DOI:
10.1039/C3AY41562G
(Paper)
Anal. Methods, 2014,
6, 282-288
Optimization of mobile phase by 32-mixture design for the validation and quantification of risperidone in bulk and pharmaceutical formulations using RP-HPLC
Received
8th September 2013
, Accepted 9th October 2013
First published on 28th November 2013
Abstract
A rapid, sensitive and optimized reversed phase-HPLC method has been developed for routine analysis of risperidone in bulk drugs and pharmaceutical formulations with UV detection at 238 nm. Chromatographic separation was performed on a Hypersil ODS C-18 column (250 × 4.6 mm, i.d., 5 μm particle size) with isocratic elution of the mobile phase containing a mixture of methanol–acetonitrile–phosphate buffer (0.02 M) (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:15, v/v/v) with a flow rate at 1.0 ml min−1 and pH 3.0. A 32 mixture design methodology was employed for the optimization and validation of the mobile phase components for a high degree of robustness i.e., peak area and retention time. The optimum mobile phase composition was a trade-off between the experimental design by graphical optimization of the technique using an overlay plot. Further, the method was validated as per the ICH guidelines using linearity, accuracy, precision, sensitivity, system suitability and selectivity as the parameters. The developed method was found to be simple, sensitive, and highly robust for routine analysis of risperidone.
:20:15, v/v/v) with a flow rate at 1.0 ml min−1 and pH 3.0. A 32 mixture design methodology was employed for the optimization and validation of the mobile phase components for a high degree of robustness i.e., peak area and retention time. The optimum mobile phase composition was a trade-off between the experimental design by graphical optimization of the technique using an overlay plot. Further, the method was validated as per the ICH guidelines using linearity, accuracy, precision, sensitivity, system suitability and selectivity as the parameters. The developed method was found to be simple, sensitive, and highly robust for routine analysis of risperidone.
1. Introduction
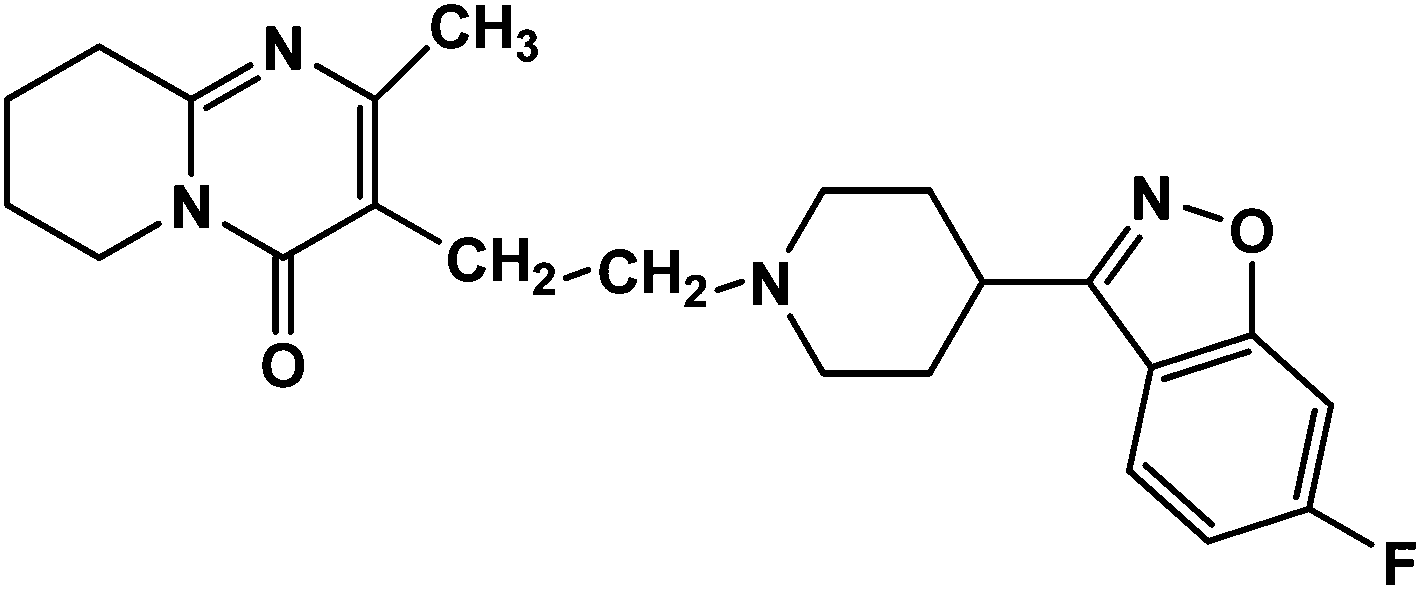
Risperidone (RIS), (Fig. 1) is designated chemically 3-[2-[4-(6-fluoro-1,2-benzisoxazol-3-yl)piperidin-1-yl]ethyl]-2-methyl-6,7,8,9-tetrahydro-4Hpyrido[1,2-a]pyrimidin-4-one with an empirical formula C23H27FN4O2 and molecular weight 410.49 g mol−1.1 This atypical antipsychotic agent is a benzisoxazole derivative and is reported to be an antagonist to dopamine D2, serotonin (5HT2), adrenergic, and histamine (H1) receptors.2 It is administered by an oral route for the treatment psychotic disorders particularly schizophrenia. HPLC methods for the determination of risperidone in bulk powder,3,4 in urine,5 in plasma and serum,6,7 and in pharmaceutical formulations8–10 have been reported.
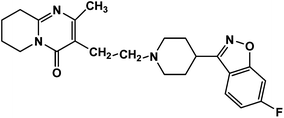 |
| | Fig. 1 Chemical structure of risperidone. | |
None of these methods have been found to be cost-effective and in terms of time economy, it takes several trial experiments to optimize the mobile phase composition. The present work aims to provide a new method to develop mobile phase composition using the most relevant experimental mixture design methodology. Rational practice of experimental mixture design in analytical method development helps to establish a robust mobile phase composition. The literature already reports the utility of experimental design methodology in LC analytical methods, such as the application of Box–Behnken design (BBD) for the optimization of mobile phase composition for drugs like amoxicillin trihydrate,11 lenalidomide,12 voriconazole,13 and fractional factorial design (FFD) and central composite design (CCD) in the screening and optimization of bioanalytical methods.14 Not only are the experimental designs restricted for process optimization, but they have also been widely explored in metabolite isolation,15 and impurity profiling using chromatographic procedures.16 Among the several types of experimental design methodologies (BBD, CCD, FFD), mixture design is the most appropriate methodology for experiments with at least two independent variables or factors having constraints and more than one response.17–19
As there is no report of an optimized analytical method using experimental design techniques, it was envisaged to develop and validate a simple and fast analytical method by HPLC to quantify risperidone by optimization of the mobile phase composition using Design Expert Software (Version 8.0.5; Stat-Ease Inc., Minneapolis, Minnesota) for purity evaluation and quantitative determination of RIS. This validation study is defined by the process by which it is established, in laboratory studies, and by the performance characteristics of the method, which should meet the requirements for the intended analytical application.20 This work describes the validation parameters stated by the ICH guidelines21 to achieve an analytical method with acceptable characteristics of suitability, reliability and feasibility.
2. Experimental
2.1 Standards and reagents
RIS (purity 99.8%) was gifted by Alkem Laboratories Ltd. (Mumbai, India) and was used as the working standard. The commercially available formulations of RIS (Respidon® tablet, Torrent, India) and an in-house developed proniosomal formulation were used for assay and quantitative determination. Potassium dihydrogen orthophosphate was of analytical grade. Methanol and acetonitrile were of HPLC grade and purchased from Merck Chemicals Ltd, (Mumbai, India). All solutions were prepared with reverse osmosis, HPLC grade double distilled water (Milipore, Mumbai, India).
2.2 Instrumentation
The HPLC system (Agilent Technologies, Waldbronn, Germany) equipped with a LC-LC-20AT pump, an SPD-20A variable wavelength detector, with Agilent LC Ezchrome software and a Rheodyne injection valve with a 20 μl loop was used for the development and evaluation of the method. The column used was a Hypersil ODS C-18 column (250 × 4.6 mm, i.d., 5 μm particle size). A mixture of methanol–acetonitrile–phosphate buffer (0.02 M) was used as the mobile phase with a flow rate of 1 ml min−1, pH 3.0 and UV detection at 238 nm. Prior to injection of the drug solution, the column was equilibrated for at least 30 min with the mobile phase flowing through the system. The data were acquired, stored and analyzed with Ezchrome software.
2.3 Method development as per mixture design
The mobile phase comprising a mixture of methanol, acetonitrile and phosphate buffer (0.02 M) was selected from preliminary studies. The concentration of mobile phase components viz. methanol (X1), acetonitrile (X2) and phosphate buffer (X3) were selected as independent variables, whereas peak area (Y1) and retention time (Y2) were chosen as response variables, parameters of robustness testing, to measure the elution performance of the mobile phase. For optimization of D-optimal mixture design a methodology was employed using Design-Expert 8.0 software (STATEASE Inc., USA). Table 1 shows the levels of mobile phase components (X1, X2 and X3), employed in the optimization at a fixed flow rate of 1 ml min−1 and pH 3.0. A total 19 experimental runs obtained from the design matrix were subjected to experiment in order to generate the response variables (Y1 and Y2), as summarized in Table 2. Statistical analysis was performed using ANOVA to compute the significant difference in the mobile phase compositions obtained from the design matrix. The response surface methodology (RSM) was employed to analyze the effect of independent variables on the responses. Further, the effect of interactions among the independent variables and responses were observed by analyzing the linear polynomial equations generated by the multiple linear regression analysis (MLRA) method. A linear model equation generated by the design is portrayed below as eqn (1). The equation indicates coefficients (β0 to β7) of various model terms including the two factor and three factor interaction terms. It helps to discern plausible interactions among the critical factors selected for the development of the HPLC method. Finally, the optimum mobile phase composition was traded-off using the numerical and graphical optimization procedure.22| | | Y = β0 + β1X1 + β2X2 + β3X3 + β4X1X2 + β5X2X3 + β6X2X3 + β7X1X2X3 | (1) |
Table 1 Selected levels of mobile phase as independent variables
| Coded factor |
Level |
X
1 (methanol %) |
X
2 (acetonitrile %) |
X
3 (PBS %) |
| −1 |
Low |
60 |
10 |
10 |
| 1 |
High |
70 |
20 |
20 |
Table 2 Experimental design matrix representing the runs of mobile phase composition and observed responses
| Experimental runs |
Methanol (X1) |
Acetonitrile (X2) |
PBS (X3) |
Peak area (Y1) |
R
t (Y2) |
| 1 |
65.00 |
20.00 |
15.00 |
301098 |
6.211 |
| 2 |
70.00 |
20.00 |
10.00 |
415045 |
6.163 |
| 3 |
65.00 |
20.00 |
15.00 |
310236 |
6.177 |
| 4 |
68.33 |
13.33 |
18.33 |
325622 |
6.193 |
| 5 |
70.00 |
10.00 |
20.00 |
398503 |
6.171 |
| 6 |
63.33 |
18.33 |
18.33 |
301275 |
6.173 |
| 7 |
66.67 |
16.67 |
16.67 |
336576 |
6.176 |
| 8 |
70.00 |
15.00 |
15.00 |
410236 |
6.166 |
| 9 |
60.00 |
20.00 |
20.00 |
309928 |
6.171 |
| 10 |
70.00 |
20.00 |
10.00 |
398850 |
6.198 |
| 11 |
68.33 |
18.33 |
13.33 |
334587 |
6.189 |
| 12 |
70.00 |
10.00 |
20.00 |
397903 |
6.168 |
| 13 |
60.00 |
20.00 |
20.00 |
309812 |
6.172 |
| 14 |
65.00 |
15.00 |
20.00 |
334577 |
6.161 |
| 15 |
65.00 |
20.00 |
15.00 |
321298 |
6.182 |
| 16 |
70.00 |
20.00 |
10.00 |
415045 |
6.185 |
| 17 |
65.00 |
20.00 |
15.00 |
329834 |
6.174 |
| 18 |
68.33 |
13.33 |
18.33 |
324533 |
6.185 |
| 19 |
70.00 |
10.00 |
20.00 |
398503 |
6.167 |
2.4 Preparation of the standard solution
A stock solution of pure drug (RIS) was prepared by dissolving 10 mg of RIS reference standard to a 10 ml volumetric flask and dissolved in methanol to make a solution of 1000 μg ml−1, which was further diluted to a lower concentration in the mobile phase as needed. From this stock solution a working standard solution of 100 μg ml−1 strength was prepared. From this solution dilutions of 1, 2, 4, 6, 8, and 10 μg ml−1 were made in 10 ml volumetric flasks with the mobile phase. The prepared dilutions were carefully aliquoted into a 20 μl syringe and injected into the system for preparation of the calibration curve.
2.5 Estimation of RIS in pharmaceutical dosage forms
The developed HPLC method was used for the determination of RIS in pharmaceutical formulations. Marketed formulations, Respidon® tablet were procured from the manufacturer and an in-house developed (proniosomal gel) formulation, were evaluated for the amount of RIS present in the formulation. Twenty tablets were weighed and powdered in a mortar and pestle. Accurately measured 100 mg of the powder equivalent to 10 mg of RIS was transferred into a 10 ml volumetric flask containing 10 ml of methanol and the contents of the flask were sonicated for 15 min, to ensure complete solubility of the drug. Finally, the volume was adjusted up to 100 ml with mobile phase. The resulting solution was thoroughly mixed and filtered through a 0.45 μm membrane filter. From this a 10 μg ml−1 concentration solution was prepared by suitable dilution. Each sample (20 μl) was analyzed in triplicate and the mean values of peak areas were determined and the drug content in the in-house developed proniosomal gel and marketed tablet were quantified using the regression equation obtained from the calibration curve.
Similarly 10 mg equivalent of risperidone gel was weighed and transferred to a 10 ml volumetric flask containing 10 ml of methanol and the contents of the flask were sonicated for 15 min, to ensure complete solubility of the drug. The same procedure was performed to prepare the 10 μg ml−1 solution.
3. Results and discussion
3.1 Method development
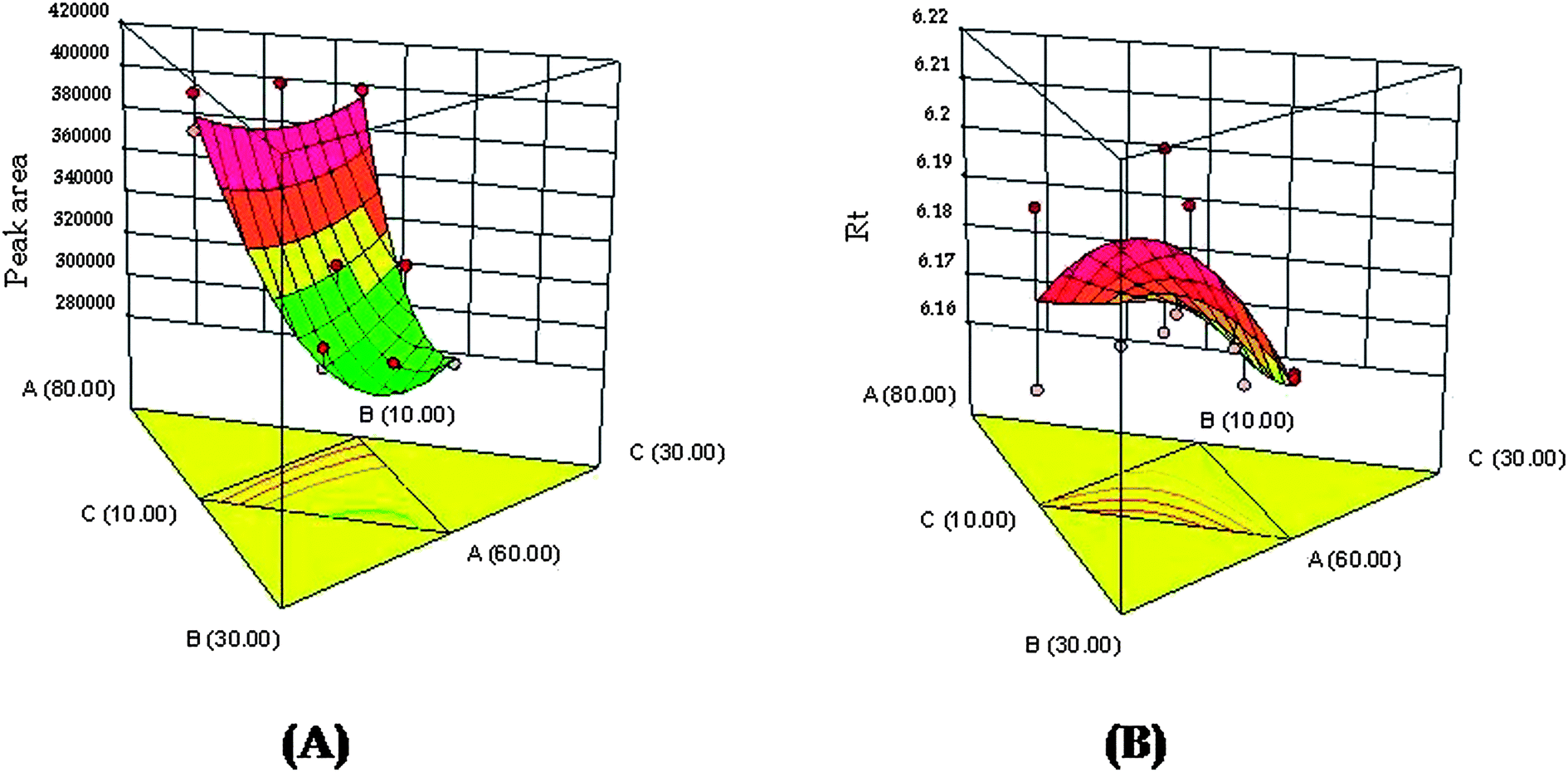
The suitability of mobile phase composition was identified by varying ratios of acetonitrile, phosphate buffer and methanol using the Design Expert software. Further, to identify the robust and optimized mobile phase composition, RSM was employed to analyze the effect of various independent variables on the selected responses. It helped in the identification of the positive and/or negative interactions amongst the independent variables. Fig. 2A and B portray the response surfaces and depict the relationship between the independent variables (X1, X2, X3) on robustness parameters i.e., peak area (Y1) and retention time (Y2). It has been observed that the response variable (Y1) is dependent on the concentration of acetonitrile and phosphate buffer as the influential factors (p-value 0.0014, significant). A curvilinear plot (Fig. 2A) was obtained indicating the proportionate increase in peak area with increasing concentration of acetonitrile and phosphate buffer. Similarly, the retention time (Y2) was found to be dependent on the concentration of acetonitrile and phosphate buffer as the influential factors in the mobile phase composition (p-value 0.0108, significant). A non-linear relationship (Fig. 2B), where the Rt was proportionately increased at initial levels and then a decreasing trend with increasing concentration of acetonitrile and phosphate buffer, was observed in the mobile phase mixture. However, methanol concentration was found to be less influential in affecting the responses parameters i.e., peak area and retention time. For a better understanding of the effect of interaction among the independent variables and responses, linear polynomial equations were studied. The linear polynomial equations generated from ANOVA are depicted below:
| Y1(peak area) = 8.071 + 0.005X1+ 2.580 + 0.005X2+ 0.005X3+ 0.005X1X2 − 9.228 + 0.005X1X3 + 0.005X2X3 |
| Y2(retention time) = 6.12X1 + 6.28X2 + 6.07X3 − 0.070X1X2 + 0.30X1X3 − 0.023X2X3 |
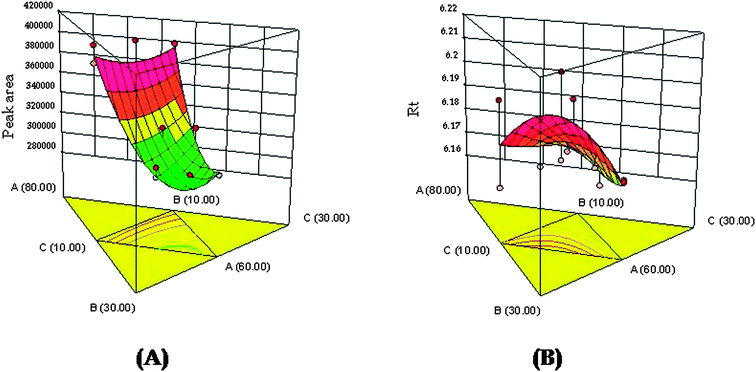 |
| | Fig. 2 (A) 3D response surface for the effect of independent variables (X1, X2, X3) on response variable Y1 (peak area); (B) 3D response surface for the effect of independent variables (X1, X2, X3) on response variable Y2 (retention time). | |
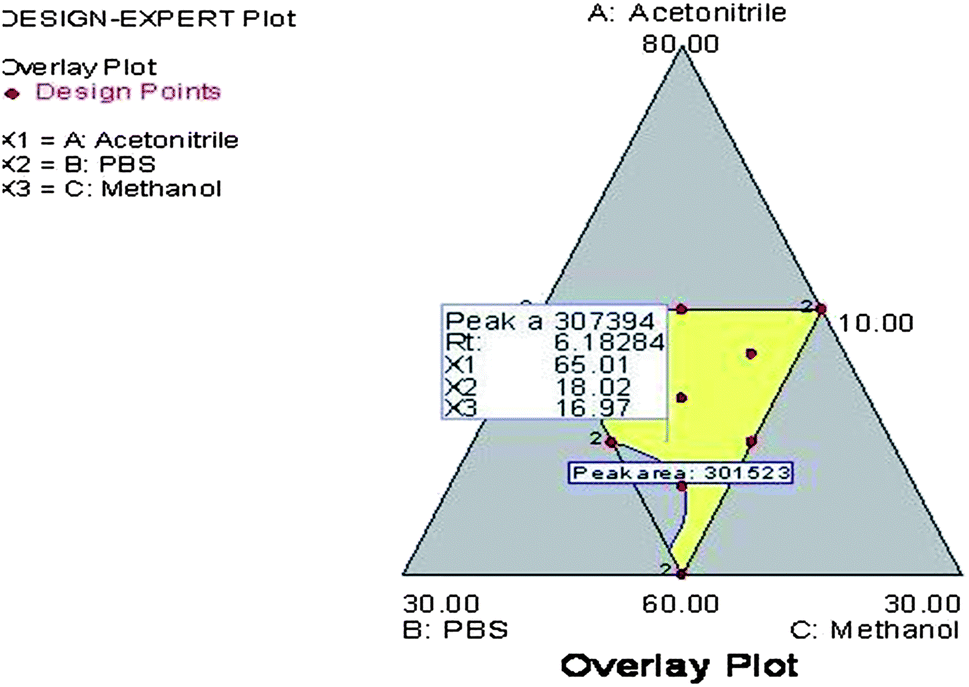
From the polynomial equations, it has been observed that all three independent variables with a positive sign indicated a positive effect of on both the observed responses. In case of response (Y1), all three interaction terms were positive which indicated the combined effect of independent variables on the peak area. However, for response (Y2), the two of the interaction terms were positive i.e., the effect of concentration of X1 and X2 and concentration of X2 and X3, whereas for the effect of concentration of X1 and X3 it was found to be negative. Further, the coefficients of independent variables (X1, X2 and X3) were found to be higher for response (Y2) compared to response (Y1). This indicated that the response (Y2) i.e., retention time is mostly influenced by all three independent variables chosen rather than response (Y1) i.e., peak area. The optimum mobile phase composition was selected from the design technique by a graphical optimization trade-off technique obtained from the overlay plot (Fig. 3). The optimized mobile phase composition contained methanol (65%), acetonitrile (20%) and phosphate buffer (15%). The chromatogram of RIS (Fig. 4) developed in the optimum mobile phase composition was obtained from experimental design. The optimized mobile phase produced good resolution in chromatographic elution. A retention time (Rt) of 6.16 min was observed with a reasonable peak area within the defined limits.
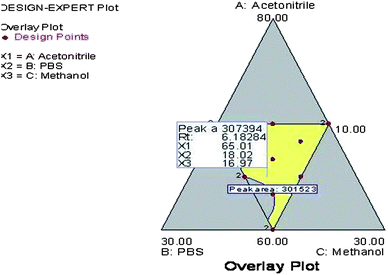 |
| | Fig. 3 Overlay plot of the experimental design indicating the optimum mobile phase composition. | |
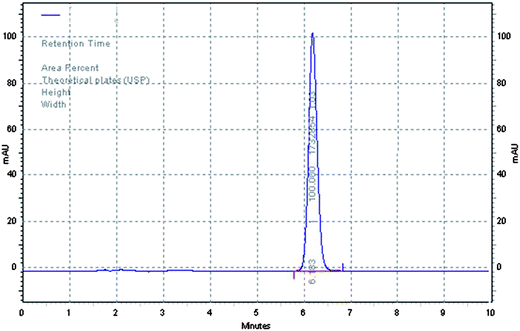 |
| | Fig. 4 Chromatograms of standard risperidone. | |
3.2 Validation
The described mobile phase composition was further subjected to validation for linearity, accuracy, precision, robustness, sensitivity, system suitability, and selectivity.
3.2.1 System suitability study.
The system suitability was assessed by six replicate analysis of 5 μg ml−1 concentration of RIS and SD (standard deviation), %RSD (relative standard deviation), SEM (standard error of mean) were determined for both peak area and retention time. The %RSD values of peak area and retention time were found to be within a 2% limit indicating the suitability of the method development (Table 4). The tailing factor and USP plate count were found to be within the limits.
3.2.2 Linearity and range.
The calibration curve constructed was evaluated by its correlation coefficient. The peak area of the drug was linear in the range of 1.0–10 μg ml−1. Standard deviations of the slope and intercept for the calibration curves generated on six different days were 0.002 and 0.012, respectively. The correlation coefficient (r2: 0.998) of the calibration curves obtained from the regression line demonstrates the excellent relationship between peak area and concentration. The statistical data of regression analysis are summarized in Table 3.
Table 3 Linear regression data for risperidone
| Statistical parameter |
HPLC |
| Concentration range (μg ml−1) |
1–10 |
| Regression equation |
215876X − 21435 |
| Correlation coefficient (r2) |
0.9986 |
| Limit of detection (LOD) (μg ml−1) |
0.17 |
| Limit of quantification (LOQ) (μg ml−1) |
0.55 |
3.2.3 Accuracy.
The accuracy study was performed by the standard addition method using three different solutions containing 3, 6 and 9 μg ml−1 of RIS. The obtained % recovery and %RSD values were within the range 99.66–100.79% and 0.44–0.98 satisfying the acceptance criteria for the study (Table 4).
Table 4 Summary of validation parameters for the proposed HPLC methoda
| Parameter |
HPLC |
|
RSD – relative standard deviation.
|
|
Accuracy
|
| Mean recovery % |
99.66–100.79% |
| RSD% |
0.44–0.98% |
| |
|
Precision (n = 3)
|
| Inter-day precision |
| RSD% |
0.58–1.56 |
| Inter-day precision |
| RSD% |
0.76–1.02 |
| |
|
System suitability
|
| RSD% |
0.22 |
| |
|
Selectivity
|
| Mean recovery % |
99.75 ± 0.28 |
| RSD% |
0.46 |
| Specificity |
Specific |
| |
|
Assay of drug
|
| RIS-tablet (Respidon) |
| Mean recovery % |
98.83 ± 0.78 |
| RSD% |
0.51 |
| RIS-proniosomal gel |
| Mean recovery % |
97.33 ± 0.62 |
| RSD% |
0.79 |
3.2.4 Precision.
Three different concentrations of RIS were assessed by assay at different time intervals in the same day (intra-day precision) and by repetition for three different days inter-day precision as per ICH guidelines. The developed method showed good precision and %RSD for intra-day and inter-day precision were in the range of 0.58–1.56% and 0.76–1.02%, which were less than 2% limit. The precision data are summarized in Table 4.
3.2.5 LOD and LOQ.
The limit of detection (LOD) and limit of quantitation (LOQ) were determined from slope (S) of the linearity plot and standard deviation of the response to the blank sample (σ), by formula LOD. The formulae used were LOD = 3.3 σ/S and LOQ = 10σ/S. The method observed LOD and LOQ value of 0.17 μg ml−1 and 0.55 μg ml−1 (Table 3) indicated that the developed method had higher sensitivity to the mobile phase composition.
3.2.6 Selectivity.
Selectivity of the proposed method was assessed by analyzing the mean percentage recovery and %RSD from five different replicates having concentrations of 10 μg ml−1 as given in Table 4. The recovery of the RIS was found to be 99.75% which shows that the method is highly selective.
3.2.7 Robustness.
The study of robustness was carried out to evaluate the influence of small but deliberate variations in the chromatographic conditions for the determination of the percentage risperidone. The factors chosen for this study were the flow rate (ml min−1), wavelength (nm) and pH of mobile phase. The influence of each of the variables was estimated as RIS % and retention time as a variables (Table 5), none of the parameters exceeded the limit and therefore showed that the study variable factors did not affect RIS %. Therefore, it can be concluded that the method is consistent vis-a-vis flow rate, wavelength, and pH.
Table 5 Chromatographic conditions investigated during robustness testing
| Variable |
Range investigated |
Risperidone% |
Retention time (min) |
Optimized value |
| Flow rate (ml min−1) |
0.8 |
99.89 |
6.11 |
1 |
| 1.0 |
100.23 |
6.12 |
| 1.2 |
100.02 |
6.14 |
| Wavelength (nm) |
235 |
99.91 |
6.10 |
238 |
| 238 |
100.86 |
6.14 |
| 241 |
99.76 |
6.13 |
| pH |
2.5 |
99.95 |
6.08 |
3 |
| 3.0 |
100.19 |
6.11 |
| 3.5 |
100.07 |
6.09 |
3.2.8 Stability.
Result obtained in the stability studies indicated that the samples were stable when kept at bench top for 12 h (short-term), and refrigerated at 4 °C for 30 days (long-term). The results of these stability studies (Table 6) indicated that the solutions were stable for the short-term as well as the long-term. The results showed that the assay of the sample does not decrease below a minimum percentage (98.56%), where the percent ratios are within the acceptance range of 90–110%.23
Table 6 Short-term and long-term stability of risperidonea
| Nominal concentration |
|
SD – standard deviation; RSD – relative standard deviation; SEM – standard error of mean.
|
|
Short-term stability
|
3
|
6
|
9
|
| Mean(n = 3) |
103.06 |
98.02 |
96.39 |
| SD |
3.73 |
3.57 |
5.79 |
| %RSD |
3.61 |
3.64 |
6.12 |
| SEM |
2.15 |
2.06 |
4.71 |
| |
|
Long-term stability
|
| Mean (n = 3) |
96.37 |
95.57 |
92.93 |
| SD |
2.89 |
5.05 |
4.61 |
| %RSD |
2.99 |
5.28 |
4.99 |
| SEM |
1.67 |
2.91 |
3.81 |
3.2.9 Analysis of the marketed tablet and gel.
The HPLC method developed is sensitive and specific for the quantitative determination of risperidone tablet and the in-house developed gel. The method has been validated for different parameters, hence it has been applied for the estimation of the drug in pharmaceutical dosage forms. Each sample was analyzed in triplicate after extracting the drug as mentioned in the sample preparation of the Experimental section. The amount of drug present (Respidon®, Torrent) was 98.83% and for the in-house developed gel was 97.23%. None of the ingredients interfered with the analyte peak when compared with the chromatogram. The spectrum of drug extracted from both formulations matched that of standard pure drug (Fig. 4) shows the purity peak. The results are given in Table 4 which shows the high percentage recoveries and low RSD (%) values.
4. Conclusion
The developed HPLC method for the estimation of risperidone in bulk drug and pharmaceutical formulations using methanol–acetonitrile–phosphate buffer (0.02 M) combination as the mobile phase showed good chromatographic separation. Application of a 32-factorial mixture design for the optimization of the mobile phase showed that a slight change in mobile phase concentration had a direct effect on peak area and retention time. Hence, special attention is required for strict monitoring of the aforementioned two factors during chromatographic testing. Upon validation system suitability, linearity, precision, accuracy, sensitivity, robustness and stability were proven to be convenient and effective for quality control as well as routine analysis of RIS in pharmaceutical dosage forms. The measured signal was shown to be precise, accurate, and linear over the concentration range tested (1.0–10.0 μg ml−1) with a good correlation coefficient of 0.9986, along with a retention time of 6.16 min making it economical due to lower solvent consumption. Thus, it could be concluded that the developed method is rapid, simple, and selective for routine analysis of RIS in bulk as well as in pharmaceutical formulations.
Acknowledgements
The author Syed Sarim Imam thanks the Indian Council of Medical Research (ICMR), India for providing financial assistance (File #45/24/2009-Nan/BMS) in the form of a senior research fellowship.
References
- L. Yaxin, T. Xing, C. Yue, Y. Zhang, Q. Feng and L. Xiumei, Eur. J. Pharm. Biopharm., 2008, 68, 422–429 CrossRef PubMed.
- S. T. Rajesh, T. J. Joseph, A. S. R. Murthy, D. V. Yadav, G. Subbaiah and R. Krishna, J. Pharm. Biomed. Anal., 2004, 36, 231–235 CrossRef PubMed.
- K. E. Goeringa, I. M. McIntyre and O. H. Drunmer, J. Anal. Toxicol., 2003, 27, 30–35 CrossRef.
- H. Thieme and H. Saches, Anal. Chim. Acta, 2003, 492, 171–186 CrossRef.
- A. Pelander, I. Ojanpera, S. Laks, I. Rasanen and E. Vuori, J. Anal. Chem., 2003, 75, 5710–5718 CAS.
- G. A. E. Balant, M. Gex Fabry, C. Genet and L. P. Balant, Ther. Drug Monit., 1999, 21, 105–115 CrossRef PubMed.
- H. Ming, S. Jian-zhong, J. U. Chen, J. Liu and H. Zhou, J. Zhejiang Univ., Sci., B, 2008, 9(2), 114–120 CrossRef PubMed.
- C. Bharathi, C. D. Krishnama, K. M. Saravana, S. Rama, V. K. Handa, D. Ramesh and A. Naidu, J. Pharm. Biomed. Anal., 2008, 46, 165–169 CrossRef CAS PubMed.
- G. Casamenti, R. Mandrioli, C. Sabbioni, F. Bugamelli, V. Volterra and M. A. Raggi, J. Liq. Chromatogr. Relat. Technol., 2000, 23, 1039–1059 CrossRef CAS PubMed.
- R. D. Zarna, R. D. Ronak, R. S. Navin, B. Jigar and P. Bhavna, Int. J. Anal. Chem., 124917 Search PubMed.
- S. Beg, K. Kohli, S. Swain and M. S. Hasnain, J. Liq. Chromatogr. Relat. Technol., 2012, 35, 393–406 CrossRef CAS.
- M. S. Hasnain, S. Rao, M. K. Singh, N. Vig, A. Gupta, A. Ansari, P. Sen, P. Joshi and S. Ali Ansari, Analyst, 2013, 138, 1581–1588 RSC.
- G. Srinubabu, C. A. I. Raju, N. Sarath and P. K. Kumar, Talanta, 2007, 71, 1424–1429 CrossRef CAS PubMed.
- L. Zivanovic, A. Licanski, M. Zecevic, B. Jocic and M. Kostic, J. Pharm. Biomed. Anal., 2008, 47(3), 575–585 CrossRef CAS PubMed.
- G. Iriarte, N. Ferreiros, I. Ibarrondo, R. M. Alonso, M. I. Maguregi, L. Gonzalez and R. M. J. Jimenez, Sep. Sci., 2006, 29(15), 2265–2283 CrossRef CAS.
- M. Medenica, B. Jancic, D. Ivanovic and A. Malenovic, J. Chromatogr., A, 2004, 1031, 243–248 CrossRef CAS PubMed.
- B. Cristiane, L. F. Maria, E. Roy, R. Jivaldo, L. B. Matos and M. Almeida, J. Braz. Chem. Soc., 2006, 17(3), 588–593 CrossRef PubMed.
- A. Alves and I. S. Scarminio, J. Sep. Sci., 2007, 30(3), 414–420 CrossRef.
- C. A. Anthony and D. T. Randall, J. Chromatogr., A, 2008, 1177, 1–11 CrossRef PubMed.
- Food and Drug Administration, 1987, Guidelines on General Principles of Process Validation, http:\\www.fda.gov/cdrh/ode/425.pdf.
-
ICH (Q2R1), Draft Guidelines on Validation of Analytical Procedures: Text and Methodology, IFPMA, Switzerland, 1995 Search PubMed.
- B. Singh, R. Kumar and N. Ahuja, Crit. Rev. Ther. Drug Carrier Syst., 2005, 22(1), 27–105 CrossRef CAS.
- R. Sistla, V. S. S. K. Tata, Y. V. Kashyap, D. Chandrasekar and P. V. Diwan, J. of Pharm. and Biomed. Anal., 2005, 39, 517–522 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.