Estimation of angiotensin peptides in biological samples by LC–MS method
Quaisar
Ali
a,
Yonnie
Wu
b,
Sourashish
Nag
a and
Tahir
Hussain
*a
aCollege of Pharmacy, Department of Pharmacological and Pharmaceutical Sciences, University of Houston, 521 Science and Research Building 2, Houston, TX 77204, USA. Fax: +1 713-743-1229; Tel: +1 713-743-1276E-mail: thussain@central.uh.edu
bMass Spec Center, Department of Chemistry and Biochemistry, Auburn University, Auburn, AL 36849, USA
First published on 15th October 2013
Abstract
The low abundance of angiotensin peptides in biological tissues such as the kidney cortex, adipose tissue, urine and plasma makes their detection and quantification a challenge. A few available methods used to quantify these peptides involve lengthy processes of sample preparation and are hardly quantitative. Here, we report a mass spectrometry approach for quantifying angiotensin peptides [Ang II, Ang-(1-7)] in the kidney cortex, epididymal white adipose tissue (eWAT), urine and plasma of male mice. Tissue homogenates, urine and plasma samples were solid-phase extracted with C18 Sep-Pak cartridges and the proteinaceous compounds were eluted off. These extracted peptide samples were separated on a C18 column with a linear acetonitrile gradient and detection was carried out using a Q-ToF mass analyzer in the ESI+-MS ion mode based on retention time, accurate mass measurement of peptides, the isotope pattern of the doubly charged molecular ion, and quantification of peak area (or ion count) when referencing to the angiotensin peptide standards. The lower limit of quantification for each angiotensin peptide was 10 pg mg−1 with the percent recovery at 100.6%. The intra-batch precision for Ang-(1-7) and Ang II were 24.0 and 12.7%, respectively, with corresponding accuracy of 84.0–123.0% and 100.2–116.0%. Using this method, we determined the levels of Ang II and Ang-(1-7) in kidney cortex, eWAT, urine and plasma. Quantification of angiotensin peptides could help target subtle therapeutic changes against pathophysiological conditions such as obesity, kidney disease and hypertension.
Introduction
Renal angiotensin (Ang) peptides are the major regulators of blood pressure and Na-homeostasis,1 while adipose angiotensin peptides have an important role in lipid metabolism.2 Ang II by acting on AT1 receptor promotes vasoconstriction and an increase in blood pressure while the action of Ang II via AT2 receptor produces opposite effects to that of AT1 receptor.1,3,4 Ang-(1-7) infusion causes vasodilation and Na-excretion.5,6 Ang II increases lipogenesis in adipose tissue7 while Ang-(1-7) promotes adipocyte differentiation, alters lipid metabolism and decreases pro-inflammatory markers.8 Since these peptides are an integral part of the renin angiotensin system and regulate such a diverse network of physiological conditions, it is imperative to study their metabolism and formation in order to understand the mechanism of several pathophysiological conditions including hypertension and obesity.Angiotensin peptides are present in tissues, plasma and urine at femtomolar levels.9 Hence, an extremely sensitive and selective method is required for their exact measurement. The available methods in the literature for the analysis of angiotensin peptides are based on high performance liquid chromatography (HPLC) followed by radioimmunoassay (RIA)10,11 using an antibody which recognizes all the peptides. These antibodies exhibit higher or equal reactivity with most of the angiotensin peptides. These methods are tedious and require much time and effort. Thus, it is highly desirable to develop a sensitive, time-efficient method which poses no health hazards.
Mass spectrometry is sensitive in detecting peptide ions. The mass resolution of the Q-ToF instrument, typically down to ppm, can separate compounds with a mass difference of 0.01 Dalton (or lower) in a biological mixture. Angiotensin peptides are mostly doubly charged ions at physiological pH when electrosprayed because of the basic residues in the sequence (for example, Ang II has ARVYVHPF), allowing their isotope pattern to be distinguished from most small molecules that are singly charged and from chemical noise, thus increasing the sensitivity and detection limit.
Solid phase extracted biological molecules from tissue lysates are a mixture of complex molecules including lipids, proteins and semi-polar small and non-polar molecules. The separation of these biological molecules on a C18 column with an acetonitrile gradient is related to their structure and hydrophobicity. Ultra-performance liquid chromatography (UPLC) using a C18 column can further reduce sample complexity and can also indicate hydrophobicity that is consistent with a target molecule. The combination of retention time with accurate mass measurement can uniquely identify an unknown molecule in complex mixtures of biological extracts.
In the present study, we have developed a novel method for the quantification of angiotensin peptides in the kidney cortex, epididymal white adipose tissue (eWAT), urine, and plasma using liquid chromatography/mass spectrometry (LC–MS). LC–MS is a highly sensitive technique and simultaneously measures angiotensin peptides [Ang II, Ang-(1-7)] in biological samples. We have evaluated this method by identifying peptides based on their molecular weight, retention time, and quantification of peak area (or ion count) when referenced to angiotensin peptide standards. This method is reproducible and enables high throughput. This method of angiotensin peptides quantification in biological tissues could be employed in research or diagnostic purposes under pathophysiological conditions and could replace the existing method which is cumbersome and creates a health hazard.
Materials
Standards for Ang II and Ang-(1-7) were purchased from Sigma Aldrich (St Louis, MO). The C18 solid phase extraction (SPE) cartridge was purchased from Waters Corporation (Milford, MA). Acetonitrile, deionized water, formic acid and all other solvents and chemicals were of HPLC grade and were purchased from Sigma or Fisher Scientific.Methods
Animals
Male mice (C57BL/6) of 15–16 weeks of age were purchased from Harlan Laboratories, Indianapolis, IN. After arrival, the animals were housed in the Auburn University animal care facility. All the animal experimental protocols were approved by the Institutional Animal Care and Use Committee at Auburn University. Metabolic cages were used to collect 24 h urine samples. After urine collections, these animals were euthanized and the kidneys and epididymal white adipose tissues (eWAT) were removed and kept frozen at −80 °C for measuring angiotensin peptides. Trunk blood was collected in EDTA tubes, centrifuged at 2000 rpm for 15 min and kept frozen for peptide analysis.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400g for 20 min. The pellet was discarded and the supernatant was added to the Sep-Pak C18 column with the remaining procedure the same as in the kidney preparation.
400g for 20 min. The pellet was discarded and the supernatant was added to the Sep-Pak C18 column with the remaining procedure the same as in the kidney preparation.
Quality control (QC) samples were prepared using the above-mentioned procedure at three levels: standard in injection solution, replicates from the same tissue donor, and unknown samples spiked with low level angiotensin (equivalent to unknown amount), and high level angiotensin (10 fold to unknown).
The ion source voltages were set at 3 kV, sampling cone at 37 V and the extraction cone at 3 V. In both modes the source and desolvation temperature were maintained at 120 °C and 225 °C, respectively, with the desolvation gas flow at 200 l h−1. The TOF MS scan was from 200 to 800 m/z at 1 s with a 0.1 s inter-scan delay using extended dynamic range acquisition with centroid data format. For real time mass calibration, direct infusion of sodium formate solution (10% formic acid–0.1 M NaOH–isopropanol at a ratio of 1:1:8) at 1 s/10 s to ion source at 1 μl min−1 was used.
:1 signal-to-noise ratio over the background. Quantification of unknowns was performed by computing the intensity of the chromatogram using either the ion count in the spectrum or the peak area displayedfor the target ion mass in the chromatogram, referenced to known amounts of standard peptides acquired under the same conditions in the same time period.
Results and discussion
Calibration curves
Using standard samples from Sigma, the calibration curves for the analysis of Ang-(1-7) and Ang II peptides by LC–MS were established under optimized conditions. For Ang-(1-7), the linear response range was from 0.8 to 80 pMol; LOD and LOQ were 0.4 and 0.8 pMol respectively (Fig. 1A.). For Ang II, the linear response range was from 0.8 to 800 pMol; LOD and LOQ at 0.4 and 0.5 pMol respectively (Fig. 1B).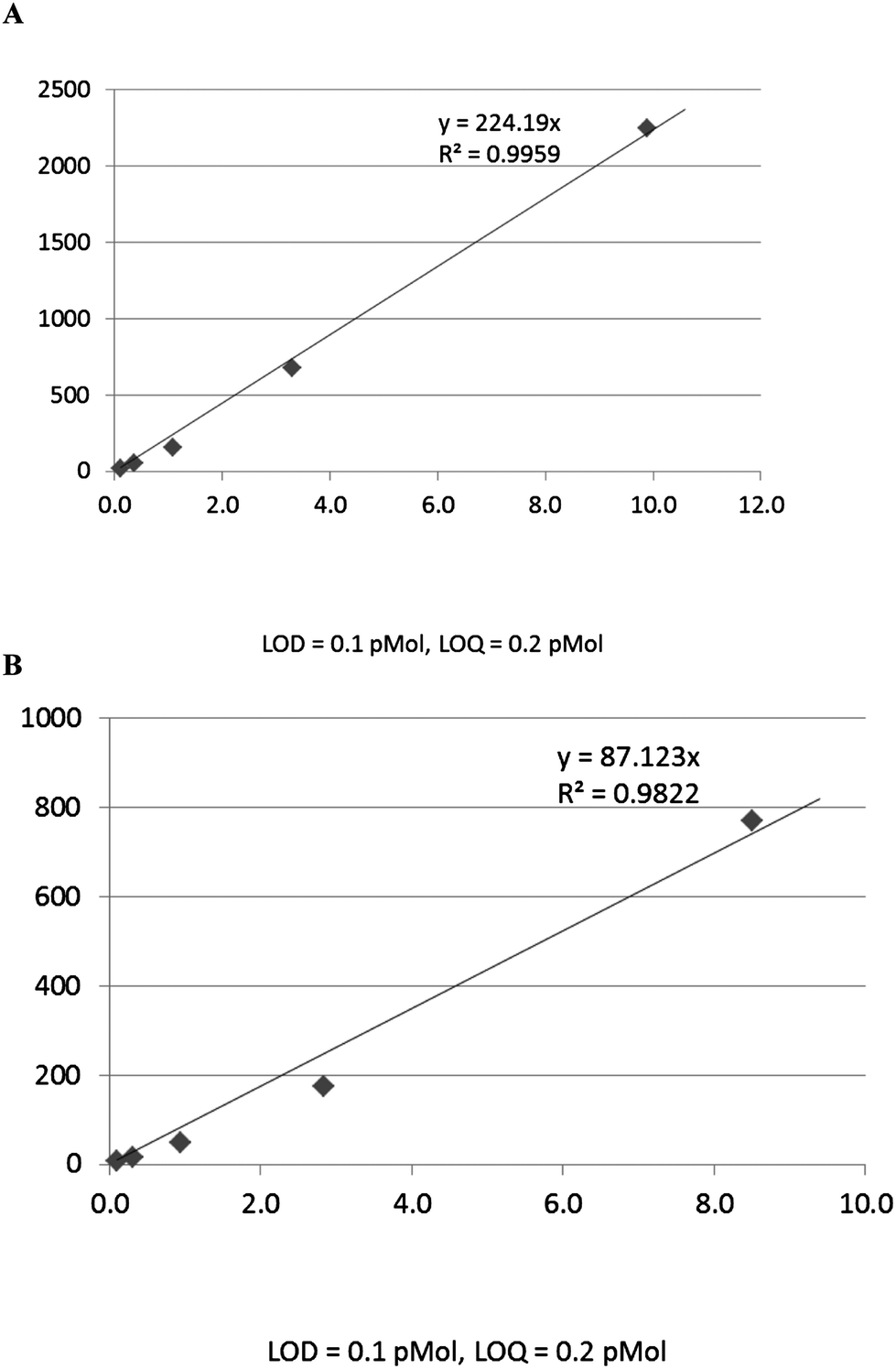 | ||
| Fig. 1 Calibration curves, (A) Ang-(1-7) standard, (B) Ang II standard, showing dynamic range, linear regression, LOD and LOQ from peak area of C18-UPLC–ESI/MS. | ||
Although electrospray is known for being semi-quantitative (as high as 30% variation for small molecular samples injected under the same conditions, but on different days),12 we found that the precision estimated from multiple injections of the same Ang II and Ang-(1-7) peptides over three different days and from injection of multiple preparations of the same concentration over three orders of magnitude was around 10%. We validated the method by analyzing samples that were prepared independently on different dates.
Loop injection vs. C18 column chromatography
Samples were delivered to the ion source by the loop injection method (the chromatogram was measured over 2 min, without a column) or by a C18 column UPLC (the chromatogram was measured over 10 min). The peak shape and retention times were different by these two methods (Fig. 2). Both methods gave almost identical results in terms of detection limit and quantification of the standard compounds. Since loop injection is high throughput and has no “wear and tear”, it is more cost-effective. However, in analyzing complex biological samples, loop injection could over-estimate the amount by including other molecules with similar m/z that otherwise could be separated by column chromatography with different retention times due to structure/hydrophobicity difference.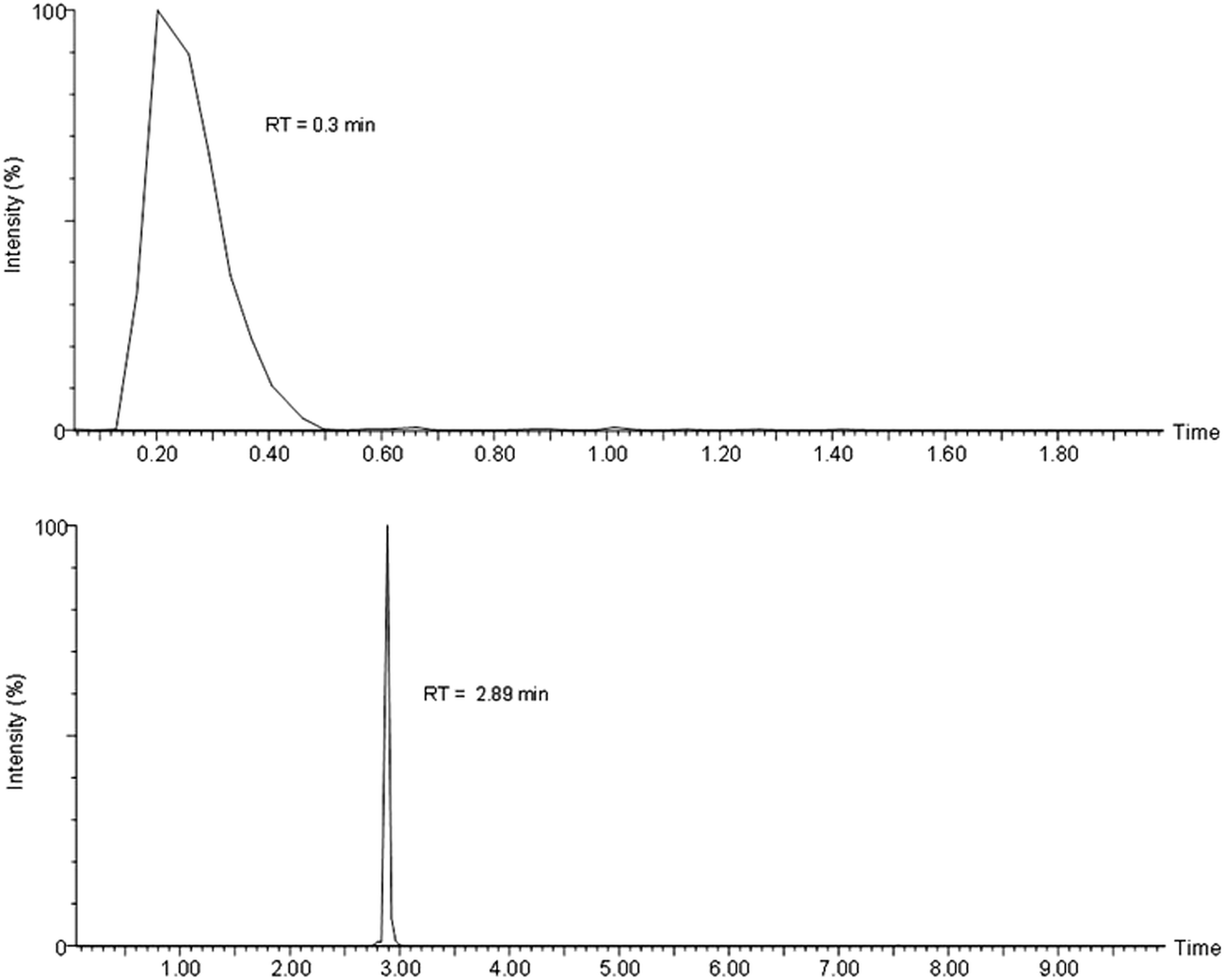 | ||
| Fig. 2 Selected ion chromatogram displays 450.2409 by the loop injection (upper panel) and the C18 UPLC (lower panel). | ||
Precision and accuracy
The precision and accuracy of the method was assessed by performing replicate analyses of samples at three levels: (1) intra-inter day variation of ESI-MS detection of angiotensin standard solutions in three orders of dynamic range (down to the detection limit), (2) replicates of the same tissue, (3) recovery of known added amounts spiked into replicate kidney tissues (which was defined as accuracy). Intra-batch precision was measured by the percentage relative standard deviation (RSD), based on replicates of 4 analyses, each with three LC–MS determinations. The upper limits of the intra-batch were considered acceptable when they were less than 20% according to the FDA guidelines.16As shown in Table 1, the precision for measuring angiotensin peptides in pure solution by ESI-MS were 1.5–12.1% in three orders of dynamic range, intra-batch precision for Ang-(1-7) and Ang II were 24.0% and 12.7% for replicate extractions from the same kidney tissue. The accuracy determined by analysing spiked replicate samples was 84.0–123.0% for Ang-(1-7) and 100.2–116.0% for Ang II. The RSD was calculated from the means and standard deviation of ion counts from each total ion count chromatogram (TIC) displaying the targeted m/z of 450.24 and 523.77 for doubly charged Ang-(1-7) and Ang II.
| Analyte | Analyses | Levels | Precision (% RSD) | Accuracy (% recovery) | Measurement (population size, n) |
|---|---|---|---|---|---|
| Ang-(1-7) | Intra-inter day variation | 7 | 1.5–10.4 | — | 84 |
| Ang II | Of standards in buffer | 7 | 5.4–12.1 | — | 84 |
| Ang-(1-7) | Extraction variation from | 1 | 24.0 | — | 12 |
| Ang II | Kidney tissue | 1 | 12.7 | — | 12 |
| Ang-(1-7) | Matrix effect from | 2 | 21.1–23.4 | 84.0–123.0 | 24 |
| Ang II | Spiked kidney extracts | 2 | 12.5–17.1 | 100.2–116.0 | 24 |
Sample stability affecting the detection limit and sensitivity
Over time, loss of angiotensin peptides in storage solution (60% acetonitrile, 1% formic acid in water) was observed. At very low quantity (a few pMol when approaching the detection limit in 10% acetonitrile, 0.1% formic acid in water), loss of sample was observed from the results of repetitive injections of the same sample in a span of minutes. Auto hydrolysis catalyzed by acid was suspected. When angiotensin peptides were incubated for 72 h at room temperature, the relative intensity of the ions normally present in the sample did not change (Fig. 3), suggesting no partial hydrolysis. Loss of formic acid in the sample over storage time may contribute to the sample loss; formic acid, being volatile, may evaporate in storage. When it was re-supplied to the same vial that was losing quantity, the signal was noticeably higher. Non-specific absorption to the container wall may be the largest contributor to the sample loss. Ang-(1-7) (Asp-Arg-Val-Tyr-Ile-His-Pro) being both polar (N-terminus) and non-polar (C-terminus) can attach to both polar surfaces (glass vial) and non-polar surfaces (plastic vial), and become unavailable for ionization. Ang II is one phenylalanine longer. Both molecules are very much detergent-like. Testing sample loss in both glass and plastic vials with different amounts of acetonitrile and formic acid, we found that sample loss was universal, the minimum being in 10% acetonitrile, 0.1% formic acid in a glass vial. At higher concentration (above 10 ng μl−1) the sample loss was insignificant. | ||
| Fig. 3 The mass spectrum of Ang-(1-7) standard. Incubation of Ang-(1-7) (80 ng) in 10% ACN, 0.1% formic acid for 72 h at room temperature, shows no obvious change in relative intensity of the peptide ions. | ||
Detection of angiotensin peptides in biological samples
Biological samples prepared from mice tissues, including kidney, adipose, urine, and plasma samples, were analyzed for angiotensin peptide content. Soluble extracts prepared in 10% acetonitrile and 0.1% formic acid were injected into the ACQUITY UPLC® BEH C18 column with a linear acetonitrile gradient. Shown in Fig. 4 is a representative chromatogram of the separation and detection of Ang-(1-7) from a mice plasma sample by C18 UPLC-ESI-MS with selected ion chromatogram displaying the ion with m/z 450.24, the inset of the figure is the spectrum of ion 450.24 at peak 3.0 min. The peak at 3.5 min included the compounds that would have interfered with Ang-(1-7) (450.242+) detection in the loop injection method (by over estimation). A MS/MS scan of the peak at 3.5 min revealed a sodiated molecule (Fig. 5); a database search (LipidMap) of the accurate mass revealed several triacylglycerolphospholipids and diacylglycerolphospholipids, matching 450.242+ when sodiated. As a membrane component, glycerolphospholipids were much more abundant than Ang-(1-7).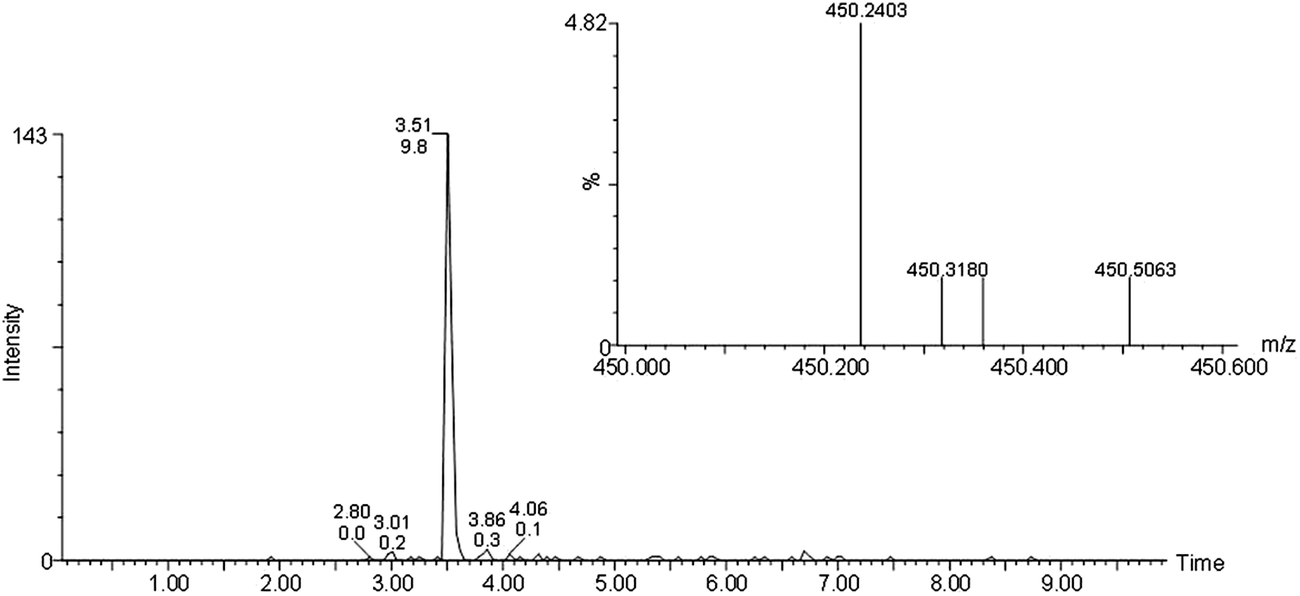 | ||
| Fig. 4 UPLC separation and MS detection of Ang-(1-7) from mice plasma sample. Selected ion chromatogram displaying 450.24. Inset: the mass spectrum of ion 450.24 from peak 3.0 min. | ||
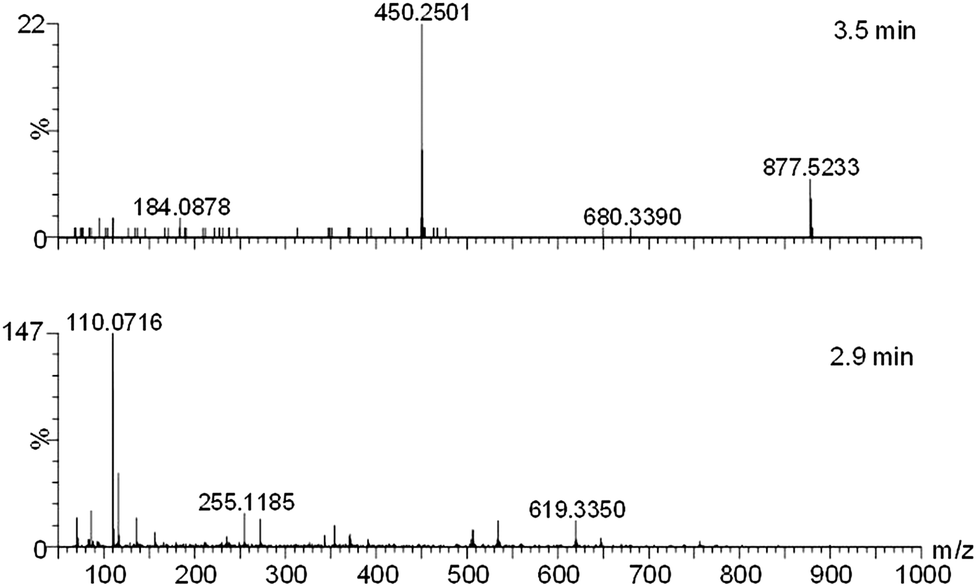 | ||
| Fig. 5 MS/MS of ion 450.242+ from 3.1 min (upper panel, showing a sodiated small molecule), and 2.9 min (lower panel, showing peptide ions). | ||
The results of estimated Ang-(1-7) and Ang II content in biological samples are summarized in Table 2. The range of Ang-(1-7) in the adipose and kidney samples was 290–350 and 300–350 fmol mg−1 tissue respectively. The concentration range in plasma and urine for Ang-(1-7) was 30–40 and 100–105 fmol μl−1 respectively. The range of Ang II in adipose and kidney samples was 120–150 and 140–200 fmol mg−1 tissue. We found that the range of Ang II in plasma and urine were 15–20 and 45–50 fmol μl−1. Earlier, we reported comparable levels of Ang II and Ang-(1-7) in the kidney of lean and obese rats.13 The difference here could be because of different species and methods (loop injection vs. C18 column) used in the present study.
Effect of biological matrix
Tissue type may play an important role in the recovery of angiotensin peptides. Ang-(1-7) is more sensitive in pure water than in the biological matrix; the quantity loss was estimated by the recovery study (in the plasma sample) where a known amount of Ang-(1-7) was added. The loss was by a factor of 10 when compared to that with pure solution at biologically relevant concentrations. The loss may due to the ionization competition, or the peptide binding to biological molecules in the extract, giving different m/z when ionized. Ideally, the standard calibration curve needs to be constructed in the same biological background as the sample in which it is detected; thus different samples (plasma, adipose and kidney tissues, urine) all need to have their own standard calibration curve. Since every tissue contains angiotensin peptides, a true calibration curve without an elevated background is difficult. With spiked samples, one could estimate the recovery due to sample interaction.Ang II has one more non-polar amino acid than Ang-(1-7), thus it is more non-polar. In the biological background, Ang II was recovered more than Ang-(1-7) in the spiked tests (data not shown). However, the ionization efficiency appears the same at high concentration (Fig. 6.), but more for Ang-(1-7) at low concentration in pure solution, which is the opposite to that in biological background where Ang-(1-7) was less sensitive, perhaps, due to the difference in their polarity.
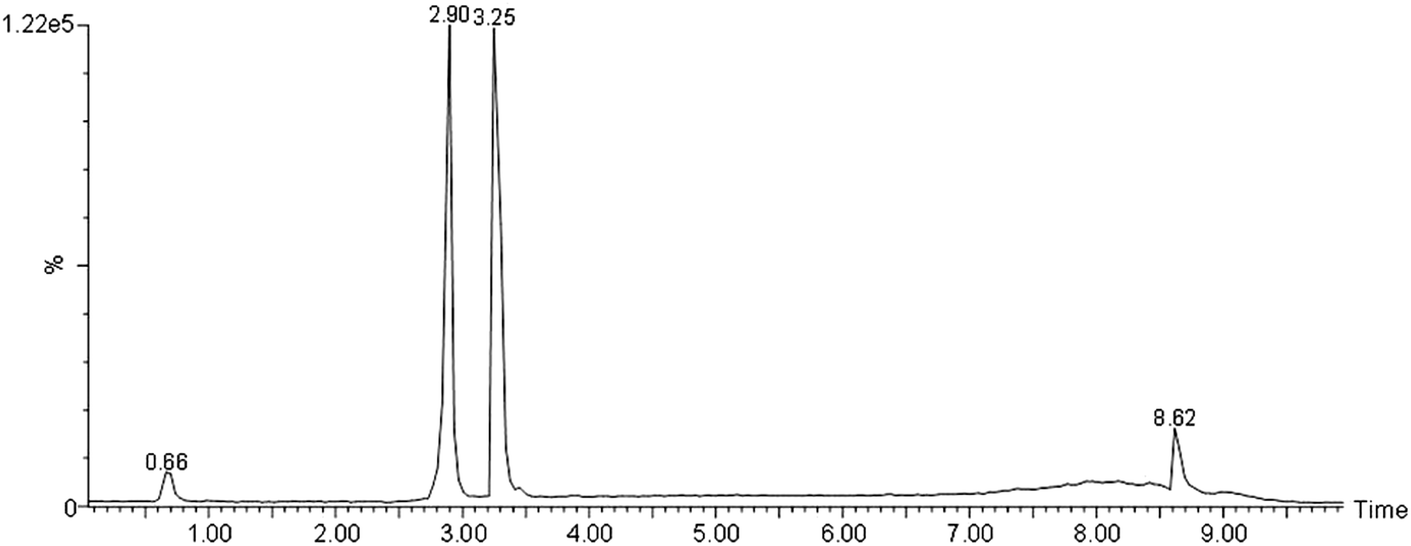 | ||
| Fig. 6 UPLC of Ang-(1-7) and Ang II standards. Their retention times were 2.9 and 3.25 min, the ionization efficiency was identical at 800 ng each in 0.5% FA, 10% ACN in glass vial. | ||
Recovery study with angiotensin peptides spiked into biological extracts at two levels revealed that the retention time was delayed by 0.04 min with the amount lost being negligible (Table 1).
Quantification by peak area vs. ion count
Quantification by combining the ion count under the peak) or by peak area gave the same results. When plotting the values derived from both methods, a positive correlation is seen (Fig. 7, standard (A), and (B) unknown of Ang-(1-7) determination), indicating that both methods can be used for quantification. The same is true for Ang II except for the unknown sample where the correlation was slightly weaker than for Ang-(1-7) for some unknown reasons.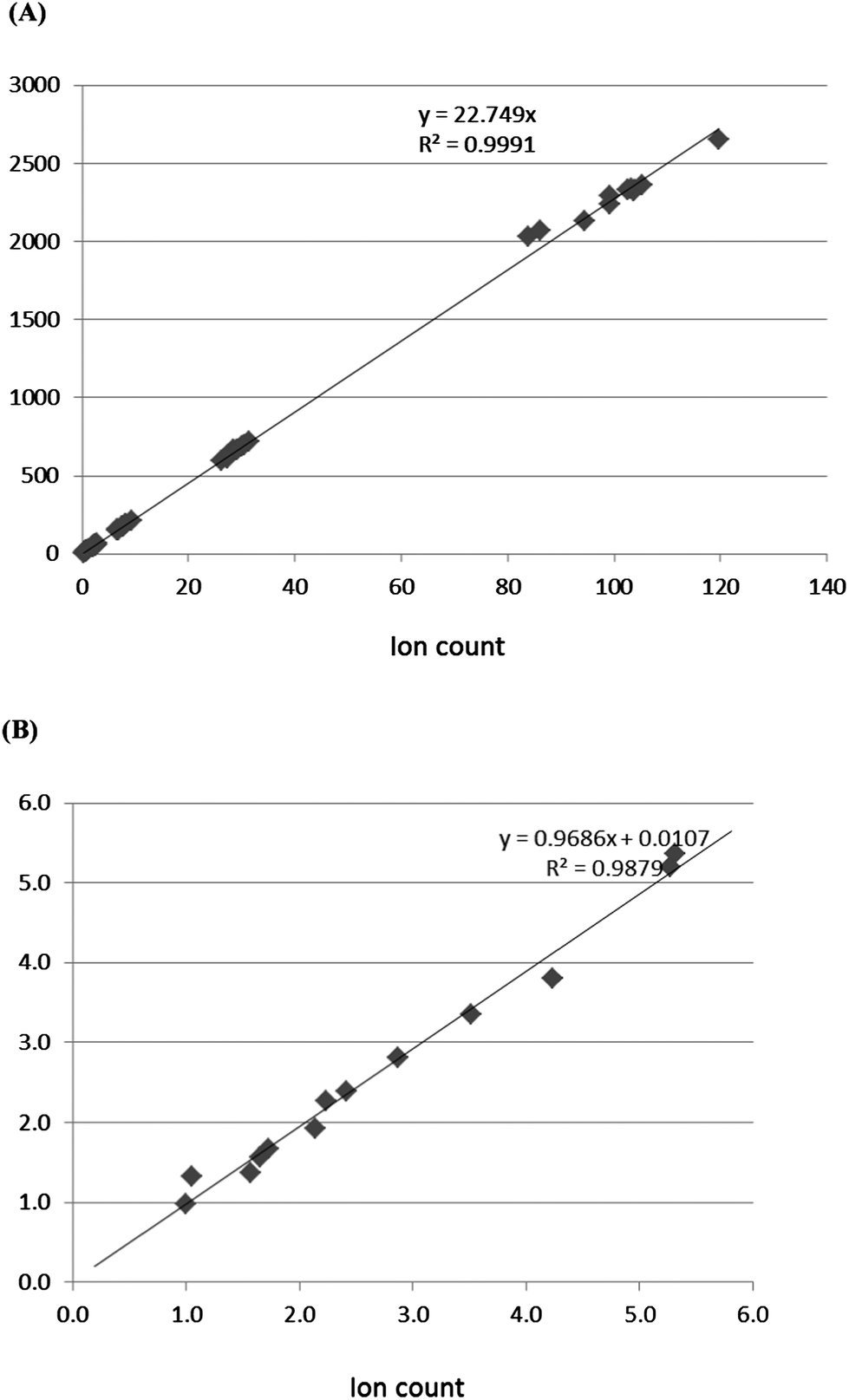 | ||
| Fig. 7 Correlation between ion count and peak area determined by quantification methods. (A) Correlation between peak area and ion count from standard Ang-(1-7) and (B) correlation between peak area and ion count from unknown Ang-(1-7). | ||
Advantages over existing methods for angiotensin peptide measurement
Several laboratories have used HPLC along with radioimmunoassay to measure angiotensin peptides levels in the kidney and plasma.9–11 Compared to the level of angiotensin fragments present in the available literature, the present study demonstrated some interesting findings and differences in the concentration of these peptides in the kidney, adipose, plasma and urine tissues. Kobori et al., and Gonzales et al.,11 reported that using their existing methodology (HPLC coupled with radioimmunoassay), the concentration of Ang II in kidney tissues was 0.25 fmol mg−1 tissue. Here we found, by using LC–MS, that the concentration of Ang II in kidney cortex was 140–200 fmol mg−1 which is approximately 400-fold higher than the reported value. Prieto et al.,10 reported that urinary Ang II in male Sprague Dawley rats was ∼1500 fmol day−1. Moreover, Park et al.14 demonstrated that plasma Ang II in mouse was 0.4 fmol μl−1. In the present study, we found that the range of urinary Ang II in mice was 9000–1000 fmol day−1 and that of plasma was 15–20 fmol μl−1. This demonstrates that the existing methods used may not be able to recover all the angiotensin peptides. Moreover, we also observed similar levels of Ang II in the eWAT which was again much higher than the reported value available in the literature.15 The existing method employs a time-consuming peptide extraction procedure by HPLC and detection by RIA, it is lengthy, costly and poses a health risk. Although mass spectrometry requires expensive instrumentation, it is automated and time-efficient; the cost of running samples is low. The most important advantage overall is that it is highly selective.Conclusion
We have presented a simple yet reliable LC–MS method for detecting low-concentrations of angiotensin peptide components with enormous regulating activity in biological tissues. Solid tissues (kidney cortex and adipose tissue) as well as fluids (plasma, urine) were analyzed for Ang II and Ang-(1-7) in a timely and efficient manner. Because of the sensitivity and resolving power of modern mass spectrometry, without much purification and enrichment wet-lab work, angiotensin peptides [Ang II, Ang-(1-7)] from crude protein extracts can be directly injected into the mass spectrometer and can be detected consistently and quantifiably. Angiotensin peptide molecules, although minute in biological samples, can clearly be detected by C18 UPLC–ESI-MS when their retention time, accurate mass, and doubly charged ion agree with standards. These results demonstrate that the accuracy and precision were within generally acceptable limits and that the quantification achieved with the current method was reproducible and accurate.Acknowledgements
The study is supported by the National Institutes of Health grant R01-DK61578. The authors would like to acknowledge Ms Fatima Qamar for proof-reading the manuscript.References
- M. de Gasparo, K. J. Catt, T. Inagami, J. W. Wright and T. Unger, Pharmacol. Rev., 2000, 52, 415–472 CAS.
- M. Iwai and M. Horiuchi, Hypertens. Res., 2009, 32, 425–427 CrossRef CAS PubMed.
- A. H. Siddiqui, Q. Ali and T. Hussain, Hypertension, 2008, 53, 256–261 CrossRef PubMed.
- Q. Ali and T. Hussain, Hypertens. Res., 2012, 35, 654–660 CrossRef CAS PubMed.
- A. M. DelliPizzi, S. D. Hilchey and C. P. Bell-Quilley, Br. J. Pharmacol., 1994, 111, 1–3 CrossRef CAS.
- M. Dilauro and K. D. Burns, TheScientificWorldJOURNAL, 2009, 9, 522–535 CrossRef CAS PubMed.
- B. H. Jones, M. K. Standridge and N. Moustaid, Endocrinology, 1997, 138, 1512–1519 CrossRef CAS.
- S. H. Santos, L. R. Fernandes, C. S. Pereira, A. L. Guimaraes, A. M. de Paula, M. J. Campagnole-Santos, J. I. Alvarez-Leite, M. Bader and R. A. Santos, Regul. Pept., 2012, 178, 64–70 CrossRef CAS PubMed.
- H. Kobori, M. Nangaku, L. G. Navar and A. Nishiyama, Pharmacol. Rev., 2007, 59, 251–287 CrossRef CAS PubMed.
- L. Liu, A. A. Gonzalez, M. McCormack, D. M. Seth, H. Kobori, L. G. Navar and M. C. Prieto, Am. J. Physiol.: Renal Physiol., 2011, 301, F1195–F1201 CrossRef CAS PubMed.
- R. A. Gonzalez-Villalobos, D. M. Seth, R. Satou, H. Horton, N. Ohashi, K. Miyata, A. Katsurada, D. V. Tran, H. Kobori and L. G. Navar, Am. J. Physiol.: Renal Physiol., 2008, 295, F772–F779 CrossRef CAS PubMed.
- C. Y. Lee, A. Lesimple, A. Larsen, O. Mamer and J. Genest, J. Lipid Res., 2005, 46, 1213–1228 CrossRef CAS PubMed.
- Q. Ali, Y. Wu and T. Hussain, Kidney Int., 2013, 84(5), 931–939 CrossRef CAS PubMed.
- S. Park, B. J. Bivona, H. Kobori, D. M. Seth, M. C. Chappell, E. Lazartigues and L. M. Harrison-Bernard, Am. J. Physiol.: Renal Physiol., 2009, 298, F37–F48 CrossRef PubMed.
- M. S. Coelho, K. L. Lopes, A. Freitas Rde, E. B. de Oliveira-Sales, C. T. Bergasmaschi, R. R. Campos, D. E. Casarini, A. K. Carmona, S. Araujo Mda, J. C. Heimann and M. S. Dolnikoff, Regul. Pept., 2010, 162, 61–67 CrossRef CAS PubMed.
- Guidance for industry-bioanalytical method validation, US Department of Health and Human Service, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM), Rockville, MD, USA. http://www.fda.gov/cvm.
| This journal is © The Royal Society of Chemistry 2014 |