Determination of impurities in solar grade silicon by inductively coupled plasma sector field mass spectrometry (ICP-SFMS) subsequent to matrix evaporation
Matthias
Balski
ab,
Heinrich
Kipphardt
*a,
Achim
Berger
a,
Sylke
Meyer
c and
Ulrich
Panne
ab
aBAM Federal Institute for Materials Research and Testing, Richard-Willstaetter-Str. 11, 12489 Berlin, Germany. E-mail: heinrich.kipphardt@bam.de; Fax: +49 30 8104 1117; Tel: +49 30 8104 1116
bHumboldt University of Berlin, Brook-Tylor-Str. 1, 12489 Berlin, Germany
cFraunhofer Center for Silicon Photovoltaics CSP, Walter-Huelse-Str. 1, 06120 Halle (Saale), Germany. E-mail: sylke.meyer@csp.fraunhofer.de; Fax: +49 345 5589 101; Tel: +49 345 5589 5116
First published on 30th August 2013
Abstract
A method for the determination of 22 trace impurities in solar grade silicon after dissolution in a mixture of HF and HNO3 and subsequent matrix evaporation is reported. The presented method involves a simple, inexpensive, one-vessel sample preparation apparatus design. The recoveries of B, Na, Mg, Al, P, K, Ca, Ti, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, As, Mo, Sb, W, and Tl at 250 μg kg−1 level are in the range of 93 to 108%. After careful selection of monitored isotopes and their respective resolutions, a sector field mass spectrometer has been used to carry out the measurements. Limits of determination down to 120 ng kg−1 have been obtained using a calibration by three-point standard addition. The method was tested on diluted NIST SRM 57b silicon powder as well as on synthetic test samples and also applied successfully on raw solar grade silicon samples in an interlaboratory comparison including NAA.
Introduction
The photovoltaic sector is a highly competitive market, where cost reduction plays a major role in current developments and research. One aspect of interest is the use of novel raw materials such as upgraded metallurgical grade (umg) silicon for the crystallization process.1,2 However, these materials can exhibit considerably higher impurity contents and variations, which in turn are the major source of defects and limited charge carrier lifetime in silicon crystals and solar cells. For a better understanding of the correlation between impurity concentration and defects, the development of new silicon materials as well as the product and production control, a reliable and accurate chemical analysis is required.The efficiency of silicon solar cells can be deteriorated by a broad range of elements, especially transition metals, at concentrations as low as 100 ng kg−1 in the case of Mo and W2. The inhomogeneous distribution of impurities present in silicon, especially in not recrystallized raw and feedstock silicon, makes the comparison of different methods difficult. Both local and volume analyses are needed to fully characterize the material. Laser ablation (LA)-ICP-MS3 requires a minimum of sample preparation, offering excellent spatial resolution down to a few micrometers and is well suited to measure the spatial distribution of analytes and the homogeneity of the sample, but is mostly limited to materials with higher impurity contents like raw or metallurgical grade silicon due to limits of determination in the μg g−1 range. Out of the techniques analytical chemistry has to offer, only a few feature the topmost aptitude in limits of determination and multi-element capability needed for analysis of re-crystallized, e.g. by the Czochralski process, solar grade silicon, of which in turn some lack the ability of easy, reliable calibration. Namely, neutron activation analysis (NAA)4–6 and glow discharge mass spectrometry (GD-MS)5,7 are relevant instrumental methods, while wet chemical methods are usually applied with ICP-MS as a detection method.
Thermal neutron activation analysis is well suited to examine impurities in semiconductor silicon. Since the cross-section of silicon for thermal neutron capture is relatively small, the sample mass can be in grams without neutron self-shielding becoming a limiting factor. Because of a low atomic number, gamma ray attenuation is not a problem. Furthermore, on activation of silicon, only one radioactive isotope 31Si is produced which has a short half-life of 2.62 h. Thus, waiting a few half-life times before starting the measurements, interfering background from the matrix can be avoided, leading to very low detection limits in the fg g−1 range for selected elements.8,9 Etching after irradiation prevents surface and saw contaminations that otherwise might falsify the results.5 The integrated measurement approach allows monitoring of the mean, bulk crystal purity. In this way, the NAA has become a kind of accepted reference analytical method especially in the semiconductor industry. However, the capabilities of this method do not comprehend the determination of all interesting elements with the desired sensitivity. Moreover, its high instrumental requirements and expenses due to the necessity of a neutron source and radiochemical laboratory make it expensive and difficult to access. Although some efforts have been made to shorten the time requirement for NAA measurements,10 the long response time of this technique makes it not suitable for regular analysis and on-line monitoring.
Especially in comparative studies, glow discharge mass spectrometry offers a very broad range of benefits for routine analysis such as short preparation and analysis time and high sample throughput. However, quantification with this method requires matrix matched standards for calibration with small uncertainty, which are not available. Recently, a study on relative sensitivity factors has been published,11 which opens the gate to quantitative analysis with small uncertainty with this technique. However it does not cover all elements of interest. The high amount of impurities in the synthetic sample used there may also alter the physical properties of silicon, notably the conductivity, compared to rather pure solar grade silicon. This might lead to falsified results especially in direct current GD-MS. Such calibration can be performed rather with techniques like pulsed and/or radiofrequency GD-MS,12 which are way less sensitive to changes in the conductivity of the samples.
Wet chemical based approaches offer a wide range of possible couplings with different methods of atomic spectroscopy for detection. Calibration of wet-chemical methods is achieved very easily by external calibration or standard addition. Attention has to be paid to the sample preparation methods, as they tend to be time-consuming and often leading to elevated blanks owing to contamination during sample handling. Analyte losses may occur through evaporation at high-temperature steps or adsorption on container walls.
A nearly universal analytical tool for inorganic trace determination is the inductively coupled plasma (ICP), often used in coupling with mass spectrometers (MS) as an analyzer and a detector. With this multi-element technique, virtually all elements of the periodic table are accessible with very low limits of determination and a wide dynamic range. One of the main limitations of this technique is the occurrence of spectral interferences due to formation of polyatomic ions or the isobaric isotopes of different elements. A dynamic reaction cell can be used to countervail the former, while for the latter, high-resolution double-focusing sector-field (SF) spectrometers are often the only practicable solution. The other limitation is the high dilution factor after sample digestion needed to minimize the matrix load in the instrument. Even with the low instrumental limits of determination of modern sector field instruments, the detection power for direct analysis of solar silicon samples is often not sufficient for many analytes because of a typical dilution factor of 1000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 necessary to achieve a tolerable matrix load of 1 g L−1 in the measurement solutions. Thankfully, silicon can easily be removed from the solution after dissolution in a mixture of hydrofluoric and nitric acids according to following reactions:
:1 necessary to achieve a tolerable matrix load of 1 g L−1 in the measurement solutions. Thankfully, silicon can easily be removed from the solution after dissolution in a mixture of hydrofluoric and nitric acids according to following reactions:
| 3Si + 4HNO3 → 3SiO2 + 4NO + 2H2O | (1) |
| 3SiO2 + 12HF → 3SiF4↑ + 6H2O | (2) |
The formed silicon tetrafluoride is highly volatile (b.p. = −95.2 °C) and hence can easily be evaporated, allowing lower dilution factors for the measurement.
To achieve this matrix evaporation, different approaches have been proposed. The simplest one is certainly evaporation in open beakers,13,14 which poses serious contamination issues for elements present in ambient air. Recently, microwave-assisted methods have become very popular for multi-element analysis15–18 especially for inert ceramics like silicon nitride. Allowing dissolution and evaporation in one vessel under heat and pressure they permit relatively short preparation times. Blank control is also much better owing to the environmental isolation of the sample. On the other hand, microwave digestion systems tend to be expensive and cleaning/maintenance is intensive for the apparatus. Moreover, loss of other elements forming volatile fluorides can occur, limiting the number of analytes that can be quantitatively determined. Since pure silicon readily dissolves in a mixture of HF/HNO3, microwave digestion has no relevant benefits or unique features over classic digestion methods.
In this paper, we present a simple, cost-effective method for the determination of impurities in high purity silicon by ICP-SFMS combining the benefits of the closed system and one-vessel approach taken from microwave-assisted digestion and the technical simplicity of conventional analysis. We developed a metal-free vaporization design with minimum air contact, small enough to be easily placed under a clean bench for better blank control. From a survey through several German photovoltaic companies along the value chain from raw silicon to solar cells about their impurities of concern, the analytes of interest are B, Na, Mg, Al, P, K, Ca, Ti, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, As, Mo, Sb, W and Tl. Our approach leads to competitive limits of determination taking into account all these analytes in one analysis.
Experimental
ICP-MS instrumentation and conditions
An ICP-SFMS of type Thermo Electron Element XR (Thermo Fisher Scientific, Dreieich, Germany), equipped with a hydrofluoric acid-resistant 100 μL self-aspiratory PFA (perfluoralkoxy copolymer) micro-concentric nebulizer (ESI, Omaha, NE, USA), a PFA spray chamber and a sapphire injector, was used. The operating parameters are given in Table 1. The instrument was tuned daily for maximum sensitivity and signal stability of 115In isotope as well as for maximum resolution. The following isotopes were monitored: 9Be, 11B, 23Na, 25Mg, 27Al, 63Cu, 89Y, 95Mo, 97Mo, 98Mo, 100Mo, 121Sb, 123Sb, 141Pr, 175Lu, 182W, 184W, 203Tl, and 205Tl in low resolution (m/Δm ≈ 300); 9Be, 10B, 11B, 23Na, 24Mg, 25Mg, 27Al, 31P, 42Ca, 44Ca, 46Ti, 47Ti, 49Ti, 52Cr, 53Cr, 55Mn, 56Fe, 59Co, 60Ni, 63Cu, 65Cu, 68Zn, 75As, and 89Y in medium resolution (m/Δm ≈ 4000); 9Be, 23Na, 39K, 66Zn, 68Zn, 69Ga, 70Ge, 71Ga, 72Ge, 75As, and 89Y in high resolution (m/Δm ≈ 10000).
| Plasma conditions | |
| Rf-power/W | 1350 |
| Coolant gas flow/L min−1 | 15 |
| Sample gas flow/L min−1 | 1 |
| Auxiliary gas flow/L min−1 | 0.95 |
| Additional gas flow/L min−1 | 0.06 |
| Data acquisition | |
| Dwell time/ms | 100 |
| No. of scans | 3 |
| No. of replicates | 3 |
| Measurement time/min per sample | 5 |
The isotopes 9Be, 89Y, 141Pr and 175Lu were used as internal standards and added to every sample and solution in an amount that the same final concentration (8 μg L−1) in the measurement solutions was achieved. The elements were chosen because they could not be found at notable concentrations in typical solar grade silicon samples, they formed no volatile fluorides, they showed good sensitivity and covered a wide mass range, ensuring a comprehensive compensation of instrument drift during measurement. The isotope 9Be was used to normalize the data for m/z: 0–31, 89Y for 32–100, 141Pr for 101–141 and 175Lu for 175–205.
Reagents and materials
High-purity water with an electrical resistivity of 18.2 MΩ cm (Milli-Q, Millipore, Bedford, MA, USA) was used throughout for preparing all solutions. The Milli-Q system was equipped with an additional boron specific filter for lowest background concentrations. Nitric acid (65% p.a.) and hydrofluoric acid (40% p.a.) were purchased from Merck, Darmstadt, Germany and further purified in-house by subboiling distillation. All elemental standard solutions except Mo and W were obtained from Merck (Certipur® ICP-MS standards). Molybdenum and tungsten Specpure® standard solutions were obtained from Alfa Aesar (Ward Hill, USA). Mannitol for boric acid determination was obtained from Merck.All standard solutions, water and acids were stored in PFA flasks and containers. Measurements were performed out of 15 mL polypropylene (PP) autosampler tubes (Greiner Bio-One, Kremsmünster, Austria) which were cleaned by a procedure based on the work of Rodushkin and co-workers.19 The tubes were rinsed thoroughly with ultrapure water, filled with 5%/5% v/v subboiling HNO3/HF (referred to the commercially available concentrated, respective 65% and 40% acids) for storage and finally rinsed again with Milli-Q water prior to use. All sample manipulations were performed within class 100 laminar flow clean benches.
Silicon volatilization and apparatus
Starting from the evaporation unit “Ultrapur” (Worldwide Analytical Systems, Kleve, Germany), which consists of a graphite block with 12 bore holes congenial for 30 mL PFA containers from Savillex (Savillex, Minnetonka, USA) on a hot plate, we changed the configuration to a vacuum evacuation design as given in Fig. 1. The apparatus consists of containers with in- and outlet, a stopcock, 1/4′′ PFA tubing leading to the storage vessels and a vacuum pump connected to these condensation vessels. The vacuum pump (PTFE membrane pump, KNF Neuberger, Freiburg, Germany) is used to keep the entire system under reduced pressure.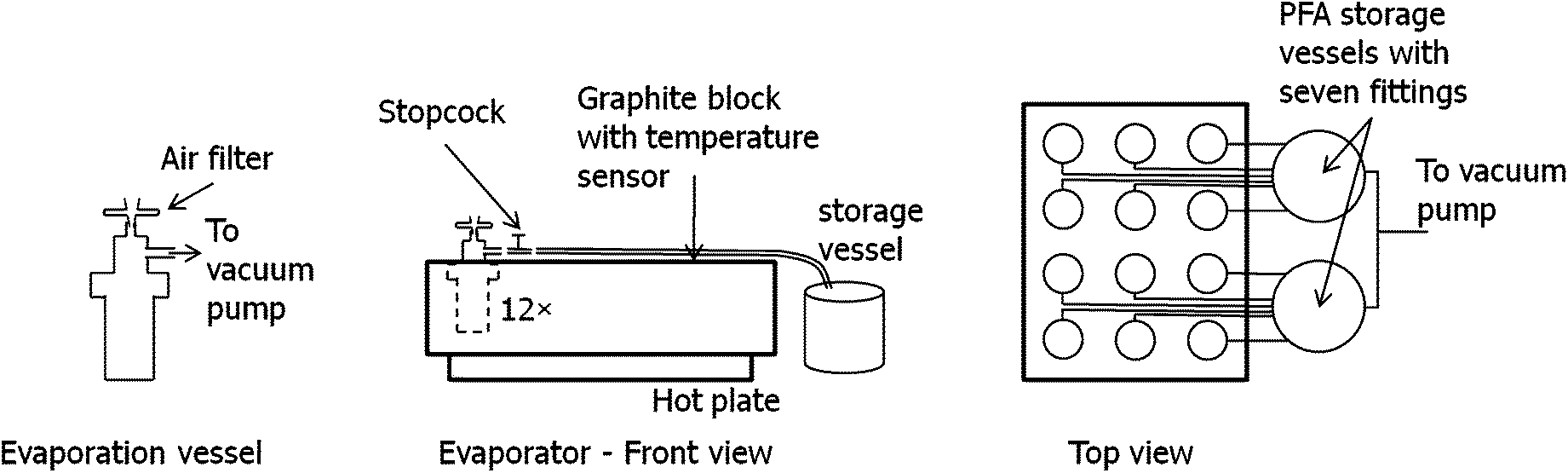 | ||
| Fig. 1 Sample preparation and evaporation apparatus. | ||
The containers are equipped with threaded caps with two 1/4′′ fittings, allowing the connection of the tubing leading to the storage vessels and vacuum pump at the outlet fitting, and an air aspiration port on the other end, thus providing a constant, moderate air flow transporting the evaporated acid and matrix to the storage vessels. Directly behind the outlet, a stopcock allows additional regulation of the air-flow if necessary. The aspiration is done through a short piece of PFA tubing, in which a syringe filter (PTFE membrane, 0.2 μm pore size, 25 mm diameter, NeoLab, Heidelberg, Germany) has been inserted to reduce the air-flow and minimize contamination risk. Experiments have shown that filters with smaller diameter show very non-uniform admission of air, leading to unequal evaporation times among the vessels. The graphite heating block was placed inside a laminar flow box to minimize the risk of contamination.
Two types of caps with fittings are commercially available for the sample containers: parallel (both fittings pointing upwards) or orthogonal configuration. Since the evaporated liquid already condenses in the tubing, the latter configuration has been chosen for this design, allowing easier drainage of the evaporated liquid towards the storage vessels through the horizontal fitting by the assistance of gravity in addition to the reduced pressure.
The storage vessels are two one-liter PFA containers with respectively six inlets and one outlet in the cap, mounted underneath the level of the evaporation vessels, bending the PFA tubing downwards for easier drainage. The evaporated acids and matrix are collected at the bottom of the vessels and can be removed after evaporation is completed. The two outlets of the storage vessels are connected to the vacuum pump via a T-piece.
All parts and tubing are made of PFA (stopcocks made of PTFE) and can easily be cleaned by evaporating clean acids through the system. Since sample preparation can be performed in 30 mL PFA containers fitting into the graphite block, only the caps have to be changed in order to switch between sample digestion and evaporation. Since the caps are not in contact with the sample solution and the evaporated acid does not flow back to the container, the system need not be cleaned between each evaporation. This increases the capacity utilization of the evaporation apparatus and shifts the critical time consuming cleaning procedures towards cheap and easy replaceable, small PFA containers.
ICP-MS sample preparation
Solid crystalline and feedstock silicon samples were provided by different companies of the German photovoltaic industry.The sample preparation procedure is outlined in Fig. 2. In each set of samples, three method blanks were analyzed together with nine samples. The blanks were treated exactly as the samples, only without addition of any silicon.
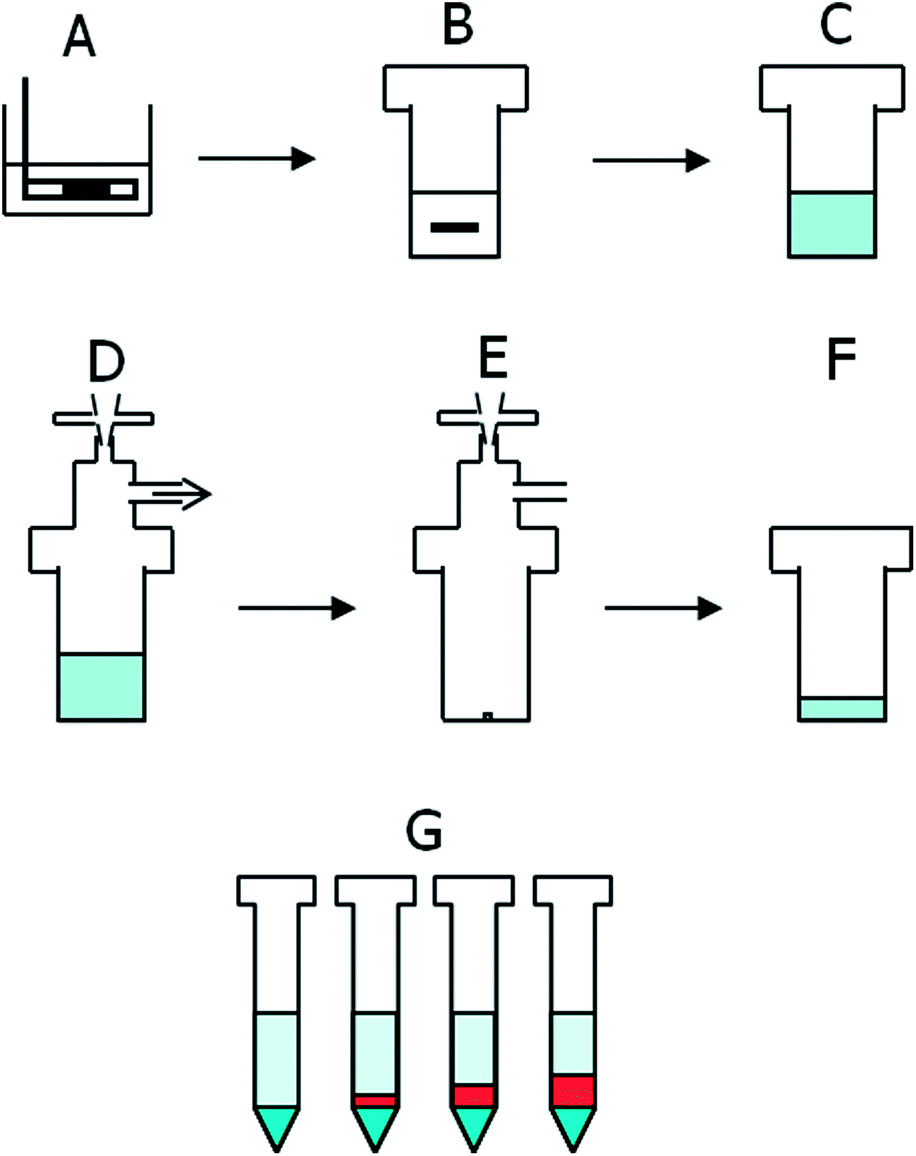 | ||
| Fig. 2 Sample preparation procedure. (A): Surface acid etching/cleaning. (B): Place silicon, HF, mannitol and internal standard into a PFA container. (C): Add HNO3 (under cooling). (D): Swap the cap to the one with an air filter (top) and connect to a vacuum pump (arrow). (E): Evaporate to dryness and swap the cap. (F): Redissolve the residue to 5 mL. (G): Prepare 4 × 1 mL aliquots, of which 1× pure sample and 3× standard addition. | ||
Silicon samples of up to 1000 mg were treated with acid etching (HNO3 + HF, 30% + 10% by volume, for ca. 10 minutes at room temperature) to remove surface contamination until any observable damage from sawing was no longer visible.
The samples were then weighed into 30 mL PFA containers with closed caps and 250 μL of an internal standard solution (Be, Y, Pr, Lu at 800 μg L−1) and 50 μL of a solution of 25 g L−1 mannitol were added. The addition of mannitol gives rise to the formation of a non-volatile complex of boron, which prevents its removal as fluoride together with silicon.20 The containers were placed in an ice bath and the total amount of 8.5 mL HF was added at once. The nitric acid was added slowly until a total addition of 4.5 mL. The reaction speed has to be controlled carefully to avoid spilling but can be automated. The addition of nitric acid takes about two hours. Cooling of the samples can be omitted with a slower addition rate of nitric acid.
The closed caps were then replaced by the evaporation caps with fittings and the containers were placed into the graphite heating block. The samples were heated to 110 °C for one hour to allow a slower and controlled evaporation of the matrix, and then further heated for 3.5 h at 150 °C to dryness. Immediate heating to 150 °C can cause clogging of the tubing.
The residues in the containers were dissolved and diluted to 5 mL using 5%/2.5% v/v HNO3/HF. These solutions were allowed to stand overnight to guarantee the total dissolution of the residues. Then, four aliquots of 1 mL each were transferred into 15 mL PP (polypropylene) tubes. One was simply filled up with Milli-Q water to a total volume of 5 mL. The remaining three were spiked with multi element standards of increasing concentration containing all elements of interest and finally filled up to 5 mL. Spiking can take place as needed according to expected concentrations or preliminary tests. The final solution contained 1% HNO3 and 0.5% HF.
The reproducibility of matrix evaporation and stability of the instrument were very good, varying less than 10% over a ten hours measurement period. Thus, if similar samples are analyzed, the sample preparation and analysis time can be considerably reduced by preparing only one standard addition for every three samples or so, relating one standard addition calibration to the following, merely diluted samples.
Recovery test
Recovery of the ICP-MS method was determined by spiking a homogeneous solution of dissolved silicon with a multi element standard. Six grams of a feedstock silicon sample were dissolved in a PFA jar and the solution was evenly partitioned in 30 mL digestion vessels. In three of these vessels, 15 μL of the 20 mg L−1 multi element standard was added prior to evaporation, leading to a spike of 10 μg L−1 in the final solution.Interlaboratory comparison of synthetic silicon containing solutions
To test the standard addition and instrumental quantification procedure, a synthetic digestion solution containing silicon as the matrix element and B, Ca, Co, Cr, Cu, Fe, K, Na, Ni, P, Ti, and Zn as analytes at “unknown” concentrations has been provided by a befriended lab and was analyzed by us, i.e., diluted 1:25 for standard addition before measurement, and the Fraunhofer Center for Silicon Photovoltaics CSP (Halle, Germany). The Fraunhofer CSP used a different approach for sample preparation, as described elsewhere.21
Test with NIST SRM 57b
The only currently available certified reference material for impurities in silicon is the NIST SRM 57b silicon powder, the concentration of the impurities being several orders of magnitude higher than in solar grade silicon. For the development of solid sampling analysis methods which are not discussed here, we prepared a silicon powder with more realistic impurity contents by grinding a feedstock silicon provided from an industrial partner in a tungsten carbide planet mill and mixing it in the ratio of 10:1 with the NIST SRM in a mixer-mill (MixerMill 8000D, SPEX SamplePrep, Metuchen, NJ, USA). This diluted powder was also used for the ICP-MS analysis. Due to the still extremely high content of impurities, only 100 μg of the powder were used for sample preparation (n = 4). Simultaneously, the pure feedstock silicon powder was analyzed (n = 2) and the content of this material was subtracted from the mixed powder. The standard addition applied in the measurement was focussed on the elements with lower content, the effective calibration range being 0–600 μg g−1 in the NIST SRM, thus comprising all elements except Al and Fe.
Interlaboratory comparison with real feedstock solar grade silicon material
Two different qualities of solar grade silicon feedstock (Material A and B) were provided by a German photovoltaic company. Subsamples of 1.5 g were cut out of the chunks, etched down to analyse mass and analyzed according to the procedure described in this paper. Determination was done in triplicate by ICP-MS at BAM and Fraunhofer CSP and in duplicate by NAA at HZB Berlin. Due to the high boron and phosphorus doping of these materials, the standard addition was performed with separate concentrations of B, P and the other elements. Phosphorus was added at final concentrations of 200, 400 and 600 ng mL−1 to the solutions of both materials. Boron was added to a final concentration of 200, 400 and 600 ng mL−1 to Material A and 20, 40, and 60 ng mL−1 to Material B. All other elements had a final concentration of 0.2, 0.4 and 0.6 ng mL−1 in the solutions of both samples.Neutron activation analysis
Neutron activation analysis has been performed using the k0 standardization. For the k0 standardization, the determination of neutron flux parameters and a comparator factor has to be performed. This is done by co-irradiating a monitor set with the samples to be analyzed. De Corte et al.22 recommended the gold + zirconium monitor method for in situ determination of neutron flux parameters.The zirconium monitor was made from a 1% Zr-solution (NIST 3169). Four zirconium standards were produced by filling app. 55 mg of the solution into quartz ampoules. The amount of the individual solution was determined gravimetrically. The ampoules were dried in an oven at 50 °C for one day and then subsequently sealed by melting.
The gold monitor was made out of a 981 mg L−1 Au-solution (PlasmaCAL, SCP Science, Champlain, NY, USA). This solution was diluted by a factor of 100 and from this dilute solution four gold standards were produced in the same manner as described for the zirconium standards.
The silicon samples of around 2.5 g were packed into an aluminium capsule. The aluminium capsule has two layers separated by a sieve. This means that in each layer one pair of samples could be packed. In addition to this pair of samples two Zr standards and two Au standards were packed. Blank positions were filled with quartz ampoules to prevent the samples from rattling. The aluminium capsule was closed with a crown cap. The bottom of the capsule and the cap have holes to allow water to flow through during irradiation to cool the samples and the quartz ampoules.
Neutron irradiation has been performed with the swimming pool reactor BER II of the Helmholtz-Zentrum Berlin für Materialien und Energie (HZB). To gain highest sensitivity, the DBVK irradiation facility has been chosen. This irradiation facility is located in the middle of the reactor core replacing one fuel element and has therefore the highest neutron flux available at the BER II reactor. The thermal neutron flux in this position is app. 1 × 1014 cm−2 s−1 and the epithermal flux is app. 9 × 1012 cm−2 s−1.
The samples were irradiated for 4.5 days and cooled down for 2 days. To remove surface contaminants, samples and quartz ampoules were etched with 25% hydrofluoric acid, rinsed three times with distilled water and dried by dipping into ethanol and acetone, respectively.
A high-purity germanium detector was used for gamma ray counting. To reduce background the detector was shielded with 10 cm of lead. The detector was connected to a Canberra (Meriden, CT, USA) gamma-spectrometer with 1.9 keV resolution at 1.33 MeV via a digital signal processor. Spectra from 50 to 2100 keV have been recorded. Counting times for each sample varied between 5 h (for two samples) and up to 24 h for the others. Due to these long counting times different cooling times for the samples accumulated, resulting in different limits of determination for some elements. Canberra Genie software was used to control the measurements and to accumulate and evaluate the spectra. The elemental concentrations including uncertainty calculations and calculation of the detection limits were done with the Kayzero for Windows software (k0 Ware, Heerlen, The Netherlands).
Results and discussion
Blank control
For reduction of contamination of the sample solutions, careful cleaning of all labware has been conducted and PFA was used where possible. The measurement tubes were cleaned as described in the methods section. Table 2 shows the effectiveness of this procedure by comparing the concentration of selected elements leached into acidified water (1% HNO3, 0.5% HF) and stored for 72 hours in cleaned or as-delivered PP tubes. Cleaning reduces the acid blanks of critical elements like P and Fe up to hundredfold.| Element | Conc./ng L−1 As-supplied, n = 7 | Conc./ng L−1 Cleaned, n = 7 |
|---|---|---|
| Na | 84 ± 26 | 10 ± 17 |
| Cu | 55 ± 12 | 3 ± 2 |
| Fe | 554 ± 479 | 6 ± 4 |
| P | 936 ± 178 | <100 |
| Ti | 54 ± 14 | 22 ± 19 |
Optimization of matrix evaporation
To minimize matrix effects in the measurements it is mandatory to optimize the matrix evaporation with respect to the reaction rate. We performed a series of tests with different amounts of nitric and hydrofluoric acids. An excess of both acids shortens the reaction time but leads to a higher amount of residue. Addition of surplus nitric acid only did not cause any adverse effects, while the addition of 10.0 instead of 8.5 mL HF to 1000 mg Si led to a cake of a mass of about 40 mg. The residue was identified by X-ray diffraction as ammonium hexafluorosilicate (NH4)SiF6. Most likely the excess of HF shifts the equilibrium of the reaction (3) to the right side, allowing cation exchange with ammonia formed by reduced nitric acid.23 Both ammonium hexafluorosilicate and hexafluorosilicic acid have decomposition temperatures above 100 °C, making it difficult to remove them from the solution.| 2HF + SiF4 ↔ H2SiF6 | (3) |
The addition of 8.5 mL HF per gram Si was a good compromise between reaction rate and evaporation with >99% Si removed.
Figures of merit
Limits of determination (LoDs) were calculated according to DIN 32645 (ref. 24) as the concentration corresponding to the signal equivalent of the 9s variation of procedure blank solutions (n = 12). Typical LoDs on sample basis are shown in Table 3 and compared to LoDs published by Ueng et al.17 In this work, the LoDs could be reduced for all elements ranging from small improvements for Co and Ni to twentyfold enhancement in the case of Fe and K. In the distinction between acid/instrument blanks on one hand and the procedure blank on the other hand, procedure determination limits have also been compared with instrumental LoDs computed from pure acid blanks to check if method LoDs are limited either by the instrument and acids used or by cross-contamination during the sample preparation procedure. For most elements, there was no significant difference between procedure and acids blanks. Exceptions are Al, Ca, Mg and Ti as shown in Table 4. Since the LoDs may vary between runs because of slight variations in the purity of acids and materials, the LoDs of each run were calculated separately from the blanks of this run.| Element | LoD/ng g−1 | LoD from[15]/ng g−1 |
|---|---|---|
| B | 22 | |
| Na | 7.1 | |
| Mg | 13 | 81 |
| Al | 11 | |
| P | 5.7 | |
| K | 7.5 | 135 |
| Ca | 53 | |
| Ti | 4.2 | |
| Cr | 0.27 | 2.7 |
| Mn | 0.21 | 1.5 |
| Fe | 1.7 | 36 |
| Co | 0.26 | 0.3 |
| Ni | 2.0 | 4.8 |
| Cu | 0.38 | 1.2 |
| Zn | 2.7 | 6 |
| Ga | 0.12 | |
| Ge | 0.16 | |
| As | 0.65 | |
| Mo | 0.25 | |
| Sb | 0.17 | |
| W | 0.37 | |
| Tl | 0.17 |
| LoD/ng g−1 for acid blank | LoD/ng g−1 for method blank | |
|---|---|---|
| Al | 6.8 | 11 |
| Mg | 2.1 | 13 |
| Ca | 18 | 53 |
| Ti | 0.8 | 4.2 |
The results of the recovery experiment, the linearity of the calibration curves (r2) and their sensitivity are shown in Table 5. All recoveries are close to 100%.
| Element | Recovery/% | Coefficient of determination | Slope/counts/ng mL−1 |
|---|---|---|---|
| B | 98 ± 3 | 1.000 | 31524 |
| Na | 98 ± 3 | 1.000 | 118073 |
| Mg | 101 ± 1 | 0.999 | 4512* |
| Al | 96 ± 5 | 1.000 | 118016 |
| P | 100 ± 3 | 0.999 | 476* |
| K | 104 ± 3 | 0.999 | 2847‡ |
| Ca | 108 ± 3 | 0.998 | 313* |
| Ti | 100 ± 11 | 0.994 | 839* |
| Cr | 103 ± 1 | 0.999 | 9265* |
| Mn | 103 ± 4 | 0.999 | 12810* |
| Fe | 103 ± 5 | 0.999 | 10858* |
| Co | 102 ± 2 | 0.999 | 12000* |
| Ni | 103 ± 3 | 0.998 | 2736* |
| Cu | 99 ± 3 | 0.997 | 101442 |
| Zn | 106 ± 1 | 0.999 | 420‡ |
| Ga | 104 ± 5 | 0.999 | 2170‡ |
| Ge | 107 ± 4 | 0.999 | 514‡ |
| As | 103 ± 2 | 0.999 | 316‡ |
| Mo | 101 ± 2 | 0.997 | 50566 |
| Sb | 93 ± 3 | 1.000 | 74408 |
| W | 96 ± 1 | 1.000 | 118541 |
| Tl | 99 ± 1 | 1.000 | 361164 |
Synthetic samples
Because preliminary tests showed a pronounced inhomogeneity of available silicon samples, we decided to test our method at first on artificial solutions like they were to expect after sample digestion and matrix evaporation. The results of the comparison with the Fraunhofer CSP are shown in Table 6. In this table, the expanded uncertainty with k = 2 based on the typical standard deviation observed for replicates is displayed. Except for the result of Na at one lab, which was due to subtraction of a too high blank value, the agreement was excellent within the uncertainty.| Reference cspiked/μg L−1 | c observed/μg L−1, CSP | c observed/μg L−1, BAM | |
|---|---|---|---|
| Ti | 4.0 ± 0.1 | 4.0 ± 3.6 | 3.8 ± 3.4 |
| Co | 3.0 ± 0.1 | 3.2 ± 0.2 | 3.0 ± 0.2 |
| Ni | 7.1 ± 0.1 | 7.0 ± 1.0 | 7.4 ± 1.1 |
| Zn | 9.0 ± 0.1 | 8.4 ± 3.4 | 10 ± 4.0 |
| Cr | 6.0 ± 0.1 | 5.8 ± 1.2 | 5.6 ± 1.1 |
| Cu | 20 ± 0.1 | 19 ± 1.9 | 20 ± 2.0 |
| Fe | 81 ± 0.6 | 82 ± 12 | 80 ± 12 |
| Na | 10 ± 0.1 | 7.9 ± 0.4 | 9.6 ± 0.5 |
| Ca | 50 ± 0.4 | 50 ± 2.5 | 50 ± 2.5 |
| K | 100 ± 1 | 83 ± 17 | 106 ± 21 |
| B | 252 ± 2 | 240 ± 36 | 244 ± 37 |
| P | 604 ± 6 | 585 ± 59 | 612 ± 61 |
Analysis of NIST SRM 57b
The very high impurity content of the NIST SRM made the direct analysis with a method as sensitive as the ICP-MS difficult, but the required sample preparation posed issues too. For example, since a WC mill with a high content of cobalt as a binding agent was used for grounding the pure silicon for solid–solid dilution, cobalt could not be measured due to very high background levels. The results for most elements are nevertheless in agreement with the certified and reference values (see Table 7). The iron concentration found is notably higher than the certified value because as described in the method section, it lies way above the calibration range, resulting in a small difference between the genuine concentration of iron in the measurement solution and the spike of the standard addition.| Element | Observed content/μg g−1 | Reference content/μg g−1 |
|---|---|---|
| a Above calibration range. | ||
| B | 12.2 ± 0.5 | 12.5 ± 2.1 |
| P | 15.8 ± 1.2 | 16.3 ± 1.5 |
| Fea | 4622 ± 770 | 3400 ± 60 |
| Ni | 18.6 ± 1.3 | 15.3 ± 1.7 |
| Cu | 16.3 ± 3.9 | 17.2 ± 5.8 |
| Cr | 20.3 ± 2.2 | 17.3 ± 3.3 |
| Ala | 1835 ± 319 | 1690 ± 220 |
| Mn | 86.0 ± 7.6 | 78.2 ± 7.2 |
| Ti | 349 ± 33 | 346 ± 49 |
Applications and interlaboratory comparison on real samples
As the figures of merit were found to be suitable for analysis, the method was compared with the ICP-MS method developed at the Fraunhofer CSP and Neutron Activation Analysis at the BER II reactor of Helmholtz-Zentrum Berlin.The results of the interlaboratory comparison on the two different feedstock silicon samples are shown in Table 8. The overall comparability of the results between the two ICP-MS methods with different sample preparation procedures and the NAA is very good and demonstrates the quality of the method presented here. Some elements like boron or phosphorus cannot be measured by NAA. Here, the two ICP-MS methods show good agreement. For a few other elements the NAA lacks the necessary limits of determination for a fair comparison. Especially for some fast-decaying isotopes, e.g. Ge and K, the LoD depends highly on the time passed between irradiation and measurement and may vary between samples by several orders of magnitude. Some elements also exhibit a vast range of content in the samples, as indicated by the uncertainty bars. The known superimposed inhomogeneity of the real material is most likely the cause for these effects.
| Element | w/μg kg−1 | w/μg kg−1 | w/μg kg−1 |
|---|---|---|---|
| ICP-MS BAM | ICP-MS CSP | NAA HZB | |
| Material A | |||
| B | 4281 ± 261 | 3816 ± 511 | |
| Na | 21 ± 8 | 266 ± 104 | 2.5 ± 0.3 |
| Mg | <4 | 31 ± 31 | |
| Al | 62 ± 16 | 83 ± 3 | |
| P | 5375 ± 508 | 4864 ± 659 | |
| K | 44 ± 54 | 24 ± 17 | <1500 |
| Ca | 99 ± 50 | 1850 ± 1784 | |
| Ti | 11 ± 4.4 | 25 ± 12 | |
| Cr | 1.1 ± 0.4 | 2.3 ± 1.2 | <0.15 |
| Mn | 0.4 ± 0.2 | 2.9 ± 2.3 | <80000 |
| Fe | 26 ± 11 | 22 ± 16 | <5 |
| Co | 0.2 ± 0.11 | 0.1 ± 0.3 | <0.008 |
| Ni | 1.2 ± 1.1 | n.d. | |
| Cu | <2 | n.d. | <3 |
| Zn | 7.8 ± 4.0 | 56 ± 62 | <0.3 |
| Ga | 0.07 ± 0.01 | n/a | <0.06 |
| Ge | n/a | 319 ± 99 | 266 ± 6 |
| As | 2.4 ± 0.1 | 1.5 ± 1.3 | 2.7 ± 0.1 |
| Mo | <0.2 | 0.5 ± 0.8 | <0.3 |
| Sb | <0.07 | 1.1 ± 1.9 | 0.02 |
| W | 0.5 ± 0.4 | 1.9 ± 2.7 | <0.05 |
| Tl | <0.04 | n.d. | |
| Material B | |||
| B | 650 ± 18 | 658 ± 20 | |
| Na | 288 ± 126 | 437 ± 171 | 254 ± 194 |
| Mg | 4.8 ± 1.8 | 23 ± 15 | |
| Al | 75 ± 13 | 139 ± 40 | |
| P | 2159 ± 55 | 1743 ± 212 | |
| K | 8.5 ± 7.5 | 22 ± 22 | <2000 |
| Ca | 30 ± 19 | 469 ± 246 | |
| Ti | 91 ± 44 | 39 ± 12 | |
| Cr | 9.1 ± 2.2 | 3.4 ± 7.5 | 9.4 ± 8 |
| Mn | 5.5 ± 2.6 | 5.9 ± 1.7 | <150 |
| Fe | 295 ± 68 | 363 ± 106 | 504 ± 433 |
| Co | 0.7 ± 0.3 | 0.73 ± 0.51 | 0.7 ± 0.6 |
| Ni | 8.3 ± 1.6 | n.d. | |
| Cu | 5.1 ± 1.8 | n.d. | <40 |
| Zn | 5.8 ± 4.0 | 40 ± 17 | <40 |
| Ga | 5.9 ± 0.4 | n/a | 4.0 ± 5.1 |
| Ge | n/a | 589 ± 126 | <60000 |
| As | 41 ± 7.7 | 15.2 ± 8.2 | 47 ± 3 |
| Mo | <0.2 | 0.53 ± 0.48 | <0.7 |
| Sb | 3.4 ± 0.1 | 2.4 ± 1.2 | 3.5 ± 0.4 |
| W | 4.3 ± 0.8 | 2.0 ± 2.0 | 1.2 ± 1.1 |
| Tl | <0.04 | n.d. | |
Conclusion
We present a simple and cost-effective sample preparation and analysis method for the determination of trace impurities in silicon. Cross-contamination is reduced by performing digestion and evaporation in a single PFA vessel. The simple and affordable apparatus is virtually maintenance-free and easily scalable. Twenty-two elements have been named as important analytes in a poll among photovoltaic companies, of which all could be analyzed in one sweep. Moreover, the sample preparation method offers improved limits of determination, a good recovery even for elements forming volatile compounds with fluoride, is easy to control and needs a minimum of attendance. The performance on synthetic and real samples shows that this method is well suited for routine analysis.Acknowledgements
Dorothea Alber and Gregor Bukalis are kindly acknowledged for performing the activation of the samples at HZB, Franziska Emmerling for the X-ray diffractometry measurements and Norbert Jakubowski for critical reading of the manuscript. This work was supported by the German Federal Ministry of Education and Research in the framework of the “Spitzencluster Solarvalley” program, project “xμ-Material” (contract no. 03SF0398G).Notes and references
- M. D. Johnston, L. T. Khajavi, M. Li, S. Sokhanvaran and M. Barati, JOM, 2012, 64, 935–945 CrossRef CAS.
- A. A. Istratov, T. Buonassisi, M. D. Pickett, M. Heuer and E. R. Weber, Mater. Sci. Eng., B, 2006, 134, 282–286 CrossRef CAS PubMed.
- J. Pisonero, I. Kroslakova, D. Gunther and C. Latkoczy, Anal. Bioanal. Chem., 2006, 386, 12–20 CrossRef CAS PubMed.
- C. C. Chu, P. Y. Chen, M. H. Yang and Z. B. Alfassi, Analyst, 1990, 115, 29–34 RSC.
- R. Kvande, L. J. Geerligs, G. Coletti, L. Arnberg, M. Di Sabatino, E. J. Ovrelid and C. C. Swanson, J. Appl. Phys., 2008, 104, 064905 CrossRef.
- D. Macdonald, A. Cuevas, A. Kinomura, Y. Nakano and L. J. Geerligs, J. Appl. Phys., 2005, 97, 033523 CrossRef.
- M. A. Martorano, J. B. F. Neto, T. S. Oliveira and T. O. Tsubaki, Mater. Sci. Eng., B, 2011, 176, 217–226 CrossRef CAS PubMed.
- M. L. Verheijke, H. J. J. Jaspers and J. M. G. Hanssen, J. Radioanal. Nucl. Chem., 1989, 131, 197–214 CrossRef CAS.
- N. B. Kim, H. W. Choi, S. K. Chun, S. Y. Cho, N. J. Woo and K. S. Park, J. Radioanal. Nucl. Chem., 2001, 248, 125–128 CrossRef CAS.
- J. Hampel, F. M. Boldt, H. Gerstenberg, G. Hampel, J. V. Kratz, S. Reber and N. Wiehl, Appl. Radiat. Isot., 2011, 69, 1365–1368 CrossRef CAS PubMed.
- M. Di Sabatino, A. L. Dons, J. Hinrichs and L. Arnberg, Spectrochim. Acta, Part B, 2011, 66, 144–148 CrossRef PubMed.
- M. Hohl, A. Kanzari, J. Michler, T. Nelis, K. Fuhrer and M. Gonin, Surf. Interface Anal., 2006, 38, 292–295 CrossRef CAS.
- J. S. Chen, H. M. Lin and M. H. Yang, Fresenius. J. Anal. Chem., 1991, 340, 357–362 CrossRef CAS.
- C. Pilger, F. Leis, P. Tschöpel, J. A. C. Broekaert and G. Tölg, Fresenius. J. Anal. Chem., 1995, 351, 110–116 CrossRef CAS.
- Y. Han, H. M. Kingston, R. C. Richter and C. Pirola, Anal. Chem., 2001, 73, 1106–1111 CrossRef CAS.
- A. C. Sahayam, S. J. Jiang and C. C. Wan, Anal. Chim. Acta, 2007, 605, 130–133 CrossRef CAS PubMed.
- R. L. Ueng, S. J. Jiang, C. C. Wan and A. C. Sahayam, Anal. Chim. Acta, 2005, 536, 295–299 CrossRef CAS PubMed.
- C. C. Wan, S. J. Jiang, M. T. You and A. C. Sahayam, J. Anal. At. Spectrom., 2005, 20, 1290–1292 RSC.
- I. Rodushkin, E. Engstrom and D. C. Baxter, Anal. Bioanal. Chem., 2010, 396, 365–377 CrossRef CAS PubMed.
- T. Ishikawa and E. Nakamura, Anal. Chem., 1990, 62, 2612–2616 CrossRef CAS.
- S. Meyer, S. Richter, M. Balski and C. Hagendorf, Photovoltaics International 14th edition, 2011, pp. 34–39 Search PubMed.
- F. De Corte and A. Simonitis, Vademecum for k0 users, DSM Research, Geleen, 1994 Search PubMed.
- M. Steinert, J. Acker, M. Krause, S. Oswald and K. Wetzig, J. Phys. Chem. B, 2006, 110, 11377–11382 CrossRef CAS PubMed.
- DIN 32645:2008-11, Deutsches Institut für Normung, Beuth, Berlin, 2008.
| This journal is © The Royal Society of Chemistry 2014 |