Investigation into dissolved neutral sugars and their microbial conversion in natural and artificially produced dissolved organic matter using ion chromatography with pulsed amperometric detection and reversed-phase liquid chromatography-high resolution mass spectrometry†
Sara
Sandron
a,
Richard
Wilson
b,
Ruth
Larragy
a,
Margaret V.
McCaul
a,
Pavel N.
Nesterenko
c,
Brian
Kelleher
a and
Brett
Paull
*c
aIrish Separation Science Cluster, National Centre for Sensor Research, School of Chemical Sciences, Dublin City University, Dublin, Ireland
bCentral Science Laboratory, University of Tasmania, Hobart, Australia
cAustralian Centre for Research on Separation Sciences, School of Chemistry, University of Tasmania, Hobart, Australia. E-mail: brett.paull@utas.edu.au; Fax: +61 (03) 6226 2858; Tel: +61 (03) 6226 6680
First published on 23rd October 2013
Abstract
Ion-exchange chromatography with pulsed amperometric detection (IEC-PAD) was employed to investigate dissolved neutral sugars and their microbial conversion in both artificially prepared dissolved organic matter (ADOM), and naturally occurring dissolved organic matter (DOM) obtained from seawater and freshwater sources. The analysis of ADOM and naturally occurring DOM samples using IEC-PAD resulted in chromatograms suggesting very similar composition, each characterised by three early eluting peaks, the latter of which being a broad co-elution of multiple compounds. For naturally occurring DOM, several sugars, including arabinose, glucose, galactose, xylose and ribose, could also be identified. The three distinctive peaks obtained from IEC-PAD of ADOM were collected and further analysed by means of reversed-phase high performance liquid chromatography-high resolution mass spectrometry (RPLC-HRMS), the latter showing that glucose was totally consumed during microbial production of ADOM and potentially transformed into higher molecular weight materials and CO2.
Introduction
Dissolved organic matter (DOM) from seawater and freshwater can be described as a mixture of organic compounds with extremely diverse chemical–physical properties and molecular weights ranging from 300 to 7000 Da. DOM typically includes amino acids, organic acids, lipids, phosphonates, carboxyl-rich alicyclic molecules (CRAM), molecules derived from linear terpenoids (MDLT) and carbohydrates.1–4Sugars, in particular, represent one of the most characterised classes of molecules within the labile portion of DOM (LDOM), with LDOM itself being subject to transformation into refractory DOM (RDOM).2 RDOM cannot be metabolised or transformed and it has been estimated that this material can be up to 6000 years old and mainly characterised as CRAM.2–4
Within DOM, it has been reported that the majority of carbohydrates seem to be complex polymeric structures (i.e. heteropolysaccharides and acyl polysaccharides), with neutral monosaccharides, such as glucose, galactose, mannose (aldohexoses), arabinose, xylose, ribose (aldopentoses), fucose and rhamnose (deoxysugars), present as the carbohydrate building blocks of cellulose, hemi-celluloses and cellular metabolites.3–6 Glucose, together with phosphorous and nitrogen, represents a crucial nutrient for microbes, and thus an important factor within in the microbial-induced remineralisation of DOM. Within this process extracellular enzymes hydrolyse substrates so they can be transported across cell membranes.7 A fraction of the carbon is incorporated into biomass, another fraction is respired to CO2, and a fraction of the carbon initially taken up is typically excreted as RDOM.
Pioneering studies on labile compounds such as glucose within marine bacteria culture media have clearly indicated that microbial processes are able to alter the molecular composition of DOM, making it resistant to further degradation or transformation processes.5 In fact, Ogawa et al. were one of the first groups to assess the microbial origin of RDOM and to kinetically monitor glucose and glutamate microbial consumption in seawater using total organic carbon (TOC) analysis.5 In such a way, the relationship between the chemical characteristics of DOM and microbial activity within a natural water source was proven, as microbes could alter the molecular structure of DOM, transforming it into refractory materials.
Sugars within seawater and freshwater can be analysed using gas chromatography (GC). However, poorly volatile carbohydrates, such as terrestrially derived polymeric materials (i.e. cellulose) are not only difficult to detect, but can also cause extensive unresolved peaks along the GC chromatogram.8–10 On the other hand, ion-exchange chromatography (IEC), and in particular anion exchange chromatography with pulsed amperometric detection (PAD), is commonly used in carbohydrate analysis, even applied to complex matrixes such as seawater.11–16 The combination of IEC with the selective nature of PAD, as pioneered by Johnson and LaCourse in the early 1990s, is now a common approach to the separation and detection of alcohols, glycols, carbohydrates, alkanolamines and amino acids.17,18
The analysis of carbohydrates in seawater using IEC-PAD can prove challenging, due to the difficulty in determining low concentrations of sugars dissolved into such a high ionic strength sample matrix. Mopper et al.14 were one of the first groups to employ IEC-PAD to directly analyse seawater. The reported chromatograms show the presence of unresolved peaks which are possibly due to the complexity of the sample matrix. Furthermore, since matrix anions were found to interfere with the analysis, a cation exchange resin was used to desalt the sample. These resins, however, also remove acidic sugars, which are therefore undetectable. Wicks et al.15 tried different desalting resins such as amino, cyano, diol, aromatic sulfonic, quaternary amine and polyethyleneimine functionalised substrates, in order to optimise the desalting procedure. However, the average recovery of neutral, amino and acidic sugars ranged from 6.5 to 19.4%, with the extracted sample composition being dependent on the employed resin. In 2000, Kaiser et al.10 processed a seawater sample without any extraction technique and further improved the limit of detection to 4 nM. This approach targeted the analysis of amino sugars only, though it is not clear to what extent the sample preparation (3 M HCl for 5 h at 100 °C) affected the nature of the sample. Such issues, as highlighted by Cheng et al.,16 can lead to an overestimation of the sugar content within the sample. In order to overcome the analytical challenge arising from the saline nature of seawater, Engel et al. employed a desalting method based upon membrane dialysis prior to IEC-PAD.19 The authors were able to target neutral, amino and acidic sugars in a single chromatographic run, and highlighted that hydrolysis conditions needed to be optimised according to the different classes of sugars.
Despite being extensively used in the separation of sugars within seawater and freshwater, IEC-PAD has never been employed to fractionate DOM itself, in order to better understand the bulk compositional analogies of different sources of DOM, or indeed artificially prepared DOM (ADOM). This approach can simultaneously be applied to help elucidate specific components of extracted DOM, notably the presence or absence of various sugars, and also provide a platform to investigate how microbes contribute to the composition of DOM. Thus herein, IEC-PAD is employed as an analytical method to help characterise DOM, and also to study the role of simple sugars in microbial processing. An ADOM sample produced by the microbial breakdown of glucose, was compared with naturally occurring DOM extracted from both seawater and freshwater. IEC-PAD was also used to fractionate the ADOM, and those fractions subsequently analysed by means of reversed-phase HPLC with high resolution mass spectrometry (RPLC-HRMS).
Experimental
Reagents
The HCl and MeOH used for DOM extractions were purchased from Sigma-Aldrich (Sigma Aldrich, Dublin, Ireland). In the preparation of ADOM, glucose, Na2HPO4 and NH4Cl were also obtained from Sigma-Aldrich (Sigma Aldrich, Dublin, Ireland). During IEC-PAD analysis, deionised water was obtained from a Milli-Q water purification system (Millipore, Watford, U.K.). For RPLC-HRMS analysis, MS grade formic acid, MeOH and deionised water were all purchased from Sigma Aldrich (Sigma Aldrich, Sydney, Australia).DOM extraction
Seawater and freshwater samples were collected in August 2011 from the Irish Sea coastline in Bray, (53°12′04″N, 6°06′41″W) and from the source of the River Shannon, Ireland (54°14′05″N, 7°55′08″W, collected in May 2011), respectively. The 10 and 60 m Irish Sea samples (54°10′08″N, 5°45′47″W) were collected on a research cruise (CV10_028) in June 2010. The samples were extracted as described in the method proposed by Dittmar et al.20 in 2008, which has also been used in other analytical studies aiming to identify DOM components.3 Briefly, 10 L of each water sample was filtered through 0.20 µm Nucleopore polycarbonate GF/F filters (Fisher Scientific, Dublin, Ireland) and acidified drop by drop to pH 2.0 with concentrated HCl. This step was performed immediately after sampling in order to preserve the sample. The water was then passed through styrene divinylbenzene (PPL) Bond Elut solid phase extraction (SPE) cartridges (1 gr, 60 mL) (Varian, Stockport, UK), with the DOM eluted within one cartridge volume of methanol, and subsequently concentrated by high vacuum evaporation. The obtained DOM (∼10 mg for every 10 L of seawater or freshwater), the appearance of which is a fluffy brownish powder, was stored at −80 °C prior to any analysis in order to preserve the sample from any degradation process or microbial activity. The DOM was then weighed (0.03 mg) and dissolved into 300 µL Milli-Q water, providing a 0.1 mg mL−1 solution. The prepared sample was sonicated for 20 minutes to fully disperse the material in solution, and then vortexed for 15 seconds to obtain a homogeneous solution (pale yellow colour). All solutions were filtered through Swinney syringe filters (13 mm diameter and 0.22 µm pore size) (Pall Corporation, Ann Arbor, Michigan, USA) to remove any non-dissolved material. The solution was then transferred into an amber vial to avoid UV degradation, prior to analysis.ADOM preparation
The experimental conditions proposed by Ogawa et al.5 were applied to prepare a seawater microbial culture (Bray, 53°12′04″N, 6°06′41″W) and a blank culture, constituted of Milli-Q water instead of seawater. All the employed glassware had been previously sterilised by heating in an autoclave. Each culture media was prepared in duplicate, consisting of 2 L of Bray seawater (or Milli-Q water in the case of the blank), 4 g L−1 of glucose as the carbon source, 6 g L−1 Na2HPO4 as the phosphorous source, and 1 g L−1 NH4CI as the nitrogen source. After two weeks incubation, both blank, and the resultant ADOM from the above cultures, were also extracted according to the method from Dittmar et al.20 In the case of ADOM, 8 mg of sample were obtained from the 2 L of seawater.IEC-PAD
The IEC-PAD was performed on a Dionex 500 system (Dionex Corp., Sunnyvale, CA) equipped with a Dionex GP50 quaternary pump, an ED40 electrochemical detector module, a Dionex ED40 compartment with a KOH eluent generator and a column compartment (Dionex LC30). The separations were performed on a CarboPac-PA1 column (4 × 250 mm, particle size 10 µm) equipped with a CarboPac-PA1 guard column, with dimensions 4 × 50 mm and particle size 10 µm (Dionex Corp., Sunnyvale, CA). Separations were carried out in gradient mode (50 to 100 mM KOH over 50 minutes). The flow rate was 1.0 mL min−1 and the injected volume 25 µL.PAD was performed with an Au working electrode (ca. 0.15 cm2), and an Ag/AgCl reference electrode (Dionex Corp., Sunnyvale, CA). The PAD reference electrode parameters were set according to Dionex optimised parameters for the analysis of carbohydrates.
RPLC-HRMS
Three characteristic peaks, which were fraction collected from the IEC-PAD separation, were concentrated by compressed air flow to a final volume of 150 µL and further analysed through RPLC-HRMS. The column used was a Nova Pak C18 (3.9 × 150 mm, 4 µm pore size, Waters, Milford, USA), on a Waters 2690 Alliance system (Waters, Milford, USA) connected to a Thermo Finnegan LTQ Orbitrap (Thermo Scientific, Scoresby, Australia). The flow rate was 0.8 mL min−1 and the gradient ran from 10% to 100% MeOH with 0.1% formic acid in 20 minutes. The mass spectra were analysed in negative mode (capillary 4500 V, collision energy 20 eV, nebuliser 10.0 psi, dry gas 5.00 L min−1, dry temperature 350 °C).Results and discussion
In previous investigations, a number of chromatographic methods have been considered as a means to fractionate the different classes of compounds within DOM. Since DOM is characteristically high in carboxylic and hydroxyl functionalities, both normal phase (NP) and RP-LC have been applied,21,22 but in both cases the complexity of the sample appeared to result in large unretained fractions, followed by very broad unresolved peaks (humps), and unavoidable irreversible absorption and sample carryover issues. More recently, Woods et al. also applied hydrophillic interaction liquid chromatography (HILIC) in combination with multi-dimensional nuclear magnetic resonance spectrometry (NMR) and MS to such samples.23,24 However, once again DOM provided a large unresolved peak, eluting across the majority of the chromatogram. For this reason HILIC, although suited for the separation of polar aliphatic compounds, was not employed within the current investigation.Herein IEC-PAD was considered a promising approach to the analysis of DOM for carbohydrate profiles. Firstly, the separation of a standard mixture of 10 sugars commonly found in DOM2,5,10,11 (namely fructose, mannitol, arabinose, glucose, galactose, xylose, sorbitol, sucrose, ribose and maltose) using IEC-PAD was carried out, using a 50 to 100 mM KOH gradient to obtain the identifying retention factors for each sugar. Standards were prepared at a concentration of 50 µg mL−1. The column employed was a PA1, which contains a PS-DVB nonporous resin bearing anion exchange functionalities (alkyl quaternary ammonium groups).25 The obtained retention data, including retention time, retention factors (k), and peak efficiencies are provided as ESI within Table S1.†
Separation of ADOM
As mentioned above, glucose represents a crucial nutrient for microbes and is a known component of LDOM. For this reason, it was decided to study the behaviour of glucose within a bacterial inocula, based upon the earlier approach of Ogawa et al.,5 but here using the IEC-PAD method to both separate and fractionate the resultant ADOM materials.Two litres of seawater from Bray were placed in a conical flask and nutrients added in order to stimulate the activity of microbes naturally present within seawater. After two weeks, the ADOM formed was extracted using the SPE procedure detailed above20 and a brown powder obtained, equalling approximately 8 mg of material. Using this extraction method, high concentrations of matrix inorganic ions were eliminated, which could otherwise affect the separation. In parallel, the same procedure was adopted for two litres of Milli-Q water, in order to obtain a blank sample, where no microbes were present to metabolise the nutrients. This time a white powder, indicating the presence of unconsumed glucose, was obtained following the extraction procedure. A 0.1 mg mL−1 solution of each sample was prepared in water and analysed using IEC-PAD.
Fig. 1 shows the chromatogram obtained for the ADOM sample (a), inset with that obtained for the glucose blank (b). The latter shows only a very broad and irregular peak, resulting from the high (excess) concentration of unconsumed glucose. In contrast the ADOM chromatogram contains no glucose, indicating that it has been consumed and converted to microbial biomass and/or CO2.
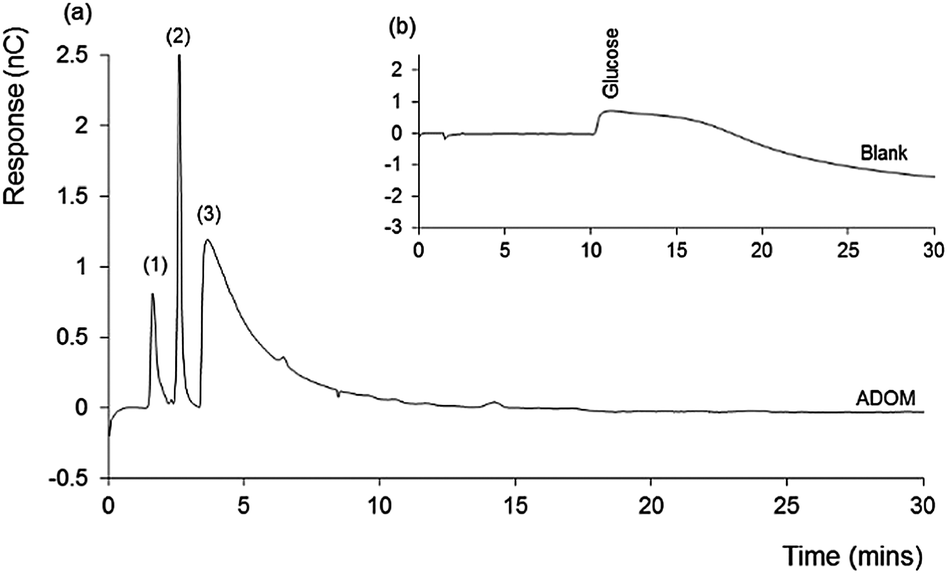 | ||
| Fig. 1 Comparison between IEC-PAD chromatograms for (a) seawater based ADOM showing three main peaks (1 to 3) and (b) the Milli-Q water blank. Flow rate = 1.0 mL min−1, with a 50 to 100 mM KOH gradient, and injected sample volume of 25 µL for a 0.1 mg mL−1 solution. | ||
A feature of the ADOM chromatogram is the presence of three separated peaks. The first two peaks were relatively sharp, with the retention time of the second peak, suggesting the presence of fructose. These peaks were followed by a significant tailed peak, eluting at approximately 6 minutes (from minute 4 to 10). The latter, by the nature of the detection method involved, must contain functional redox active groups, and the peak shape suggests a number of coeluting species.
Natural seawater and freshwater DOM
Natural DOM from seawater and freshwater was also analysed by IEC-PAD to compare profiles, both with each other, and with that obtained for the ADOM. Average absolute recovery for the natural DOM samples using the extraction procedure applied herein,20 has been found to be ∼10 mg of DOM per 10 L of water sample, notably lower than that observed for the glucose incubated ADOM sample.Fig. 2 shows the chromatograms obtained for both the (a) coastal seawater and (b) freshwater DOM samples, both of which are overlaid with a standard chromatogram for selected sugars. The chromatograms share a similar profile, with three significant peaks, the first two of which are both sharp and well defined, followed by a broad and tailing peak, dominating the two chromatograms. The similarity between these chromatograms supports the accepted theory that suggests the major components within DOM are consistent for both marine and freshwater derived samples.1,2
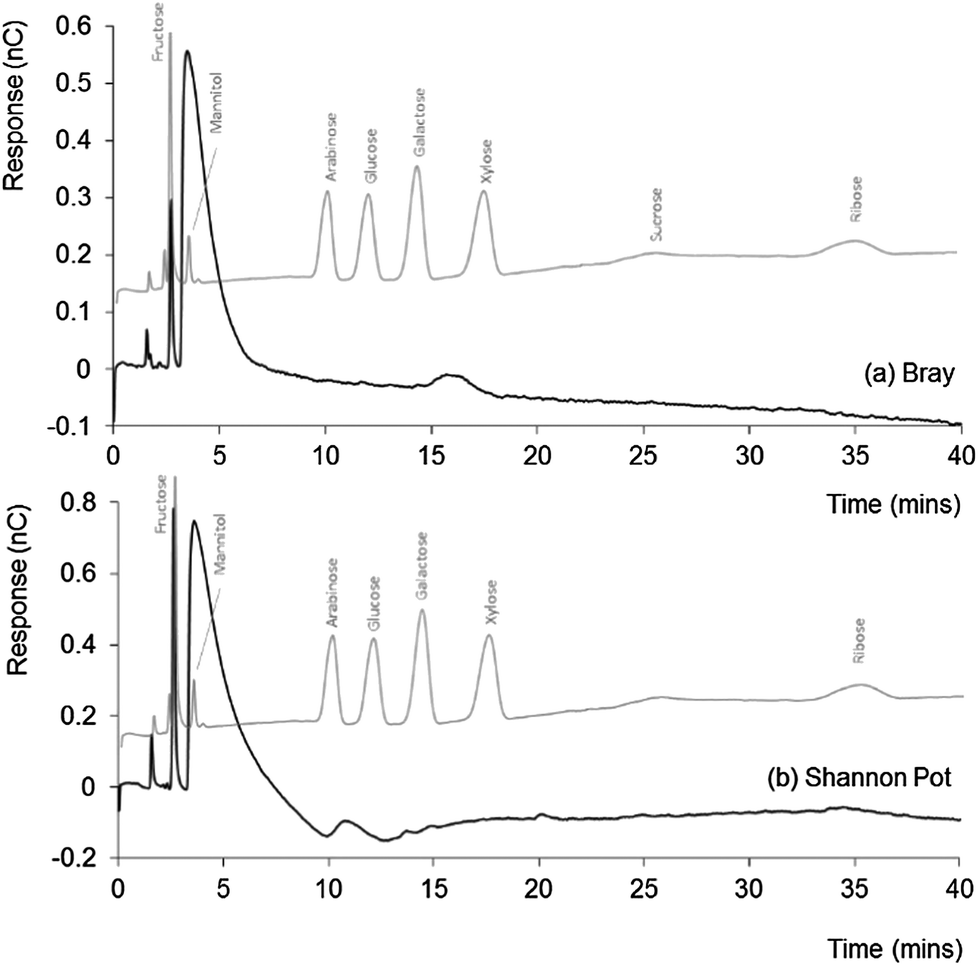 | ||
| Fig. 2 IEC-PAD chromatograms obtained for (a) Bray (seawater) and (b) Shannon Pot (freshwater) samples, overlaid with standard chromatograms for selected sugars. Other conditions as Fig. 1. | ||
The chromatograms obtained for the two DOM samples (and indeed the ADOM sample) are also significant, as to-date the fractionation of DOM into resolved or partially resolved peaks, which can easily be collected and characterised by a second off-line chromatographic dimension, has proven elusive. Size exclusion chromatography (SEC) is commonly employed as the chromatographic tool to fractionate DOM, however, DOM separations achieved to-date using SEC have been disappointing, with eluent conditions (i.e. phosphate buffers) often not allowing further characterisation by means of MS.26–28
All three early peaks appeared larger within the freshwater sample, which also showed a series of smaller peaks distinguishable from the baseline noise but too small to identify. Taking aside the early peak, which shares elution time with fructose, there was little to indicate the presence of other sugars within either the coastal seawater or freshwater DOM samples, but a small peak eluting at a similar time to that of galactose within the freshwater (Shannon Pot) sample. Since the Bray sample was collected from the shoreline and the Shannon Pot is an inland pool surrounded by agricultural land and woodland, it is reasonable to think that these water sources are probably heavily affected by materials of terrestrial origin and are characterised by a high microbial activity. Hence, the presence of galactose would not be unexpected here, as it is a constituent and the hydrolysis product of galactan, a polymer present in hemicellulose, the main constituent of plant cell walls.29
As emphasised by Hedges et al., low sugar concentrations are usually related to an enhanced presence of humic-like substances, as products of microbially degraded leaf materials solubilised in water.30 Therefore, the general absence of detectable sugars, in particular glucose, is perhaps not surprising, as this nutrient is readily consumed in such samples, not only by microbes naturally occurring in seawater and freshwater, but also by species such as algae, which commonly accumulate on the shore line.31
Irish Sea DOM from the depth of 10 m and 60 m
In order to better understand the significance of the above findings, two further DOM samples were collected from the Irish Sea, at a site well removed from the coast, and sampled from the same seawater column at two depths (10 and 60 m). The chromatograms obtained for these samples are shown in Fig. 3.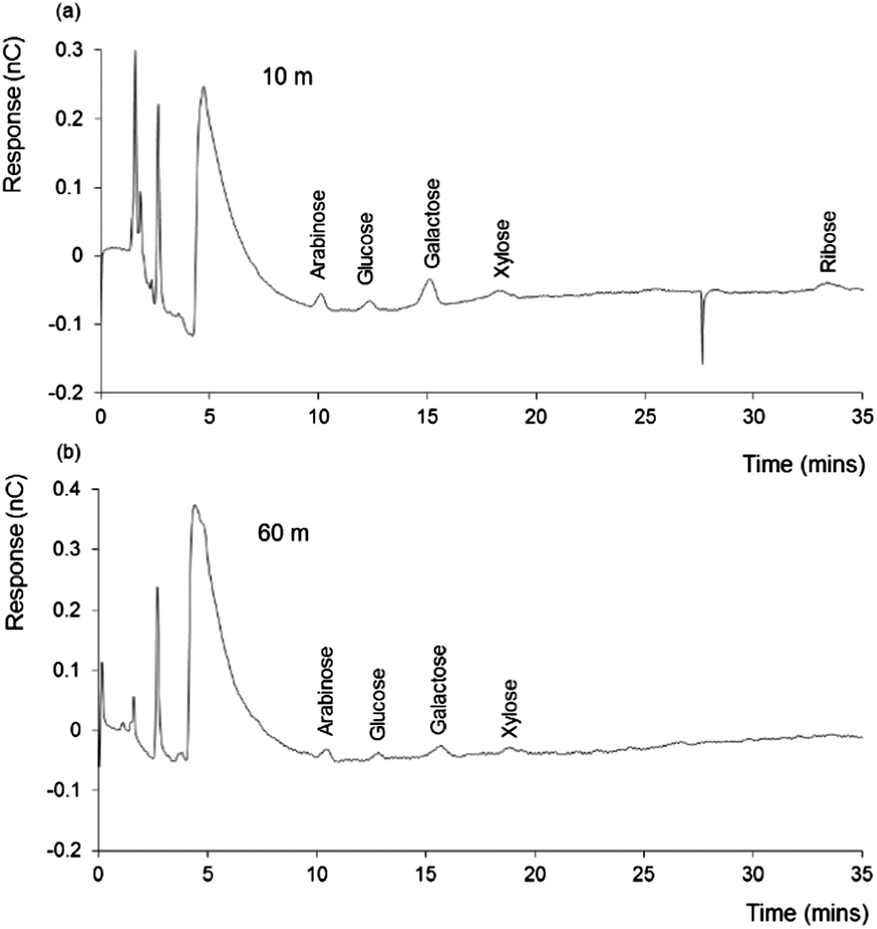 | ||
| Fig. 3 IEC-PAD chromatograms obtained for 10 m and 60 m depth Irish Sea DOM samples. Other conditions as Fig. 1. | ||
Once again, a distinctive series of peaks is evident within both samples, although noticeably smaller than those seen with either the ADOM sample, or the freshwater and coastal seawater DOM samples. However, from both samples, peaks corresponding to the retention times of arabinose, glucose, galactose, xylose and ribose (for 10 m sample only), were clearly present. Each of these peaks appeared greater in the 10 m sample, particularly galactose, which may suggest a higher influence from plant derived materials (i.e. hemicellulose), which is reasonable for a sample collected at the top of the water column. Xylose and arabinose also suggest a terrestrial influence on these sample, as these sugars can also be found as hemicellulose building blocks.
Ribose, which appears to be present within the 10 m sample at a retention time ∼35 minutes (Fig. 3(a)), can be related to the presence of RNA and other derivatives (i.e. ATP). As described by Cowie et al.,32 the distribution of microbially derived aldoses are often related to the ribose content within seawater. According to this study, ribose concentration is directly proportional to the concentration of such aldoses.
Microbes within seawater are prone to phage attacks, which can cause the release of cellular materials,7 including RNA etc. Such processes represent a potential source of ribose within this water source. The chromatograms of both 10 m and 60 m samples (Fig. 2 and 3) indicate the presence of various neutral sugars, whereas these peaks are missing from the coastal and freshwater samples. As suggested earlier, a possible reason for these differences is lower microbial activity within the off-shore samples.
To confirm the peak at 11.2 minutes as glucose, a seawater derived DOM sample was spiked with increasing concentrations of glucose: 3.75 µg mL−1, 7.5 µg mL−1 and 15.0 µg mL−1. The results of this standard addition can be seen as Fig. 4. As shown the addition of glucose, even at excess concentration, did not affect the chromatography of the three early eluting peaks, and further confirmed that all the glucose present during the incubation of the ADOM was consumed. Considering the peak height data for the samples at a concentration of 3.75 µg mL−1 (0.3871 nC) and 7.5 µg mL−1 (0.9132 nC), and 0.03 nC for the unspiked DOM sample, a concentration of glucose of ∼3.6 µM was calculated for the extracted DOM powder, which is in broad agreement with values reported in previous studies.16,18,33
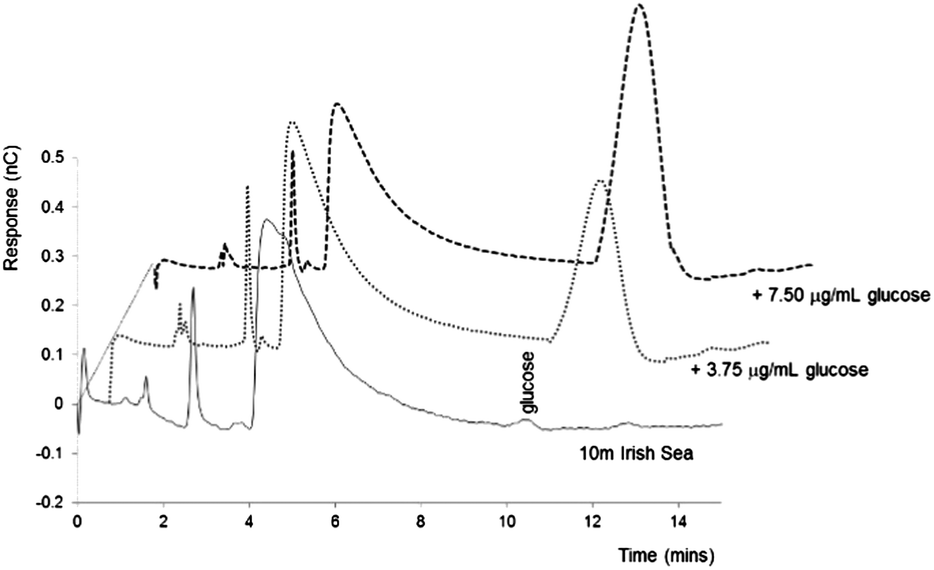 | ||
| Fig. 4 IEC-PAD chromatograms obtained for 10 m Irish Sea DOM sample spiked with increasing concentrations of glucose. Other conditions as Fig. 1. | ||
ADOM analysis using RPLC-HRMS
As the chromatograms from the naturally occurring DOM samples were similar to those obtained for the ADOM sample (Fig. 1), the three dominant peaks eluting from the ADOM sample (retention times: peak (1) 1.6 min, peak (2) 3.6 min, and peak (3) eluting from 4 to 9.5 minutes) were collected, concentrated by evaporation and further analysed using RPLC-HRMS in negative mode (negative ionisation mode was chosen because of the high concentration of KOH present in the collected peaks after concentration). Each of the three samples were analysed using RPLC-HRMS in triplicate to assess the intra-sample variability. For each collected IEC-PAD peak, 153 to 180 distinctive features (signals three times more intense than the baseline) were identified by HRMS. To identify differentially-abundant features between the IEC-PAD fractions, XCMS on-line was used.34,35 This software uses a run alignment algorithm to match and compare the intensity of LC-MS features between sample runs.The mirror plots (Fig. 5) display ions in which the intensities are altered between two different samples according to statistical thresholds set by the user (in this analysis a ≤0.005 p-value). Features that are down-regulated (represented in red colour) are marked as circles on the bottom of the plot, whereas features that are up-regulated (in green colour) are represented as circles on the top. The size of each circle corresponds to the logarithm of the fold change of the feature: larger circles correspond to peaks with greater fold differences. The colour intensity represents the p-value: the brighter the circles, the lower the p-value.35,36 Furthermore, if the features match those present on Metlin databank, the circles are outlined in black.34
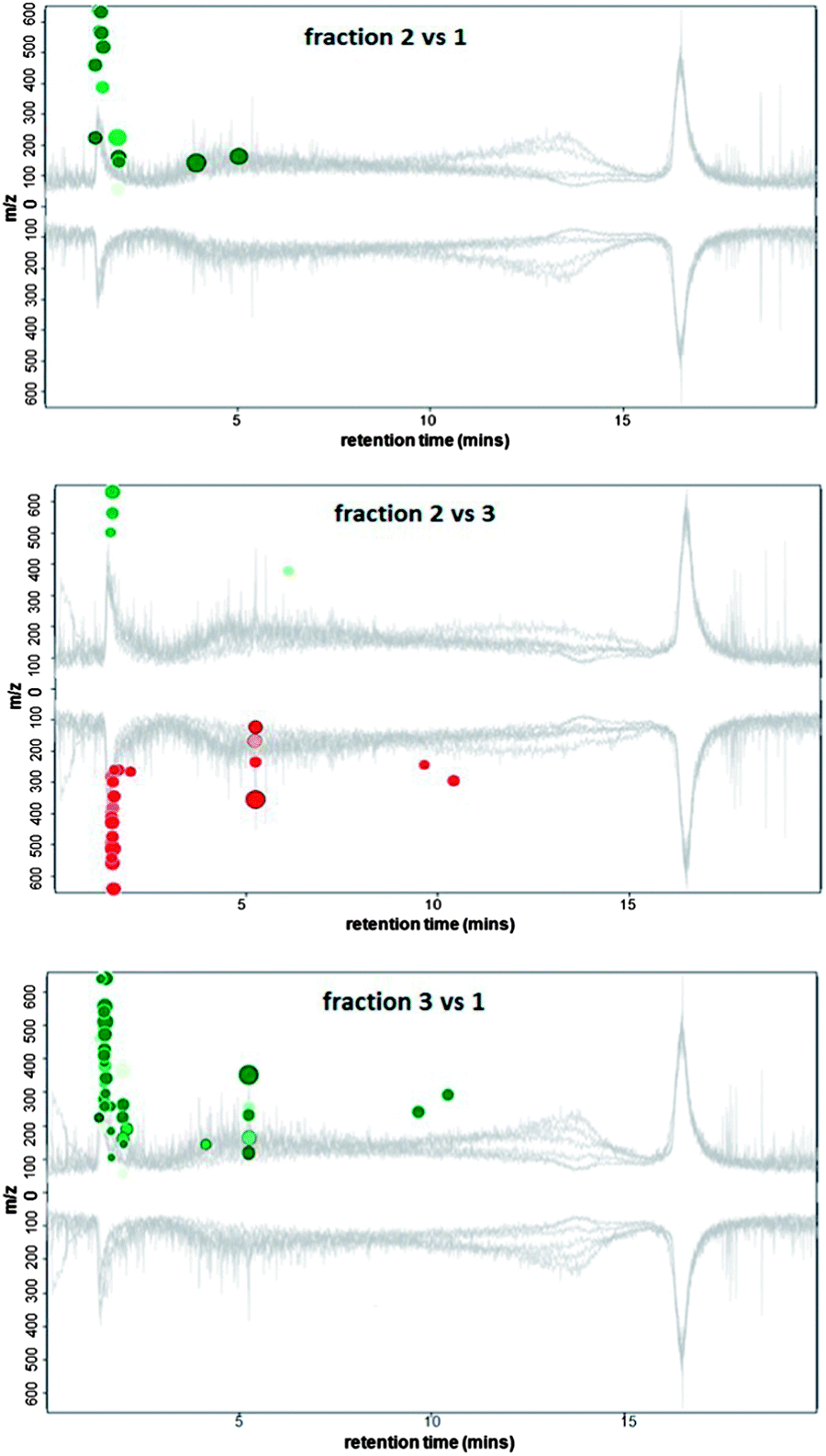 | ||
| Fig. 5 Mirror plots representing the comparison between the three collected peaks and the markings for their unique features. Flow rate 0.8 mL min−1, capillary 4500 V, collision energy 20 eV, nebuliser 10.0 psi, dry gas 5.00 L min−1. | ||
From the plots reporting the comparison between the three chromatographic peaks (Fig. 5), it can be seen that all the chromatograms appear to be similar. This is as expected, as not only does each fraction contain a great deal of co-eluting material (much of which is not detected using PAD), but also due to the fact that most of the sample data is hidden by the baseline noise resulting from the solvent contribution. Thus sample chromatographic features have to be obtained from subtracting any interference from contaminants present along the chromatogram (Fig. S1 in ESI,† illustrates this with the most intense ions at m/z 91.0032 and 112.9850 being due to solvents, which are present from the start to the end of the run).
The most distinctive features detected within each of the three collected peaks were found at the beginning of the RP-LC chromatogram (Fig. 5 and ES1†). Therefore, it can be assumed such compounds are relatively polar and thus poorly retained on the C18 stationary phase.
Amongst all the most dominant features which differentiate the three peaks collected from IEC, just 5 matched Metlin databank (Table 1).
| Peak | RT (mins) | m/z | Metlin match |
|---|---|---|---|
| a Flow rate 0.8 mL min−1, capillary 4500 V, collision energy 20 eV, nebuliser 10.0 psi, dry gas 5.00 L min−1, dry temperature 350 °C. | |||
| 2 | 1.3 | 225.0606 | Hexahydroxyheptanoic acid |
| 2 | 3.9 | 145.0500 | Adipic acid |
| 3 | 2.1 | 191.0190 | Citric acid |
| 3 | 4.1 | 145.0500 | Adipic acid |
| 3 | 5.2 | 121.0289 | Benzoic acid |
Although the identity of the compounds in Table 1 would require further confirmation via the injection of standards, the match for hexahydroxyheptanoic acid and adipic acid (present both from peaks 2 and 3, see Table 1) support the assumption that the compounds eluting as these poorly retained peaks are alkylic materials, with polar functional groups. Such compounds are not naturally occurring and may represent contamination arising from the sampling procedures. The same considerations may be applied to benzoic acid, however, this compound can also be naturally found in seawater as related to lignin-like materials, further emphasising the terrestrial origin of the sample.36,37
In order to assess the presence of contaminants from the sample extraction and preparation, investigations with protocol blanks did not reveal the presence of the aforementioned anthropologically derived species. The match for citric acid is quite interesting as this molecule is naturally occurring, therefore further investigation needs to be carried out to confirm its identity and source within DOM. Its presence may be related to the citric acid cycle, which covers a key role in cellular metabolism.
According to the m/z detected within the three peaks collected from the IEC separation, all the compounds are within a low molecular weight range (none of the m/z exceeds 530). The peak characterised by the highest number of distinctive features was predictably peak 3 (Fig. 5), the most representative of which are listed within Table 2.
| # | m/z | RT (mins) |
|---|---|---|
| a Flow rate 0.8 mL min−1, capillary 4500 V, collision energy 20 eV, nebuliser 10.0 psi, dry gas 5.00 L min−1, dry temperature 350 °C. | ||
| 1 | 451.1201 | 1.35 |
| 2 | 225.0606 | 1.36 |
| 3 | 461.149 | 1.36 |
| 4 | 529.1357 | 1.38 |
| 5 | 89.0242 | 1.67 |
| 6 | 128.0347 | 1.88 |
| 7 | 191.019 | 2.05 |
| 8 | 145.05 | 4.12 |
| 9 | 165.0185 | 5.23 |
| 10 | 166.0219 | 5.25 |
| 11 | 121.0289 | 5.25 |
| 12 | 233.0054 | 5.25 |
| 13 | 353.026 | 5.25 |
| 14 | 173.081 | 5.71 |
| 15 | 187.0967 | 6.57 |
| 16 | 242.175 | 9.66 |
| 17 | 91.0035 | 12.01 |
| 18 | 311.1674 | 14.38 |
| 19 | 339.1986 | 14.48 |
| 20 | 325.183 | 15.1 |
| 21 | 339.1987 | 16.08 |
| 22 | 311.1674 | 16.11 |
Given the m/z range of the compounds detected from RPLC-HRMS, which represent compounds with a higher molecular weight if compared to the starting material (glucose) it is reasonable to suppose that the ADOM sample is characterised by two possible by-products: one derived from the degradation of microbial cells and a second one indicating the presence of refractory and more complex materials, presumably from the consumption and transformation of glucose from the microbes naturally present in seawater.
Conclusions
For the first time, IEC-PAD was employed as an alternative approach to the separation and fractionation of DOM, providing three distinct fractions, which, in the case of ADOM, were then characterised using RPLC-HRMS. Thus the approach provided a platform to further investigate the nature of ADOM and naturally occurring DOM from freshwater, coastal and off-shore seawater sources.Interestingly, all the analysed samples from different water sources or depths, and the ADOM sample, show very similar IEC-PAD chromatograms, which could support the conclusion that the bulk of the material separated and detected using IEC-PAD share analogous classes of compounds, together with sugars that can assess the terrestrial influence of the sample. This represents a further confirmation of the findings from Ogawa et al.,5 who firstly assessed the microbial origin of the refractory portion of DOM. Further studies are currently underway to improve upon the fractionation methods developed herein, coupled directly with various modes of both GC- and LC-MS, for more comprehensive fractional characterisation.
Acknowledgements
The authors would like to thank Science Foundation Ireland (Grant Number 08/SRC/B1412) for research funding under the Strategic Research Cluster programme, together with the FP7-PEOPLE-2010-IRSES Marie Curie Action “International Research staff Exchange Scheme” for funding of this research collaboration within the “Materials and Advanced Sensor Knowledge Exchange (MASK)” grant. B. Paull would also like to acknowledge the Royal Society of Chemistry ‘Journal Grants for International Authors’ scheme for supporting this collaborative research.Notes and references
- K. Mopper, A. Stubbins, J. D. Ritchie, H. M. Bialk and P. G. Hatcher, Chem. Rev., 2007, 107, 419–442 CrossRef CAS PubMed.
- D. A. Hansell and C. A. Carlson, Biogeochemistry of Marine Dissolved Organic Matter, Elsevier, 2002 Search PubMed.
- N. Hertkorn, M. Harir, B. P. Koch, B. Michalke, P. Grill and P. Schmitt-Kopplin, Biogeosci. Discuss., 2012, 9, 745–833 CrossRef.
- N. Hertkorn, R. Benner, M. Frommberger, P. Schmitt-Kopplin, M. Witt, K. Kaiser, A. Kettrup and J. I. Hedges, Geochim. Cosmochim. Acta, 2006, 70, 2990–3010 CrossRef CAS PubMed.
- H. Ogawa, Y. Amagai, I. Koike, K. Kaiser and R. Benner, Science, 2001, 292, 917–920 CrossRef CAS PubMed.
- B. Lam, A. Baer, M. Alaee, B. Lefebvre, A. Moser, A. Williams and A. J. Simpson, Environ. Sci. Technol., 2007, 41, 8240–8247 CrossRef CAS.
- N. Jiao, F. Azam and S. Sanders, Microbial carbon pump in the ocean, Science/AAAS, Washington, DC, 2011, DOI:10.1126/science.opms.sb0001.
- P. M. Mendeiros and B. R. Simoneit, J. Chromatogr. A, 2007, 1141, 271–278 CrossRef PubMed.
- C. Panagiotopulos and R. Sempéré, Limnol. Oceanogr.: Methods, 2005, 3, 419–454 CrossRef.
- K. Kaiser and R. Benner, Anal. Chem., 2000, 72, 2566–2572 CrossRef CAS.
- N. H. Borch and D. L. Kirchman, Mar. Chem., 1997, 57, 85–95 CrossRef CAS.
- R. D. Rocklin and C. A. Pohl, J. Liq. Chromatogr., 1983, 6, 1577–1590 CrossRef CAS.
- Y. C. Lee, Anal. Biochem., 1990, 189, 151–162 CrossRef CAS.
- K. Mopper, C. A. Schultz, L. Chevolot, C. Germain, R. Revuelta and R. Dawson, Environ. Sci. Technol., 1992, 26, 133–138 CrossRef CAS.
- R. J. Wicks, M. A. Moran, L. J. Pittman and R. E. Hodson, Appl. Environ. Microbiol., 1991, 57, 3135–3143 CAS.
- X. Cheng and L. A. Kaplan, J. Chromatogr. Sci., 2003, 41, 434–438 CAS.
- D. C. Johnson and W. R. La Course, Anal. Chem., 1990, 62, 589A–597A CAS.
- W. R. La Course and D. C. Johnson, Anal. Chem., 1993, 65, 50–55 CrossRef CAS.
- A. Engel and N. Händel, Mar. Chem., 2011, 127, 180–191 CrossRef CAS PubMed.
- T. Dittmar, B. P. Koch, N. Hertkorn and G. Kattner, Limnol. Oceanogr.: Methods, 2008, 6, 230–235 CrossRef CAS.
- A. J. Simpson, L. H. Tseng, M. J. Simpson, M. Spraul, U. Braumann, W. L. Kingery, B. P. Kelleher and M. H. Hayes, Analyst, 2004, 129, 1216–1222 RSC.
- B. P. Koch, K. U. Ludwichowski, G. Kattner, T. Dittmar and M. Witt, Mar. Chem., 2008, 111, 233–241 CrossRef CAS PubMed.
- G. C. Woods, M. J. Simpson and A. J. Simpson, Water Res., 2012, 46, 3398–3408 CrossRef CAS PubMed.
- G. C. Woods, M. J. Simpson, P. J. Koerner, A. Napoli and A. J. Simpson, Environ. Sci. Technol., 2011, 45, 3380–3386 Search PubMed.
- V. R. Preedy, Dietary Sugars: Chemistry, Analysis, Function and Effects, Royal Society of Chemistry Publishing, Cambridge, U.K., 2012 Search PubMed.
- M. Yan, G. Korshin, D. Wang and Z. Cai, Chemosphere, 2012, 87, 879–885 CrossRef CAS PubMed.
- N. Her, G. Amy, D. Foss, J. Cho, Y. Yoon and P. Kosenka, Environ. Sci. Technol., 2002, 36, 1069–1076 CrossRef CAS.
- M. B. Muller, D. Schmitt and F. H. Frimmel, Environ. Sci. Technol., 2000, 34, 4867–4872 CrossRef.
- C. Piccini, D. Conde, J. Pernthaler and R. Sommaruga, Photochem. Photobiol. Sci., 2009, 8, 1321–1328 CAS.
- J. I. Hedges, G. L. Cowie, J. E. Richey, P. D. Quay, R. Benner, M. Strom and B. R. Forsberg, Limnol. Oceanogr., 1994, 39, 743–761 CrossRef CAS.
- R. F. Vaccaro and H. W. Jannasch, Limnol. Oceanogr., 1966, 11, 596–607 CrossRef CAS.
- G. L. Cowie and J. I. Hedges, Geochim. Cosmochim. Acta, 1984, 48, 2075–2087 CrossRef CAS.
- S. J. Goldberg, C. A. Carlson, M. Brzezinski, N. B. Nelson and D. A. Siegel, Geophys. Res. Lett., 2011, 38, 1–7 CrossRef.
- R. Tautenhahn, G. J. Patti, D. Rinehart and G. Siuzdak, Anal. Chem., 2012, 84, 5035–5039 CrossRef CAS PubMed.
- C. A. Smith, E. J. Want, G. O'Maille, R. Abagyan and G. Siuzdak, Anal. Chem., 2006, 78, 779–787 CrossRef CAS PubMed.
- M. A. Nanny and N. Ratasuk, Water Res., 2002, 36, 1572–1584 CrossRef CAS.
- J. L. Weishaar, G. R. Aiken, B. A. Bergamaschi, M. S. Fram, R. Fujii and K. Mopper, Environ. Sci. Technol., 2003, 37, 4702–4708 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ay41549j |
| This journal is © The Royal Society of Chemistry 2014 |