Quantification of the arylesterase activity of paraoxonase-1 in human blood
Clara G.
Dias
a,
Joana R.
Batuca
a,
Aline T.
Marinho
a,
Umbelina
Caixas
ab,
Emília C.
Monteiro
a,
Alexandra M. M.
Antunes
c and
Sofia A.
Pereira
*a
aCentro de Estudos de Doenças Crónicas (CEDOC), NOVA Medical School, Campo dos Mártires da Pátria 130, 1169-056 Lisboa, Portugal. E-mail: sofia.pereira@fcm.unl.pt; Tel: +351 21 880 30 00
bCentro Hospitalar de Lisboa Central (CHLC), EPE, Lisboa, Portugal
cInstituto Superior Técnico, Centro de Química Estrutural (CQE), Universidade de Lisboa, Lisboa, Portugal
First published on 1st November 2013
Abstract
Paraoxonase-1 (PON1) is known as a free-radical scavenging system associated with circulating serum high-density lipoprotein (HDL). PON1 catalyzes the hydrolysis of multiple compounds such as arylesters, lactones and hydroperoxides. The arylesterase (AREase) activity of PON1 is involved in the detoxification of lipid peroxides, which are related to several clinical conditions. Therefore, the possibility of measuring the AREase activity in routine clinical studies would be advantageous. The AREase activity was obtained by monitoring the formation of acetic acid, upon the hydrolysis of phenyl acetate, using 10 μL of sample. The method accuracy was higher than 90% and intra-assay and inter-assay precisions were 96% and 95%, respectively. The method validation supported that this analytical procedure is suitable for use in human serum and heparinized plasma samples, while ethylenediaminetetra-acetic acid (EDTA)-containing samples should be avoided. The methodology herein described constitutes an easy, fast and reliable method for assessing the AREase activity of PON1. This method can be easily implemented as a clinical analytical tool and is also suitable for research purposes.
1. Introduction
The research interest on paraoxonase-1 (PON1) has grown over the past few years, in particular due to its potential as a biomarker of disease status.1,2 PON1 is a calcium-dependent enzyme that belongs to the A-esterase class.3 This enzyme is synthesized mainly by the liver and secreted into the circulation, where is it associated with the high-density lipoprotein (HDL).PON1 behaves as a human body endogenous free-radical scavenging system, contributing to the detoxification of organophosphate compounds and carcinogenic lipid soluble radicals from lipid peroxidation.4,5 Moreover, the enzyme catalyzes the hydrolysis of multiple compounds such as arylesters, lactones and hydroperoxides.6,7 PON1 also exerts antioxidant and antiatherogenic properties, as it protects low-density lipoproteins (LDL) and HDL from oxidative modifications.8
Currently, there are 3 main activities identified for PON1 which might explain its antioxidant and anti-inflammatory potential. The enzyme was firstly found to have paraoxonase (POase) activity, reflecting its ability to catalyze the hydrolysis of paraoxon, an insecticide that gave rise to the enzyme's name, hence protecting against xenobiotic toxicity.1 The detoxification of lipid peroxides by PON1 is possible via its arylesterase (AREase) activity. Furthermore, its lactonase activity was recently discovered, which protects against homocysteine thiolactone toxicity6 as well as the fact of being involved in the metabolism of certain drugs.9
Increasing evidence has been opening out the PON1 link to several pathological conditions. Due to its antioxidant and anti-inflammatory properties, the enzyme might have a role in lipid metabolism and the protection against atherosclerosis.10,11 Hence, PON1 could be involved in cardiovascular disease and ischemic stroke.12,13 PON1 has also been studied in other contexts such as chronic renal failure/impairment, infection (tuberculosis, human immunodeficiency virus infection), type 2 diabetes and metabolic syndrome.14 Furthermore, this enzyme is also involved in several neurodegenerative disorders such as Alzheimer15 and Parkinson16 diseases, autism17 and in a large number of cancer types including breast,18 prostate4 and central nervous system.19
Despite most of the studies exploring the role of PON1 in disease used the POase activity as a biomarker for the enzyme status,20,21 this activity does not reflect the real physiological activity of PON1. Thus, it is critical to start looking at its remaining activities, AREase and lactonase, which have been proved to be more physiological.10
Several studies indicated that the AREase activity best reflects the antioxidant activity of PON1, as it is responsible for the detoxification of oxidized lipids.10 Similar to the POase activity, the AREase activity of PON1 was also found to be stabilized by the apolipoprotein A-1 (ApoA-1), a major player in cholesterol homeostasis.22 Additionally, in contrast to the POase activity, the measurement of the AREase activity is not influenced by genetic polymorphisms.23
The AREase activity has shown to be reduced in a wide range of clinical conditions associated with inflammation and oxidative stress. For instance, in patients with coronary artery disease, both POase and AREase activity were found to be significantly lower as compared to controls.24 Also, in gastroesophageal cancers, a drop in the activity was detected and was well correlated with the extent of circulating inflammatory markers, including C-reactive protein and IL-6.25 Moreover, even in an animal model of intestinal nematode Nippostrongylus brasiliensis infection, the activity was low, which again correlated with pro-inflammatory cytokines such as IL-1, IL-6 and TNF-α.26 The AREase activity was also studied in several cancer types, including lung cancer,27 gastrointestinal tumors25 and ovarian malignancy.28 All the performed studies reported lower activity levels in comparison with healthy controls. Regarding neurological disorders, the AREase activity was found to be significantly low in autistic17 and schizophrenic29 patients.
In light of this evidence, there is a need to develop a rapid and economic method for the measurement of the AREase activity, which is the main goal of the present study.
2. Results and discussion
2.1. Method validation
| Standard (mM) | Accuracy (%) | Intra-assay precision (%) | Inter-assay precision (%) |
|---|---|---|---|
| LLOQ (5.50) | 90 | 94 | 92 |
| QC1 (12.58) | 103 | 96 | 96 |
| QC2 (16.78) | 102 | 95 | 96 |
| HLOQ (26.21) | 100 | 98 | 97 |
2.1.4.1. Intra-assay precision. The values obtained for the intra-assay precision were higher than 94% (Table 1).
The AREase activity of the serum sample from a healthy volunteer was 115.0 ± 3.30 kU L−1, and the intra-assay precision was 97%.
2.1.4.2. Inter-assay precision. The inter-assay precision was 95% (Table 1). Moreover, the serum sample of the healthy volunteer had an AREase activity of 114.9 ± 7.67 kU L−1, and the inter-assay precision was 93%.
2.2. Method application
In the current study, the 3 types of samples (serum, heparinized plasma and plasma collected with EDTA) obtained from five healthy volunteers were used to quantify the AREase (Section 2.2.1) and POase activities (Section 2.2.2). The five volunteers were all caucasians, with ages between 22 and 27 years old and four were females.| Sample type | Activity (kU L−1) | p-value* | Inhibition (%) |
|---|---|---|---|
| a NS: non-significant; NA: not applicable; * Student's t-test. | |||
| Serum | 105.5 ± 34.4 | — | — |
| Heparinized plasma | 106.8 ± 32.2 | NS | NA |
| EDTA plasma | 43.46 ± 10.7 | 0.009 | 55 ± 17 |
The lowest activity was obtained from the samples of the male volunteer.
Furthermore, there was a direct correlation between the AREase activity of serum and heparinized plasma (Pearson r = 0.999, p < 0.0001). However, no association was found between the AREase activity measured in serum and in plasma collected with EDTA. Likewise, no correlation was found between the AREase activity of heparinized and EDTA plasma samples.
Moreover, the AREase activity inhibition by EDTA ranged from 30 to 70% in comparison with the AREase activity in serum samples. This inhibition was not dependent on the AREase activity.
| Sample type | Activity (U L−1) | p-value* | Inhibition (%) |
|---|---|---|---|
| a NA: not applicable; * Student's t-test. | |||
| Serum | 243.1 ± 41.0 | — | — |
| Heparinized plasma | 228.2 ± 36.5 | 0.031 | NA |
| EDTA plasma | 152.4 ± 61.1 | 0.014 | 38 ± 19 |
A significant decrease was observed in the POase activity assessed in heparinized and EDTA samples, in comparison with the POase activity obtained in serum samples (Student's t-test, p = 0.031 and p = 0.014, respectively). Additionally, the POase activity in plasma collected with EDTA was also lower than the one obtained in heparinized plasma samples (t-test, p = 0.015).
The POase activity from serum was positively associated with the POase activity of heparinized plasma samples (Pearson r = 0.972, p = 0.006). Nevertheless, this association was not observed for EDTA samples, neither between heparinized and EDTA-containing plasma samples.
The inhibition of the POase activity by EDTA was also assessed, and ranged from 5 to 50% in comparison with the POase activity obtained in serum samples. This inhibition was not activity-dependent.
In the present study, a simple, fast and inexpensive method suitable for the measurement of the AREase activity of PON1 enzyme in human blood was developed and validated. This method is capable of measuring the AREase activity in several samples simultaneously, using a very small amount of biological fluid and remaining solutions. Furthermore, as the assay is not performed at a UV but at a visible spectrum range (405 nm), the use of quartz microplates is not required. And hence, the enzymatic kinetics can be performed in a spectrophotometer available at any research/clinical facility.
The development of microplate-based methods allowed the high-throughput measurement of PON1 activity using paraoxon as the substrate.30,31 As such, the same can be applied for the AREase activity, avoiding in turn the use of higher amounts of both biological sample and remaining reagents.
The current available methods for the assessment of the AREase activity of PON1 monitor the formation of phenol at an UV range. However, the quantification of acetic acid instead of phenol allows the use of simple titration based methods that can be monitored at a visible range, thus the use of sophisticated and expensive equipment is unnecessary, which might not be available in clinical routine labs. Our enzymatic assay is based on a method initially proposed by Sharp and Rosenberry (1982) for the measurement of the kinetic properties of acetylcholinesterase with its physiological substrate, acetylcholine.32 As in the hydrolysis of acetylcholine, the hydrolysis of phenyl acetate by PON1 produces acetic acid in stoichiometric amounts to the substrate degradation. Therefore, by including a pH indicator dye such as phenol red, the color change resulting from the production of acetic acid can be monitored spectrophotometrically in the visible range and be directly related to the AREase activity.
However, the use of phenol, which in turn is toxic and photoreactive, can be set as the major drawback of the proposed method. The use of toxic substrates is a handicap of PON1 measurement as paraoxon is also toxic.
The method herein proposed relies on the ability of PON1 to hydrolyze phenyl acetate into acetic acid and phenol. Using phenol red as a pH indicator dye, it was possible to monitor the change in color from red to yellow upon the formation of acetic acid in a linear way.
Spectrophotometric assays require the use of a substrate containing a chromophore.32 Whereas the chromophores of both phenyl acetate and phenol do not allow monitorization at visible wavelengths, the production of acetic acid can be coupled to the change in color of phenol red. Typically, to observe a maximal change in absorbance using a pH indicator, the reaction mixture must be unbuffered, which turns the system extremely prone to environmental factors.33 Sharp and Rosenberry also addressed this issue and stated that if a pH indicator dye and a nonchromophoric buffer salt were chosen in a way that their pKa values were not extremely different from each other, as it is the case of phenol red and HEPES buffer, the change in absorbance reported by a given concentration of a pH indicator can be proportional to the change in acid concentration.32 Moreover, the use of a pH value of 8.0 for PON1 activity assessment has been widely used,34,35 as it is thought to be the optimal pH for the enzyme.36,37 Also, this pH condition was also proved to be ideal for measuring rates of hydrolysis of phenyl acetate.38
The activities obtained for the serum samples of the five healthy volunteers were consistent with the ones already reported.39
This method has applicability in plasma and serum samples, and since PON1 enzyme is associated with circulating HDL, blood would be the main fluid of interest. However, this method could possibly be adapted for other sample types, such as cell culture supernatants and even cerebrospinal fluids, which further show its applicability for clinical and research purposes.
The enzyme activities measured in plasma samples are often lower than those measured in serum samples, mainly due to the ability of fibrin clots to retain a certain amount of water, resulting in a higher concentration of analytes in serum, relative to plasma samples.40,41 Although, we showed that this method is suitable for the AREase activity in serum and heparinized plasma samples. To the best of our knowledge, the relationship between serum and heparinized plasma samples was only demonstrated for the POase activity.42,43 The POase activity is inhibited on heparinized plasma samples showing a handicap of using the protocol with plasma mixed with serum samples. Whereas serum is the preferred type of sample used for the measurement of the POase activity, previous reports classified the effect of lithium-heparin on POase activity measurement as negligible.43 Likewise, several groups have reported studies on POase activity in lithium-heparin-treated samples, and the results were lower but consistent with those obtained in serum samples.44,45 The AREase activity differs importantly as it is not influenced by the lithium-heparin-treatment of the samples.
On the other hand, the present data showed that EDTA-containing samples should be avoided. Both AREase and POase activities of PON1 require calcium for their activity.46 The use of the anticoagulant EDTA is well known to be unsuitable for the POase activity assay.47,48 In fact, EDTA-plasma samples had lower POase activity than serum samples by 38%, and was consistent with previous published studies using the same sample type, with a mean inhibition of 41%.42 Moreover, a consistent inhibition percentage was also found in studies using the purified PON1 enzyme.49 This decreased activity was not dependent on the activity per se, nor related to the activity in serum or heparinized samples. Given that PON1 requires calcium for both activity and stability, the presence of calcium chelators (e.g. EDTA and citrate) as anticoagulants is expected to inhibit enzymatic activities. Therefore, the inhibition of PON1 by EDTA has been reported for the measurement of the activities of the enzyme using a variety of substrates.47 Herein it was for the first time found that this inhibition is even more pronounced for the AREase activity in human samples.
As such, in studies where PON1 is involved, the blood collection conditions should be carefully defined and the use of plasma samples collected with EDTA should be avoided.
Finally, the AREase and POase activities were not related in the 3 different blood sampling conditions tested. Despite the data being conflicting on this issue,28,50,51 structure–activity studies might give some clues for this evidence. Firstly, a histidine dyad composed of His115 and His134 was suggested to be directly involved in the catalytic mechanism of PON1 for both ester (e.g. phenyl acetate) and phosphotriester (e.g. paraoxon) hydrolysis. Mutagenesis experiments support this mechanism although it was later found that these mutants were probably misfolded and, therefore, inactive,36 thus undermining these results. Subsequently, Khersonsky and Tawfik (2006) showed, by site-directed mutagenesis, that the lactonase and AREase activities were both mediated by the His115–His134 dyad and that notably, the POase activity, which is a promiscuous activity of PON1, is mediated by other residues.52 This evidence might explain the differences found in POase and AREase activities, and the absence of a relationship between them. Moreover, this could also explain the different effects of lithium and EDTA on both activities.
3. Experimental
3.1. Method development
Despite the methods already available for this purpose,30,31 they do not fulfill our aims of easy application in clinical setting. The rationale for the method developed herein is taking advantage of the acetic acid production by hydrolyses of phenyl acetate, a substrate of the AREase activity of PON1 (Fig. 1). The acetic acid formation can be monitored by the color variation of the titration with phenol red reagent. Briefly, a molecule of phenyl acetate is hydrolyzed into phenol and acetic acid. Hence, the acetic acid is produced in stoichiometric amounts to the substrate hydrolysis. This reaction can be monitored spectrophotometrically, at 405 nm, by the color change of the phenol red reagent.
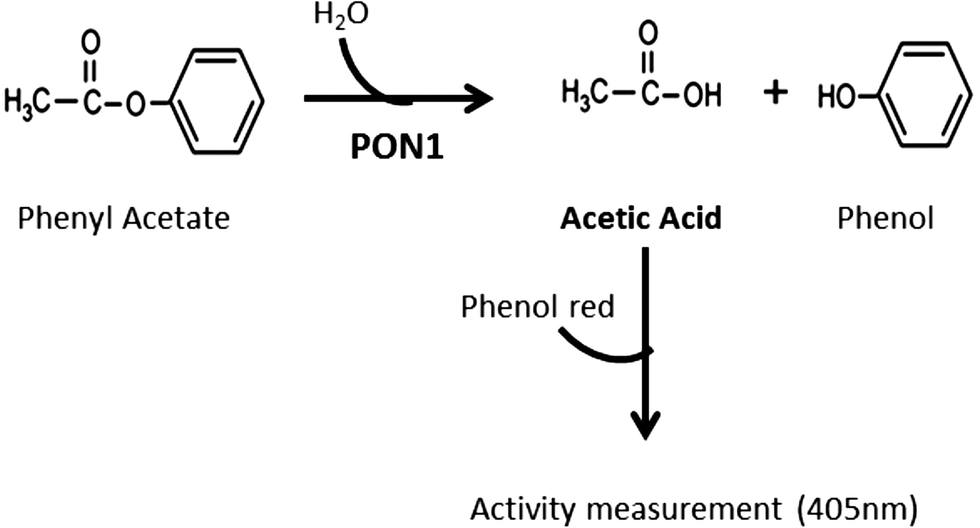 | ||
| Fig. 1 Method rationale: hydrolysis of phenyl acetate by PON1 and its monitoring for the assessment of the AREase activity. | ||
3.1.2.1. Stock solutions. The stock solutions were prepared by dissolving appropriate amounts of acetic acid (M&B Laboratory Reagents) and phenol (Fluka) in freshly prepared HEPES (Roth) buffer (2.0 mM, at pH 8.0), containing CaCl2 (BDH Chemicals Ltd Pool England) (1.0 mM) and albumin from bovine serum (BSA) (Roth) (0.005%). Ideally, these solutions should be used right after their preparation and should be prepared in 2 mL tubes covered in aluminum, since phenol is a photoreactive compound.
3.1.2.2. Standards preparation. For the preparation of the standards, the stock solutions were diluted in physiological serum in order to obtain six standards. Furthermore, a standard only with physiological serum and HEPES buffer (blank sample) was also prepared in order to correct the non-enzymatic hydrolysis of phenyl acetate.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 in physiological serum and 10 μL were added to each well. The samples and the previously prepared standards were incubated at 37 °C, during 10 minutes, where upon 190 μL of freshly prepared HEPES buffer (2.0 mM, at pH 8.0) containing CaCl2 (1.0 mM), BSA (0.005%), phenol red (Fluka) (106 μM) and 5.0 mM phenyl acetate (Fluka) (5.0 mM) were added to each well. The absorbance at 405 nm was measured on a microplate reader (Biotrack II plate reader, Amersham Biosciences). The activity was directly obtained from the calibration curve (with concentrations ranging from 5.50 mM to 26.21 mM) and expressed as kU L−1, defined as the amount of enzyme producing 1 mM of acetic acid per minute. All samples/standards were analyzed in triplicate.
:5 in physiological serum and 10 μL were added to each well. The samples and the previously prepared standards were incubated at 37 °C, during 10 minutes, where upon 190 μL of freshly prepared HEPES buffer (2.0 mM, at pH 8.0) containing CaCl2 (1.0 mM), BSA (0.005%), phenol red (Fluka) (106 μM) and 5.0 mM phenyl acetate (Fluka) (5.0 mM) were added to each well. The absorbance at 405 nm was measured on a microplate reader (Biotrack II plate reader, Amersham Biosciences). The activity was directly obtained from the calibration curve (with concentrations ranging from 5.50 mM to 26.21 mM) and expressed as kU L−1, defined as the amount of enzyme producing 1 mM of acetic acid per minute. All samples/standards were analyzed in triplicate.
3.2. Method validation
The validation criteria were defined according to guidebooks, regarding the validation of bioanalytical methods.53 For all validation purposes, each sample was analyzed in triplicate.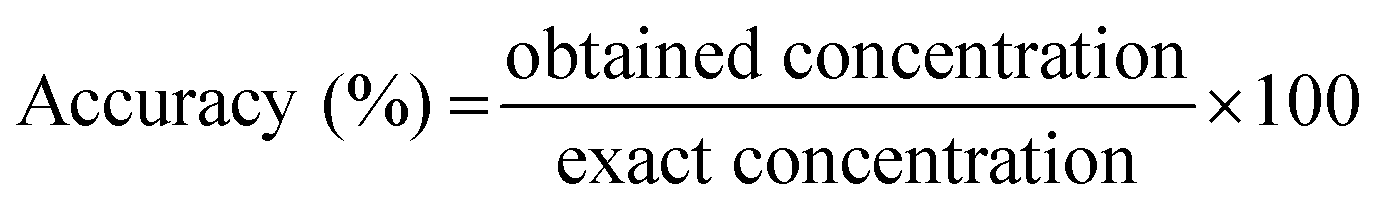 | (1) |
3.2.4.1. Intra-assay precision. The intra-assay precision was evaluated by analyzing six aliquots of the LLOQ, QC1, QC2 and HLOQ. These aliquots were analyzed on the same run. The calculation of the intra-assay precision was performed assuming that its value would ideally be 100%.
Hence, the intra-assay precision was obtained by subtracting the variation coefficients (CV) of the analyzed aliquots, according to eqn (2).
| Intra-assay precision (%) = 100 − CV | (2) |
Samples obtained from a healthy volunteer were also analyzed.
3.2.4.2. Inter-assay precision. For the study of this parameter, the same samples described in Section 3.2.4.1 were analyzed, albeit these analyses were performed in different runs. The inter-assay precision was calculated using eqn (3). Samples obtained from a healthy volunteer were also analyzed.
| Inter-assay precision (%) = 100 − inter-assay CV | (3) |
3.3. Method application
The current work was conducted in accordance with the principles of the Declaration of Helsinki and the informed consent was obtained from all the volunteers.
3.4. Statistical analysis
Statistical analysis was performed using GraphPad Prism version 5.0.55 Data were expressed as mean ± standard deviation (SD), unless otherwise stated.4. Conclusions
In summary, a reproducible, reliable and suitable method for the monitoring of the AREase activity of PON1 in human blood is herein described for the first time. The method allows the assessment of the AREase activity in a large number of samples in a short period of time which is especially advantageous for clinical applications. As far as we know, this study reports for the first time, the effect of different blood collection types on the AREase activity.Acknowledgements
Thanks are due to Inês Fasutino (NOVA Medical School) for technical support and to the Portuguese Foundation for Science and Technology (FCT) for financial support (EXPL/DTP-FTO/0204/2012).References
- L. Costa, T. Cole, A. Vitalone and C. Furlong, Clin. Chim. Acta, 2005, 352, 37–47 CrossRef CAS PubMed.
- S. Searles Nielsen, B. A. Mueller, A. J. De Roos, H. M. Viernes, F. M. Farin and H. Checkoway, Environ. Health Perspect., 2005, 113, 909–913 CrossRef.
- W. N. Aldridge, Biochem. J., 1953, 53, 110–117 CAS.
- M. Marchesani, A. Hakkarainen, T. K. Tuomainen, J. Kaikkonen, E. Pukkala, P. Uimari, E. Seppälä, M. Matikainen, O. P. Kallioniemi, J. Schleutker, T. Lehtimäki and J. T. Salonen, J. Natl. Cancer Inst., 2003, 95, 812–818 CrossRef CAS PubMed.
- H. G. Davies, R. J. Richter, M. Keifer, C. A. Broomfield, J. Sowalla and C. E. Furlong, Nat. Genet., 1996, 14, 334–336 CrossRef CAS PubMed.
- H. Jakubowski, J. Biol. Chem., 2000, 275, 3957–3962 CrossRef CAS PubMed.
- B. N. La Du, in Pharmacogenetics of Drug Metabolism, ed. W. Kalow, Pergamon Press, New York, NY, 1992, pp. 51–91 Search PubMed.
- M. I. Mackness, S. Arrol, C. Abbott and P. N. Durrington, Atherosclerosis, 1993, 104, 129–135 CrossRef CAS.
- K. Tougou, A. Nakamura, S. Watanabe, Y. Okuyama and A. Morino, Drug Metab. Dispos., 1998, 26, 355–359 CAS.
- M. Rosenblat, L. Gaidukov, O. Khersonsky, J. Vaya, R. Oren, D. Tawfik and M. Aviram, J. Biol. Chem., 2006, 281, 7657–7665 CrossRef CAS PubMed.
- A. Tward, Y. Xia, X. Wang, Y. Shi, C. Park, L. Castellani, A. Lusis and D. Shih, Circulation, 2002, 106, 484–490 CrossRef CAS.
- M. C. Garin, R. W. James, P. Dussoix, H. Blanché, P. Passa, P. Froguel and J. Ruiz, J. Clin. Invest., 1997, 99, 62–66 CrossRef CAS PubMed.
- B. Voetsch, K. S. Benke, B. P. Damasceno, L. H. Siqueira and J. Loscalzo, Stroke, 2002, 33, 1459–1464 CrossRef.
- J. Marsillach, S. Parra, N. Ferré, B. Coll, C. Alonso-Villaverde, J. Joven and J. Camps, in The paraoxonase: their role in disease development and xenobiotic metabolism, ed. B. Mackness, M. Mackness, M. Aviram and C. Paragh, Springer, Dordrecht, 2008, pp. 187–198 Search PubMed.
- X. M. He, Z. X. Zhang, J. W. Zhang, Y. T. Zhou, M. N. Tang, C. B. Wu and Z. Hong, Chin. Med. J., 2006, 119, 1204–1209 CAS.
- E. Zintzaras and G. M. Hadjugeorgiou, J. Hum. Genet., 2004, 49, 474–481 CrossRef PubMed.
- S. P. Pasca, B. Nemes, L. Vlase, C. E. Gagyi, E. Dronca, A. C. Miu and M. Dronca, Life Sci., 2006, 78, 2244–2248 CrossRef CAS PubMed.
- V. L. Stevens, C. Rodriguez, A. L. Pavluck, M. J. Thun and E. E. Calle, Cancer Epidemiol., Biomarkers Prev., 2006, 15, 1226–1228 CAS.
- A. M. Kafadar, A. Ergen, U. Zeybek, B. Agachan, C. Kuday and T. Isbir, Cell Biochem. Funct., 2006, 24, 455–460 CrossRef CAS PubMed.
- S. A. Pereira, J. R. Batuca, U. Caixas, T. Branco, J. Delgado-Alves, I. Germano, F. Lampreia and E. C. Monteiro, Br. J. Clin. Pharmacol., 2009, 68, 891–897 CrossRef CAS PubMed.
- Y. U. Soyoral, M. Aslanb, H. Emreb, H. Begenikb, F. M. Erdurb, A. Turkelb, S. Selekc and R. Erkoca, Atherosclerosis, 2011, 218, 243–246 CrossRef CAS PubMed.
- R. C. Sorenson, C. L. Bisgaier, M. Aviram, C. Hsu, S. Billecke and B. N. La Du, Arterioscler., Thromb., Vasc. Biol., 1999, 19, 2214–2225 CrossRef CAS.
- H. Eckerson, C. Wyte and B. La Du, Am. J. Hum. Genet., 1983, 35, 1126–1138 CAS.
- R. Gamboa, J. C. Regalado, C. Huesca-Gómez, C. Posadas-Romero, J. Verdejo Paris, G. Vargas-Alarcón and O. Pérez-Méndez, Arch. Cardiol. Mex., 2008, 78, 360–368 CAS.
- M. Krzystek-Korpacka, D. Boehm, M. Matusiewicz, D. Diakowska, K. Grabowski and A. Gamian, Clin. Biochem., 2008, 41, 804–811 CrossRef CAS PubMed.
- A. S. Farid, K. Nakahara, N. Murakami, T. Hayashi and Y. Horii, Am. J. Trop. Med. Hyg., 2008, 78, 770–776 CAS.
- E. T. Elkiran, N. Mar, B. Aygen, F. Gursu, A. Karaoglu and S. Koca, BMC Cancer, 2007, 7, 48 CrossRef PubMed.
- H. Camuzcuoglu, D. T. Arioz, H. Toy, S. Kurt, H. Celik and O. Erel, Gynecol. Oncol., 2009, 112, 481–485 CrossRef CAS PubMed.
- A. Sarandol, S. Kirli, C. Akkaya, N. Ocak, E. Eroz and E. Sarandol, J. Psychopharmacol., 2007, 21, 857–863 CrossRef CAS PubMed.
- M. Naderi, M. Hashemi, F. Komijani-Bozchaloei, A. Moazeni-Roodi and M. Momenimoghaddam, Pathophysiology, 2011, 18, 117–120 CrossRef CAS PubMed.
- J. Beltowski, G. Wójcicka, M. Mydlarczyk and A. Jamroz, Pol. J. Pharmacol., 2002, 54, 143–150 CAS.
- T. R. Sharp and T. L. Rosenberry, J. Biochem. Biophys. Methods, 1982, 6, 159–172 CrossRef CAS.
- P. A. Benkovic, M. Hegazi, B. A. Cunningham and S. J. Benkovic, Biochemistry, 1979, 18, 830–836 CrossRef CAS.
- R. Sharma, B. Singh and M. Mahajan, Kaohsiung J. Med. Sci., 2007, 23, 225–231 CrossRef CAS.
- I. Seres, G. Paragh, E. Deschene, T. J. Fulop and A. Khali, Exp. Gerontol., 2004, 39, 59–66 CrossRef CAS PubMed.
- M. Harel, A. Aharoni, L. Gaidukov, B. Brumshteun, O. Khersonsky, R. Meged, H. Dvir, R. B. G. Ravelli, A. McCarthy, L. Toker, I. Silman, J. L. Sussman and D. S. Tawfik, Nat. Struct. Mol. Biol., 2004, 11, 412–419 CAS.
- O. Khersonsky and D. S. Tawfik, Biochemistry, 2005, 44, 6371–6382 CrossRef CAS PubMed.
- R. J. Richter, G. P. Jarvik and C. E. Furlong, Circ.: Cardiovasc. Genet., 2008, 1, 147–152 CrossRef CAS PubMed.
- A. F. Hernández, F. Gil, E. Leno, O. López, L. Rodrigo and A. Pla, NeuroToxicology, 2009, 30, 628–635 CrossRef PubMed.
- M. Mackness, Atherosclerosis, 1998, 136, 195–196 CrossRef CAS.
- M. Guthold, W. Liu, B. Stephens, S. Lord, R. Hantgan, D. Erie, R. Taylor and R. Superfine, Biophys. J., 2004, 87, 4226–4236 CrossRef CAS PubMed.
- M. Araoud, F. Neffeti, W. Douki, A. Kenani and M. Najjar, Asian Biomed., 2011, 5, 217–224 CAS.
- N. Ferre, J. Camps, J. Marsillach, B. Mackness, M. Mackness, B. Coll, M. Tous and J. Joven, Clin. Chem., 2005, 51, 922–923 CAS.
- G. Jarvik, N. Tsai, L. McKinstry, R. Wani, V. Brophy, R. Richter, G. Schellenberg, P. Heagerty, T. Hatsukami and C. Furlong, Arterioscler., Thromb., Vasc. Biol., 2002, 22, 1329–1333 CrossRef CAS.
- J. Martín-Campos, J. Julve, J. Escolà, J. Ordóñez-Llanos, J. Gómez, J. Binimelis, F. González-Sastre and F. Blanco-Vaca, J. Lipid Res., 2002, 43, 115–123 Search PubMed.
- C. L. Kuo and B. N. La Du, Drug Metab. Dispos., 1998, 26, 653–660 CAS.
- E. Erdös and L. Boggs, Nature, 1961, 190, 716–717 CrossRef.
- M. Mackness, Biochem. Biophys. Res. Commun., 1998, 242, 249 CrossRef CAS PubMed.
- L. Golmanesh, H. Mehrani and M. Tabei, J. Biochem. Biophys. Methods, 2008, 70, 1037–1042 CrossRef CAS PubMed.
- D. N. Nevin, A. Zambon, C. E. Furlong, R. J. Richter, R. Humbert, J. E. Hokanson and J. D. Brunzell, Arterioscler., Thromb., Vasc. Biol., 1996, 16, 1243–1249 CrossRef CAS.
- H. Camuzcuoglu, H. Toy, M. Vural, A. Camuzcuoglu, A. Taskin and H. Celik, Turk. J. Med. Sci., 2011, 41, 185–191 CAS.
- O. Khersonsky and D. S. Tawfik, J. Biol. Chem., 2006, 281, 7649–7656 CrossRef CAS PubMed.
- A. G. González and M. Á. Herrador, TrAC, Trends Anal. Chem., 2007, 26, 227–238 CrossRef PubMed.
- J. R. Batuca, P. R. Ames, D. A. Isenberg and J. Delgado Alves, Ann. N. Y. Acad. Sci., 2007, 1108, 137–146 CrossRef CAS PubMed.
- H. Motulsky, GraphPad Prism Version 5.0 Statistic Guide, GraphPad, Prism Software Inc, San Diego, CA, 2007 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |