Gas chromatographic determination of three chlorophenols in toilet paper by ultrasonic assisted extraction and synchronous derivative dispersive liquid–liquid microextraction
Miao
Zhang
a,
Jing
Cheng
*a,
Min
Wu
a,
Ting
Du
a,
Xiaohua
Wang
a and
Min
Cheng
*b
aKey Laboratory of Pesticide and Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, Wuhan 430079, China. E-mail: chengjingok@mail.ccnu.edu.cn; Tel: +86-27-67867961
bSchool of Mechanical Science and Engineering, Hua Zhong University of Science and Technology, Wuhan 430074, China. E-mail: 494350301@qq.com; Fax: +86-27-87543670; Tel: +86-27-87543770
First published on 18th October 2013
Abstract
A simple and sensitive method for the analysis of three chlorophenols (CPs) in toilet paper has been developed for the first time. Acetone was used as a solvent to extract the three CPs from toilet paper assisted by ultrasound irradiation. When the extraction process was finished, 0.5 mL acetone extract was subjected to synchronous derivative dispersive liquid–liquid microextraction (DLLME). During this process, the analytes were rapidly transferred from the acetone extract to another extraction solvent (chlorobenzene) for further clean-up and enrichment. Acetone could also act as a dispersant during the DLLME process, which combined the advantages of ultrasonic assisted extraction and DLLME appropriately. Different factors affecting the extraction efficiency and derivatization step were carefully optimized. Under optimal conditions, the limits of detection for 2,4-dichlorophenol, 2,4,6-trichlorophenol and pentachlorophenol were 1.5 μg kg−1, 0.75 μg kg−1, 0.25 μg kg−1, respectively. Satisfactory linear ranges were observed from 5–500 μg kg−1, 2.5–250 μg kg−1, 1–20 μg kg−1, respectively. Recoveries of three CPs in toilet paper were in the range 70.9–118%. The optimized method was successfully applied to three different toilet paper samples, and pentachlorophenol has been detected in one of the tested toilet papers at a concentration level of 1.2 μg kg−1.
1. Introduction
Most of us cannot imagine living without toilet paper. It is used not only for bathroom hygiene, but for nose care, wiping up spills, removing makeup, and small bathroom cleaning chores. Toilet paper is generally made from new or “virgin” paper, using a combination of softwood and hardwood trees. Softwood trees such as Southern pines and Douglas firs have long fibers that wrap around each other, which gives the paper strength. Hardwood trees like gum, maple and oak have shorter fibers that make a softer paper. Toilet paper is generally a combination of approximately 70% hardwood and 30% softwood.1 Wood decomposes easily when exposed to air and water, so large quantities of higher chlorophenols (CPs) are used in pressure treatment in the wood preservation industry for the protection of wood and wood-based products.2However, CPs have been confirmed to possess carcinogenic and immunosuppressive properties. Owing to their toxicity, both the US Environmental Protection Agency (EPA)3 and the European Community (EC)4 have included some CPs in their list of priority pollutants. CP treated wood may be an important source of CP residues in toilet paper, showing therefore a potential risk for humans. To achieve the necessary trace levels of sensitivity for CPs when analyzing toilet paper samples, extraction, clean-up and preconcentration techniques were needed.
Ultrasonic radiation is of great help in the pre-treatment of solid samples as it can facilitate and accelerate operations when it is used to extract organic and inorganic compounds. Ultrasound-assisted extraction (UAE) results in increased solubility, diffusivity, and pressures, which favors penetration and transport, at the interface between an aqueous or organic solution subjected to ultrasonic energy and a solid matrix, and further increases extraction efficiencies. In many situations, UAE is an expeditious, inexpensive and efficient alternative to conventional extraction techniques and in some cases, even to supercritical fluid and microwave-assisted extraction, as demonstrated by applications to both organic and inorganic analytes in a wide variety of samples.5–7 In the present study, UAE was used to extract CPs from toilet paper samples.
A variety of sample pre-treatment techniques have been applied for CPs in various samples: classical techniques, such as liquid–liquid extraction (LLE)8 and solid-phase extraction (SPE),9,10 stir bar sorptive extraction (SBSE),11 and to an even greater extent, solid-phase microextraction (SPME).12,13 However, from the point of the subsequent purification and enrichment steps following the UAE procedure, the recently emerged dispersive liquid–liquid microextraction (DLLME) technique is the most appropriate clean-up and preconcentration procedure. DLLME was developed by Assadi and his co-workers.14 It is based on the use of a ternary component solvent system. Some new pre-treatment technologies were developed based on DLLME, including dispersive liquid–liquid microextraction based on solidification of floating organic droplet (DLLME-SFO),15 ultrasound-assisted surfactant-enhanced emulsification microextraction (UASEME),16 and solvent-based demulsified dispersive liquid–liquid (SD-DLLME),17 ionic liquid-based dispersive liquid–liquid microextraction (IL-DLLME),18 ionic liquid/ionic liquid dispersive liquid–liquid microextraction (IL/IL-DLLME),19,20 and temperature-controlled ionic liquid dispersive liquid phase micro-extraction (TILDLME).21,22 All of the above mentioned methods based on DLLME were developed in aqueous solution, and the purpose of the present work is to apply a DLLME based method to a solid matrix. In this method, the target analytes were extracted from the solid matrix (toilet paper) by ultrasound radiation, and the extract can be used as disperser solvent in the next DLLME procedure for further enrichment and purification.23 The derivatization reaction was almost simultaneous with the DLLME process. The synchronous derivative DLLME possesses simplicity of operation, rapidity, low sample volume and cost.
Many analytical instruments have been used for the analysis of trace-level chlorophenols, including high-performance liquid chromatography (HPLC),24,25 capillary electrophoresis (CE),26 and gas chromatography (GC).27,28 Among these methods, GC-ECD is the most often used because of its high sensitivity and high-resolution power towards halogen-containing compounds. Since chlorophenols are high polarity and low volatility compounds, it is usually necessary to perform a prior derivatization step before GC determination. A large number of derivatization reagents, such as diazomethane,29 pentafluorobenzyl bromide,30 methyliodide,31 trimethylsilyl-N,N-dimethylcarbamate (TMSDMC),32N-(tert-butyldimethylsilyl)-N-methyl trifluoroacetamide (MTBSTFA)33 and acetic anhydride,34–36 have been used for this purpose. However, the use of acetic anhydride in alkaline conditions is the most popular since the derivatization reaction can take place directly in the aqueous phase at room temperature in a few minutes (even in water),37 and acetic anhydride is cheaper than the other derivatization reagents.
The aim of this study was to develop a sensitive and rapid method termed ultrasonic assisted extraction-synchronous derivative DLLME for the analysis of three chlorophenols at trace levels in toilet paper using GC-ECD. To the best of our knowledge, this is the first paper that has focused on the analysis of chlorophenols residues in toilet paper.
2. Materials and methods
2.1. Reagents and materials
2,4-Dichlorophenol (2,4-DCP, 98%), 2,4,6-trichlorophenol (2,4,6-TCP, 98%) and pentachlorophenol (PCP, 98%) were obtained from the Environmental Protection Monitoring Research Institute, Ministry of Agriculture (China). The 100 μg mL−1 individual stock solutions of the above-mentioned three analytes were prepared in methanol and stored in the refrigerator. Working solutions were obtained by appropriate dilution of the stock standard solution. Acetic anhydride, anhydrous potassium carbonate, sodium chloride were of analytical grade and supplied by Zhanyun Chemical Co, Ltd, Shanghai, China. HPLC-grade acetone, acetonitrile and methanol were obtained from Tedia Company Inc. (OH, USA). Three different brands of toilet paper were obtained from a supermarket in Wuhan, China.2.2. Apparatus
An ultrasonic water bath with temperature control (Kunshan Ultrasonic Instrument Co., Ltd., Jiangsu, China) was applied to extract CPs from toilet paper samples. Hamilton syringe (Bonaduz, Switzerland) was used to inject organic solvent and derivatization reagent into aqueous samples and inject 1.0 μL of sample into GC system. The centrifugation process was proceeded with an 80-2 centrifuge (Changzhou Guohua Electric Appliance Co., Ltd., Jiangsu, China). Sample analysis was carried out on an Agilent 6890 GC (Agilent Technologies, Palo Alto, CA, USA) equipped with an electron capture detector (ECD) and a split/splitless injector. The analytes were separated on a DB-5MS capillary column (30 m × 0.25 mm I.D., 0.25 μm film thickness). The oven temperature was programmed as follows: initially at 100 °C (3 min), raised to 170 °C at 8 °C min−1 and held at 170 °C for 2 min. The carrier gas was nitrogen (purity 99.9995%), at a flow rate of 1.0 mL min−1. The inlet temperature was set at 220 °C with the splitless mode selected, the temperature of the detector was set at 300 °C and N2 was used as make-up gas at a flow rate of 40 mL min−1.2.3. UAE-synchronous derivative DLLME procedures
As shown in Fig. 1, 8 mL acetone was added to 0.5 g spiked toilet paper samples in a conical flask. The sealed conical flask was placed under ultrasonic irradiation at 25 °C for 20 min. After completion of ultrasound-assisted extraction, the toilet paper and acetone extract were separated through filtration. 0.5 mL acetone extract was taken out and injected rapidly into a 5 mL centrifuge tube containing 4.5 mL 0.5% K2CO3 aqueous solution. Then, 20 μL chlorobenzene (extraction solvent) and 30 μL acetic anhydride (derivatization reagent) were also immediately injected into the above-mentioned 5 mL centrifuge tube. The tube was then shaken gently by hand for 2 minutes. A cloudy solution was formed and the tube was then centrifuged for 5 min at 4000 rpm. 1.0 μL of the sedimented organic phase was collected and injected into GC-ECD.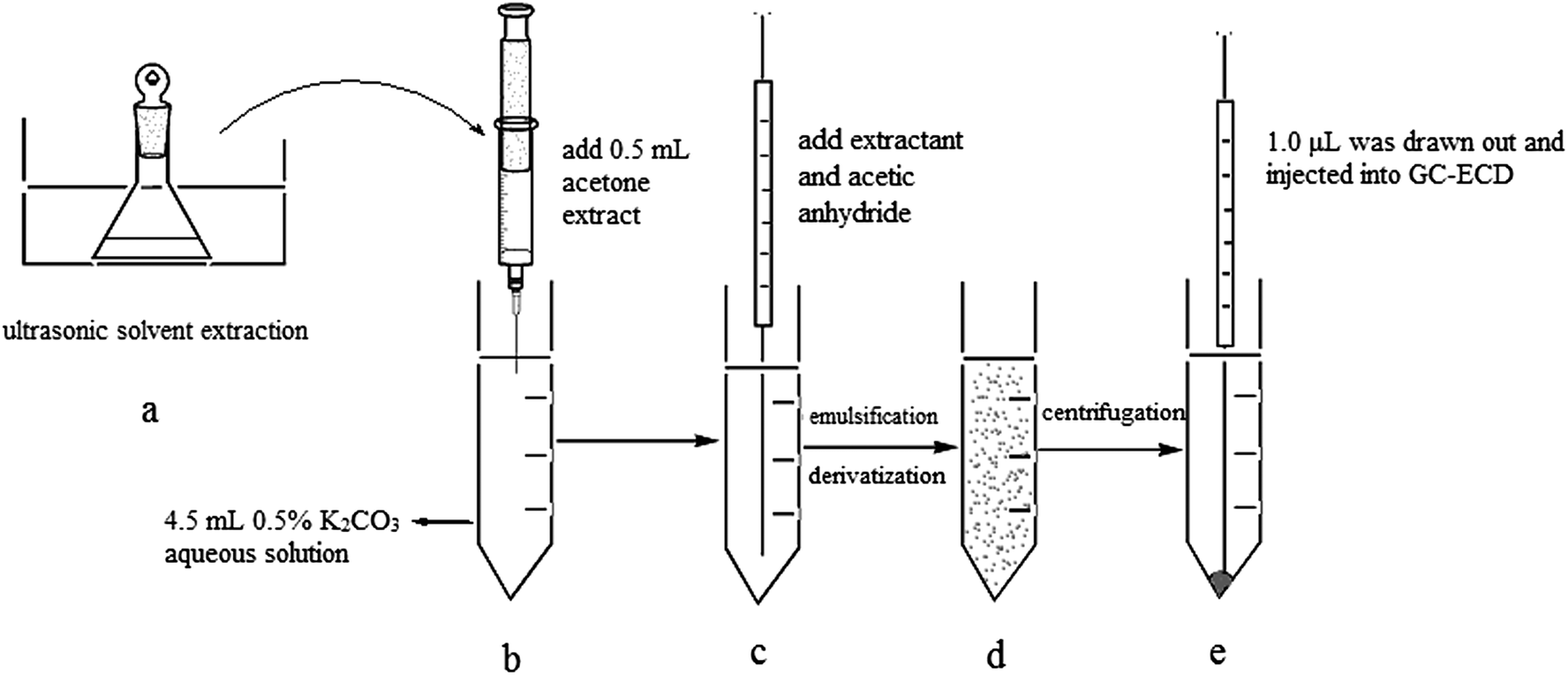 | ||
| Fig. 1 Schematic of the different steps in UAE-synchronous derivative DLLME: (a) 8 mL acetone solution was added to 0.5 g toilet paper, and was put under ultrasonic radiation at 80 kHz for 20 min; (b) 0.5 mL acetone extract was added to a 5 mL centrifugation tube containing 4.5 mL 0.5% K2CO3 aqueous solution; (c) 20 μL chlorobenzene and 30 μL acetic anhydride were rapidly injected into the above-mentioned centrifugation tube; (d) the synchronous derivative DLLME process; (e) 1.0 μL sedimented phase was collected and injected for GC-ECD analysis. | ||
3. Results and discussion
The possibility of combination of UAE and synchronous derivative DLLME coupled to GC-ECD for extraction, preconcentration, clean-up and analysis of three CPs from toilet paper was investigated. Several critical variables affect the UAE-synchronous derivative DLLME performance, including type and volume of ultrasonic extraction solvent, type and volume of extraction solvent for DLLME, volume of dispersive solvent for DLLME, amount and volume of the derivatization reagent, pH value and ionic strength, which were systematically investigated.3.1. Selection of type and volume of extraction solvent for UAE
The qualities of the ultrasonic extraction solvent for toilet paper should be that it is a good extractor and miscible with water, and has low volatility and toxicity. Of the three solvents with these properties selected (acetonitrile, methanol and acetone), acetone was chosen because it acted well and has low toxicity as an extraction solvent for toilet paper (step 1) and as a dispersive solvent in synchronous derivative DLLME (step 2) (Fig. 2).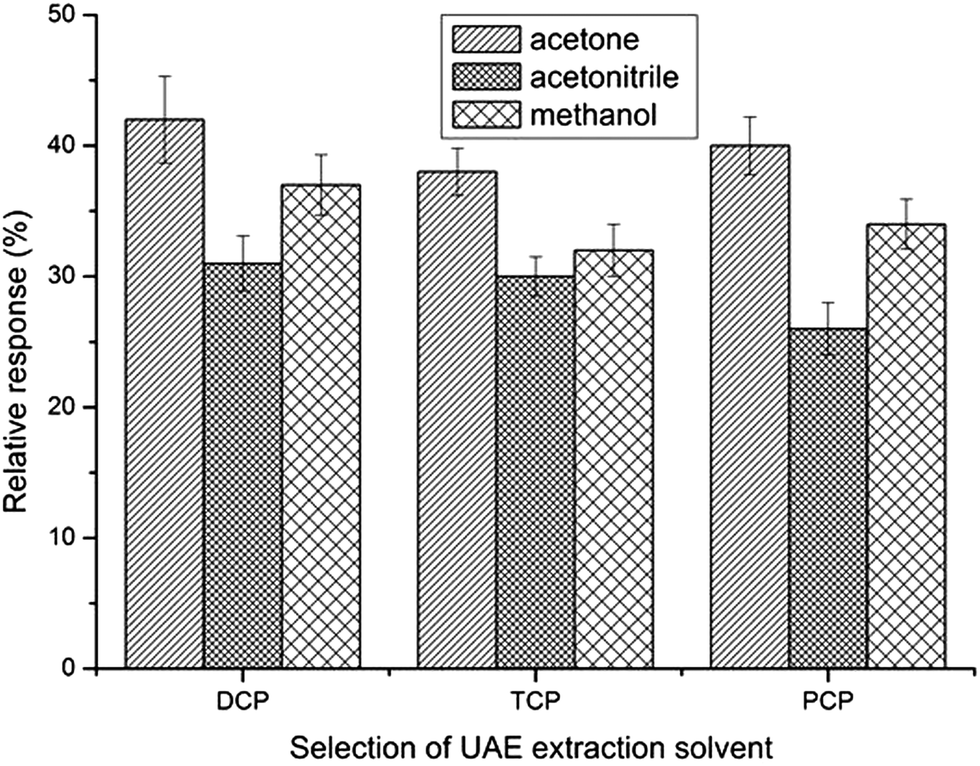 | ||
| Fig. 2 Selection of UAE solvent. UAE conditions: temperature: 25 °C; ultrasound frequency: 80 kHz; ultrasonic extraction time: 30 min. CPs concentration: 50 μg kg−1. DLLME with simultaneous derivatization conditions: 4.5 mL 0.5% K2CO3 aqueous solution; acetone extract: 0.5 mL; extraction solvent volume: 30 μL chlorobenzene; volume of acetic anhydride: 30 μL. No salt addition; centrifugation time: 5 min. | ||
The efficiency of ultrasonic extraction increases with the increase of extraction solvent volume. However, the analytes are diluted as a result of the increase in the volume of the ultrasonic extraction solvent. To find the most suitable extraction volume, different volumes (8, 15 and 20 mL) were evaluated. As can be seen from Fig. 3, relative response was the highest when using 8 mL acetone as extraction solvent. Extraction solvent volumes smaller than 8 mL were also tested, and results showed it led to very small extract volumes (due to the volatility of acetone), which were not enough to achieve a stable dispersive system in the DLLME step and thus, lead to poor relative response. Consequently, 8 mL acetone was selected for further studies.
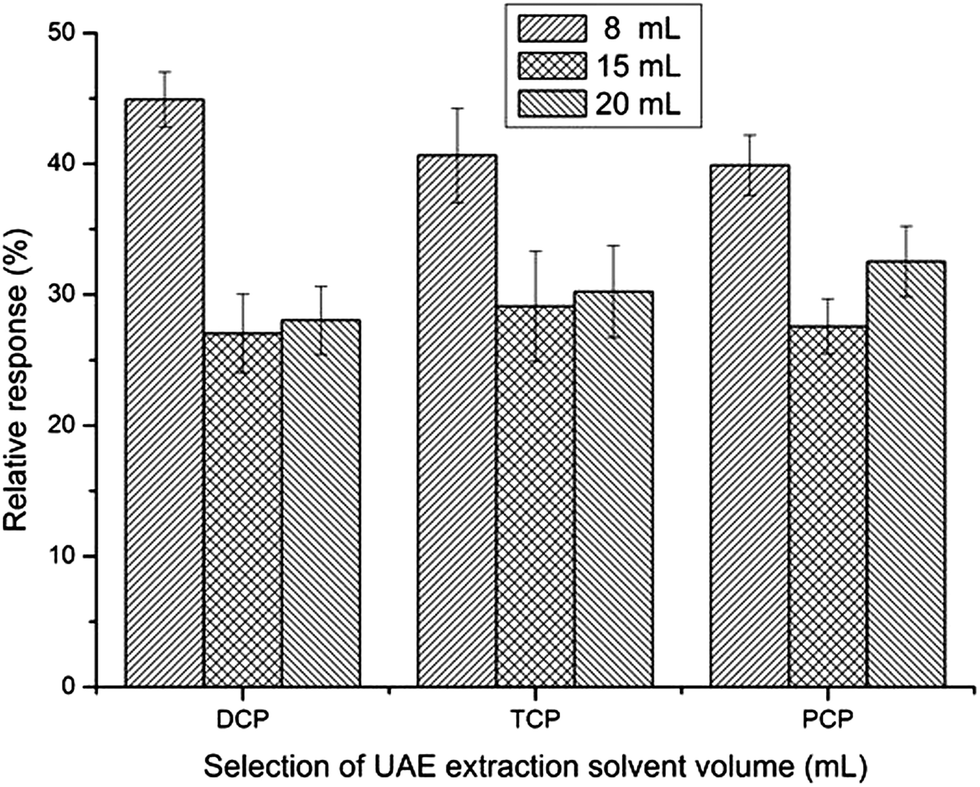 | ||
| Fig. 3 Selection of UAE solvent volume. UAE conditions: extraction solvent: acetone; temperature: 25 °C; ultrasound frequency: 80 kHz; ultrasonic extraction time: 30 min. CPs concentration: 50 μg kg−1. DLLME with simultaneous derivatization conditions: 4.5 mL 0.5% K2CO3 aqueous solution; acetone extract: 0.5 mL; extraction solvent volume: 30 μL chlorobenzene; volume of acetic anhydride: 30 μL. No salt addition; centrifugation time: 5 min. | ||
3.2. Ultrasonic extraction time
Ultrasonic extraction can make collapsing cavitation bubbles which help to disrupt the saturated boundary layer surrounding the particles of toilet paper samples, and bring fresh solvent to the surface of the toilet paper. This favors the mass transfer of the analytes from toilet paper into the sample solution leading to an enhancement of sensitivity.38 The effect of ultrasonic extraction time was examined in the range of 10 min to 40 min. It was observed that the relative response increased with the increase of extraction time from 10 min to 20 min, and almost kept constant with the increase of extraction time from 20 min to 40 min. Therefore, 20 min was selected and used for further experiments.3.3. Selection of the type and volume of extraction solvent for DLLME
For DLLME (step 2), the extraction solvent should have a ow solubility in water and a higher density than water. It should also be possible to extract the analytes well, and its chromatographic peak should be easily distinguishable from that of the target compounds. In this work, four different organic solvents were evaluated chlorobenzene, chloroform, tetrachloromethane and tetrachloroethane. Fig. 4 shows that chlorobenzene had the best relative response of all the three CPs. Therefore, chlorobenzene was selected as the extraction solvent in the subsequent experiments.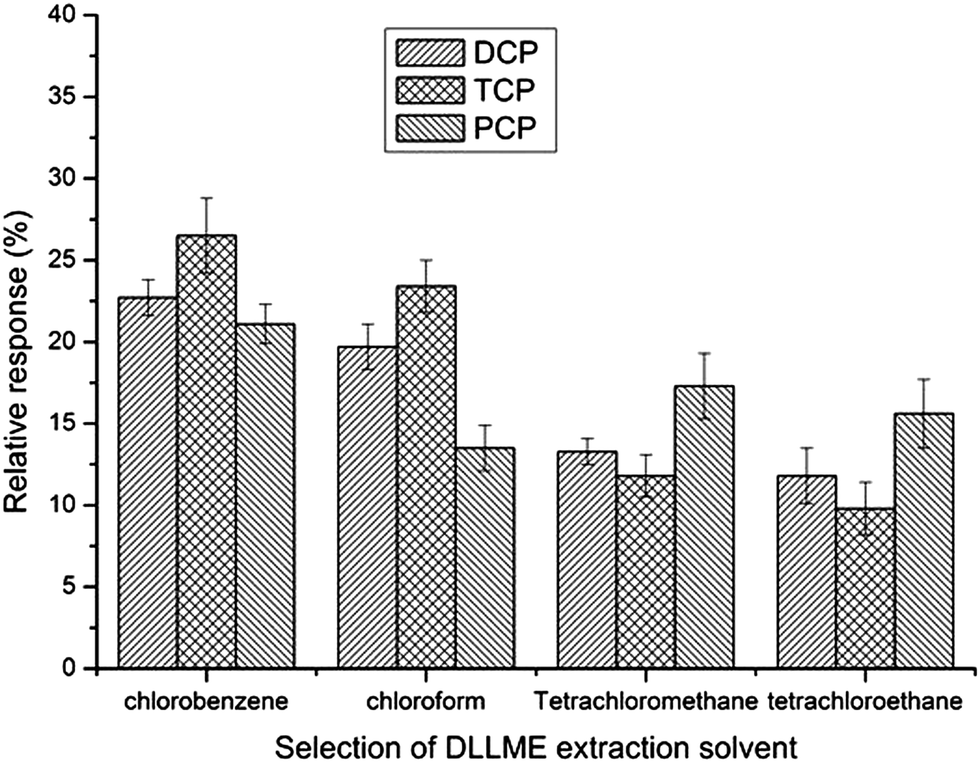 | ||
| Fig. 4 Selection of DLLME extraction solvent. Extraction conditions were as described in Fig. 3 except that the volume of acetone was 8 mL and ultrasonic assisted extraction time was 20 min. | ||
To study the effect of the volume of the extraction solvent on the performance of the procedure, the volume of chlorobenzene was varied in the range from 20 μL to 50 μL. To make sure that the sedimented phase volume was enough for GC injection, the extraction phase volume needed to be 20 μL or more. As can be seen from Fig. 5, when the volume of the extraction solvent was 20 μL (the sedimented phase was about 10 μL) the relative response reached its apex. Thus, 20 μL chlorobenzene was chosen for further studies.
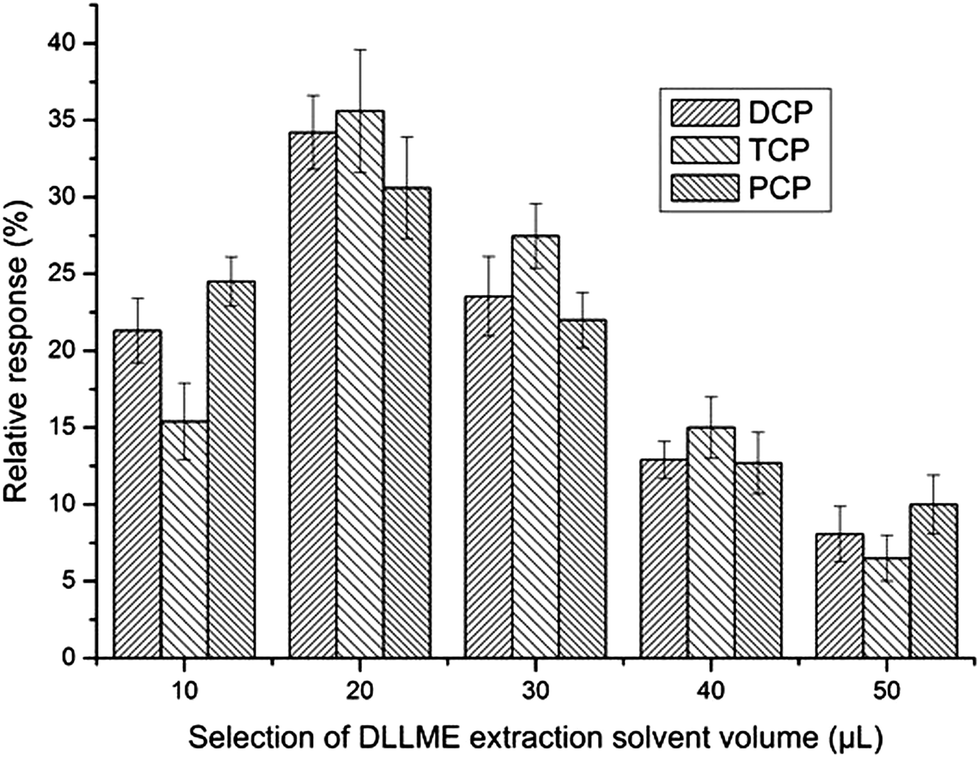 | ||
| Fig. 5 Selection of DLLME extraction solvent volume. Extraction conditions were as described in Fig. 4 except that the extraction solvent was chlorobenzene. | ||
3.4. Selection of dispersant volume for DLLME
The dispersant should be miscible with both the organic solvent and water, so acetone usually acts as a dispersant in the DLLME procedure, and especially, acetone was used as an extractant in the aforementioned UAE procedure. To make the DLLME procedure easier, the acetone extract made by UAE was selected as the dispersant in the DLLME procedure rather than adding another solvent.39 After selecting the acetone extract as the dispersant, its volume should be optimized. At low volume, acetone cannot disperse the extraction solvent properly and the cloudy solution does not completely form. However, at high volume, the solubility of analytes in water increases, which will result in a decrease of sensitivity. To obtain the optimized volume of acetone, the effect of its volume on sensitivity was investigated in the range of 400–800 μL. As can be seen from Fig. 6, the relative response increased with the increase of the volume of acetone when it was less than 500 μL. Reduction of extraction efficiency was observed after the volume of acetone exceeded 500 μL. So, 500 μL was chosen as the optimum volume.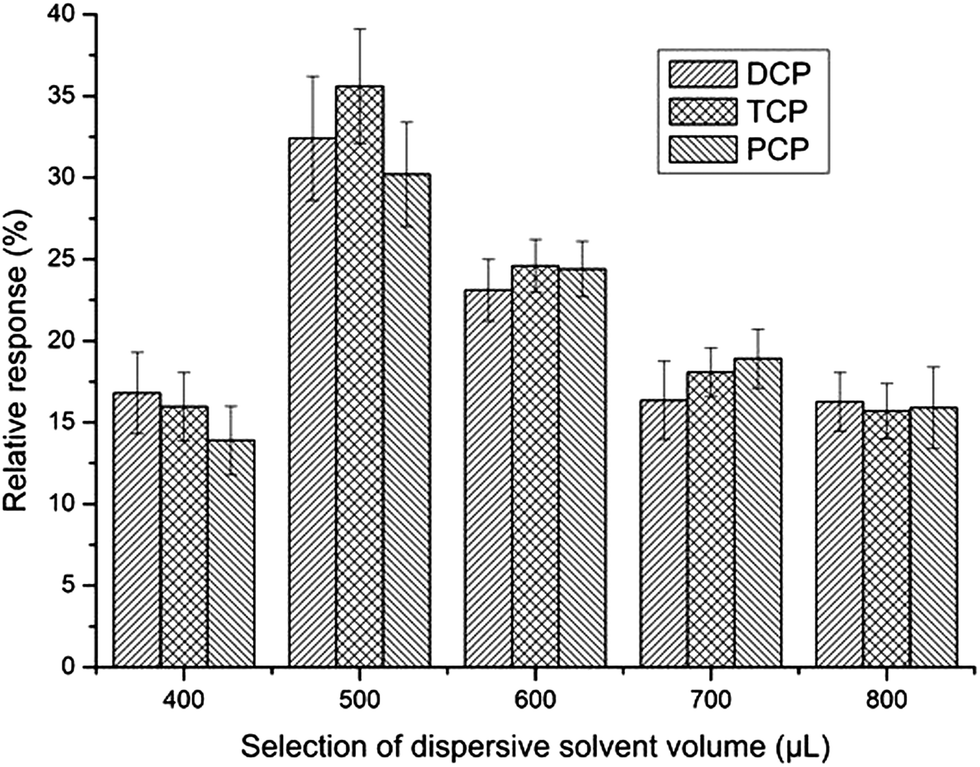 | ||
| Fig. 6 Selection of dispersive solvent volume. Extraction conditions were as described in Fig. 5 except that the volume of chlorobenzene was 20 μL. | ||
3.5. Effect of synchronous derivative reaction for DLLME
In order to detect CPs in toilet paper better, experiments were carried out to select the optimum conditions for synchronous derivatization. Different concentrations of potassium carbonate ranging between 0 and 1.5% (w/v) were studied, which corresponded to pH values of 5 to 12. Best sensitivity was attained for all compounds when the concentration of potassium carbonate was 0.5% (w/v), which resulted in a pH value of 10. With respect to the derivatization reagent, concentrations ranging from 0.4 to 1.0% (v/v) were assayed, the best signals being obtained at 0.6% (v/v) concentration.Once the conditions for the derivatization stage had been selected, the influence of changing the ionic strength of the matrix was studied by adding different amounts of sodium chloride ranging from 0 to 0.5 g to the solution containing 0.5 mL of acetone extract while keeping the other conditions constant. The experimental results showed that increasing the amount of salt from 0% to 10% (w/v) resulted in reduced relative response. This might be caused by the decreased solubility of extraction solvent with the increase of ionic strength. So, further extraction was performed in the absence of any salt.
3.6. Method validation
The analytical parameters including linear ranges, limits of detection (LODs), repeatability, recoveries were studied under the optimal conditions to evaluate the practicality of the proposed UAE-DLLME method. The results are listed in Table 1. The linear ranges for 2,4-DCP, 2,4,6-TCP and PCP were 5–500 μg kg−1, 2.5–250 μg kg−1 and 1–20 μg kg−1, respectively. The correlation coefficients were from 0.9968–0.9992. The limits of detection (LODs), defined as a signal-to-noise of three were varied from 0.25 μg kg−1 to 1.5 μg kg−1. The reproducibility of the proposed method was determined by evaluating the intra-day and inter-day precision. The relative standard deviations (RSDs, n = 6) were not higher than 9.6% .3.7. Comparision of the proposed method with other published methods
Compared to the other extraction methods for analysis of the three chlorophenols from a solid matrix, UAE-DLLME-GC-ECD possessed the shortest extraction time, as shown in Table 2. Additionally, the LODs of the proposed method were significantly lower than that of MAE- LC-MS, and nearly identical to that of LLE-GC-ECD, UAE-SBSE-GC-MS and QuEChERS-GC-MS.| Analyte | Method | Sample | LOD (μg kg−1) | Extraction time | Ref. |
|---|---|---|---|---|---|
| DCP | UAE-SBSE-GC-MS | Soil | 0.8 | 6.5 h | 40 |
| MAE- LC-MS | Soil | 60 | >40 min | 41 | |
| UAE-DLLME-GC-ECD | Toilet paper | 1.5 | 25 min | This work | |
| TCP | LLE-GC-ECD | Cork | 0.5 | >1 h | 42 |
| QuEChERS-GC-MS | Soil | 3 | >1 h | 43 | |
| UAE-SBSE-GC-MS | Soil | 0.7 | 6.5 h | 40 | |
| MAE- LC-MS | Soil | 30 | >40 min | 41 | |
| UAE-DLLME-GC-ECD | Toilet paper | 0.75 | 25 min | This work | |
| PCP | LLE-GC-ECD | Cork | 1.7 | >1 h | 42 |
| QuEChERS-GC-MS | Soil | 0.5 | >1 h | 43 | |
| UAE-SBSE-GC-MS | Soil | 0.2 | 6.5 h | 40 | |
| MAE- LC-MS | Soil | 20 | >40 min | 41 | |
| UAE-DLLME-GC-ECD | Toilet paper | 0.25 | 25 min | This work |
3.8. Real samples analysis
Under optimal conditions the proposed method was applied for the determination of three CPs in toilet paper samples. The utility of this method was evaluated by recovery studies. The recoveries of all the three CPs from three toilet paper samples at spiking levels of three different concentration levels were in the range of 70.9–118.5%. Results are shown in Table 3. No 2,4-DCP and 2,4,6-TCP were found in the three toilet paper samples while PCP was found in one type of toilet paper sample. The chromatograms of (a) the blank toilet paper sample and (b) the spiked toilet paper sample treated with this method are shown Fig. 7.| Analyte | Code name of toilet paper sample | Real (μg kg−1) | Added (μg kg−1) | Found (μg kg−1) | Relative recovery (%) |
|---|---|---|---|---|---|
| a ND, not detected. | |||||
| 2,4-DCP | 1 | NDa | 10 | 9.46 | 94.6 |
| 50 | 36.8 | 73.7 | |||
| 100 | 71.3 | 71.3 | |||
| 2 | NDa | 10 | 8.20 | 82.0 | |
| 50 | 37.2 | 74.4 | |||
| 100 | 88.2 | 88.2 | |||
| 3 | NDa | 10 | 76.5 | 76.5 | |
| 50 | 41.6 | 83.3 | |||
| 100 | 79.3 | 79.3 | |||
| 2,4,6-TCP | 1 | NDa | 5 | 4.20 | 84.1 |
| 25 | 25.9 | 103 | |||
| 50 | 44.8 | 89.6 | |||
| 2 | NDa | 5 | 3.90 | 77.6 | |
| 25 | 22.1 | 88.5 | |||
| 50 | 48.7 | 97.3 | |||
| 3 | NDa | 5 | 36.8 | 73.5 | |
| 25 | 21.3 | 85.4 | |||
| 50 | 47.6 | 95.3 | |||
| PCP | 1 | NDa | 2 | 1.52 | 76.2 |
| 10 | 10.5 | 106 | |||
| 20 | 19.8 | 99.1 | |||
| 2 | 2 | 1.42 | 70.9 | ||
| 10 | 11.0 | 110 | |||
| 20 | 23.7 | 118 | |||
| 3 | 1.2 | 2 | 2.82 | 81.1 | |
| 10 | 8.42 | 72.2 | |||
| 20 | 21.8 | 103 | |||
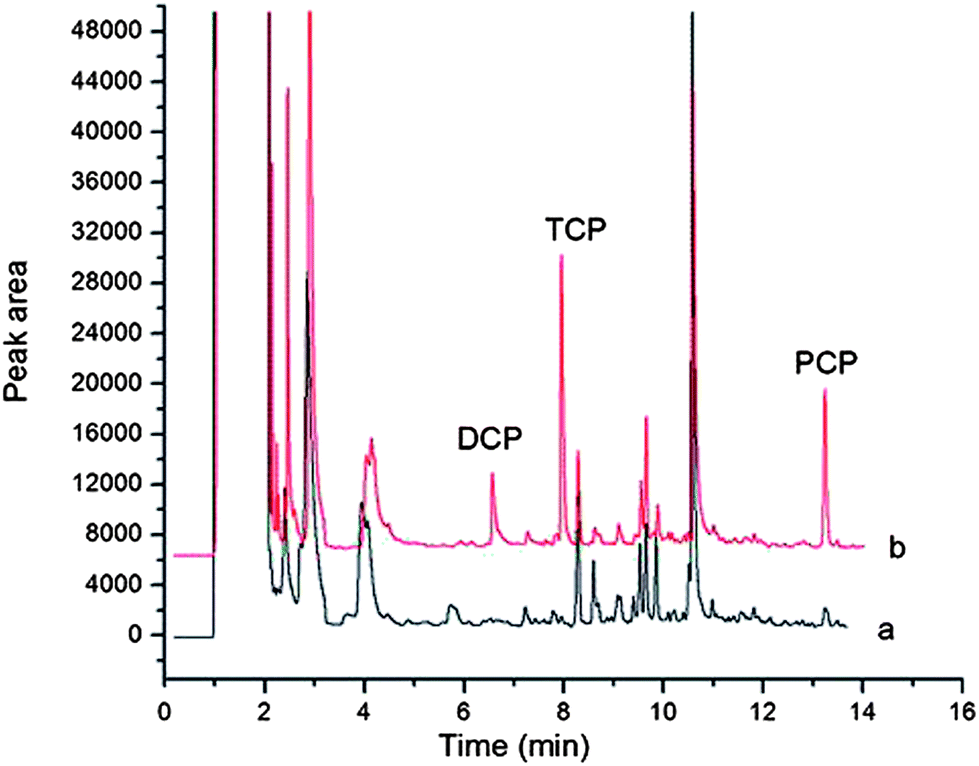 | ||
| Fig. 7 Chromatograms of (a) the blank toilet paper sample and (b) the spiked toilet paper sample treated with this method. The samples were spiked with three CPs at 2.5 μg kg−1 for DCP, 5 μg kg−1 for TCP and 8 μg kg−1 for PCP, respectively. | ||
4. Conclusions
This paper describes for the first time a method to determine three CPs that are used as preservatives in toilet paper. UAE followed by synchronous derivative DLLME was used to deal with the toilet paper samples and GC-ECD was used to detect the three chlorophenol derivatives. Several factors affecting these procedures were optimized systematically. Pentachlorophenol was successfully found in one toilet paper sample. The results indicated the proposed method was simple, fast, sensitive, and inexpensive for monitoring CPs residues in toilet paper.References
- I. Merrit, The comforts of home, Clarkson Potter, New York, 1999 Search PubMed.
- D. Jean-Marc, Rapid Determination of Nineteen Chlorophenols in Wood, Paper, Cardboard, Fruits, and Fruit Juices by Gas Chromatography/Mass Spectrometry, J. AOAC Int., 2001, 84, 853–860 Search PubMed.
- US Environmental Protection Agency (EPA), Ground water and drinking water: list of drinking water contaminants and MCLs. http://www.epa.gov/safewater/mcl.html.
- The list of priority substances in the field of water policy and amending directive, Council directive 2455/2001/ECC, Official Journal L331, 2001, pp. 1–5.
- J. L. Gómez-Ariza, E. Morales, R. Beltrán, I. Giráldez and M. Ruiz-Benítez, Ultrasonic treatment of molluscan tissue for organotin speciation, Analyst, 1995, 120, 1171–1174 RSC.
- J. Mierzwa, Y. C. Sun and M. H. Yang, Determination of Co and Ni in soils and river sediments by electrothermal atomic absorption spectrometry with slurry sampling, Anal. Chim. Acta, 1997, 355, 277–282 CrossRef CAS.
- K. Ashley, R. N. Andrews, L. Cavazos and M. Demange, Ultrasonic extraction as a sample preparation technique for elemental analysis by atomic spectrometry, J. Anal. At. Spectrom., 2001, 16, 1147–1153 RSC.
- A. Peña-Neira, B. Fernández de Simón, M. C. García-Vallejo, T. Hernández, E. Cadahía and J. A. Suarez, Presence of cork-taint responsible compounds in wines and their cork stoppers, Euro. Food Res. Technol., 2000, 211, 257–261 CrossRef.
- A. Martínez-Uruñuela, I. Rodríguez, R. Cela, J. M. González-Sáiz and C. Pizarro, Development of a solid-phase extraction method for the simultaneous determination of chloroanisoles and chlorophenols in red wine using gas chromatography–tandem mass spectrometry, Anal. Chim. Acta, 2005, 549, 117–123 CrossRef PubMed.
- S. Insa, E. Anticó and V. Ferreira, Highly selective solid-phase extraction and large volume injection for the robust gas chromatography–mass spectrometric analysis of TCA and TBA in wines, J. Chromatogr., A, 2005, 1089, 235–242 CrossRef CAS PubMed.
- J. Llorca-Pórcel, M. Martínez-Parreño, E. Martínez-Soriano and I. Valor, Analysis of chlorophenols, bisphenol-A, 4-tert-octylphenol and 4-nonylphenols in soil by means of ultrasonic solvent extraction and stir bar sorptive extraction with in situ derivatisation, J. Chromatogr., A, 2009, 1216, 5955–5961 CrossRef PubMed.
- F. Bianchi, M. Careri, A. Mangia and M. Musci, Optimization of headspace sampling using solid-phase microextraction for chloroanisoles in cork stoppers and gas chromatography–ion-trap tandem mass spectrometric analysis, J. Sep. Sci., 2003, 26, 369–375 CrossRef CAS.
- A. Martínez-Uruñuela, J. M. González-Sáiz and C. Pizarro, Optimisation of the derivatisation reaction and subsequent headspace solid-phase microextraction method for the direct determination of chlorophenols in red wine, J. Chromatogr., A, 2004, 1048, 141–151 CrossRef PubMed.
- M. Rezaee, Y. Assadi, M. R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, Determination of organic compounds in water using dispersive liquid–liquid microextraction, J. Chromatogr., A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- M. I. Leong and S. D. Huang, Dispersive liquid–liquid microextraction method based on solidification of floating organic drop combined with gas chromatography with electron-capture or mass spectrometry detection, J. Chromatogr., A, 2008, 1211, 8–12 CrossRef CAS PubMed.
- P. Liang, G. Liu, F. Wang and W. Wang, Ultrasound-assisted surfactant-enhanced emulsification microextraction with solidification of floating organic droplet followed by high performance liquid chromatography for the determination of strobilurin fungicides in fruit juice samples, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2013, 926, 62–67 CrossRef CAS PubMed.
- C. K. Zacharis, P. D. Tzanavaras and K. Roubos, Solvent-based de-emulsification dispersive liquid–liquid microextraction combined with gas chromatography–mass spectrometry for determination of trace organochlorine pesticides in environmental water samples, J. Chromatogr., A, 2010, 1217, 5895–5900 CrossRef PubMed.
- S. Wen, J. Wu and X. Zhu, Room temperature ionic liquid-based dispersive liquid–liquid microextraction combined with flame atomic absorption spectrometry for the speciation of chromium(III) and chromium(VI), J. Mol. Liq., 2013, 180, 59–64 CrossRef CAS PubMed.
- R. S. Zhao, X. Wang, F. W. Li, S. S. Wang, L. L. Zhang and C. G. Cheng, Ionic liquid/ionic liquid dispersive liquid–liquid microextraction, J. Sep. Sci., 2011, 34, 830–836 CrossRef CAS PubMed.
- R. S. Zhao, X. Wang, L. L. Zhang, S. S. Wang and J. P. Yuan, Ionic liquid/ionic liquid dispersive liquid–liquid microextraction, a new sample enrichment procedure for the determination of hexabromocyclododecane diastereomers in environmental water samples, Anal. Methods, 2011, 3, 831–836 RSC.
- Q. X. Zhou, H. H. Bai, G. H. Xie and J. P. Xiao, Temperature-controlled ionic liquid dispersive liquid phase microextraction, J. Chromatogr., A, 2008, 1177, 43–49 CrossRef CAS PubMed.
- F. Kamarei, H. Ebrahimzadeh and Y. Yamini, Optimization of temperature controlled ionic liquid dispersive liquid phase microextraction combined with high performance liquid chromatography for analysis of chlorobenzenes in water samples, Talanta, 2010, 83, 36–41 CrossRef CAS PubMed.
- Y. W. Zhou, L. T. Han, J. Cheng, F. Guo, X. R. Zhi, H. L. Hu and G. Chen, Dispersive liquid–liquid microextraction based on the solidification of a floating organic droplet for simultaneous analysis of diethofencarb and pyrimethanil in apple pulp and peel, Anal. Bioanal. Chem., 2011, 399, 1901–1906 CrossRef CAS PubMed.
- N. Sharma, A. Jain, V. K. Singh and K. K. Verma, Solid-phase extraction combined with headspace single-drop microextraction of chlorophenols as their methyl ethers and analysis by high-performance liquid chromatography-diode array detection, Talanta, 2011, 83, 994–999 CrossRef CAS PubMed.
- L. Guo and H. K. Lee, Ionic liquid based three-phase liquid–liquid–liquid solvent bar microextraction for the determination of phenols in seawater samples, J. Chromatogr., A, 2011, 1218, 4299–4306 CrossRef CAS PubMed.
- J. Zhang, T. Sao and H. K. Lee, Development and application of microporous hollow fiber protected liquid-phase microextraction via gaseous diffusion to the determination of phenols in water, J. Chromatogr., A, 2006, 1121, 10–15 CrossRef CAS PubMed.
- J. A. Padilla-Sánchez, P. Plaza-Bolaños, R. Romero-González, A. Garrido-Frenich and J. L. M. Vidal, Application of a quick, easy, cheap, effective, rugged and safe-based method for the simultaneous extraction of chlorophenols, alkylphenols, nitrophenols and cresols in agricultural soils, analyzed by using gas chromatography–triple quadrupole-mass spectrometry/mass spectrometry, J. Chromatogr., A, 2010, 1217, 5724–5731 CrossRef PubMed.
- S. Insa, V. Salvadó and E. Anticó, Assays on the simultaneous determination and elimination of chloroanisoles and chlorophenols from contaminated cork samples, J. Chromatogr., A, 2006, 1122, 215–221 CrossRef CAS PubMed.
- EPA Method 8041, Phenols by Gas Chromatography: Capillary Column Technique, US Environmental Protection Agency, Washington, 1995, pp. 1–28 Search PubMed.
- F. Sacher, F. T. Lange, H. J. Brauch and I. Blankenhorn, Pharmaceuticals in groundwaters analytical methods and results of a monitoring program in Baden-Wurttemberg, Germany, J. Chromatogr., A, 2001, 938, 199–210 CrossRef CAS.
- I. Cruz and D. E. Wells, Determination of Pentachlorophenol by Exhaustive Methylation and Capillary Gas Chromatography in Sewage Sludge, Contaminated Water and Suspended Particulates, Int. J. Environ. Anal. Chem., 1992, 48, 101–113 CrossRef CAS.
- Á. Kovács, A. Kende, M. Mörtl, G. Volk, T. Rikker and K. Torkos, Determination of phenols and chlorophenols as trimethylsilyl derivatives using gas chromatography–mass spectrometry, J. Chromatogr., A, 2008, 1194, 139–142 CrossRef PubMed.
- J. B. Quintana, R. Rodil, S. Muniategui-Lorenzo, P. López-Mahía and D. Prada-Rodríguez, Multiresidue analysis of acidic and polar organic contaminants in water samples by stir-bar sorptive extraction–liquid desorption–gas chromatography–mass spectrometry, J. Chromatogr., A, 2007, 1174, 27–39 CrossRef CAS PubMed.
- N. Fattahi, Y. Assadi, M. R. M. Hosseini and E. Z. Jahromi, Determination of chlorophenols in water samples using simultaneous dispersive liquid–liquid microextraction and derivatization followed by gas chromatography-electron-capture detection, J. Chromatogr., A, 2007, 1157, 23–29 CrossRef CAS PubMed.
- N. Campillo, R. Peñalver and M. Hernández-Córdoba, Evaluation of solid-phase microextraction conditions for the determination of chlorophenols in honey samples using gas chromatography, J. Chromatogr., A, 2006, 1125, 31–37 CrossRef CAS PubMed.
- A. Martínez-Uruñuela, J. M. González-Sáiz and C. Pizarro, Optimisation of the derivatisation reaction and subsequent headspace solid-phase microextraction method for the direct determination of chlorophenols in red wine, J. Chromatogr., A, 2004, 1048, 141–151 CrossRef PubMed.
- G. Gatidou, N. S. Tomaidis, A. S. Stasinakis and T. D. Lekkas, Simultaneous determination of the endocrine disrupting compounds nonylphenol, nonylphenol ethoxylates, triclosan and bisphenol A in wastewater and sewage sludge by gas chromatography–mass spectrometry, J. Chromatogr., A, 2007, 1138, 32–41 CrossRef CAS PubMed.
- H. Li, Z. H. Zhang, S. P. Tang, Y. N. Li and Y. K. Zhang, Ultrasonically assisted acid extraction of manganese from slag, Ultrason. Sonochem., 2008, 15, 339–343 CrossRef CAS PubMed.
- A. R. Fontana, N. B. Lana, L. D. Martinez and J. C. Altamirano, Ultrasound-assisted leaching-dispersive solid-phase extraction followed by liquid–liquid microextraction for the determination of polybrominated diphenyl ethers in sediment samples by gas chromatography–tandem mass spectrometry, Talanta, 2010, 82, 359–366 CrossRef CAS PubMed.
- J. Llorca-Pórcel, M. Martínez-Parreño, E. Martínez-Soriano and I. Valor, Analysis of chlorophenols, bisphenol-A, 4-tert-octylphenol and 4-nonylphenols in soil by means of ultrasonic solvent extraction and stir bar sorptive extraction with in situ derivatisation, J. Chromatogr., A, 2009, 1216, 5955–5961 CrossRef PubMed.
- M. C. Alonso, D. Puig, I. Silgoner, M. Grasserbauer and D. Barcelo, Determination of priority phenolic compounds in soil samples by various extraction methods followed by liquid chromatography–atmospheric pressure chemical ionisation mass spectrometry, J. Chromatogr., A, 1998, 823, 231–239 CrossRef CAS.
- S. Insa, V. Salvadó and E. Anticó, Development of solid-phase extraction and solid-phase microextraction methods for the determination of chlorophenols in cork macerate and wine samples, J. Chromatogr., A, 2004, 1047, 15–20 CrossRef CAS PubMed.
- J. A. Padilla-Sáncheza, P. Plaza-Bolaños, R. Romero-González, A. Garrido-Frenicha and J. L. M. Vidal, Application of a quick, easy, cheap, effective, rugged and safe-based method for the simultaneous extraction of chlorophenols, alkylphenols, nitrophenols and cresols in agricultural soils, analyzed by using gas chromatography–triple quadrupole-mass spectrometry/mass spectrometry, J. Chromatogr., A, 2010, 1217, 5724–5731 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |