted to pH = 7.5 using 1 M HCl. TBE buffer (Tris 89 mM, boric acid 89 mM, EDTA 2 mM) with 10% glycerol (Invitrogen, Grand Island, NY, purity ≥ 99.5%) was used as CE running buffer. 8% linear polyacrylamide (LPA) (MW = 600000–1000000, Polysciences, Inc, Warrington, PA) in TBE buffer with 10% glycerol was prepared and filtered through a 0.45 μm membrane filter. Poly(ethylene oxide) (PEO) was dissolved in deionized water to 0.2% by mass with 0.1 M HCl in a water bath at 95 °C with stirring. All buffer solutions were prepared using nuclease free water (Integrated DNA technologies, Coralville, IA) and filtered through a 0.2 μm membrane before use. All other reagents and chemicals were from Sigma-Aldrich (St. Louis, MO).
Fabrication
Fabrication of the microfluidic dialysis device was performed following a previously described procedure.86 A two-step soft lithography process was used.87 An SU8 master was fabricated and multiple PDMS replicas were then cast from this master. The chip geometry was designed using CAD software which was then used to print a transparency film mask. Standard photolithography procedures were used to make the SU8 master. The master was pre-treated with fluorinated silanes (UCT, Bristol, PA). Degassed PDMS prepolymer (10:1 ratio) was then poured onto the master and baked at 65 °C for 3 hours. The cured PDMS was removed from the master. A second PDMS layer was made using the same process. The resulting channels were 250 μm wide and 110 μm deep. The total channel length was 17 cm with an overlapped channel length of 15.3 cm. Inlet and outlet holes were punched into one PDMS layer using a piece of 21 gauge stainless steel tubing (McMaster-Carr, Chicago, IL). Both PDMS layers were sonicated in dilute HCl for 10 minutes to reactivate prior to bonding.88 A 12 mm × 20 mm × 6 μm polycarbonate membrane with 800 nm pore diameter (Sterlitech Corp., Kent, WA) was placed on one layer of PDMS over the overlapped area of the channel. The PDMS layers were O2 plasma treated at 600 mTorr and 75 W for 10 s. The two layers were then quickly aligned and bonded irreversibly. The entire assembly was placed in an oven at 65 °C for 2 hours to complete the bonding process. Two glass substrates were used to support the device and eight screws on the edges of the substrates were finger tightened to seal the membrane and the PDMS slabs. 0.5′′ long pieces of stainless steel tubing with the same inner and outside diameter as a 23 gage needle (New England Small Tube Corp., Litchfield, NH) were inserted into in the inlet and outlet holes of the device allowing connections to be made with Tygon tubing (0.02 ID, 0.06 OD, Small parts, Inc, Miramar, FL). Buffer was pumped into the device using a syringe pump (Harvard Apparatus, Holliston, Massachusetts).
Instrumentation and data collection
A Synergy 2 multi-mode microplate reader (BioTek, Winooski, VT) mounted with a Tungsten Halogen lamp was used for fluorescence intensity measurements (λex = 485 nm, λem = 528 nm). Fluorescence data were recorded and processed using Gen5 Reader Control and Data Analysis software (BioTek, Winooski, VT). Capillary electrophoresis (CE) was performed on a P/ACE MDQ system (Beckman Coulter Inc., Fullerton, CA) equipped with a laser induced fluorescence (LIF) detection module (λex = 488 nm, λem = 520 nm). 32 Karat Software (Beckman Coulter Inc., Fullerton, CA) was used for collecting and recording data.Sample preparation
Two complementary 80 nt ssDNA sequences (5′-biotin-biotin-TT CAC GGT AGC ACG CAT AGG CGA TCA TAA AGC CTC CAT AGC ATT GTA CGT CGA AAC CTG CCA TCT GAC CTC TGT GCT GCT-FAM-3′ and 5′-AGC AGC ACA GAG GTC AGA TGG CAG GTT TCG ACG TAC AAT GCT ATG GAG GCT TTA TGA TCG CCT ATG CGT GCT ACC GTG AA-3′) were synthesized by Integrated DNA technologies (Coralville, IA) and annealed to generate dsDNA1 manually in the following steps. Both ssDNA sequences were dissolved in Nuclease Free Duplex Buffer (Integrated DNA technologies, Coralville, IA) at concentrations of 100 μM. Equal volumes of each ssDNA solution were mixed together and heated to 94 °C for 2 min followed by gradual cooling to room temperature. The resulting dsDNA1 product was diluted in TE buffer to 10 μM and stored at −20 °C for future use. A second pair of 80 nt ssDNA sequences (5′-biotin-biotin-TT CAC GGT AGC ACG CAT AGG CGA TCA TAA AGC CTC CAT AGC ATT GTA CGT CGA AAC CTG CCA TCT GAC CTC TGT GCT GCT-3′ and 5′-FAM-AGC AGC ACA GAG GTC AGA TGG CAG GTT TCG ACG TAC AAT GCT ATG GAG GCT TTA TGA TCG CCT ATG CGT GCT ACC GTG AA-3′) were synthesized by Integrated DNA technologies (Coralville, IA) and annealed to generate dsDNA2 using the same procedure. The only difference between dsDNA1 and dsDNA2 was the location of the FAM label allowing the path of each sequence to be tracked through the device independently depending on the specific experiment (see Fig. 1).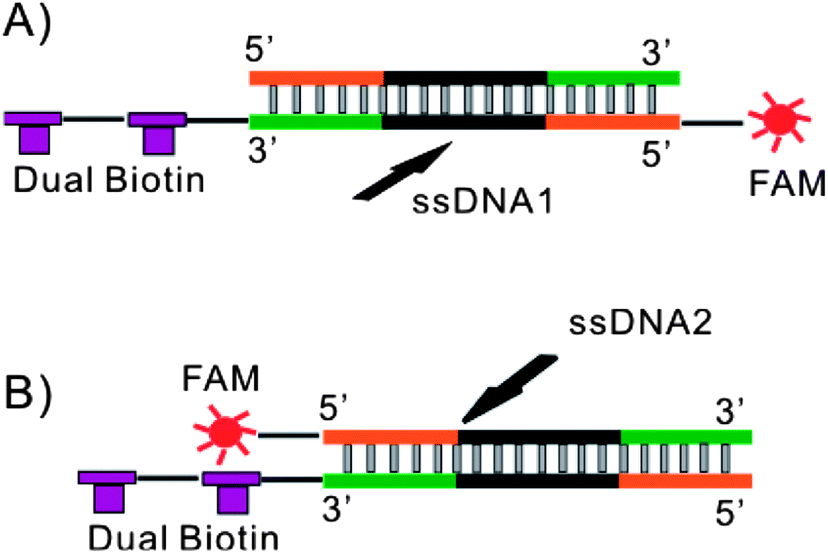 | ||
| Fig. 1 Schematic demonstrating the labeling configuration of (A) dsDNA1 and (B) dsDNA2. In dsDNA1 dual biotin and FAM were labeled on the same strand allowing the complementary strand to be followed using fluorescence. In dsDNA2 dual biotin and FAM were labeled on opposite strands allowing the forward sequence to be followed. | ||
Binding dsDNA to beads
A 150 μl suspension of streptavidin-coated polystyrene beads was pelleted by centrifugation (15000 rpm for 5 min) to remove the storage buffer. The beads were resuspended in washing buffer followed by pelleting (15000 rpm for 5 min) three times. After the final pelleting beads were resuspended in 300 μl binding buffer. 200 pmol dsDNA in 300 μl nuclease-free water was mixed with the bead suspension and incubated at room temperature for 15 min. The supernatant was collected after centrifugation and the fluorescence intensity was measured to determine how much free dsDNA existed in the supernatant. By comparing how much dsDNA was added with how much dsDNA remained in solution after incubation, the amount of dsDNA immobilized onto the beads could be determined. Washing buffer was used to remove any unbound dsDNA with two centrifugation, resuspension cycles. Finally the beads were resuspended in TE buffer with a dsDNA concentration of 100 nM.
ssDNA generation by alkaline denaturation
The dsDNA-bead suspension was mixed with a 1:1 volume of freshly prepared NaOH solution. This mixture was continuously pumped into the sample channel of the microfluidic dialysis device (see Fig. 2). A 1:1 mixture of TE buffer and NaOH was pumped into the buffer channel of the device at the same flow rate. The buffer composition on each side the membrane was kept the same to simplify the fluorescence analysis. Since the buffers were identical the ratio of DNA in each channel was simply the ratio of observed fluorescence intensities. The distribution of each of the two ssDNA in each channel was determined at six different NaOH concentrations (12.5 mM, 18.75 mM, 25 mM, 50 mM, 100 mM and 300 mM). Concurrent flow was adopted in the device (see Fig. 2) to eliminate pressure differences across the membrane which caused beads to be trapped in the pores. The flow rate in each channel was set to be 0.2 ml h−1 which corresponded to 2.2 mm s−1 and a 70 s equilibration time.
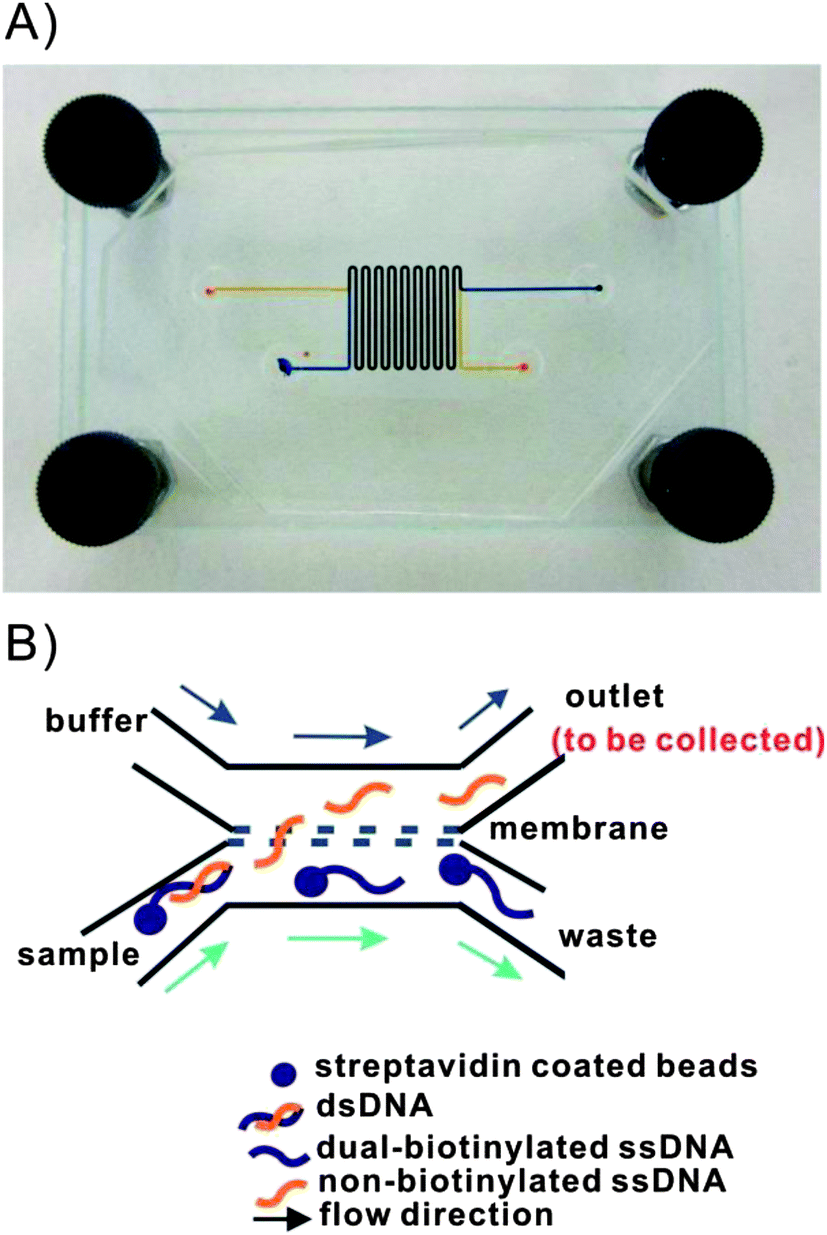 | ||
| Fig. 2 (A) Image of the microfluidic dialysis device. Two 250 μm wide × 110 μm deep PDMS channels were separated by a polycarbonate membrane (800 nm pore size). The total channel length was 17 cm with a 15.3 cm long overlapping region. Channels were filled with food coloring to facilitate visualization of the sample and buffer channels. (B) Schematic of the anticipated flow paths of the denatured ssDNA in the microfluidic dialysis device. The forward sequence is freed from the bead during denaturation and is small enough to diffuse across the membrane. The biotinylated complementary sequence remains immobilized on the bead and is carried to the waste channel. | ||
An analogous manual method for isolating ssDNA was performed in parallel as a point of reference. Aliquots of the dsDNA-bead suspension were transferred into seven centrifuge tubes and six tubes were spiked with an equal volume of NaOH (12.5 mM, 18.75 mM, 25 mM, 50 mM, 100 mM and 300 mM). The control experiment was performed by mixing the dsDNA-beads suspension with an equal volume of TE buffer. After incubating for 10 min at room temperature, the solution was centrifuged at 15000 rpm for 5 min and the supernatant was collected. CssDNA2/CssDNA1 was determined using fluorescence. All experiments were conducted three times.
Determining the effect of flow rate
Equal volumes of dsDNA-bead suspension and 25 mM NaOH solution were mixed and the resulting solution was pumped into the sample channel of the device. Equal volumes of TE buffer and 25 mM NaOH were mixed and the resulting solution was pumped into the buffer channel of the device. CssDNA2/CssDNA1 in the outlet channel was calculated at five different flow rates (0.5 ml h−1, 0.3 ml h−1, 0.2 ml h−1, 0.1 ml h−1 and 0.05 ml h−1). Samples were collected from the outlet of the buffer channel after 15–20 min from the moment of adding NaOH into dsDNA-bead suspension for consistency.Determining the effect of incubation time
Equal volumes of the dsDNA-bead suspension and 25 mM NaOH solution were mixed and the resulting solution was pumped into the sample channel of the device as the sample. Equal volumes of TE buffer and 25 mM NaOH were mixed and the resulting solution was pumped into the buffer channel of the device. Aliquots were collected from the outlet of the buffer channel every 6 min starting at 8 min after NaOH was added to the dsDNA-bead suspension. Separate experiments were conducted for dsDNA1 and dsDNA2 allowing the distribution of both the forward and complementary sequences to be determined.Capillary electrophoresis (CE) characterization
A 40 cm-long, 75 μm i.d., 360 μm o.d. uncoated fused-silica separation capillary (Polymicro Technologies, Phoenix, AZ) was used. Electroosmotic flow (EOF) was suppressed by coating the capillary surface with PEO according to Iki and Yeung's method.89 The capillary was rinsed for 10 minutes each of 0.1 M NaOH, H2O, 1 M HCl, 0.2% PEO/0.1 M HCl and TBE buffer containing 10% glycerol. Barron's method for separating ssDNA and dsDNA was adopted to confirm the purity of the ssDNA isolation.90,91 8% w/v LPA (MW 600000–1000000) in TBE buffer containing 10% glycerol was used as the sieving matrix for separating 80 nt ssDNA and 80 bp dsDNA. Glycerol has previously been reported to improve resolution of ssDNA and dsDNA.92,93 Linear polyacrylamide (LPA) was filtered through a 0.45 μm membrane filter and degassed by centrifuging at 12000 rpm for 1 min. All the other solutions were filtered through a 0.2 μm membrane filter. LPA was loaded into the 40 cm capillary in the MDQ capillary electrophoresis system at 20 psi for 20 min. Samples were injected using 2 kV for 15 s. The separation was performed under the following conditions: 10 kV voltage (reverse polarity) thermostatted to 15 °C with laser induced fluorescence (LIF) detection at 488/520 nm. TBE buffer containing 10% glycerol was used as the separation buffer. Before performing CE separations, the LPA-loaded capillary was conditioned under 10 kV voltage (reverse polarity) overnight in order to maintain a consistent gel structure for improved run-to-run reproducibility.
Results and discussion
Design considerations
An important consideration in a continuously flowing device such as the one described here is the potential for beads to sediment over time. In particular, if beads settle to the bottom of the syringe it becomes difficult to introduce a consistent concentration of beads into the device, especially over longer time periods. The settling velocity is related to the balance of viscous drag and gravitational force. According to Stoke's law, which is derived by solving the general Navier–Stokes equation at the Stokes flow limit for small Reynolds numbers:94–97where Vm is the maximum settling velocity (cm s−1), g is the acceleration of gravity (980.7 cm s−2), η0 is the viscosity of the external phase in poises (g cm−1 s−1), ρs is the density of a solid sphere (g cm−3), ρm is the density of the medium (g cm−3) and d is the mean diameter of the suspended particles (cm). The rate of sedimentation is determined by the density and the diameter of the particles. Therefore, choosing the appropriate size and material of the beads is crucial to make long term operation of the device feasible. According to the manufactures' data, streptavidin-coated polystyrene beads have a lower density than comparable silica or magnetic beads. Polystyrene (PS) beads are available in a series of diameters ranging from low nm to high μm. For the current application beads should have a diameter larger than the membrane pore diameter while maintaining a low density to keep the beads in suspension for long periods. To maximize ssDNA recovery across the membrane a relatively large pore size was chosen (800 nm). Taking this into consideration, 1.26 μm streptavidin coated polystyrene beads were chosen to immobilize dsDNA.
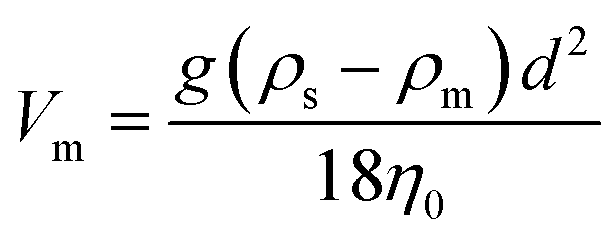
Alkaline denaturation was chosen to isolate ssDNA due to its simplicity. This method makes integration of heating elements into the device unnecessary. Similarly, reestablishing re-naturing conditions is as simple as neutralizing the solution with no further purification necessary. A potential drawback to the alkaline denaturation approach is the addition of NaOH could also denature streptavidin, disrupting its interaction with biotin and releasing dsDNA from the beads. To address this a dual-biotin label was used to increase the stability of the streptavidin–biotin interaction. Similar approaches have proven successful in stabilizing streptavidin–biotin interactions through multiple PCR cycles.98 Ideally a single dsDNA would be used with a fluorescent label attached to each strand allowing the path of each sequence to be tracked through the device simultaneously. In practice synthetic limitations, the potential for signal cross talk and impact on quantum yield made using two dsDNA's, each with a single label to track either the forward or complementary strand, a much more reliable approach (see Fig. 1).
NaOH concentration
Alkaline treatment is known to be a reliable and convenient way to denature double-stranded DNA in a mechanism related to the deprotonation of guanine and cytosine.99–102 However, if the denaturation conditions are too harsh, the streptavidin–biotin interaction may be disrupted,103–105 allowing dsDNA to cross the membrane into the outlet channel. Therefore, careful consideration must be given to optimize the denaturation procedure to ensure effective recovery of the desired ssDNA strand.Six different concentrations of NaOH were assessed. In each case solution from the outlet of the buffer channel was collected and its fluorescence measured to determine the concentration of each strand of DNA that crossed the membrane (see Fig. 3). In ssDNA2 the forward sequence was fluorescently labeled. Fig. 3A shows that the forward sequence was efficiently denatured and crossed the membrane at NaOH concentrations of 25 mM or higher. In dsDNA1 the fluorescent label was on the biotinylated complementary strand. Overall the amount of complementary DNA crossing the membrane was low, indicating that the biotin–streptavidin interaction remained effective even in the presence of NaOH. A trend of increasing complementary DNA crossing the membrane with increasing NaOH was observed. Fig. 3B plots the ratio of forward and complementary sequences that crossed the membrane. A clear optimum is observed at 25 mM NaOH. dsDNA is not effectively denatured at lower NaOH concentrations. At higher NaOH concentrations the forward sequence is already completely denatured but the biotin–streptavidin interaction is increasingly disrupted. At 25 mM NaOH the concentration of forward sequence ssDNA was 22-fold higher than the observed complementary sequence resulting in a purity of >95%.
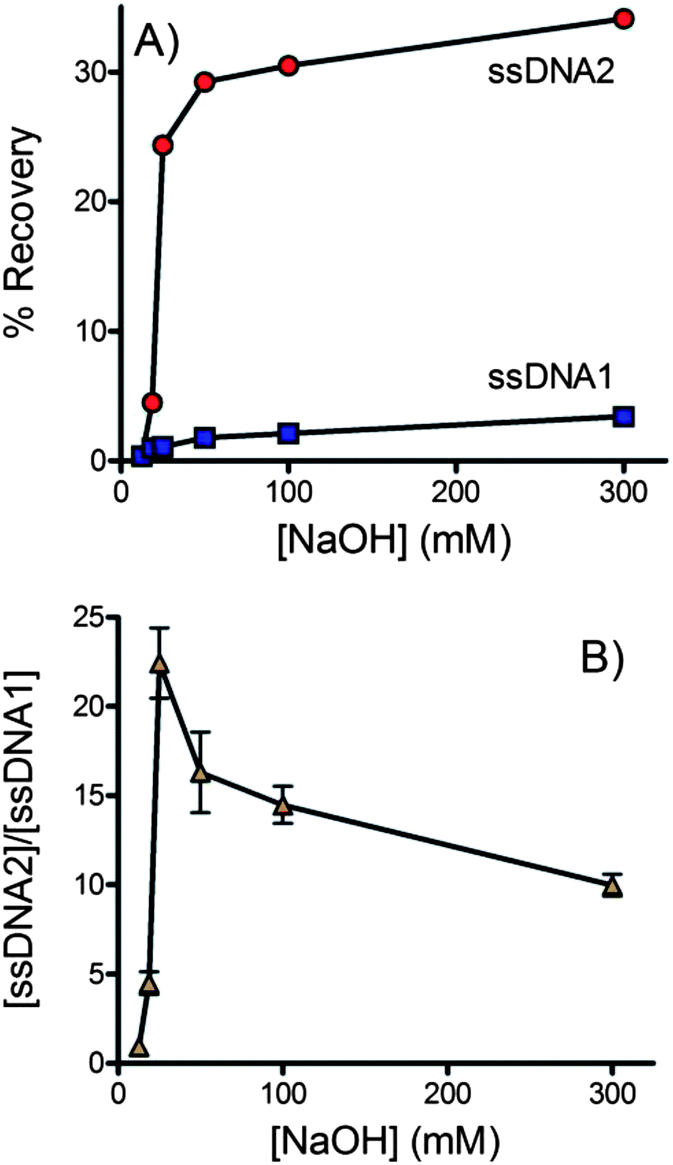 | ||
| Fig. 3 Effect of NaOH concentration on ssDNA isolation. Solution was collected from the outlet of the buffer channel and fluorescence was measured to quantify the DNA that crossed the membrane. (A) The forward sequence is labeled in dsDNA2 (red circles) and the biotinylated complementary sequence is labeled in dsDNA1 (blue squares). (B) The ratio of the forward and complementary ssDNA sequences ([ssDNA2]/[ssDNA1]) that crossed the membrane into the buffer channel. >95% purity was achieved at 25 mM NaOH. All experiments were performed in triplicate. In some cases error bars were smaller than the symbols. The flow rate was 0.2 ml h−1 and buffer contained 10 mM Tris, 1 mM EDTA, 150 mM NaCl and 0.002% Triton X-100. | ||
Flow rate
Flow rate through the device impacts the equilibration time and therefore recovery of ssDNA across the membrane. The overlapping region of the channels is 15.3 cm long. Equilibration time therefore ranges from 30.6 s to 4.2 min for flow rates of 0.5 ml h−1 to 0.06 ml h−1 (5 mm s−1 to 0.61 mm s−1).Fig. 4A and B show the impact of flow rate on the % recovery of the forward and complementary DNA sequences across the membrane. As expected the recovery of both sequences trends lower as flow rate increases and equilibration time decreases. A concurrent flow configuration was chosen for this application. Concurrent dialysis is limited to a maximum % recovery of 50% since complete equilibration is achieved at this point. Counter current devices are able to achieve nearly 100% recovery.86 Unfortunately, counter current devices generate a pressure differential across much of the membrane. In the current application it was found that this pressure differential was enough to trap passing beads into the pores, effectively blocking ssDNA transport within a short period of time. Bead adsorption onto the membrane was minimal when a concurrent flow configuration was adopted. At 0.06 ml h−1 the % recovery of the forward sequence reached 42 ± 3%, 84% of the theoretical maximum of a concurrent flow device. At a more practical 0.2 ml h−1, the % recovery was 24 ± 0.2%, 48% of the theoretical maximum.
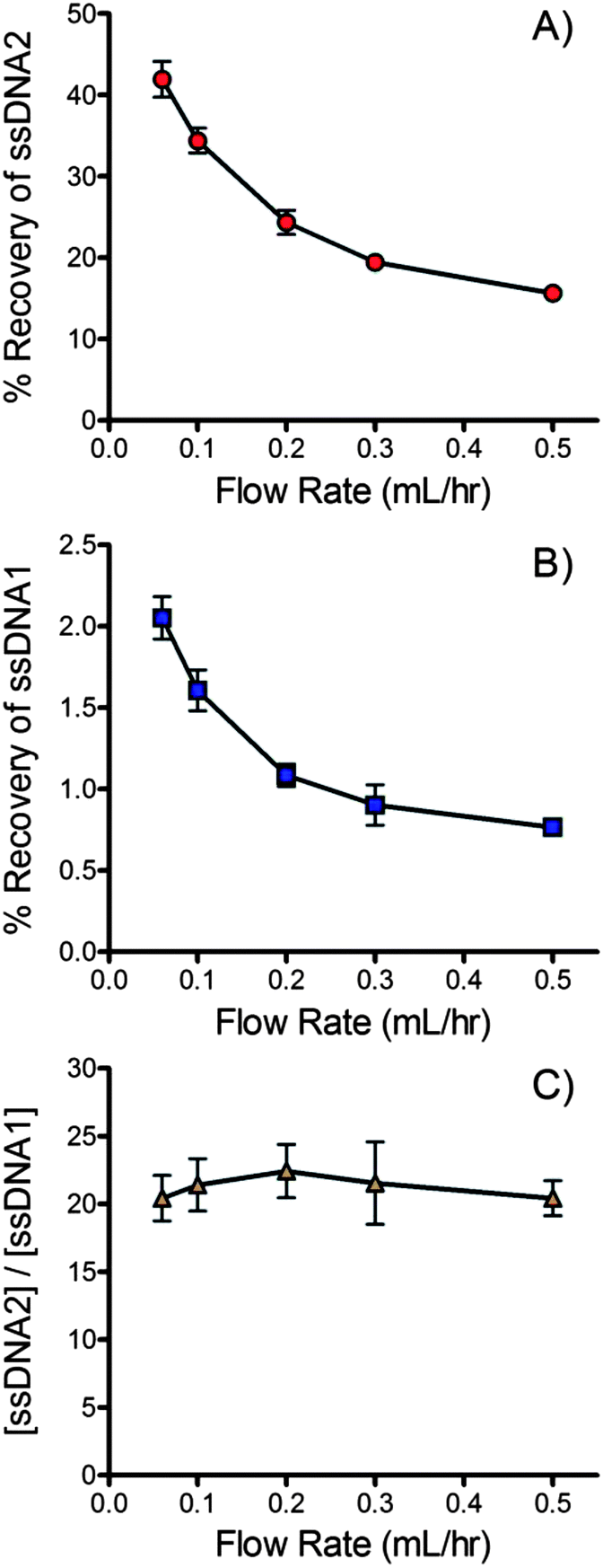 | ||
| Fig. 4 Effect of flow rate on % recovery of (A) the forward sequence (ssDNA2) and (B) the biotinylated complementary sequence (ssDNA1). (C) Ratio of forward and complementary sequences ([ssDNA2]/[ssDNA1]) that crossed the membrane. All data were recorded in triplicate. 25 mM NaOH was used for denaturation and the buffer contained 10 mM Tris, 1 mM EDTA, 150 mM NaCl and 0.002% Triton X-100. | ||
It should be noted that similar trends in recovery were observed for the forward and complementary sequences (see Fig. 4A & B). Fig. 4C plots the ratio of the forward and complementary sequences that crossed the membrane at different flow rates. Although the recovery of each sequence decreased with increasing flow rate the purity of the recovered ssDNA was largely unaffected since the recovery of each sequence was affected equally.
Incubation time
The incubation time between the addition of NaOH and delivery into the device is an important variable. This time must be long enough to achieve complete denaturation of the dsDNA but not be so long as to promote disruption of the biotin–streptavidin interaction. The effect of incubation time on the amount of the forward sequence ssDNA2 and the complementary sequence ssDNA1 is shown in Fig. 5. No trend in the recovery of the forward sequence was observed (see Fig. 5A) suggesting that complete denaturation of the dsDNA occurred within several minutes. Conversely, the amount of complementary DNA crossing the membrane increased linearly with time, suggesting a constant, continual denaturation of streptavidin was occurring with prolonged exposure to NaOH. Fig. 5C shows the ratio of forward and complementary sequences that cross the membrane. Over longer periods the purity of the recovered ssDNA decreases due to the slow release of the complementary sequence. Combined this data suggests that beads should be delivered to the device immediately after addition of NaOH. Future devices would benefit from incorporation of an online mixing tee to ensure fast, reproducible, delivery of beads into the device after addition of NaOH.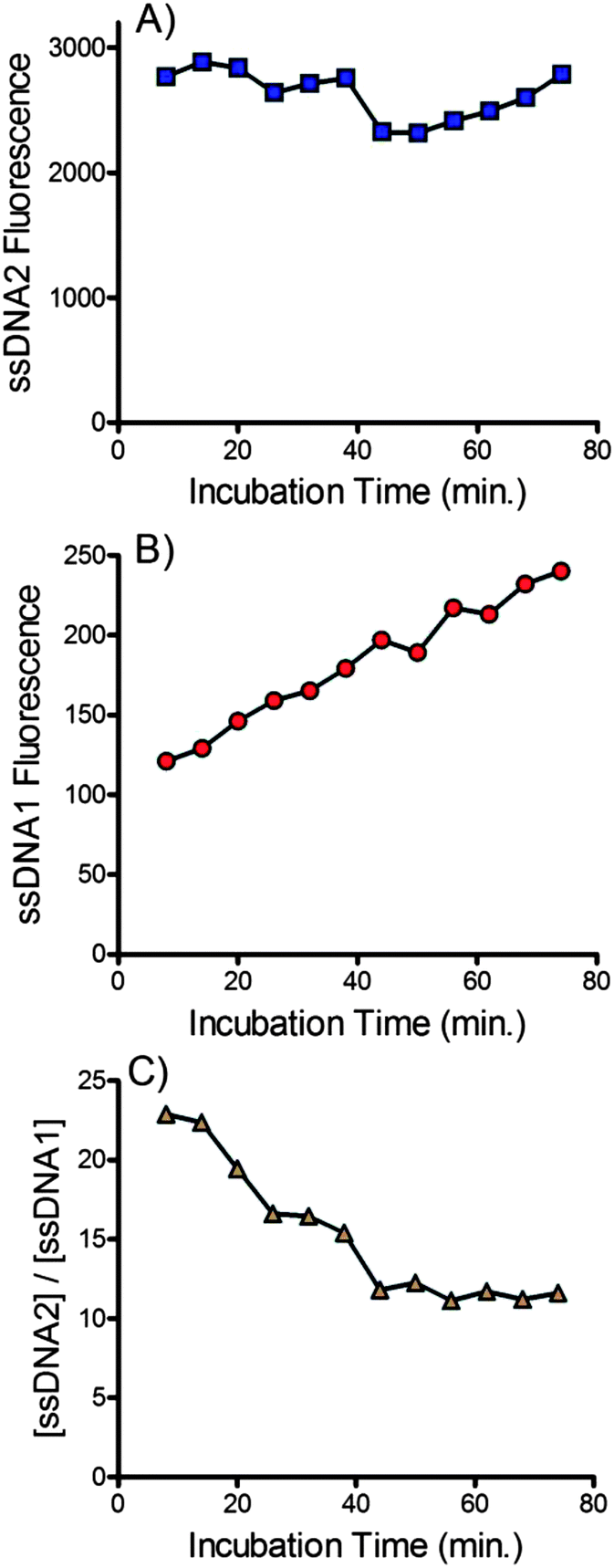 | ||
| Fig. 5 Effect of incubation time on amount of (A) forward sequences (ssDNA2) and (B) biotinylated complementary sequences (ssDNA1) that cross the membrane into the buffer channel. (C) Effect of incubation time on the ratio of forward and complementary sequences ([ssDNA2]/[ssDNA1]) that crossed the membrane. 25 mM NaOH was used for denaturation and the buffer contained 10 mM Tris, 1 mM EDTA, 150 mM NaCl and 0.002% Triton X-100. The flow rate was 0.2 ml h−1. | ||
Comparison with manual methods
The purity of the ssDNA recovered using the flow through, microfluidic dialysis device was compared with ssDNA recovered using an analogous manual method. For the manual method dsDNA-bead suspension was incubated with NaOH at room temperature for 10 minutes. The beads were pelleted using centrifugation and the amount of labeled DNA in the supernatant was assessed using fluorescence.Fig. 6A compares the ratio of forward and complementary sequence DNA obtained by each method across a range of NaOH concentrations. Overall, the microfluidic device performed similarly to the manual procedure. The microfluidic device does consistently outperform the manual procedure at NaOH concentrations ≥50 mM. The additional steps of the manual procedure make it difficult to isolate the ssDNA quickly, likely giving rise to increased disruption of the biotin–streptavidin interaction, releasing the complementary sequence. Integration into a single, flow-through microfluidic device would allow these steps to be performed more efficiently, potentially improving purity. Fig. 6B compares the % recovery of the microfluidic and manual approaches. The manual approach significantly outperforms in this regard. This is not surprising considering that the concurrent approach used in the microfluidic device described here is limited to a maximum recovery of 50%. Further development of counter current devices will be necessary to match the % recovery of the manual procedure.
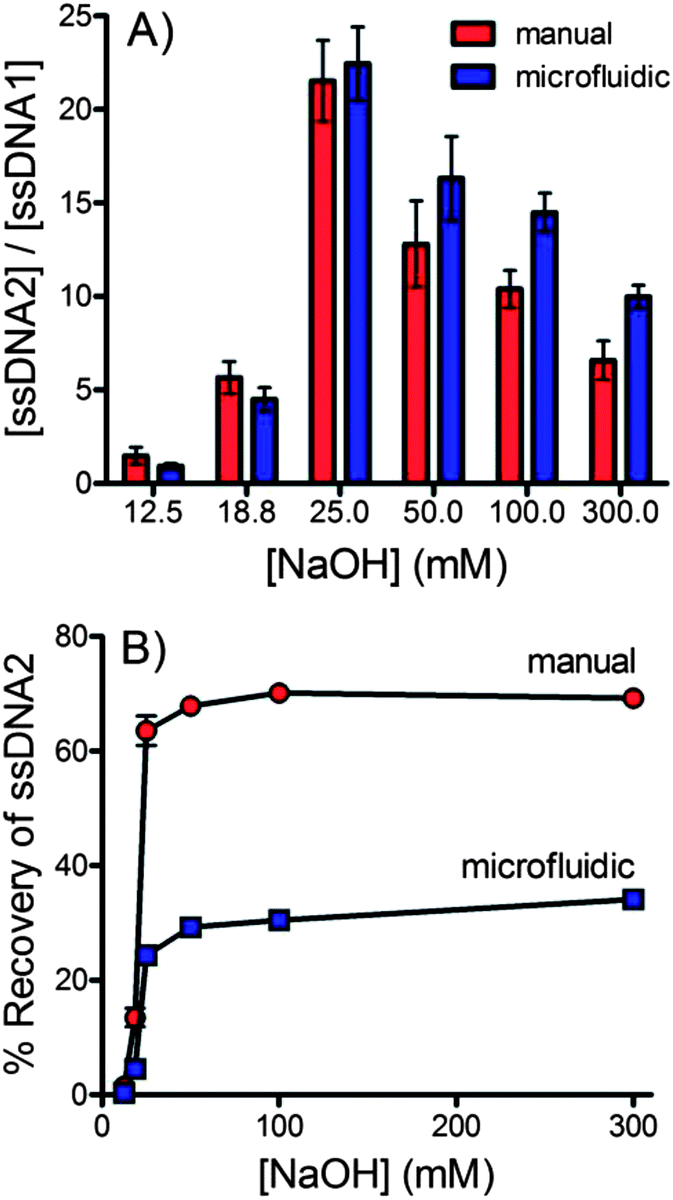 | ||
| Fig. 6 (A) A comparison of the ratio of forward and complementary sequences ([ssDNA2]/[ssDNA1]) isolated using the manual (red) and continuous flow microfluidic (blue) techniques. (B) A comparison of the % recovery of ssDNA2 sequences isolated using the manual (red) and continuous flow microfluidic (blue) techniques. | ||
CE characterization
CE was used to confirm that the isolated DNA was indeed single stranded. This confirmation is important since if NaOH significantly denatures streptavidin on the timescale of the isolation, the dsDNA released from the beads would be small enough to cross the membrane. Fig. 7A shows electropherograms of fluorescently labeled single stranded and double stranded DNA. Clear resolution is observed between the two peaks suggesting that CE is an appropriate assay for confirming the relative abundance of ssDNA and dsDNA.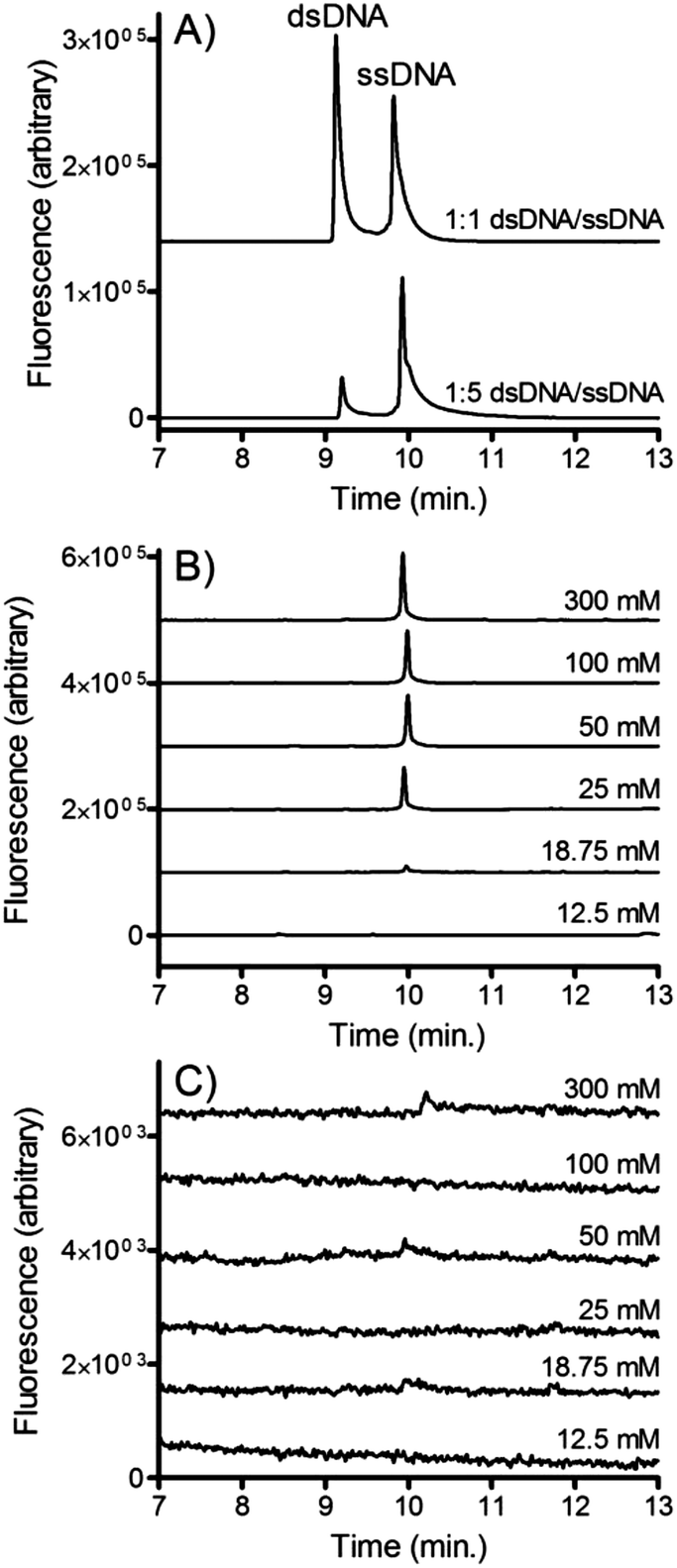 | ||
| Fig. 7 (A) CE-LIF analysis of ssDNA and dsDNA. In the top trace 100 nM dsDNA2 was mixed with an equal volume of 100 nM ssDNA2. In the bottom trace 20 nM dsDNA2 was mixed with an equal volume of 100 nM ssDNA2. (B) dsDNA2-bead and (C) dsDNA1-bead suspensions were incubated with varying concentrations of NaOH and pumped into the microfluidic dialysis device at 0.2 ml h−1. Solution from the buffer outlet channel was collected, neutralized and analyzed using CE-LIF. | ||
Solutions of dsDNA were incubated with varying concentrations of NaOH and pumped into the microfluidic dialysis device. Aliquots were collected from the outlet of the buffer channel and analyzed using CE. Fig. 7B shows the electropherograms that were observed when dsDNA2 (i.e. the forward sequence is fluorescently labeled) is pumped into the device. Similar to the fluorescence experiments, at low NaOH concentrations no DNA crosses the membrane into the buffer channel, confirming that the forward sequence remained bound to the bead through its complementary sequence. At NaOH concentrations greater than 25 mM a clear ssDNA peak is observed. This confirms that the forward sequence is freed from the beads and is able to cross the membrane. dsDNA was not observed suggesting that denaturation of the streptavidin was minimal.
Fig. 7C show the electropherograms that were observed when dsDNA1 (i.e. the biotinylated complementary strand is fluorescently labeled) is introduced into the device. In contrast to dsDNA2, very little DNA crosses the membrane at any NaOH concentration. This confirms that the complementary sequence largely remains bound to the bead, even at NaOH concentrations where significant concentrations of the forward sequence is collected.
Conclusions
We have demonstrated the application of a microfluidic dialysis device for the isolation of a specific ssDNA strand from dsDNA. This is an important step of the SELEX process for isolating aptamers as well as many other biotechnology procedures. NaOH denaturation was optimized to maximize recovery of the forward sequence while minimizing release of the complementary strand. The purity of the correct forward sequence ssDNA collected reached >95% under optimized conditions, higher than that obtained using an analogous manual procedure. Yield was closely tied to flow rate through the device with 24% recovery achieved at 0.2 ml h−1. This relatively low throughput does restrict the potential of the device somewhat. Further development of membrane materials with higher pore densities that are robust enough to survive the fabrication and denaturation procedures would allow reasonable recoveries to be achieved at higher flow rates.It should be noted that the major innovation reported here is not in regards to the performance of the device itself. In many ways the observed performance is similar or even somewhat worse that what can be performed using currently available manual approaches. This is not surprising for a technique such as this that is amenable to manual intervention. The advantage of the microfluidic dialysis approach described here is the potential for further integration. The flow through design provides simple operation without requiring any special timing or valving. In particular the microfluidic dialysis design is especially well suited for integration with other flow through devices such as μFFE aptamer selection82 and flow through PCR.85 Combining these microfluidic modules suggests a unique approach towards designing a fully integrated microfluidic SELEX device.
Acknowledgements
This research was supported by the National Institutes of Health (GM063533). Fabrication was performed using facilities at the University of Minnesota Nanofabrication Center, a NNIN funded site.References
- J. F. Lee, G. M. Stovall and A. D. Ellington, Curr. Opin. Chem. Biol., 2006, 10, 282–289 CrossRef CAS PubMed.
- P. Wang, Y. Yang, H. Hong, Y. Zhang, W. Cai and D. Fang, Curr. Med. Chem., 2011, 18, 4169–4174 CrossRef CAS.
- S. M. Nimjee, C. P. Rusconi and B. A. Sullenger, Annu. Rev. Med., 2005, 56, 555–583 CrossRef CAS PubMed.
- J.-H. Lee, M. D. Canny, A. De Erkenez, D. Krilleke, Y.-S. Ng, D. T. Shima, A. Pardi and F. Jucker, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18902–18907 CrossRef CAS PubMed.
- E. W. M. Ng, D. T. Shima, P. Calias, E. T. Cunningham, D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5, 123–132 CrossRef CAS PubMed.
- T. C. Chu, F. Shieh, L. A. Lavery, M. Levy, R. Richards-Kortum, B. A. Korgel and A. D. Ellington, Biosens. Bioelectron., 2006, 21, 1859–1866 CrossRef CAS PubMed.
- J. R. Collett, E. J. Cho, J. F. Lee, M. Levy, A. J. Hood, C. Wan and A. D. Ellington, Anal. Biochem., 2005, 338, 113–123 CrossRef CAS PubMed.
- K. Dominik, E. N. Yin-Shan and D. T. TShima, Biochem. Soc. Trans., 2009, 37, 1201–1206 CrossRef PubMed.
- J. Hesselberth, M. P. Robertson, S. Jhaveri and A. D. Ellington, Rev. Mol. Biotechnol., 2000, 74, 15–25 CrossRef CAS.
- E. N. Brody, M. C. Willis, J. D. Smith, S. Jayasena, D. Zichi and L. Gold, Mol. Diagn., 1999, 4, 381–388 CrossRef CAS.
- R.-M. Kong, X.-B. Zhang, Z. Chen and W. Tan, Small, 2011, 7, 2428–2436 CAS.
- K. J. Odenthal and J. J. Gooding, Analyst, 2007, 132, 603–610 RSC.
- S. Jhaveri, M. Rajendran and A. D. Ellington, Nat. Biotechnol., 2000, 18, 1293–1297 CrossRef CAS PubMed.
- H. Wang, R. Yang, L. Yang and W. Tan, ACS Nano, 2009, 3, 2451–2460 CrossRef CAS PubMed.
- M. J. A. Shiddiky, A. A. J. Torriero, Z. Zeng, L. Spiccia and A. M. Bond, J. Am. Chem. Soc., 2010, 132, 10053–10063 CrossRef CAS PubMed.
- Y. Peng, X. Wang, Y. Xiao, L. Feng, C. Zhao, J. Ren and X. Qu, J. Am. Chem. Soc., 2009, 131, 13813–13818 CrossRef CAS PubMed.
- W.-J. Zhou, A. R. Halpern, T. H. Seefeld and R. M. Corn, Anal. Chem., 2012, 84, 440–445 CrossRef CAS PubMed.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS PubMed.
- M. Groth, K. Huse, K. Reichwald, S. Taudien, J. Hampe, P. Rosenstiel, G. Birkenmeier, S. Schreiber and M. Platzer, Anal. Biochem., 2006, 356, 194–201 CrossRef CAS PubMed.
- T. Hultman, S. Stahl, E. Hornes and M. Uhlen, Nucleic Acids Res., 1989, 17, 4937–4946 CrossRef CAS PubMed.
- S. Stahl, T. Hultman, A. Olsson, T. Moks and M. Uhlen, Nucleic Acids Res., 1988, 16, 3025–3038 CrossRef CAS PubMed.
- S.-H. Chou, K.-H. Chin and A. H.-J. Wang, Nucleic Acids Res., 2003, 31, 2461–2474 CrossRef CAS PubMed.
- C. Tuerk, S. MacDougal and L. Gold, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 6988–6992 CrossRef CAS.
- D. J. Schneider, J. Feigon, Z. Hostomsky and L. Gold, Biochemistry, 1995, 34, 9599–9610 CrossRef CAS.
- R. F. Macaya, P. Schultze, F. W. Smith, J. A. Roe and J. Feigon, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3745–3749 CrossRef CAS.
- N. Jing, R. F. Rando, Y. Pommier and M. E. Hogan, Biochemistry, 1997, 36, 12498–12505 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850–852 CrossRef CAS PubMed.
- M. Aguade, W. Meyers, A. D. Long and C. H. Langley, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4658–4662 CrossRef CAS.
- M. Orita, H. Iwahana, H. Kanazawa, K. Hayashi and T. Sekiya, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 2766–2770 CrossRef CAS.
- A. Okamoto, K. Tanaka, T. Fukuta and I. Saito, J. Am. Chem. Soc., 2003, 125, 9296–9297 CrossRef CAS PubMed.
- M. Liu, M. Yuan, X. Lou, H. Mao, D. Zheng, R. Zou, N. Zou, X. Tang and J. Zhao, Biosens. Bioelectron., 2011, 26, 4294–4300 CrossRef CAS PubMed.
- M. Ji, P. Hou, S. Li, N. He and Z. Lu, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2004, 548, 97–105 CrossRef CAS PubMed.
- N. Pourmand, E. Elahi, R. W. Davis and M. Ronaghi, Nucleic Acids Res., 2002, 30, e31 CrossRef PubMed.
- R. Sen, H. D. Ishak, D. Estrada, S. E. Dowd, E. Hong and U. G. Mueller, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17805–17810 CrossRef CAS PubMed.
- T. Hultman, S. Stahl, E. Hogan and M. Uhlen, Nucleic Acids Res., 1989, 17, 4937–4946 CrossRef CAS PubMed.
- M. Murphy, H. Hammond and C. Caskey, Methods Mol. Biol., 1996, 65, 163–176 CAS.
- J. Wang, D. Onoshima, M. Aki, Y. Okamoto, N. Kaji, M. Tokeshi and Y. Baba, Anal. Chem., 2011, 83, 3528–3532 CrossRef CAS PubMed.
- S. C. Donhauser, R. Niessner and M. Seidel, Anal. Chem., 2011, 83, 3153–3160 CrossRef CAS PubMed.
- M. Famulok and G. Mayer, Acc. Chem. Res., 2011, 44, 1349–1358 CrossRef CAS PubMed.
- M. Syed and S. Pervaiz, Oligonucleotides, 2010, 20, 215–224 CrossRef CAS PubMed.
- S. D. Mendonsa and M. T. Bowser, J. Am. Chem. Soc., 2005, 127, 9382–9383 CrossRef CAS PubMed.
- R. K. Mosing, S. D. Mendonsa and M. T. Bowser, Anal. Chem., 2005, 77, 6107–6112 CrossRef CAS PubMed.
- M. Famulok, Curr. Opin. Struct. Biol., 1999, 9, 324–329 CrossRef CAS.
- D. Shangguan, Y. Li, Z. Tang, Z. C. Cao, H. W. Chen, P. Mallikaratchy, K. Sefah, C. J. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS PubMed.
- C. Berens, A. Thain and R. Schroeder, Bioorg. Med. Chem., 2001, 9, 2549–2556 CrossRef CAS.
- M. Famulok, J. Am. Chem. Soc., 1994, 116, 1698–1706 CrossRef CAS.
- M. Sassanfar and J. W. Szostak, Nature, 1993, 364, 550–553 CrossRef CAS PubMed.
- P. Burgstaller and M. Famulok, Angew. Chem., Int. Ed. Engl., 1994, 33, 1084–1087 CrossRef.
- G. Mayer, M.-S. L. Ahmed, A. Dolf, E. Endl, P. A. Knolle and M. Famulok, Nat. Protoc., 2010, 5, 1993–2004 CrossRef CAS PubMed.
- Q. Deng, I. German, D. Buchanan and R. T. Kennedy, Anal. Chem., 2001, 73, 5415–5421 CrossRef CAS.
- V. Pavlov, Y. Xiao, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2004, 126, 11768–11769 CrossRef CAS PubMed.
- S. Tombelli, M. Minunni and M. Mascini, Biomol. Eng., 2007, 24, 191–200 CrossRef CAS PubMed.
- Y. Fichou and C. Ferec, Trends Biotechnol., 2006, 24, 563–570 CrossRef CAS PubMed.
- R. Walder, J. Hayes and J. Walder, Nucleic Acids Res., 1993, 21, 4339–4343 CrossRef CAS PubMed.
- K. Williams and D. Bartel, Nucleic Acids Res., 1995, 23, 4220–4221 CrossRef CAS PubMed.
- L. J. Nordstrom, C. A. Clark, B. Andersen, S. M. Champlin and J. J. Schwinefus, Biochemistry, 2006, 45, 9604–9614 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Anal. Bioanal. Chem., 2005, 383, 83–91 CrossRef CAS PubMed.
- U. Gyllensten and H. Elrlich, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 7652–7656 CrossRef CAS.
- C. Boiziau, E. Dausse, L. Yurchenko and J.-J. Toulme, J. Biol. Chem., 1999, 274, 12730–12737 CrossRef CAS PubMed.
- E.-i. Fukusaki, T. Kato, H. Maeda, N. Kawazoe, Y. Ito, A. Okazawa, S.-i. Kajiyama and A. Kobayashi, Bioorg. Med. Chem. Lett., 2000, 10, 423–425 CrossRef CAS.
- S. Y. Low, J. E. Hill and J. Peccia, Biochem. Biophys. Res. Commun., 2009, 378, 701–705 CrossRef CAS PubMed.
- S. Javaherian, M. U. Musheev, M. Kanoatov, M. V. Berezovski and S. N. Krylov, Nucleic Acids Res., 2009, 37, e62 CrossRef PubMed.
- T. Reske, M. Mix, H. Bahl and G.-U. Flechsig, Talanta, 2007, 74, 393–397 CrossRef CAS PubMed.
- M. Citartan, T.-H. Tang, S.-C. Tan and S. Gopinath, World J. Microbiol. Biotechnol., 2011, 27, 1167–1173 CrossRef CAS.
- R. G. Higuchi and H. Ochman, Nucleic Acids Res., 1989, 17, 5865 CrossRef CAS PubMed.
- K. Dastjerdi, G. H. Tabar, H. Dehghani and A. Haghparast, Biotechnol. Appl. Biochem., 2011, 58, 226–230 CrossRef CAS PubMed.
- R. Wilson, Nucleic Acid Ther., 2011, 21, 437–440 CrossRef CAS PubMed.
- M. Espelund, R. Stacy and K. Jakobsen, Nucleic Acids Res., 1990, 18, 6157–6158 CrossRef CAS PubMed.
- H. Dwivedi, R. Smiley and L.-A. Jaykus, Appl. Microbiol. Biotechnol., 2010, 87, 2323–2334 CrossRef CAS PubMed.
- D. A. Daniels, H. Chen, B. J. Hicke, K. M. Swiderek and L. Gold, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15416–15421 CrossRef CAS PubMed.
- Y. S. Kim, C. J. Hyun, I. A. Kim and M. B. Gu, Bioorg. Med. Chem., 2010, 18, 3467–3473 CrossRef CAS PubMed.
- D. Liu, in Handbook of nucleic acid purification, CRC Press, Boca Raton, 2009, ch. 23 Search PubMed.
- M. Heller and A. Guttman, in Integrated microfabricated biodevices, ed. R. Ramanujam, W. Sacks and J. Kang, Marcel Dekker, Inc., New York, 2002, pp. 71–84 Search PubMed.
- C. Marimuthu, T.-H. Tang, J. Tominaga, S.-C. Tan and S. C. B. Gopinath, Analyst, 2012, 137, 1307–1315 RSC.
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- A. Arora, G. Simone, G. B. Salieb-Beugelaar, J. T. Kim and A. Manz, Anal. Chem., 2010, 82, 4830–4847 CrossRef CAS PubMed.
- S.-m. Park, J.-Y. Ahn, M. Jo, D.-k. Lee, J. T. Lis, H. G. Craighead and S. Kim, Lab Chip, 2009, 9, 1206–1212 RSC.
- X. Lou, J. Qian, Y. Xiao, L. Viel, A. E. Gerdon, E. T. Lagally, P. Atzberger, T. M. Tarasow, A. J. Heeger and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2989–2994 CrossRef CAS PubMed.
- J. Qian, X. Lou, Y. Zhang, Y. Xiao and H. T. Soh, Anal. Chem., 2009, 81, 5490–5495 CrossRef PubMed.
- M. Jing and M. T. Bowser, Lab Chip, 2011, 11, 3703–3709 RSC.
- C.-J. Huang, H.-I. Lin, S.-C. Shiesh and G.-B. Lee, Biosens. Bioelectron., 2012, 35, 50–55 CrossRef CAS PubMed.
- B. S. Ferguson, S. F. Buchsbaum, J. S. Swensen, K. Hsieh, X. Lou and H. T. Soh, Anal. Chem., 2009, 81, 6503–6508 CrossRef CAS PubMed.
- M. Hashimoto, P. C. Chen, M. W. Mitchell, D. E. Nikitopoulos, S. A. Soper and M. C. Murphy, Lab Chip, 2004, 4, 638–645 RSC.
- Y. X. Sheng and M. T. Bowser, Analyst, 2012, 137, 1144–1151 RSC.
- J. McDonald, D. Duffy, J. Anderson, D. Chiu, H. Wu, O. Schueller and G. Whitesides, Electrophoresis, 2000, 21, 27–40 CrossRef CAS.
- B. Jo, L. Van Lerberghe, K. Motsegood and D. Beebe, J. Microelectromech. Syst., 2000, 9, 76–81 CrossRef CAS.
- N. Iki and E. S. Yeung, J. Chromatogr., A, 1996, 731, 273–282 CrossRef CAS.
- C. N. Hestekin, J. S. Lin, L. Senderowicz, J. P. Jakupciak, C. O'Connell, A. Rademaker and A. E. Barron, Electrophoresis, 2011, 32, 2921–2929 CrossRef CAS PubMed.
- I. V. Kourkine, C. N. Hestekin, B. A. Buchholz and A. E. Barron, Anal. Chem., 2002, 74, 2565–2572 CrossRef CAS.
- M. N. Albarghouthi, B. A. Buchholz, P. J. Huiberts, T. M. Stein and A. E. Barron, Electrophoresis, 2002, 23, 1429–1440 CrossRef CAS.
- C. D. O'Connell, D. H. Atha, M. C. Oldenburg, J. Tian, M. Siebert, R. Handrow, K. Grooms, L. Heisler and M. de Arruda, Electrophoresis, 1999, 20, 1211–1223 CrossRef CAS.
- H. H. Steinour, Ind. Eng. Chem., 1944, 36, 618–624 CrossRef CAS.
- H. Lamb, Hydrodynamics, Cambridge University Press, 6th edn, 1994 Search PubMed.
- R. Buscall and L. R. White, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 873–891 RSC.
- J. F. Richardson and W. N. Zaki, Chem. Eng. Sci., 1954, 3, 65–73 CrossRef CAS.
- D. Dressman, H. Yan, G. Traverso, K. W. Kinzler and B. Vogelstein, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8817–8822 CrossRef CAS PubMed.
- M. Ageno, E. Dore and C. Frontali, Biophys. J., 1969, 9, 1281–1311 CrossRef CAS.
- H. C. Bimboim and J. Doly, Nucleic Acids Res., 1979, 7, 1513–1523 CrossRef PubMed.
- G. Luck, C. Zimmer, G. Snatzke and G. Söndgerath, Eur. J. Biochem., 1970, 17, 514–522 CrossRef CAS.
- R. Barber, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1971, 238, 60–66 CrossRef CAS.
- A. Holmberg, A. Blomstergren, O. Nord, M. Lukacs, J. Lundeberg and M. Uhlén, Electrophoresis, 2005, 26, 501–510 CrossRef CAS PubMed.
- P. Weber, D. Ohlendorf, J. Wendoloski and F. Salemme, Science, 1989, 243, 85–88 CAS.
- V. Bulmus, Z. Ding, C. J. Long, P. S. Stayton and A. S. Hoffman, Bioconjugate Chem., 1999, 11, 78–83 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |