Non-eroding drug-releasing implants with ordered nanoporous and nanotubular structures: concepts for controlling drug release
Moom
Sinn Aw
,
Mima
Kurian
and
Dusan
Losic
*
School of Chemical Engineering, The University of Adelaide, Engineering North Building, SA 5005, Adelaide, Australia. E-mail: dusan.losic@adelaide.edu.au; Fax: +61 8 8313 4373; Tel: +61 8 8313 4648
First published on 14th November 2013
Abstract
To address the limitations of systemic drug delivery, localized drug delivery systems (LDDS) based on nano-engineered drug-releasing implants are recognized as a promising alternative. Nanoporous anodic alumina (NAA) and nanotubular titania (TNT) fabricated by a simple, self-ordering electrochemical process, with regard to their outstanding properties, have emerged as one of the most reliable contenders for these applications. This review highlights the development of new LDDS based on NAA and TNT, focusing on a series of strategies for controlling their drug release characteristics that are based on: modification of their nanopore/nanotube structures, altering internal chemical functionalities, controlling pore openings by biopolymer coatings and using polymeric micelles as drug nano-carriers loaded within the implants. Several new strategies on externally triggered stimuli-responsive drug release for LDDS are also reviewed, and their significance toward the development of advanced smart implants for localized therapy is discussed. Finally, the review is summarized with conclusions and future prospects in this research field.
 Moom Sinn Aw | Moom Sinn Aw was born in 1986 in Malaysia. She received her B. Chem. Eng. (Honours) degree from the University of Adelaide (2008), Australia. She completed her PhD at the Ian Wark Research Institute, the University of South Australia (2012), with her thesis titled, ‘Nano-engineered porous materials for controlled drug delivery’ followed by a short postdoctoral position at the School of Chemical Engineering, the University of Adelaide. She is currently employed as a postdoctoral researcher at the National Institute of Chemistry, Ljubjlana, Slovenia, working on catalysts synthesised from nanomaterials for renewable energy production. |
 Mima Kurian | Mima Kurian was born in 1990 in Kerala, India. She obtained her Masters degree in Technology (Nanotechnology) from the Amity Institute of Nanotechnology of the Amity University, India (2013). She joined the Losic Nanoscience Research Group at the School of Chemical Engineering at the University of Adelaide in 2012 to undertake her Masters thesis, where she worked on local drug delivery systems based on titania nanotube implants in wires for ex vivo studies in mice. Her research interests are focused on developing nanoscaled systems for local and targeted drug delivery and tissue regeneration. |
 Dusan Losic | Dusan Losic is an Australian Future Fellow and Research Professor at the School of Chemical Engineering, the University of Adelaide where he is leading the Nano research group. He completed his PhD in Nanoscience and Nanotechnology at Flinders University (2003) where he worked as a Research Fellow and followed it with a 5 year ARC Research Fellowship (2007) at the Ian Wark Research Institute at the University of South Australia. His Multidisciplinary Nanotechnology research involves fundamental, engineering and applied aspects across several disciplines including chemistry, materials science, engineering, biology and medicine working on diverse research topics (molecular separations, biosensing and drug delivery). |
1. Introduction
To address problems associated with conventional drug delivery (DD) related to limited drug solubility, overdose, short circulating time, poor biodistribution, lack of selectivity, side effects, toxicity and unfavorable pharmacokinetics/pharmacodynamics, considerable research has been directed in the last two decades toward the development of new and more efficient drug delivery systems (DDS).1–3 Nearly 90% of currently used drugs are hydrophobic, which cannot dissolve in the blood and this reduces their pharmacological efficiency. As a result, their delivery route is particularly challenging.4,5 On the other hand, some bioactive agents such as proteins, nucleic acids, enzymes and genes administered through oral or intravenous routes can be easily degraded by metabolism and are unable to reach the designated targeted sites.6–8 Local drug delivery systems (LDDS) are considered a promising alternative to systemic routes and conventional DDS to address many of these limitations, especially by means of local implantation.3,5,9 LDDS at the earlier stages of development were made of biodegradable implants based on poly(methyl methacrylate) (PMMA),10 poly(lactic acid) (PLA),11 poly(glycolic acid) (PLGA) matrices,12 collagen,13 hyaluronan,14 chitosan, fibrin,15 silk, hydroxyapatite (HAP),16 ceramics and injectable calcium phosphate cements. They have been used for the delivery of bioactive agents in bone repair or tissue engineering.17,18 Unfortunately, many disadvantages are inherent in these systems, for instance, uncontrolled burst release of therapeutics and the failure to achieve sustained release over a long period of time, or in the desired dosage. These issues are mainly due to the amorphous state of the biomaterials, or their non-ordered porous structures.19,20 Hence, it is necessary to devise an LDDS which can safely deliver drugs with predictable release kinetics and in adequate quantity prescribed to treat various diseases (e.g. cancers, pathogenic infections and inflammation), all of these being particularly critical in orthopedics. Moreover, degenerative bone diseases such as osteomyelitis and osteoarthritis are globally known problems that are prevalent,21–23 not to mention the surging number of implant surgeries which can cause severe post-surgical complications (e.g. bone marrow swelling during bacterial infection, and blood supply reduction caused by the compression of blood vessels in post-implant surgeries), thereby summoning the urgent need to control drug dosing and duration.24–26To overcome these existing problems, new implants for LDD with ordered nanoporous and nanotubular structures were explored using nanotechnology based approaches.27 The application of nanotechnology in medicine, referred to as “nanomedicine” is recognized as an emerging field with enormous potential for the development of new and advanced LDDS.28 Some of the most significant examples are nanoporous anodic alumina (NAA) and titania nanotubes (TNT), two new emerging biomaterials synthesized by a lithography-free, top-down nano-engineering process. In addition to their biomedical applications, they are applied in many research areas which have attracted more than 1000 publications in the last seven years.29 Their fabrication process is based on a low-cost, simple, time effective, scalable, self-ordering electrochemical anodization, widely employed in the metal processing industry. By controlling the relevant parameters such as electrolyte, voltage, time, temperature and current, oxide layers comprised of nanopore and nanotubular structures with controllable dimensions can be fabricated on the surface of transition and valve metals.30–32 These nanomaterials, especially TNTs, have been explored for DD applications owing to their versatile and unique properties, such as excellent biocompatibility, non-toxicity, stability, mechanical robustness, chemical resistivity and inertness, high surface area, controllable nanotube thickness (1–300 μm) and diameters (10–300 nm), simple low-cost fabrication on bulk titanium (Ti) and Ti alloys in the form of sheets, wires, plates or screws.27,33–35
The drug release mechanism from these non-eroding implants is elucidated by a diffusion mediated process, which is driven by a concentration gradient that permits the transport and release of drug molecules from the nanopores. Unlike previously devised polymer based implants such as poly(glycerol methacrylate) (PGMA) hydrogels, problems arising from implant swelling or disintegration are actually irrelevant, because TNT is stable and imposes no side effects on drug release rates or mechanisms. Nonetheless, the transport of drugs is no longer controllable once the implant is inserted inside the body.36 Therefore, it is vital to devise appropriate strategies to modulate their drug release by regulating the diffusion mechanism. Additionally, the criteria that have to be considered for good therapeutic efficiency include: the right selection of drugs, optimized drug dosage, constant drug release rate, release duration, implant size, morphology and the nature of the implant site. These factors are important because despite the progress in implant technology, there are still unresolved issues in the design of various biomaterials pertaining to their clinical applications, such as non-sustainable capping of the drug inside the implant, poor drug retention within the biomaterials, fragility that causes drug leakage, implant-to-tissue instability and side effects due to implantation. Moreover, prolonged and sustained release within the proper therapeutic window of the involved drugs, which can effectively overcome the short half-lives of drug, is yet to be seen in current LDDS.37
With regard to this matter, strategies were explored by several groups and hence the aim of this paper is to review approaches for controlling drug release from non-eroding drug-releasing implants based on self-ordered nanoporous and nanotubular structures. We herein present the basic concept and several examples from our work, as well as from other groups, on the development of these strategies. A variety of approaches designed to control the diffusion of drug molecules from these implants with the aim of achieving extended and sustained drug release are discussed. These include structural tuning and chemical modification of nanopores and nanotubes, as well as the application of biodegradable biopolymer coating and nano-carriers such as polymeric micelles. Several new concepts for externally triggered stimuli-responsive drug release by using magnetic, ultrasonic and radiofrequency (RF) sources are discussed. These stimuli are able to produce alternative drug release pathways that could circumvent the diffusion mechanism instead. This review is concluded with an overview of recent developments and the prospective outlook for future trends in this field.
2. Ordered nanoporous and nanotubular implants based on NAA and TNTs
Nanoporous anodized alumina (NAA) and titania nanotubes (TNT) are emerging as two of the most prominent drug-releasing implants in LDD.27 Depending on the metal type, electrolyte, current and voltage, two different oxide growth morphologies, i.e. nanoporous aluminium oxide (Al2O3) and nanotubular titanium dioxide (TiO2) can be obtained by a self-ordering electrochemical anodization process as shown in Fig. 1. They can be easily fabricated via a simple two-step anodization to form self-assembling and highly ordered nanopores or nanotube structures with controllable pore diameter (10–400 nm), inter-pore distance and pore length (1–300 μm), uniform structures, high volume (>20 m3 g−1), high surface area (180–250 m2 g−1), high aspect ratio (1000–2000) and porosity (1010 pores cm−1).32,38–41 In fact, their size, porosity and geometry can be modulated at a nanometer scale via computer-controlled anodization parameters, such as electrolyte composition, pH, temperature, anodization time, current and voltage.31,42–44 These nanostructures can also be fabricated on three dimensional (3D) non-planar and curved surfaces, such as thin, long surgical wires for bone fixture, due to their malleability, facilitating insertion into the body during implant surgeries. In both cases, the fabrication process of NAA and TNT based implants is scalable and inexpensive, which are based on low cost materials and equipment, favoring their commercial mass production. Their potential and application as LDDS have been demonstrated in previous reports using many drugs as examples, all of which show considerably high level of drug loading and therapeutic efficiency.45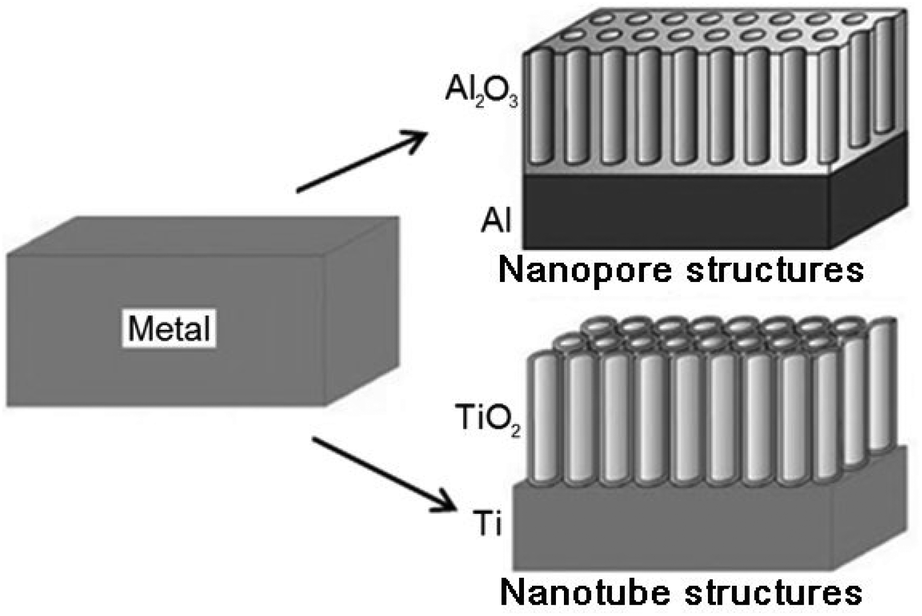 | ||
| Fig. 1 Schematic diagram showing the self-ordering anodization on selected metals (Al and Ti) to produce nanoporous and nanotubular metal oxides (Al2O3 and TiO2) with highly-ordered and self-aligning uniform structures. These two materials were explored as non-eroding drug-releasing implants for LDD applications. Reprinted with permission from ref. 43. Copyright 2009, The Royal Society of Chemistry (RSC). | ||
Compared with other LDDS systems, two substrates based on mesoporous silica prepared by organic synthesis and porous silicon fabricated by electrochemical process were most widely explored in the last several years.46–48 These materials, due to their unique features, such as biocompatibility, controllable pore structure, tunable surface chemistry, high surface area, high drug loading capability, chemical resistivity and mechanical rigidity, offer many advantages to be applied as drug carriers for the development of advanced drug delivery systems including LDDS.49–51 However, despite the advanced synthesis of various silica mesoporous materials, including the most popular series such as MCM-41 and SBA-15, their synthetic protocols are complicated, time-consuming, and expensive, and involve high energy consumption, toxic materials and waste. Another silicon based material, the porous silicon (PSi) with less ordered pore structures, which is a very similar material to NAA and TNT, is demonstrated to be an excellent substrate for DD applications. However, the main disadvantage of pSi is the chemical instability and mechanical fragility of its porous structure which disqualify them from functioning as implants.52,53
3. Strategies to control drug release from non-eroding nanoporous structures
Drug release from nanoporous or nanotubular structures in contact with physiological solutions is governed by a diffusion mechanism as schematically presented in Fig. 2a. The release process is attributed to the mass transfer of drug molecules from nanopores, which is mainly a diffusion-controlled release process, considering that the drug is completely soluble in the physiological solution without precipitation at particular concentrations.54 This process is influenced by a number of factors, for instance, the molecular size of drugs, interfacial interaction between the drug molecules and the implant surface, the dissolution rate of drugs, diffusion coefficient, pore or tube dimensions, pH of medium, polarity, or ionic charge of the pore surface.55 Depending on these conditions, Fig. 2b displays the possibility of obtaining different drug release profiles from nanoporous implants, i.e. burst release, zero-ordered, combined first and zero-ordered, and stimuli-responsive drug release from the nanostructures. They comprise either free or restricted diffusion with respect to time. Zero-order type release is the most desirable release strategy as it yields the highest dosing stability and resembles the ideal situation whereby the drug is released at a uniform and constant rate independent of time and concentration.56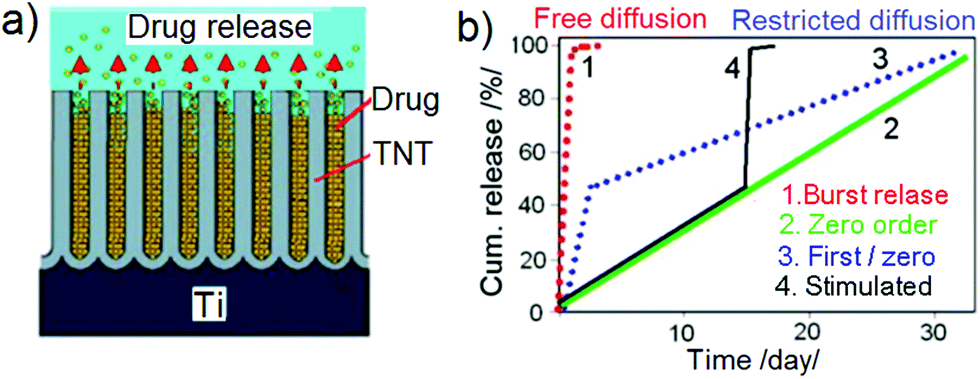 | ||
| Fig. 2 (a) Schematic diagram of drug release via diffusion mechanism from the pore structures to bulk solution (phosphate buffered saline, PBS medium), (b) conceptual graphs presenting cumulative release curves of free and restricted diffusion with respect to time, showing different characteristics of release, i.e. burst, zero-order (ideal case), combined first and zero-order, and finally, stimuli-responsive drug release from TNT. | ||
Numerous studies were focused on exploring these parameters and better understanding drug release processes from non-eroding nanopore structures that were mainly based on mesoporous silica. Vallet-Regí's group and several others have extensively reviewed the application of mesoporous silica as a DD platform for in vitro and in vivo DD with respect to various modifications on the silica surface and its influence on the drug release performance.57–60 Many examples of advanced DD combined with targeting agents such as proteins, peptides, antibodies and folic acids were demonstrated using these strategies for specific therapies.61,62 Furthermore, the outer surfaces of these mesoporous silica can be functionalized with selective chemical moieties to fine-tune their surface charge, or they can be adhered with coatings such as polyethylene glycol (PEG) or polymers sensitive to certain stimulants. More complex systems for stimulated and controlled drug release using nano-caps that can be opened and closed upon a trigger from pH, redox potential, thermal energy, or biomolecular interactions were also demonstrated using these DDS.63–65
An important aspect in the study of drug release mechanisms and the rate of release from non-eroding porous structures such as NAA and TNT is mathematical modeling. These models can be used to predict drug release behavior and the performance of implantable LDDS. The basic model is described by the diffusion process according to Fick's first law at the bulk level.66 Degradation of platforms and drug release from unstable surface-eroding matrices are ruled out, since TNT and NAA are finite materials with fixed dimensions. Typically, the diffusion process is described through Fickian equations for the evaluation of drug release kinetics from implants by estimating the concentration change with respect to time due to diffusion. Furthermore, several release models were developed and specifically applied for mesoporous materials including: Weibull, Higuchi, zero-order, first-order and Korsmeyer–Peppas (eqn (1)–(6)). These models are excellent tools to assist in the development of DDS by predicting the drug release kinetics and mass transfer from the non-eroding nanostructures.67
| Fickian diffusion : dΦ/dt = D(d2Φ/dx2) | (1) |
| Zero order: Qt = Q0 + k0t | (2) |
| First order: lnQt = lnQ0 − k1t | (3) |
| Higuchi: Qt = kHt0.5 | (4) |
| Weibull: log[−ln(1 − m)] = β log(t − Ti) − log a | (5) |
| Korsmeyer–Peppas: Mt/M∞ = ktn | (6) |
The governing parameters are: Φ, concentration with respect to volume or mole; t, time; D, diffusion coefficient; x, length or thickness of nanostructures; Qt, amount of drug released in time t; Q0, initial amount of drug in the drug-releasing implants; k0, k1, kH, release rate constants; m, accumulated fraction of the drug; β, shape parameter; a, scale parameter; Ti, location parameter; Mt/M∞, fraction of drug released at time t; k, the release rate constant; n, the release exponent used to characterize different release for cylindrical shaped matrices.
These parameters can be varied and used to develop strategies for controlling the release rate and also to achieve the optimal dosage within the therapeutic window and required timeframe, taking into account the drug activity and bioavailability. These requirements are different for different therapies and disease conditions, and may vary from on-demand, stepwise, multi-drug delivery, delayed, time-programmed, sequential to switchable release behaviors. Therefore, it is important to practically design an optimal system with release properties corresponding to one of these possible cases to control the diffusion of drugs.
To achieve these conditions, structural as well as surface modifications have to be applied in the design of the drug-releasing implant with nanopore structures. Strategies explored by our group toward such advanced implants are shown in Fig. 3. The schematic diagrams showing a single nanopore or nanotube structure subjected to a variety of modifications for controlling drug release are depicted. We explored several strategies, such as: (a) controlling pore dimensions (diameter and length), (b) controlling surface chemistry by a specific functional group with different properties and interactions with drugs, (c) controlling pore openings by depositing a layer of biopolymer via plasma polymerization, (d) controlling drug release by the degradation of biopolymer film that was deposited on the implant surface covering the nanopores, (e) using polymeric micelles as drug nano-carriers loaded inside the nanotubes and releasing single or multiple drugs with delayed release, and (f) several stimulated drug release strategies using external sources, including magnetic field, ultrasound and radiofrequency (RF). In the following sections, we present these strategies and discuss their advantages and disadvantages toward controlling the drug release rate from nanotubes and nanopores.
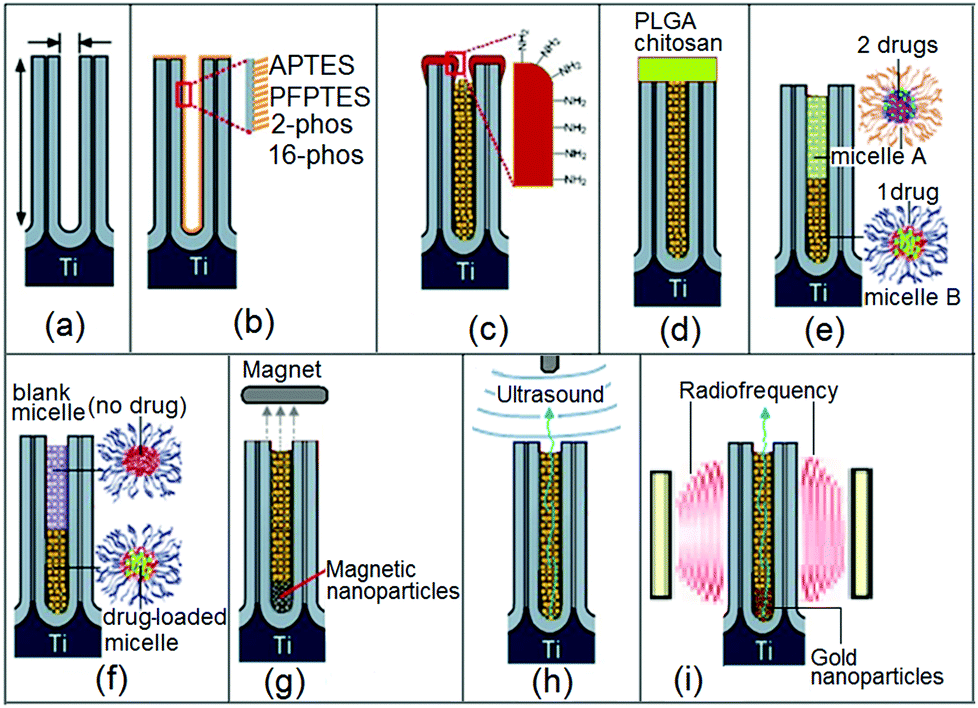 | ||
| Fig. 3 Strategies for controlling drug release from nanoporous and nanotubular drug-releasing implants using: (a) variations in the pore size and length; (b) surface chemistry (hydrophobic, hydrophilic, charged); (c) pore opening reduction by plasma polymerization; (d) pore closing by biodegradable film (PLGA or chitosan); (e) using drug nano-carriers (micelles) for multidrug delivery; (f) delayed/sequential drug release of drugs/drug-carriers; (g) external stimuli release using magnetic field and magnetic nanoparticles; (h) ultrasound; or (i) radiofrequency with gold nanoparticles. Only single nanopore/nanotube is shown to present an array of these nanostructures generated on the implant surface of NAA or TNTs. | ||
3.1. Altering drug release by controlling pore dimensions
As previously stated, the diffusion rate of drug molecules from nano-confined structures such as nanopores, according to Fick's law, depends on their dimensions.68,69 Since the pore size in these confined nano-channels can be controlled precisely at nanoscale, this strategy is primarily devised to control the drug release. Hence, it is expected that by changing the nanopore dimensions, it would be possible to tune their release performance.To explore the influence of pore dimensions on the drug release pattern, we studied NAA with pore diameters ranging from 65 to 160 nm; and TNT with nanotube lengths from 25 to 100 μm. The entire size of these implants (including the metal base of Al and Ti metal foils, respectively) is: 12 mm (width) × 12 mm (length) × 0.25 mm (thickness). To prepare NAA with different pore diameters but the same length, a pore widening procedure was applied, using 0.1 M phosphoric acid for 10 min, 50 min, 85 min and 120 min to create a range of pore diameters, i.e. 65 nm, 90 nm, 120 nm and 160 nm, respectively. Their structures and pore dimensions were confirmed in SEM images as depicted in Fig. 4a. Typical NAA pore structures from the top surface are presented, with the inset showing a photo of real NAA implant fabricated on Al foil. The pore diameters of NAA were controlled by a chemical dissolution process (pore widening) and Fig. 4b shows the graph of pore diameters versus treatment time. Pore diameters from 65 nm to 160 nm were found to linearly correspond to the pore widening time (10 min to 120 min). All prepared samples had a constant thickness (pore length) of 20 μm. A typical structure of TNT is presented in Fig. 4c, in which a cross-sectional SEM image is shown, with the inset depicting the actual TNT implant fabricated on a Ti foil. The length of the nanotubes is controlled by anodization time, and Fig. 4d shows the graph of TNT lengths (25 μm, 50 μm, 75 μm and 100 μm) which are found to be linearly dependent on the anodization time (i.e. 30 min, 60 min, 90 min and 120 min). All TNT pore diameters are held constant at 110 nm.
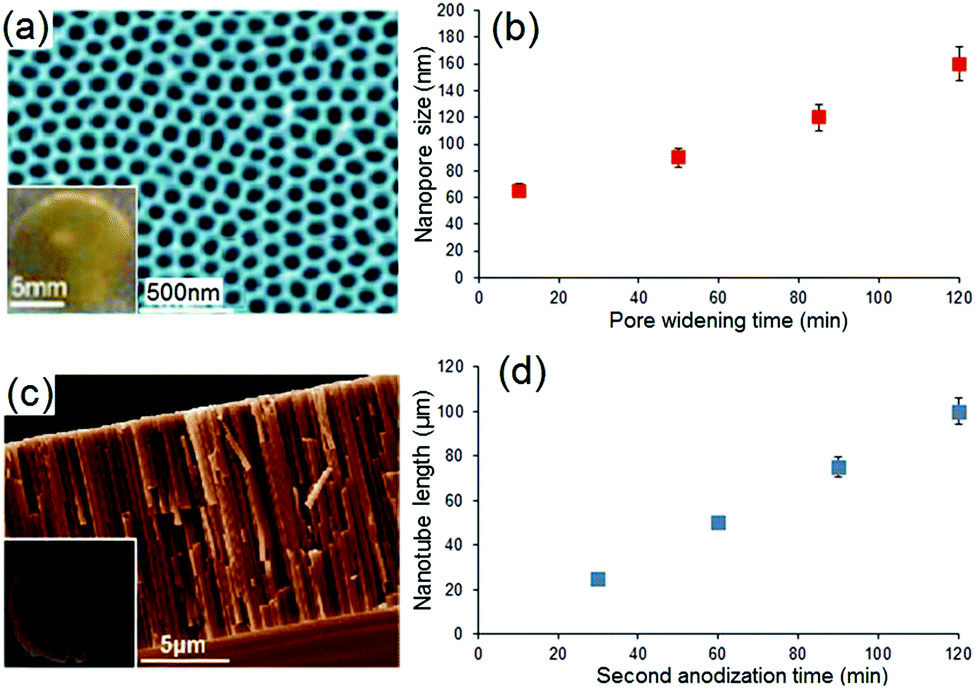 | ||
| Fig. 4 (a) SEM image of a typical surface of NAA showing the nanopore structure, with inset displaying the whole NAA sample fabricated on Al foil. (b) Graph showing the influence of the pore widening time (10–120 min) on the pore diameters (65–160 nm) of NAA using 0.1 M phosphoric acid. (c) Cross-sectional SEM image of a TNT layer fabricated on Ti foil showing the vertical nanotube structures, with inset showing the photo of real TNT implant. (d) Graph showing the influence of anodization time (30–120 min) on the nanotube length (25–100 μm) of TNT. (Reproduced with permission from ref. 27, Copyright Informa Healthcare). | ||
Drug release studies on these NAA and TNT samples were performed using indomethacin (ind), a water insoluble anti-inflammatory drug, as a model drug. From thermogravimetric analysis (TGA), it was confirmed that NAA with varied pore sizes of 65 nm to 160 nm gave rise to drug loading of 19% to 26%, and with drug release extending from 5 to 6 days. TNT has similar drug loading capacity, ranging from 15% to 26%, with longer drug release, i.e. from 6 to 23 days. These results are as expected and comparable with the literature since larger pore size and longer tube length offer more space and volume to load drugs, and with higher drug amounts stored inside the implants, release time was prolonged, indicating their proportionality to their dimension.70 The graph plotted (data not shown) to correlate drug storage capacity to the variation of dimension also revealed that the drug mass which can be accommodated in NAA is linearly proportional to their pore diameters. Meanwhile, it was shown that shorter TNT (with length of 25 μm and 50 μm) offers lower volume for drug containment (15–17 wt%) compared with longer TNT (75 μm and 100 μm). However, we observed that there are some limitations to this correlation when considering the longer nanopore and nanotube structures. This is because there is no comparable increase in the loading capacity with greater tube length. Drug molecules most likely cannot reach the deeper ends of very long, narrow capillary-like nanotubes during the loading process due to boundary limits from its geometrical confinement and possible air entrapment occupying the bottom of the channels. Table 1 presents the data of drug storage percentage and specific drug loading.
| NAA pore size (nm) | Drug storage capacity (wt%) | Specific drug loading (mg cm−2) |
|---|---|---|
| 65 | 18.9 | 1.90 |
| 90 | 20.1 | 2.48 |
| 120 | 22.3 | 2.64 |
| 160 | 26.0 | 2.97 |
| TNT tube length (μm) | Drug storage capacity (wt%) | Specific drug loading (mg cm−2) |
|---|---|---|
| 25 | 15.4 | 1.98 |
| 50 | 17.3 | 2.02 |
| 75 | 25.8 | 2.86 |
| 100 | 25.4 | 2.53 |
Drug release profiles for NAA with different nanopore dimensions and constant pore lengths are depicted in Fig. 5a. The graphs present cumulative drug release (%) with respect to release time (days), with insets showing the burst release related to release time (hours). These results show that the release time was slightly reduced due to the widening of pore diameters. Two phases of drug release were observed in Fig. 5a; firstly, an early or burst release for 0–3 h that spiked up to 65–78% at the beginning, followed by a prolonged, slower release that lasted 5 or 6 days. The large initial burst indicates a massive amount of drug eluted out of the system, implying that the physical containment of the drug inside the nanostructure is weak. Larger pore diameter suggests greater space for molecular transport, as it leads to greater contact for drug and the aqueous medium, resulting in lesser impedance for drug diffusion to the bulk solution. The smaller pore size (65 nm and 90 nm) could better engage the surface interactions between the drug molecules and the pore walls, which in turn amplify the capillary effects resulting in slower release. The difference with respect to cumulative release percentage of drugs and the time required for the release completion from NAA is not statistically significant. However, the reduction to considerably smaller pore diameters (10 nm to 20 nm) is expected to further reduce drug release time and drug loading capacity.
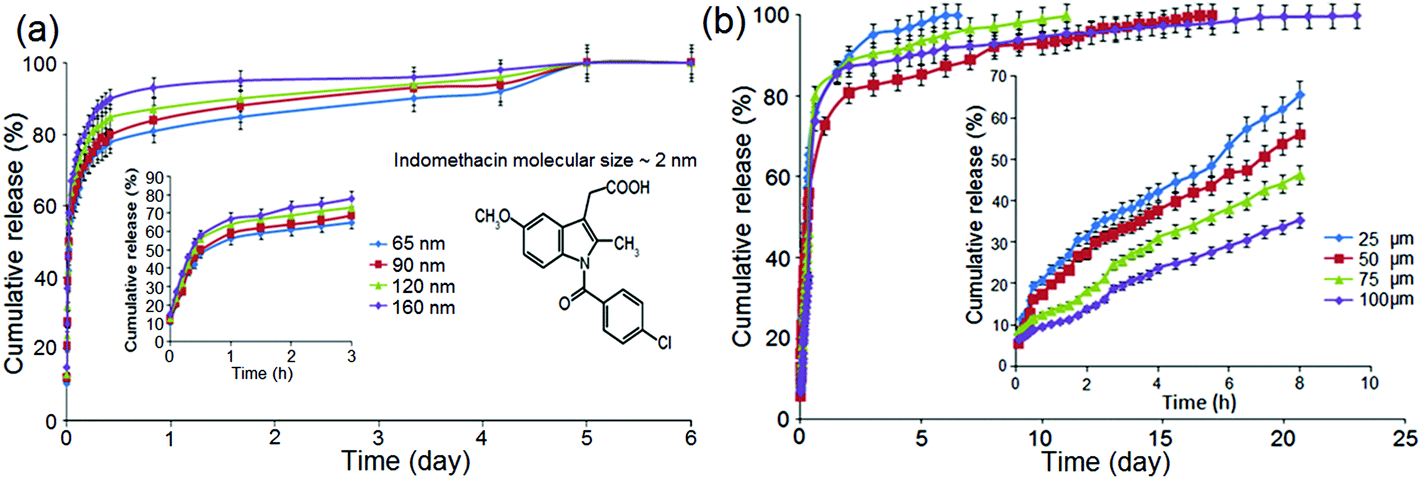 | ||
| Fig. 5 Graph showing the cumulative release (%) of indomethacin (poorly soluble drug) from: (a) NAA with different pore diameters (65 nm to 160 nm) versus release time (days); (b) TNT with different nanotube length (25 μm to 100 μm) versus release time (days). Insets of both graphs show the initial burst for the first 6 h. | ||
The influence of pore dimensions on drug release characteristics has been reported in the literature, where varied drug release profiles were obtained due to variation in the pore size of mesoporous silica matrices that possess unidirectional channels.71,72 A similar structural modification approach is applied for the fine-tuning of molecular transport by modifications in the pore diameters and chemical selectivity of NAA, where molecular separation in membranes was demonstrated.73,74 This tunability of nanotubes/pore dimensions can be tailored to meet specific therapeutic requirements, depending on drug types, implant size, and the type of organs (bone or cancer) to be targeted.75
Drug release profiles of indomethacin loaded in TNT implant with a constant pore diameter of 110 nm and different nanotube lengths (25–100 μm) are shown in Fig. 5b. These results show that the release time was extended due to an increase in the nanotube length. The graph shows a longer release range that spans 6–24 days. The release profile is again biphasic with a fast (burst) phase over the first 6 h; followed by an overall release in the next 1–3 weeks. The burst release of 32–64% is attributed to the high initial concentration of drug at the top of the TNTs surface. Interestingly, it was found that the longer the nanotube structures are, the lower the burst release. This observed extended drug release is supported by the fact that drug was trapped deeper inside the vertical channels, and therefore needed a longer time to diffuse out as capillary effects were more efficacious.
These results are in agreement with previous studies on NAA and TNT, confirming the influence of pore dimensions on drug loading (paclitaxel) and drug release performances.76 For example, Gong et al. demonstrated the controlled release of fluorescein isothiocyanate and dextran conjugates from NAA, proving their release could be readily controlled depending on the pore size of NAA.77 A similar influence on pore dimensions has also been reported in the literature when using mesoporous silica matrices as the drug releasing platform.78 One example is templated MCM-41, where its size difference was reflected on the ibuprofen diffusion rate.75,79,80
The drug release rate can be modeled by best fitting curves according to eqn (7) below:
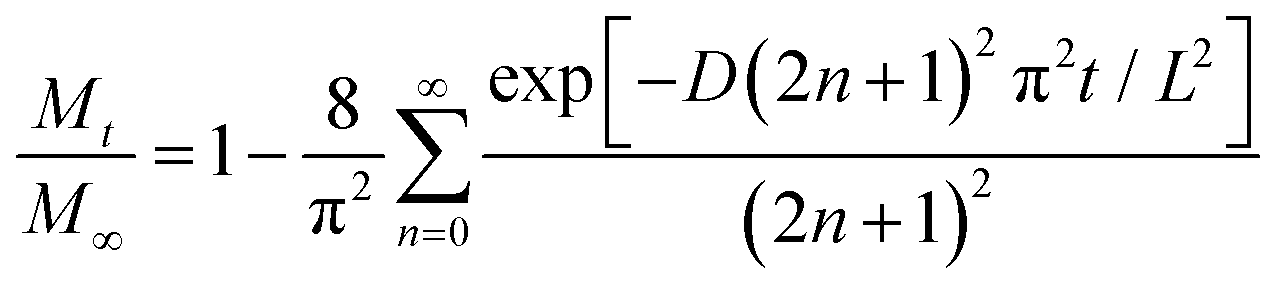 | (7) |
In conclusion, these studies showed that there are some limitations resulting from the structural modification approach due to a trade-off between loading capacity and release performance, which need to be optimized depending on the required therapy, dosage, properties of drug molecules, time release etc. These results showed that by altering pore diameters by <10–20 nm, it is possible to manipulate drug loading and release by 10–20%. The limitations of using larger pore diameters are seen, because drug molecules can diffuse very fast from nanoporous structures (as the size of molecules is considerably smaller compared with nanopores). Using nanopores with higher aspect ratio is found to be beneficial to achieving a longer drug release. These results confirmed that the optimization of pore dimensions in implants is essential to meet specific requirements such as total loaded drug, drug dosage and the release time. It can be concluded that NAA and TNT implants were explored for the loading of drugs, for instance, anticancer drugs, antibiotics, anabolics, peptides and morphogenetic proteins to demonstrate their potential applications for localized bone and cancer therapies.
3.2. Altering drug release by surface functionalization of nanopores
The next approach for tuning the drug release characteristics of non-eroding LDDS was based on the surface functionalization of nanopore and nanotube structures. The aim is to introduce chemical modifications inside nanopores by imparting hydrophobic and hydrophilic surface properties to dynamically change the interaction with drug molecules which will in turn alter drug loading and release kinetics. This concept is demonstrated on porous silica particles used as drug-carriers, whereby hydrophobic silica matrices loaded with poorly water soluble drug (ibuprofen) led to lower diffusion rates in simulated biological fluids (gastric and intestinal) and aqueous medium such as in our case (i.e. phosphate buffered saline solution (PBS) at pH = 7.2).80,82,83To explore the impact of surface functionalization on drug release characteristics of NAA and TNT, typical surface modifications commonly used for the functionalization of oxidized surface were applied based on self-assembled monolayers (SAM) of organo-silanes and organic acids.84 In our work, we explored (i) organosilanes, i.e. 3-aminopropyl triethoxysilane (APTES) and penta-fluorophenyldimethylchlorosilane (PFPTES); and (ii) phosphonic acids, i.e. 2-carboxyethyl-phosphonic acid (2-phos) and 16-phosphono-hexadecanoic acid (16-phos). The scheme of modified NAA and TNT structures is presented in Fig. 6a,b. The advantages of SAM are: simple and versatile process, good stability of prepared monolayer, flexibility to use different functional groups and tunable interfacial and binding properties for the internal surface of nanopores.85,86 We investigated the effects of these internal surface modifications (hydrophilic, hydrophobic and charged properties) on drug release characteristics of a water insoluble drug (indomethacin) from both NAA and TNT implants.
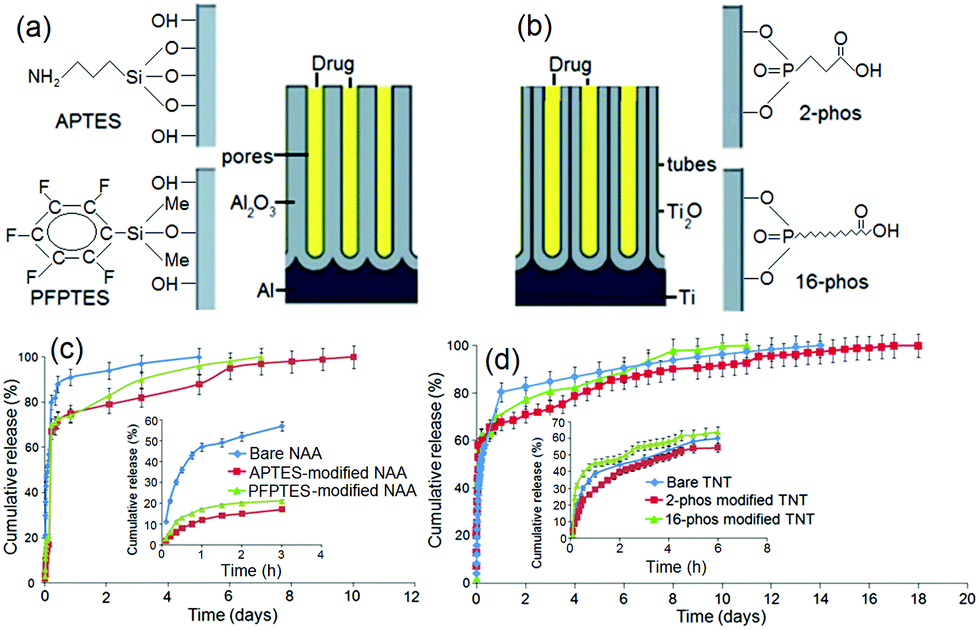 | ||
| Fig. 6 Schemes showing the concept of chemical modification: (a) on the surface of NAA by organosilanes using 3-aminopropyl triethoxysilane (APTES) and penta-fluorophenyldimethylchlorosilane (PFPTES), (b) on TNT by phosphonic acid using 2-carboxyethyl-phosphonic acid (2-phos) and 16-phosphono-hexadecanoic acid (16-phos), (c) the release graphs showing the release profile of insoluble drug indomethacin (ind) from bare (unmodified) NAA, APTES- and PFPTES-modified NAA surface, (d) drug release from 2-phos, 16-phos-modified TNT and the control sample (unmodified, bare TNT). | ||
Drug release profiles of these studies are summarized in Fig. 6c,d. The first observation is that the diffusion of drugs was faster for hydrophobic PFPTES-modified nanopores than the one with APTES-modified nanopores, which is hydrophilic and positively charged. For PFPTES, the aromatic fluorosilane ring is the functional moiety that creates hydrophobicity on the NAA surface. Compared to bare NAA, acid-functionalized NAA favors longer release, suggesting that highly electronegative monovalent fluoride and chloride anions have a great affinity for the indomethacin drug. The ionic charges and polarity of PFPTES supersede its hydrophobic side, thus advocating a slower and longer release time. Similar observations are also confirmed on TNT using different surface chemistries that were negatively charged. For TNT, the difference of release time amongst bare, hydrophobic and hydrophilic TNT surfaces is 3–4 days. In terms of sustained release, the performance in ascending order is: 16-phos TNT < bare TNT < 2-phos TNT, indicating that the hydrophilic surface supports a longer release, as the migration of drug molecules from inside the 16-phos TNT was noticeably faster.
The mechanistic transport of drugs in porous media can be underpinned by theoretical concepts such as the Kelvin equation.87 Drug loading onto the nanopores or nanotubes occurs mainly via drug adsorption by capillary action, with the latter mechanism induced by highly curved menisci of nanopore structures. The loading was also partly due to surface effects that were magnified at nanoscale, occurring inside the cylindrical thin and long nanopores and nanotubes.88 By functionalizing amino-rich APTES onto NAA and carboxyl-terminated 2-phos onto TNT, better drug adsorption to nanotube walls was achieved due to their hydrophilic properties, thereby improving drug loading capacity by 30–36 wt% as compared to their unmodified counterparts. For drug loading carried out in surface-modified NAA and TNT, considering APTES (hydrophilic) and PFPTES (hydrophobic) as modifying agents that were covalently bound to the interior of the NAA walls, drug loading was found to be more effective with the modified surface as opposed to the unmodified counterpart, as these samples elicit extended release duration, i.e. 40% and 100% longer for PFPTES and APTES-modified NAA, respectively. APTES was able to improve the loading of indomethacin by nearly 8.1% over PFPTES due to the functional amines in APTES. They can be efficiently bound by attractive intermolecular forces to the polar regions of the drug. Comparing previous reports using mesoporous alumina with a wide pore size distribution (2–7 nm) functionalized with hydrophilic and hydrophobic moieties for modulating the delivery of ibuprofen (model drug), our drug loading was found to be relatively similar (14–22%) to their hydrophobic NAA (∼20%), but ours yielded longer release (measured in days) (5–10 days) instead of hours (>5 h).89 This significant difference in release duration is mainly by virtue of the vast difference in pore size, which in our case, is an order of magnitude higher (80–110 nm).
The advantage of having the silane group in APTES is that, it is highly reactive and easily renders the nanopore surface hydrophilic. Likewise, the reactive groups in other functionalizing agents can also impart different properties onto these implant surfaces.90,91 The fact that APTES protonates at neutral pH in PBS is very useful for drugs with similar properties. In the literature, chemical functionality was proven to be a governing factor for enhanced drug loading and release; for example, adenosine triphosphate (ATP) molecules were more strongly physisorbed to organically functionalized mesoporous channels than vancomycin drug molecules due to difference in their chemical properties and drug-to-medium interaction.92–94
In conclusion, these results confirmed the importance of interfacial, chemical and surface charge properties of nanopores and nanotubes for their drug loading and drug-releasing characteristics. The properties of drug molecules (size, charge, and binding affinity) should also be considered to impart appropriate surface chemistry for nanoporous implants to achieve desired, optimized outcomes (drug loading and release). Hence, a surface modification strategy is useful for designing advanced implants with optimized performance. Nevertheless, it is still limited to achieving ultra-long sustained drug-releasing conditions (>3 months).
3.3. Extended drug release by controlling pore opening via plasma polymerized biopolymer coating
In previous sections, we learned that the drug release rate from nanoporous structures is directly proportional to the pore dimensions (diameter and length) and their surface chemistry. However, both methods have limitations in achieving a long and sustained drug release. Hence, to overcome this problem, we came up with the idea of using biopolymer coating on the top surface of NAA and TNT to specifically reduce the pore opening at the top. The concept is presented in Fig. 7a,b.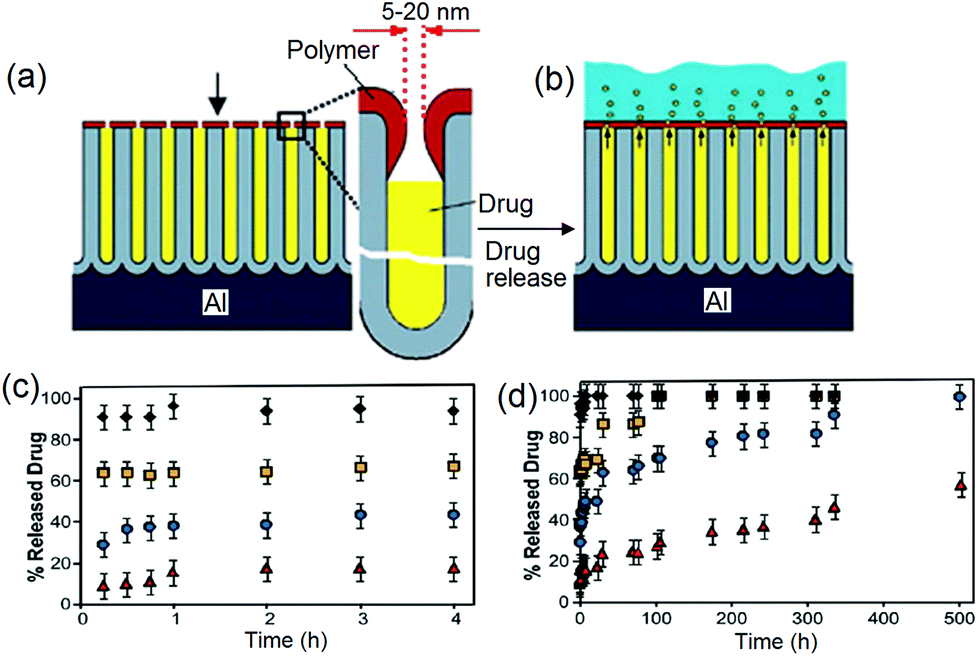 | ||
| Fig. 7 (a) Scheme depicting the concept for controlling drug release from nanopores by reduction of the pore opening (5–20 nm) on the top of NAA using plasma polymerization of biopolymer (polyallylamine); (b) suppressed diffusion of drug molecules from the reduced nanotube structures into the PBS (drug release medium); (c) controlled release of model drug vancomycin from plasma-modified NAA for 4 h and (d) 500 h. The pore opening is precisely controlled by plasma polymerization time from 50 s (squares), 120 s (circles), 200 s (triangles) and the control sample (no plasma deposition, diamonds). Reprinted with permission from ref. 98, copyright the Royal Society of Chemistry (RSC). | ||
The strategy devised for pore size reduction on the nanopore opening was aimed at imparting an ultrathin plasma polymer coating over the top surface. It is proposed that a thin poly(allylamine) (PAa) layer can be deposited by means of plasma polymerization of its organic monomer (allylamine) onto NAA. Its highly polar nature due to the presence of primary amines can be used to manipulate drug release. Plasma polymerization is an appealing approach, because it is a one-step, neat, simple (no preparation of solutions required), fast (only a couple of minutes needed), scalable, direct and dry technique.95
The drug release profiles from NAA for samples with and without plasma polymer film deposited on the top surface for a period of 50 s, 120 s and 200 s are presented in Fig. 7c,d. Clearly, increasing the time of deposition (film thickness) leads to slower drug release. From the uncoated NAA sample, the drug was completely released within 45 min. Depositing PAa for 50 s extends the release up to approximately 200 h, whereas a deposition for 120 s extends the release to almost 500 h. When PAa is deposited for 200 s, only half the amount of drug was released within 500 h. Reported methods for controlled drug release by the alteration of pore dimension can extend the diffusion of small molecules for up to 2–3 weeks, and in the case of the larger ones by up to a month.96 Therefore, it is encouraging that more effectively controlled drug release over an extended period of time is achieved using our method, i.e. only 50% of the total payload was released after 3 weeks. The controlled release kinetics is fitted into a zero-order equation, which is the best fitting model for the graph.
Furthermore, the zero-order release constant, k0 (eqn (2)), follows the same rank order as plasma polymer deposition time; the trend is such that with increasing deposition time, the zero-order release constants exponentially decrease. Zero-order release kinetics is a desirable release pattern, which is typical for reservoir transdermal delivery systems but rarely reported for porous devices.97 This result confirmed that it is possible to achieve zero-order release kinetics from porous structures by significant reduction of the pore opening, which is by the controlled deposition of a plasma polymer layer. This concept was proved on NAA and TNT with examples of more than 10 different drugs with various size and properties, including anti-inflammatory drugs, namely, indomethacin, antibiotics (such as gentamicin and vancomycin), anticancer drugs (such as doxorubicin and paclitaxel), proteins, bovine serum albumin, bone morphogenetic proteins (BMPs), drug nano-carriers (micelles) and functionalized metal oxide nanospheres (gold nanoparticles) etc.98–101
Additionally, this concept was proved in a previous report to provide zero-order release of bovine serum albumin (BSA) and human growth hormone (hGH) by single-file diffusion from cylindrical nanoporous block copolymer nano-channels (of 15 nm pore size) with polysulfone as the supporting membrane (with 200 nm pore size). The report shows consistent in vivo release of hGH from this nano-device implanted in Sprague–Dawley (SD) rats, showing the viability for long-term controlled delivery of therapeutics in cylindrical nano-channels for DD.102 Moreover, sizable micro and nanopore diameters of silicon-derived membranes applied as biocapsules were investigated by Dejai's group. A similar strategy was applied using biopolymer layers on NAA and TNT for structural modification, which was successfully used for adjustable insulin secretion from glucose stimulated biocapsules in nutrient exchange within islet cells, featuring steady-state, zero-ordered characteristics.103 The advantage of this modification lies in the fact that there are an unlimited number of polymers that can be used for coating which can be carefully selected to impart other desirable properties to the implant, such as better integration to reduce implant rejection in vivo.104 Most medical implant procedures, for instance, hip replacements, dental or vascular stents and vessel diseases such as restenosis require a long-term drug therapy to control blood clotting or decrease inflammation.105,106 Bacterial infection and poor osseo-integration identified as the major reasons for orthopedic implant failures can also be tackled through such surface modifications. However, the limitation of this system is that a direct correlation of film thickness and plasma conditions requires calibration. Even though the plasma deposition system equipped with a vacuum pump is not complicated and is commercially available, it requires considerable financial investment ($5000–$10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and some technical knowledge that is not available in hospitals.107
000) and some technical knowledge that is not available in hospitals.107
To address the limitations of the plasma method related to facility costs and technical complexity, another but significantly simpler method was explored to coat nanoporous implants with degradable biopolymers. This method is based on one-step solution casting (dip-coating) to cover the pore openings of the nanopore/nanotube structures on the top surface. The polymer film thickness and polymer degradability were explored as the method to control the drug release rate.108,109 Two biocompatible and biodegradable polymers, i.e. poly(lactic-co-glycolic acid) (PLGA) and chitosan, were selected as they are also widely used for DD applications and are known to offer antibacterial and osseo-integration properties. TNT implants were loaded with indomethacin as the poorly soluble drug model to prove the concept, although there are no limitations in terms of drugs that could be delivered by means of this approach (Fig. 8).110
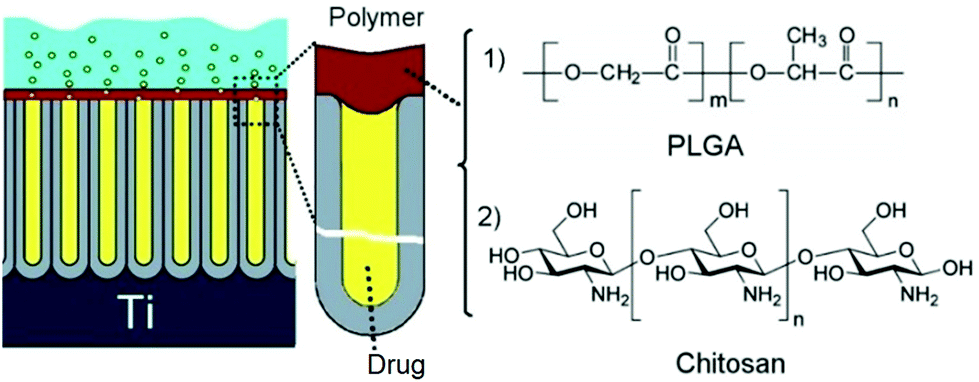 | ||
| Fig. 8 Schematic diagram of TNT implant loaded with drugs where the nanotubes were covered with ultrathin film of biodegradable polymer (PLGA or chitosan) using a simple dip-coating process. (Reprinted with permission from ref. 111. Copyright Elsevier 2012.) | ||
SEM images (Fig. 9) show the characterization of TNT morphology after biopolymer coating (PLGA and chitosan) with top and cross-sectional SEM images to ascertain their adherence onto the TNT implant. A visible and translucent polymer layer can be detected in Fig. 9a,b, showing the entire top surface shielded by a layer of chitosan and PLGA, respectively. No open pores were observed as they were fully covered. Although their presence has been proved (Fig. 9c,d), the thickness of these polymers was difficult to determine from SEM. Therefore, the ellipsometry method was used instead to determine their thickness (with chitosan measured at 200–300 nm thick, and PLGA 300–400 nm thick).
 | ||
| Fig. 9 SEM images of dip-coated biopolymer films deposited on top of the TNTs surface: (a) chitosan (inset showing closed-up view) and (b) PLGA with corresponding cross-sectional image for (c) chitosan and (d) PLGA. (Reprinted with permission from ref. 111. Copyright 2012 Elsevier.) | ||
The release characteristics of indomethacin from TNT implants with and without PLGA and chitosan are displayed in Fig. 10. The drug release graph shows a dual-phase release which consists of an initial burst during the first 6 h, followed by an overall extended release. Release characteristics show a high percentage (77%) of the drug released in the burst phase for uncoated TNT implants (control sample), followed by a much lower release over the subsequent 4–5 days. Upon coating with thin or thick chitosan and PLGA layer, a reduction of pore diameters (60–80 nm) allows a significant decrease of the burst release by 50–80%. Specifically, comparing thin chitosan-coated TNT to bare (uncoated) TNT, burst release was significantly reduced from 77% to 40%, whilst its overall release increased from 4 to >9 days. However, burst drug release for the thin PLGA layer was less reduced (to 57%), compared to 77% for bare TNT. Contrarily, the overall release was significantly extended (19 days) as compared to chitosan (9 days). Thus, it is shown that the thin PLGA polymer layer gave rise to greater initial burst and extended release. Meanwhile, the thin chitosan coating resulted in a lower burst and shorter overall release. The drug release characteristics of TNT deposited with thick chitosan and PLGA were different from the thin coated ones. For example, the thick PLGA resulted in a significant burst reduction (<12%). Both chitosan and PLGA showed an extended overall drug release over 30 days, confirming the potential of this approach to achieve a prolonged and sustained drug release for effective anti-infection bone therapy.111
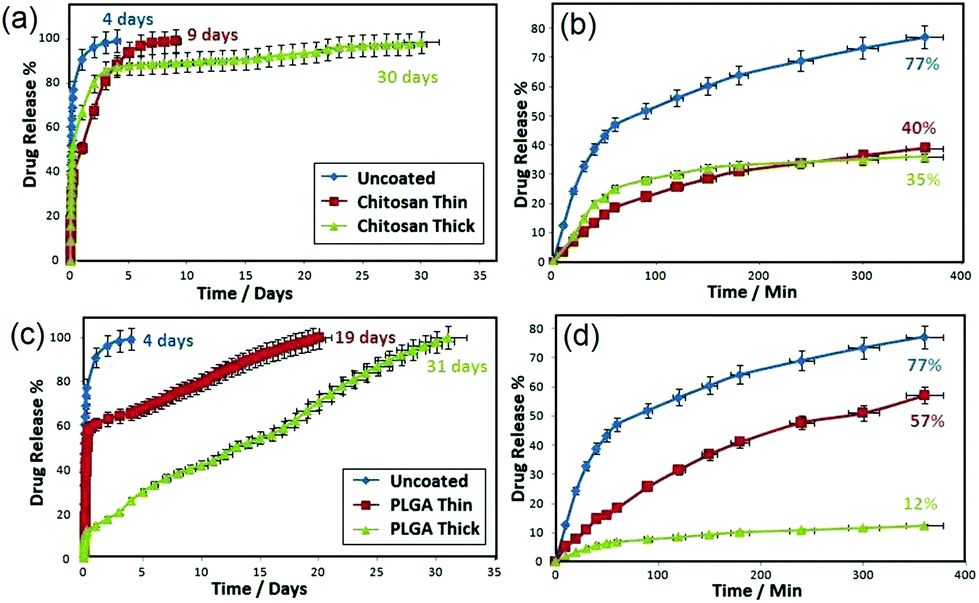 | ||
| Fig. 10 Comparative drug release graphs of anti-inflammatory drug (indomethacin) from polymer-coated TNT implants. (a, b) Overall and burst drug release from uncoated and coated TNT implants with thin and thick chitosan layer, and (c, d) overall and burst drug release from uncoated and coated TNT implants with thin and thick PLGA layer. Reprinted with permission from ref. 111. Copyright 2012, Elsevier. | ||
In conclusion, these results showed that by controlling the thickness of biopolymer film, it is possible to tune the drug-releasing profile with a significantly suppressed burst release and extend the overall release. Also, drug release characteristics can be governed by the approach used for coating (e.g. dip-coating or spin-coating), using polymers with different degradation rates and chemical compositions.112 This design of drug-releasing implants is particularly attractive for the local delivery of antibiotics for 6–8 weeks elution in post-surgical implant surgeries. Past studies have shown that systemically delivered gentamicin has fewer side effects and limitations in promoting bone healing over the course of several weeks.113 In addition to the delivery of therapeutics with small size of molecules which usually takes 7–14 days, these surface modified TNTs can also be applied for drugs with larger molecules and proteins, such as bovine serum albumin (BSA), where a longer drug release for >30 days is observed.114 TNT was previously explored as drug nano-reservoirs with layer-by-layer assembled gelatin/chitosan coatings for the entrapment of bone morphogenetic protein 2 (BMP-2).115 Controlled drug release from the TNT, as well as its capability of stimulating monogenic responses of mesenchymal stem cells (MSCs) and promoting the osteoblastic differentiation of MSCs are also confirmed.116,117
Apart from the advantages in extending drug release, the adhesion and proliferation of human osteoblastic cells (HOS) on TNT implants from our previous studies showed that biopolymer coated TNT enhances cell attachment, especially with chitosan coated TNT implants, since positively-charged chitosan chains have a high density of amino groups which can easily bond with the negatively charged cell membrane. This confirms that polymer-coated TNT enhances osteoblast integration to implants. The prolonged release kinetics help minimize peak plasma levels and the risk of adverse reactions, reduce the frequency of re-dosing and at the same time improve patient compliance.118,119 The dip-coating method for the preparation of non-eroding drug-releasing implants is a very simple and facile process with low cost facility and no specific training requirements. We believe it is achievable in a clinical environment, whilst plasma deposition is relatively more sophisticated and warrants more complex protocols and higher facility costs.
This approach of extended drug release using coatings can be compared to other LDDS reported in the literature.120–122 One example is mesoporous silica nanoparticle (MSNP) LDDS equipped with a polyethyleneimine (PEI) coating to release rifampin into M. tuberculosis-infected macrophages.123 The MSNP releases high concentrations of anti-tuberculosis drugs intracellularly and after it was PEI-coated, it showed much higher loading of rifampin than uncoated MSNP and significantly greater efficacy against M. tuberculosis-infected macrophages.
3.4. Extended drug release using drug nano-carriers for drug encapsulation
The majority of published studies have considered porous therapeutic implants containing sensitive drugs and proteins. These drugs can easily degrade or precipitate during in vivo release, such as in the case of highly lipophilic and potent anticancer drugs. Therefore, it is important to consider their protection, both inside and outside the implant. Encapsulating potent drugs in nano-carriers such as micelles prior to their loading in TNT has been proposed as a prospective solution.124 The nano-carriers are expected to have a smaller diameter compared to pore diameters of the implants. By this approach, we aimed to achieve LDDS based on NAA and TNT with considerably extended drug-release using polymeric micelles as nano-carriers to encapsulate drugs. The concept is presented in Fig. 11a. Polymeric micelles were chosen to consolidate our DDS instead of other drug carriers, e.g. dendrimers, fullerenes, nanogels, spheres, viral vectors and virus-like particles (VLP), because they can circumvent host defenses and extravagate from the vascular system, accumulate in tissues with increased vascular permeability such as that at infarct zones for passive targeting.125–127 They can enhance the therapeutic effects of drugs due to suitable and prolonged drug interactions in targeted organs, cells or malignant tumors with reduced toxicity.128 Moreover, long circulation times with stealth effect ensue from steric resistance imparted by their hydrophilic shell and smaller size (∼15–80 nm), unlike liposomes which are 2–3 times larger. Micelles are also less vulnerable and not susceptible to deformation upon stress when compared to liposomes. Micelle is effective in delivering poorly soluble drug via lipid conjugation or dissolution in the micellar core, with increased stability in aqueous milieu above their solubility limit.129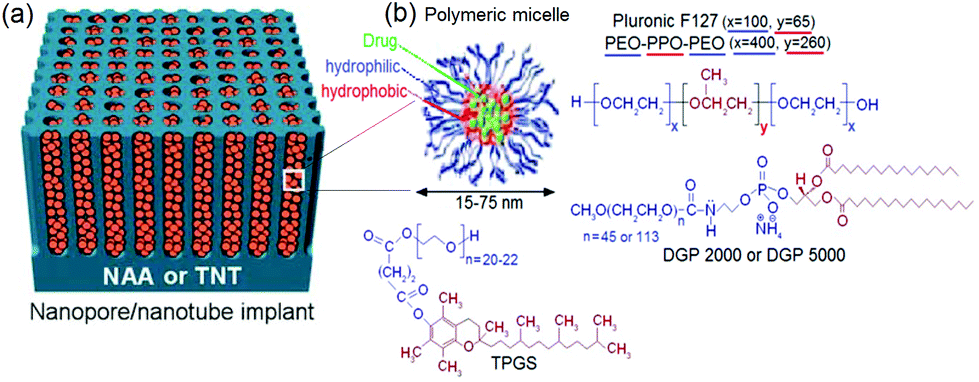 | ||
| Fig. 11 (a) Micelles encapsulated inside nanopores of NAA or TNT structures. (b) Scheme of amphiphilic micelles (drug carrier), comprising an inner core for encapsulating poorly soluble drug, and outer shell/corona (hydrophilic (blue) tail and hydrophobic (red) head), with chemical structures of micelles used in this work shown and color-coded. Reprinted with permission from ref. 113. Copyright 2011, The Royal Society of Chemistry (RSC). | ||
Four polymeric micelles, i.e. Pluronic F127®, d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) and two types of PEGylated phospholipids: 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly ethylene glycol)-2000] (DGP 2000) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly ethylene glycol)-5000] (DGP 5000) micelles were explored in our work. Their chemical structures are illustrated in Fig. 11b. Pluronic polymers with PEO–PPO–PEO organic chains that have different numbers of tri-block monomers were used. They consisted of copolymers – lipophilic alkyl tail (PPO) and hydrophilic polar head (PEO). Previous studies showed that doxorubicin encapsulated in mixed micelles of Pluronic demonstrated vital promises in phase II study in patients with advanced esophageal carcinoma.130 TPGS – a vitamin E derivative, was selected as it has been widely used as an emulsifier, solubilizer, bioavailability enhancer, an excipient and a drug carrier for lipid-based drug formulation.131 It is similar to other micellar amphiphiles, with excellent incorporation in membranes for biological activities. TPGS can greatly improve drug encapsulation efficiency (up to 100%), inhibit P-glycoprotein mediated drug efflux and increase the bioavailability of anticancer drugs.132,133 As micelles can evade the reticulo-endothelial system (RES), anticancer drugs encapsulated in micelles have nearly threefold lower toxicity level than orally or intravenously administered free drug, whilst preserving their potent anti-tumor effects.134 The PEGylated phospholipids (DGP 2000 and DGP 5000) are more robust micelles with NH-derivatives and they have broad commercial applications.135
Release characteristics of indomethacin (ind) loaded in different types of micelles (drug nano-carriers) from NAA and TNT are shown in Fig. 12. Results show an extended drug-micelle release for 8–21 days from NAA and TNT. All micelles exhibit a burst release phase in the first 6 h, followed by a slow release over 1–3 weeks. It was noticed that micelle size and charge did not influence the release behavior, considering the same size and charge range for all micelles except PEO–PPO–PEO. The main difference was observed in the release characteristics of large PEO–PPO–PEO micelles (75 nm). In fact, larger micelles exhibit faster release. It can be suggested that smaller micelles have stronger inter-micellar interactions within nanopores/nanotubes, which impede faster diffusion. When comparing diffusion coefficients of other forms of drug vehicles, such as liposomes within hydrogels, our system based on micelles for releasing ind from bare NAA and TNT provides more favorable drug release kinetics, since the values are lower. Drug diffusion coefficients (ind), k were: 1.12 × 10−8 m2 s−1 for NAA and 5.96 × 10−9 m2 s−1 for TNT, whereas corresponding effective diffusion coefficients from hydrogels were: 7.82 × 10−10 m2 s−1 and 7.98 × 10−10 m2 s−1, respectively; whilst effective diffusion coefficients from liposome-containing hydrogels were: 4.82 × 10−10 m2 s−1 for lidocaine hydrochloride and 4.305 × 10−10 m2 s−1 for dihydroquercetin.136 The advantage of this DDS is that release kinetics is dependent on and thus can be manipulated via geometric parameters, such as the pore size (diameter) of these implants.
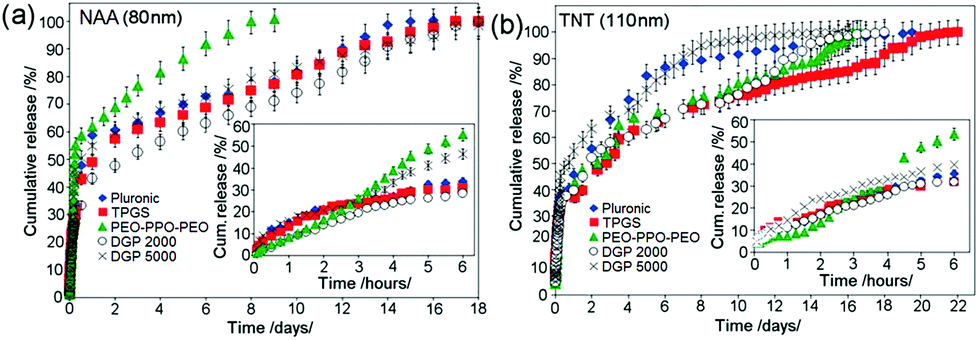 | ||
| Fig. 12 (a) Comparative drug release of polymeric micelles used (Pluronic F127®, TPGS, PEO–PPO–PEO, DGP 2000 and DGP 5000) as drug nano-carriers from NAA with 80 nm pore diameter and (b) TNT with 110 nm pore diameters. The insets show the burst release for the first 6 h for NAA and TNT respectively. Reprinted with permission from ref. 113. Copyright 2011, The Royal Society of Chemistry (RSC). | ||
4. Advanced drug release strategies with multidrug, sequential and delayed drug release
The use of conventional systemic therapy based on a single therapeutic agent is shown not to be fully successful to treat complex diseases that require more than a type of drug for treatment, such as in the case of cancers, HIV/AIDS, malaria, multiple sclerosis, hypertension, infection, cardiovascular and metabolic diseases, immune and psychiatric conditions.137–139 The administration of a single drug cannot target different key signal transduction pathways all at once, or eliminate specific tumors with particular types of therapeutics delivered together. To tackle this problem, a new concept of applying polymeric micelles loaded with several drugs was explored, such as multi-drug nano-carriers loaded inside TNT in order to design advanced multi-drug releasing implants. This approach is expected to be promising as the advantages of multidrug therapy against single-drug therapy have already been proved using traditional medicine. The additional synergistic effects could offer specific advantages to cure the complex nature of malignancy and proliferation of tumor cells and tissues, which otherwise is not possible by means of single-drug treatment which is also associated with severe side effects. Hence, the administration of multiple pharmaceutical agents displaying different toxicity profiles is proposed to be more efficient. Previous reports on mixed drugs were reported using polymer microcapsules, combination tablets, nanoparticles and silicon micro-machines,140 layer-by-layer polymer gels,141 microcapsules,142 adhesive transdermal patches and implant developed from 3D printing technology.143 Most of these approaches show some limitations due to the instability of drug vectors or complex host design, inability to regulate drug release and failure to accommodate multiple drugs safely or discharge more than one drug type in situ and within the right timing.144Hence, an advanced drug release strategy that is capable of delivering several drugs: (i) simultaneously, (ii) one after another, or (iii) by delayed release, by withholding them in storage to be dormant for a pre-programmed time to permit a later release is proposed. Firstly, to develop multiple DD concept based on TNT, we have designed our implants for LLD with bi- or tri-drug co-delivery of selected drugs. The concept is presented in Fig. 13. The system is built on the combination of unique geometrical features of nanotubes and polymeric micelles as drug carriers specifically placed in layered fashion inside the nanotubular structures (Fig. 13a,b). The idea is based on the formation of two or more immiscible layers of drug carriers to create a series of sequential release in a time controlled manner. Drug carriers that have opposite interfacial properties (hydrophobic and hydrophilic), which are regular and inverted polymeric micelles, respectively, are selected as the most suitable candidates for this purpose. This combination can provide a desired formation of several polymer layers inside the nanotubes without inter-mixing. The immiscibility of these layers of drug carriers is proposed to generate a unique release pattern from nanotubes in successive and independent steps where the numbers of released drugs, their quantity, release time and release order can be controlled by the loading conditions, TNT dimensions and surface properties of the nano-carriers. Indomethacin (ind) (water insoluble anti-inflammatory drug) and gentamicin (gen) (water soluble antibiotics) were used as model drugs, namely, ind in regular micelles (TPGS) and gen in inverted micelles (DGP 2000), respectively. Inverted micelles were synthesized such that their opposite core–shell configuration and interfacial chemistry permit the dissolution of gen inside their hydrophilic center (drug cargo). A third drug was introduced as well, to accentuate the meaning of “multiple”. Itraconazole (itr), a water insoluble anti-fungal drug was encapsulated in TPGS micelles: ind-TPGS and itr-TPGS (1 or 2 drugs in 1 micelle) and ind-itr-TPGS. Inverted micelles were loaded first at the bottom of the TNT, followed by regular micelles on the top. Different mass ratios of these micelles were employed to investigate their effects on drug release.145
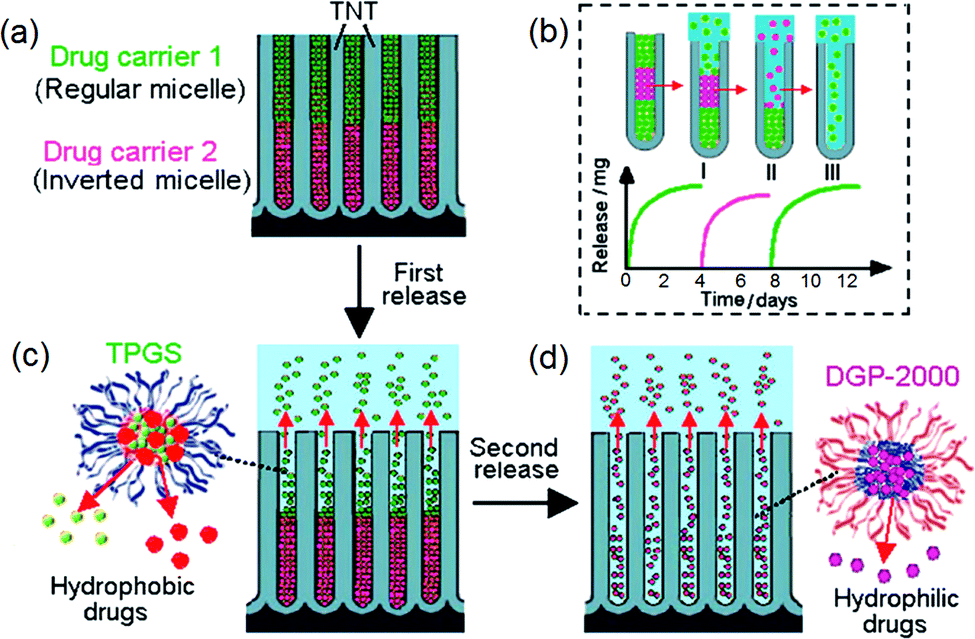 | ||
| Fig. 13 Scheme of local drug delivery with multiple drug release using titania nanotube arrays (TNT), showing: (a) TNT loaded with two types of polymeric micelles, a regular micelle (TPGS) encapsulated with two hydrophobic drugs (indomethacin and itraconazole) on the top and inverted micelle (DGP 2000) encapsulating hydrophilic drug (gentamicin) on the bottom; (b) the release in sequential order of the micelles loaded in two or three layers; (c) the release of nano-carrier with multi-drug from the first layer (TPGS), followed by (d) the release of DGP-gentamicin from the bottom layer in the second step. Reprinted with permission from ref. 121. Copyright 2012, The Royal Society of Chemistry (RSC). | ||
A typical release graph showing the TNT based multiple-drug delivery system loaded with two layers of drug carriers encapsulating three drugs are shown in Fig. 14. It presents the sequential release of regular micelle (TPGS) encapsulated with two hydrophobic drugs (ind and itr) loaded on the top, followed by the release of an inverted micelle (DGP) encapsulated with hydrophilic drugs (gen) loaded at the bottom. This sequential release is synchronized into two steps over a period of 10 days (5 days each), confirming the independent release of 2 + 1 drugs loaded within two carriers. Very sharp transition between releases of these two drug carriers was observed, showing that the release of the second drug occurred only after the drug loaded into the top carrier was completely released. This release pattern is explained by a high repulsive force that existed between the polymeric micelles having hydrophilic (TPGS) and hydrophobic outer layers (DGP 2000) which separate them into two different layers, resulting in their release profiles being independent and distinct from each other. The sequence of their release can be swapped by opposite loading, showing the flexibility of these systems in releasing hydrophobic and hydrophilic drugs in a different order. Nevertheless, there is a strict protocol in performing the loading of hydrophobic and hydrophilic drug carriers in subsequent order to prevent their inter-mixing inside the nanotubes.
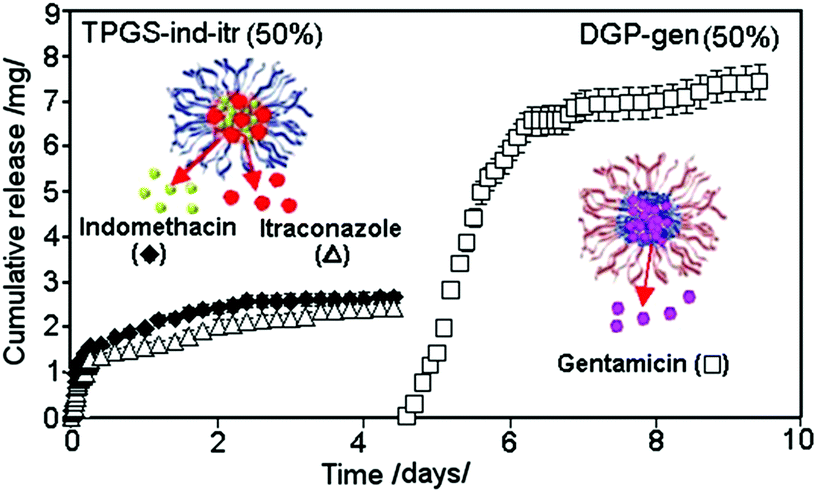 | ||
| Fig. 14 Sequential and multiple drug release from TNT implant loaded with two drug carriers, showing the first release from the top layer, with regular micelle (TPGS) encapsulating two hydrophobic drugs (ind and itr), followed by the second release after 5 days from the bottom layer loaded with inverted micelle (DGP 2000) encapsulating hydrophilic drug (gen). Reprinted with permission from ref. 121. Copyright 2012, The Royal Society of Chemistry (RSC). | ||
To prove that it is possible to control the sequence or time release of individual layers (drugs), we prepared several compositions using TPGS-ind and DPG-gen drug-loaded polymeric micelles with different loading proportions (25:75; 50:50, 75:25). Drug release graphs are summarized in Fig. 15. These show that the time of sequential release of two drug carriers is directed by their proportion inside the TNT. By changing the ratio of micelles, it was shown that the release time for each drug can be adequately controlled, e.g. for the second drug from 2 h to 6 h or 4 days, respectively, based on their ratio and amount loaded inside the TNTs. These results imply that the two micelles, i.e. TPGS with an outer hydrophilic layer and DGP 2000 with an outer hydrophobic layer were not intermixed inside the nanotubes. Their release is independent of each other, with the release outcome mimicking that of a nanoparticle with layer-by-layer drug loading. Sustained release with zero and first order kinetics were seen for both, which are desirable for combined immediate and long-term delivery in bone therapies.146
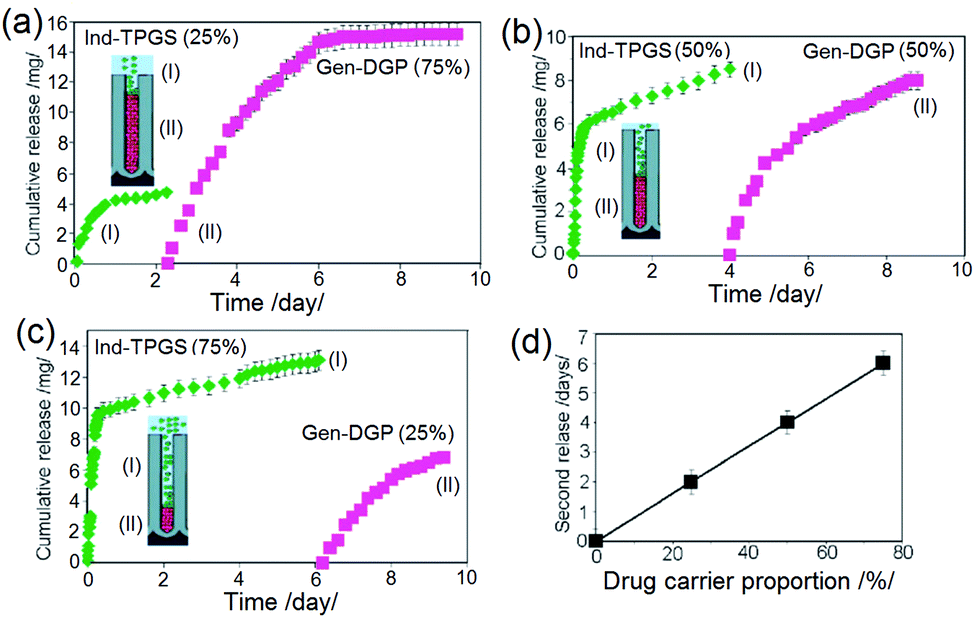 | ||
| Fig. 15
In vitro drug release characteristics of two drug carriers, including TPGS loaded with indomethacin and DGP 2000 loaded with gentamicin which demonstrated sequential drug release from TNT implants. Three different proportions of micelles inside the nanotubes, with ratios of (a) 1:1 (b) 3:1 (c) 1:3 were used to control the drug release rate and release time. (d) Graph depicting the linear time dependence of release for the second drug with respect to the proportion of drug carrier (micelles inside the TNT implants). Reprinted with permission from ref. 121. Copyright 2012, The Royal Society of Chemistry (RSC). | ||
Another approach based on micelles and TNT is a time-delayed, programmable LDD to provide localized delivery at lagged times (drug released at a later stage).147 The design for this approach closely resembled that of multidrug DDS mentioned previously and is presented in Fig. 16a. The only difference is that the micelles in this case comprise of a combination of unloaded (no drug) empty (blank) micelles, plus single drug-loaded micelle (1 drug for 1 micelle). Delayed DD was experimented using ind in Pluronic, TPGS and PEO–PPO–PEO micelles released from TNT. The ratios of empty micelles to drug loaded micelles (1:3, 1:1, 3:1) were designed such that the upper TNT channels were filled with empty micelles, whilst lower regions in the TNTs were filled with drug-encapsulating micelles. The drug release graphs from this system are presented in Fig. 16. Drug release pattern can be described in a biphasic model, whereby an initial burst occurred in the first 6–8 h (empty micelles, no drugs), followed by 7–10 days of slower release (depending on the “empty to drug-filled micelles” ratio, drugs were only released in the last few days). Empty micelles without drugs at the upper part of TNT can be perceived as a suppressing agent to eliminate burst release, as they act as a diffusion barrier, and produce no drug release for the first few days, thus providing a delayed DD. By intentionally postponing the release using this approach, therapeutic effects can subsequently occur at a later stage. This strategy is particularly important for designing drug-eluting implants in orthopedics and bone therapies, for treatments that require the release of drugs at a desirable postponed time after surgeries to suppress bacterial/viral infections or to enhance bone integration.
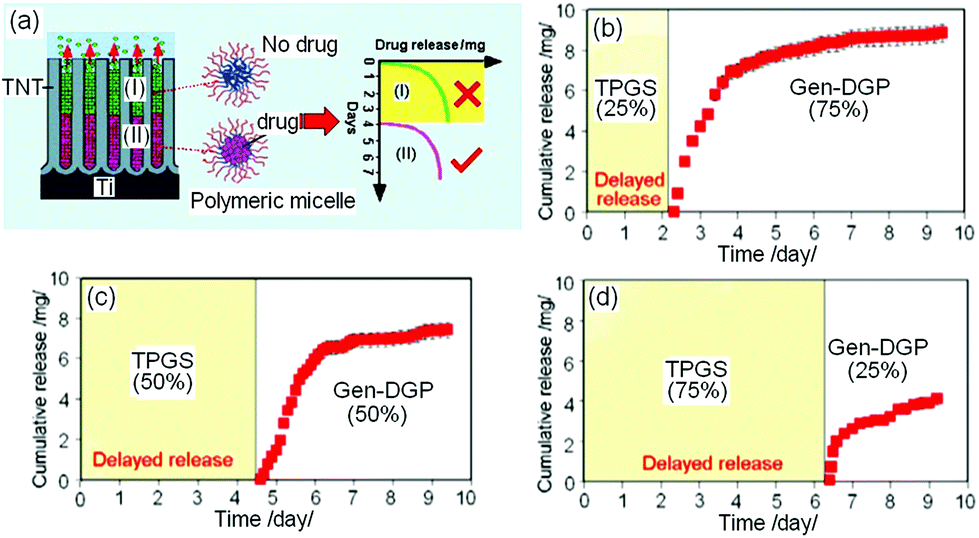 | ||
| Fig. 16 (a) Scheme showing the delayed release from TNTs with empty polymeric micelles at the top layer of the nanotubes carrying no drugs; whilst the immiscible, bottom layer of micelles (of another type) were encapsulated with drugs, giving rise to the delayed release of therapeutics. Release characteristics shows the tunable delayed time by controlling the ratio of empty micelles (blank TPGS) and drug-loaded micelles (DGP-gen) with ratios of (b) 1:3 (c) 1:1, (d) 3:1. Reprinted with permission from ref. 145. Copyright 2012, John Wiley & Sons, Inc. | ||
A similar concept with multi-drug and sequential release was explored on other LDD vehicles such as multifunctional core–shell mesoporous silica prepared via photo-induction. A successful sequential release of various bioactive compounds can equally be accomplished, in which this system permits the channeling of drugs into living cells in vivo from lipid-covered mesoporous silica nanoparticles with covalently bound photosensitizers as membrane-opening agents.148
5. External stimuli for responsive and on-demand drug delivery
All of the previously demonstrated LDD strategies are focused on providing extended and optimized drug release for long-term therapies. However, their applications are inappropriate in critical conditions where high concentrations of drug are immediately required, such as during situations where sudden viral attack, unexpected onset of inflammation, osteomyelitis or septic arthritis occur. Therefore, to address these emergency conditions, the ability to control the delivery of therapeutics from open-looped, externally triggered delivery system is recommended. Many investigations were explored to increase the therapeutic efficacy in the required timeframe, which is sometimes narrow and involves high dosage with precise timing, or that is needed only for a short period to treat urgent diseases or unpredictable pathogenic attack.149,150 So far no solutions have been found to deliver pre-determined doses of drugs that can be administered instantly upon demand from implants with controlled pharmacokinetic parameters. Drug vehicles which are potentially useful for this purpose, including nano-carriers such as hydrogels,151 dendrimers,152 cyclodextrins,153 and quantum dots,154 however, could not fully meet the criteria, due to instability of the substrates, ease of contamination, possible defective formation, complex and long synthesis process under restricted conditions. Herein, we show several concepts for non-intrusive and controllable drug release from TNT using (1) magnetic field, (2) ultrasound and (3) radiofrequency as the external trigger for stimuli-responsive LDD in our implants.5.1. Magnetically triggered drug release
A strategy for prompt release of drugs based on using magnetic field as an external stimulus for triggering magnetic nanoparticles (MNPs) loaded inside the TNT (beneath the layer of micelles with encapsulated drug) was proposed. The conceptual scheme of magnetically stimulated release is shown in Fig. 17a. The system comprised of TNT loaded with drug encapsulated polymeric micelles at the top acting as drug carriers and magnetic nanoparticles (NP) at the bottom of the nanotubes. The stimulus-release concept is based on applying a magnetic field to induce the movement of magnetic particles from the bottom which forced the drug-loaded micelles to be released from the implant into PBS whenever a magnetic field is applied. Iron oxide magnetic nanoparticles with their outer layer modified with dopamine functioning as a soft interfacial cushion to improve the biocompatibility of the MNPs were loaded inside the TNT. Three drug-loaded micelles (Pluronic, TPGS and PEO–PPO–PEO) were explored to study the concept of magnetically triggered DDS. The in vitro stimulated release pattern is shown in Fig. 17b,c.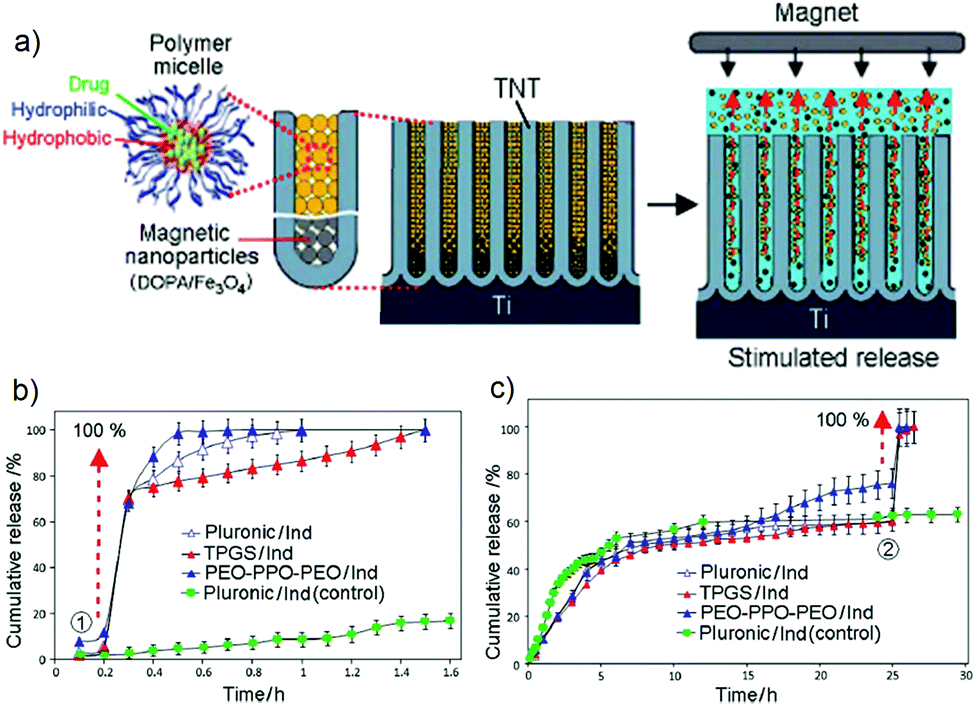 | ||
| Fig. 17 (a) Scheme of the magnetic stimuli-responsive drug release from TNTs which integrates polymeric micelles as drug carriers, incorporated with poorly soluble drugs and magnetic nanoparticles loaded at the bottom of the nanotubular structures. The release is achieved by applying an external magnetic field. (b) Release profile of magnetic field triggered release of drug (ind) encapsulated polymeric micelles (TPGS, Pluronic and PEO–PPO–PEO) at different stages of release process, i.e. at the beginning (0.25 h), and (c) during 50–60% of release (25 h). Non-triggered release with Pluronic-ind was used as the control for comparison. Reprinted with permission from ref. 155, Copyright 2012, The Royal Society of Chemistry (RSC). | ||
In the first case, when the magnetic field was applied during the much earlier stage of release, 100% drug release occurred within the normal burst release period (Fig. 17b). The graph confirms that immediate 100% release of all three drug nano-carriers was achieved within 1–1.5 h after a magnetic field was induced. Minor differences in release graphs observed may be attributed to the non-uniformity in the type (size) of the micelles used. The cumulative drug concentration released from the control sample via unforced diffusion from the nanotubes reached only less than 20% under the same condition. To prove that triggered release can be performed during arbitrary times of release from the TNT, in which the actual normal (unforced) release usually takes 10–25 days, a second experiment was performed by applying a magnetic field after 50–60% of drug carriers were released from the nanotubes (Fig. 17c). This graph again confirms that 100% release of loaded drug carriers from TNT was achieved within a short time upon exposure to a magnetic field. Thermogravimetric analysis (TGA) proved that after magnetic triggering, the nanotubes were completely emptied at the end of the release experiment. Even though the distribution of magnetic nanoparticles inside the TNT was not determined, these results indicate that micelles are located at the top and magnetic NP at the bottom of the nanotubes.155 TNT are used here as model structures, but the approach is generic and can be applied to other materials with nanoporous structures as well.
Other governing factors for controlling triggered release in this particular type of LDDS include magnetic field strength, direction, inherent NP properties (depending on the type, structure, size and chemical composition used), the amount of MNPs and micelles loaded at the bottom and the top of TNT.156 Sources to enhance DD based on physical modulation that is similar to this, such as magnetic nanoparticles and micro-needles, have been explored for switchable DDS.157–159 However, there remain minor complications and safety concerns arising from foreign source that are likely to cause accidental release or leakage of drugs from these systems.160 Therefore, to devise an improved and at the same time, non-intrusive stimulus-responsive system, an alternative source was otherwise explored for stimuli responsive DDS, namely for safe and prompt release of drugs at the site of action.
5.2. Ultrasonically triggered drug release
To address the limitations of magnetic-field stimulated release that may result as a consequence of uncontrolled release triggered by existing magnetic fields from the environment; we explored the application of local ultrasonic field which is expected to be a more reliable method. This technology has already been widely used in medical diagnostics and its benefits include allowing the body to receive a diagnostic signal from echogenic contrast agents and facilitate guided movement of nanoparticles based DDS into cells for drug/gene release.161,162 The concept of using ultrasonic waves (USW) as the trigger for stimulus-responsive LDDS combining TNT implants is presented in Fig. 18a. The principle of this proposed concept for ultrasound-mediated drug-micelles release was based on exerting oscillating pressure waves from a probe (Sonotrode) inserted in the medium (PBS at pH 7.2) close to the drug-micelles loaded TNT. Indomethacin, a non-steroidal anti-inflammatory drug, was used as the model for water insoluble drug encapsulated in TPGS micelles. Several USW parameters were explored to control the drug-micelles release, including pulse length, pulsation time, amplitude and power intensity. The amount of released drug carriers from TNT into PBS during the generation of USW was monitored closely in vitro, and measurements were obtained using UV-Vis spectroscopy and TGA. Fig. 18b shows the in vitro release profiles of drug-micelles from TNT implants triggered by USW using different pulse lengths (long or short pulse waves), generated based on different number of pulses per unit time, i.e. 1, 5, 10 and 15 pulses per min, in comparison with the free diffusion release without trigger (control sample). Nevertheless, these two external stimulants dramatically altered the sustained release pattern (previously obtained by imparting a drastic, quick and immediate drug-micelle release), whereby drug-micelle release can be arbitrarily completed from as brief as 10 min to as long as 2 h, depending on USW parameters (amplitude, frequency, and pressure) and the timeframe (arbitrary moment throughout the drug-micelle release process for USW field to be applied) on the LDDS. Also, USW was found to enable stepwise and pulsatile drug release (graph not shown due to data congestion). Interestingly, micellar size characterization and dynamic light scattering (DLS) studies revealed that, neither the externally applied magnetic field nor USW had sufficient rigor to cause rupture of the micelles to release drugs whilst inside or outside the nanotubes,163 contrary to the past literature report which claimed that ultrasonic irradiation had caused sequestration of drugs in tumors.164–166 These different types of releases, i.e. rapid-rising (magnetic field) or cyclic, pulsatile/stepwise (USW-mediated) characteristics stimulated by USW, are potentially useful for urgent remedial measures during medical emergencies and applicable as orthopedic implants in bone therapies, stents or brain therapy. Further studies on ex vivo and in vivo models using different drug-releasing implants and model drugs are required to provide more information on the practical applications of this method.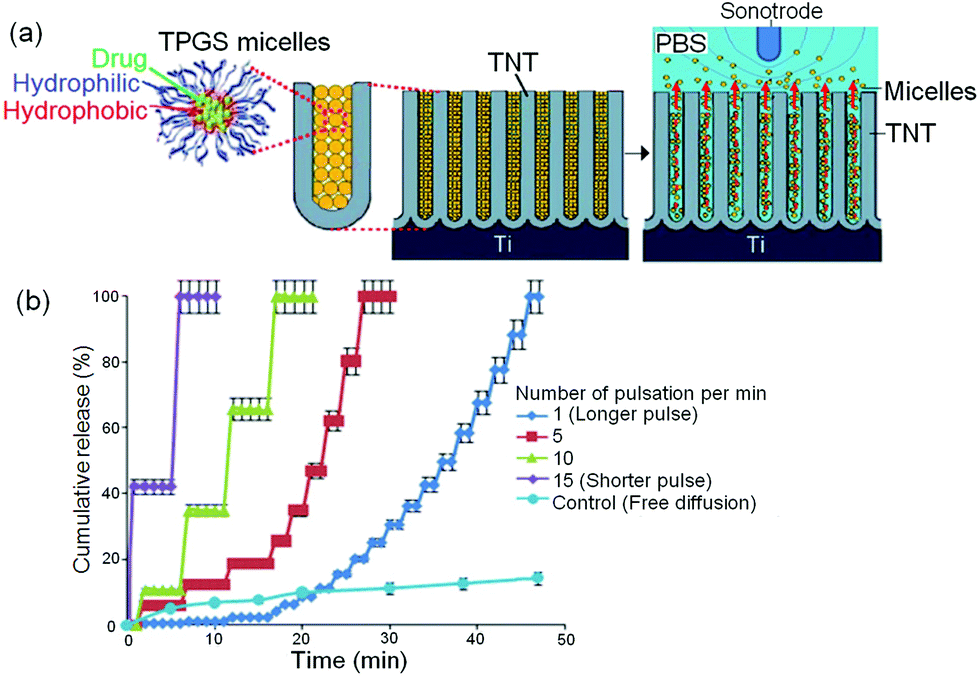 | ||
| Fig. 18 (a) Scheme of ultrasound-stimulated drug delivery (DD) based on titania nanotube (TNT) implants and polymeric micelles as drug carriers. (b) Release profiles of ultrasound stimulated release of drug-micelles from TNT implants, showing the effects of pulse length varied by the number of pulsation, i.e. 1 (long), 5, 10, 15 (short) pulse per min generated by the ultrasonic device. Reprinted with permission from ref. 163. Copyright 2012, Elsevier. | ||
This concept of ultrasonic-driven DD has been applied in other types of nano-carriers, for example, liposomes – a LDD vehicle that is thermosensitive, with the aid of MRI contrast agent such as iron oxide nanoparticles that are loaded with drugs (therefore allowing their tracking and/or visualization by MRI).167,168 An external thermal source derived from high intensity ultrasound controlled by MR temperature mapping demonstrated a viable, non-invasive technique to generate local hyperthermia for drug release. Nevertheless, the subject on drug vehicle stability for liposomes, factors that could affect release rate and drug amount inside the thermo-sensitive magneto-liposomes remains controversial and thus has to be thoroughly investigated before it can become a tool for image-guided local drug delivery.
5.3. Radiofrequency triggered drug release
To use a more convenient and non-invasive source of external stimulus, radiofrequency-responsive release is yet another very attractive strategy amongst the various stimuli-responsive DDS approaches available. This technology is closely related to its existing application in biomedical radiography. The concept of LDD using this strategy is shown in Fig. 19. Radiofrequency (RF) is used as a trigger for the release of polymeric micelles and drugs from TNT, with gold nanoparticles (AuNPs) acting as a stimulant and an energy transmitter (Fig. 19a). Radiofrequency refers to the electrical oscillations in the frequency range of 3 kHz–300 GHz, which is non-invasive and non-ionizing and hence safer for use, as compared to laser or infrared modules for transmitting and receiving electromagnetic wave signals. They are better energy absorbers and can travel through a larger distance than the rest.169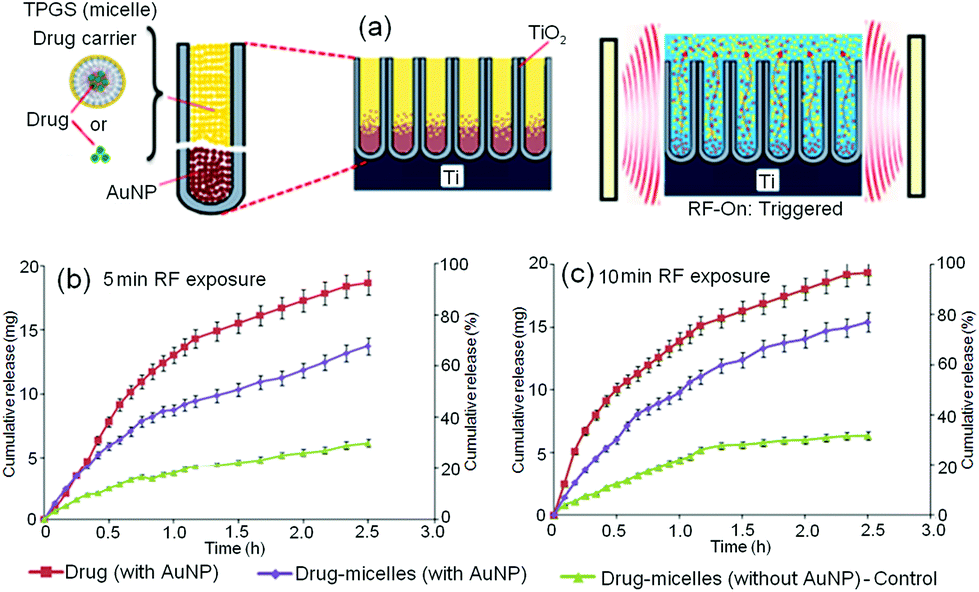 | ||
| Fig. 19 (a) Scheme of non-invasive and on-demand triggered release from drug-eluting TNT implants using radiofrequency (RF) and gold nanoparticles (AuNPs). TNT were used as a model of drug-releasing implant for in vitro release studies triggered by RF, demonstrated using drug (indomethacin) and drug carriers (TPGS – polymeric micelles loaded with drug). (b) Profiles of RF-triggered release of drug (indomethacin encapsulated TPGS) from TNT with and without AuNPs as energy transducer in comparison to the control (non-trigger) sample. Release profiles for different exposure times: (b) 5 min and (c) 10 min respectively. Reprinted with permission from ref. 169. Copyright 2012, Future Medicine Ltd, UK. | ||
Under an externally applied RF field, the polymeric micelles demonstrated an abrupt in vitro release as compared to slower release under no-trigger conditions (control sample). Fig. 19b,c shows the drug–drug nano-carrier release for both cumulative percentage (%) and mass (mg) with respect to time from the TNT implant subjected to different RF exposure times, such as 5 min and 10 min. In the first case, RF was applied to the samples (drug-micelles without AuNPs – control, drug-micelles with AuNPs and drug with AuNPs) at the beginning of the release. The graph confirms that immediate and near complete (90–100%) release of all three samples was achieved within 1–3 h after RF-induced heating of the gold nanoparticles loaded and stored at the bottom of the TNT. A difference in release was observed, with drug together with AuNPs loaded inside TNTs released the fastest, followed by drug-micelles together with AuNP, and lastly, drug-micelles without AuNPs producing the slowest release. Also, by increasing the RF exposure time from 5 to 10 min, there is an increase in the rate and quantity of drug and drug-micelles released into the PBS. Gold nanoparticles are good thermal transducers to transfer radiofrequency energy to the drug/drug-loaded micelle and trigger the release of micelle/free drug to stimulate drug release. Contrarily, the control sample (without AuNPs) did not show any acceleration in the drug release process. Drug and drug-micelle release in the control sample occurred very slowly through natural free diffusion without any stimulation over a period of ∼2 weeks. This unforced release from nanotubes reached around 30% as compared to the RF-induced samples within the 3 h experiment. In practice, the control sample took about 15–20 days to complete release (data not shown due to incompatible time scale in weeks).
The localized heating of AuNPs occurs in a manner similar to the phenomenon observed in RF-induced hyperthermia and thus has much potential for application in the ablation of cancer cells at the implant location.170,171 The power channeled from the generator and transferred to our samples was merely 20 W (low range), and hence is completely safe as it did not damage or alter the micelle structure or the TNT implant. The increase in temperature was only applicable to the AuNPs, which implicitly contributed to the accelerated diffusion and subsequent release of therapeutics. This reflects its selectivity in stimulation and non-invasiveness. Therefore, this remotely triggered and safe DDS is excellent for precise targeting with a good control in dosage for stimulated release within a short time, and it can be implemented at any time when treatment is required. Although the method is demonstrated using two cases, such as water insoluble drugs (indomethacin) and drug carriers (TPGS micelles), it is a generic approach and there is no limitation for other therapeutics (e.g. antibiotics, anti-inflammatory drugs, anti-cancer etc.) to be applied, in which bioactive compounds can be locally released to address emergency conditions, where immediate release of high concentrations of therapeutic agent is required at unsolicited times.
6. Ex vivo study of drug release characteristics
In previous strategies, all studies were performed in vitro using phosphate buffered saline (PBS) as the drug accepting medium for the NAA and TNT implants. This is, however, considerably different from clinical practice where real bone tissues and real biological environment are examined. The delivery of drugs to specific skeletal sites is a challenge and because of this, bone diseases are a major health problem worldwide. They have a highly deleterious effect on both the qualities of life for patients and financial health expenses, representing >10% of annual health care cost in developed countries.172 A number of therapeutic approaches have been established to treat bone diseases, and these are dominated by the use of systemic drug administration. However, conventional systemic drug therapies in general and in bone have yet to eliminate low efficacy, overcome poor bioavailability and biodistribution, drug overdose and toxicity in non-target tissues.173,174 To address these issues and to perform drug release experiment in real bone environment, we designed an ex vivo set-up using a 3D bone bioreactor and TNT-Ti as the drug-loaded implants (with the Ti wire measuring at 10 mm in length, TNT fabricated with its pore width at 140 nm, tube length at 50 μm). Rather than using implants with planar surface (Ti foil), here we used TNTs prepared on Ti wires, which are deemed a better modality instead, because they are geometrically more favorable and convenient for insertion into bones. A Ti wire with TNTs on a curved surface is used to devise a viable implant that occupies relatively much lesser space, with long thin geometrical extension that is accessible to narrow areas. These Ti wires, similar to K-wires used in standard orthopedic surgeries, are tiny, compact, prodding devices that enable the loading, storage and delivery of drugs in bones, as well as for extended drug release.175Bone is a complex porous material consisting of a solid bone matrix and pore spaces. There are three major types of bone cells – osteoblasts, osteoclasts and osteocytes. To date, it is not known how drug release in solution relates to drug release in bone tissues, given that cancellous bone is a highly hydrated tissue, with substantial interstitial fluid surrounded by marrow with a high fat content.176 Hence, we sought the use of an ex vivo bone bioreactor, comprising a trabecular bone explant as an alternative. Several three-dimensional (3D) bone models have been developed to study the biological aspect of bone cell behavior and bioengineering of bone tissues, including the Zetos™ system. The Zetos™ is an exclusive system jointly devised by David Jones (Department of Experimental Orthopedics and Biomechanics, Philipps University of Marburg, Marburg, Germany) and Everett Smith (Medical Sciences Center, Madison, WI) to enable real-life simulation of cancellous bone.177–179 The purpose is to maintain the bones outside the body for approximately 3 weeks by continuous perfusion with nutrient-containing culture medium and daily loading with physiologically relevant strains mimicking that of their biological conditions. The Zetos™ bone bioreactor was used for ex vivo study of drug distribution in bone, for the first time, to demonstrate the application of TNT as drug-releasing implants for LDD. The TNT on Ti wires implants were loaded with fluorescence dye (rhodamine B (RhB)) as a model drug and inserted into the center of trabecular bone discs (Fig. 20a–c), which were placed inside the bone bioreactor with continuous release of RhB monitored by repetitive capturing of the bone images using The Xenogen IVIS® 100 Bio Photonic Imaging® (Fig. 20d) microscopy.
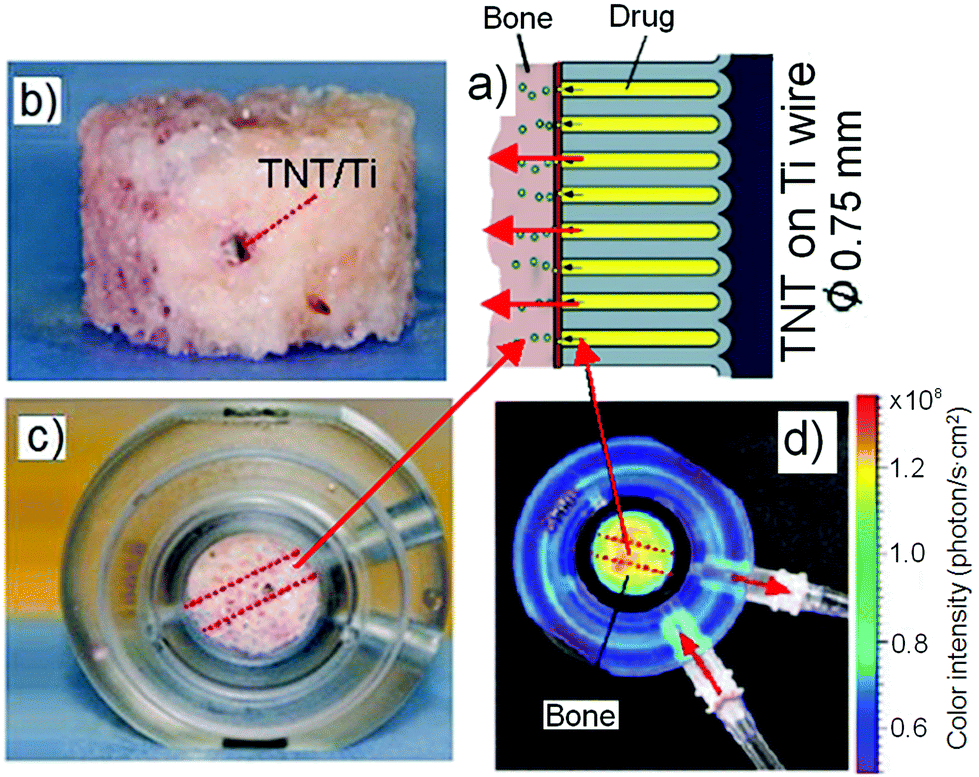 | ||
| Fig. 20 (a) Schematic diagram of in situ drug release from a TNT/Ti wire drug-releasing implant in a trabecular bone core model; (b) real image of trabecular bone model with embedded TNT/Ti wire implant at the center of the trabecular bone core (side view); (c) trabecular bone core and the embedded TNT/Ti wire implant fastened inside the bone chamber; (d) bioluminescent image of the bone core and the embedded implant inside the perfusion chamber to probe the concentration of the model drug (rhodamine B) released from the implant. Reprinted with permission from ref. 175. Copyright 2012, Dove Medical Press Ltd. | ||
Results show an increasing concentration of the model drug within the 3D bone matrix. A series of drug concentration profiles in bone were obtained from these images and summarized in Fig. 21, which show the distribution of drug concentration in bones across all directions (x, y, and z axes) from the TNT on Ti wire implant. The graphs show significant changes in drug (RhB) concentration in bone over time, confirming that both processes, i.e. the diffusion of drug molecules from the TNT implants and distribution into bone have occurred. Initially, the release of drug molecules followed burst release kinetics, and the highest concentration was observed at locations close to the implant surface, with zero concentration at greater distances from the implant. Over time, as more drug molecules were released, the concentration increased further from the implant, showing their distribution across the bone tissue in all directions (results for only two directions, vertical and horizontal, are presented). After 4 and 5 days of release, the highest concentration was observed at the outer locations of the bone, which was 3.5–5 mm away from the surface of the implant, showing a concentration of nearly 15–20 mg. After 5 days of release, there was no measurable drug remaining in the TNT/Ti wires, as confirmed by TGA, which is consistent with the diffusion of drug molecules into the bone tissue. The graphs also show that the spatial distribution of drug in bone is not uniform, which is likely explained by the variable nature of the internal bone microarchitecture and the influence of culture medium flow inside the bone chamber. This method has the potential to provide new knowledge to understand drug distribution in the complex bone microenvironment and to considerably improve existing technologies for local administration of drugs in bone, including solving some critical problems in bone therapy and orthopedic implants.180
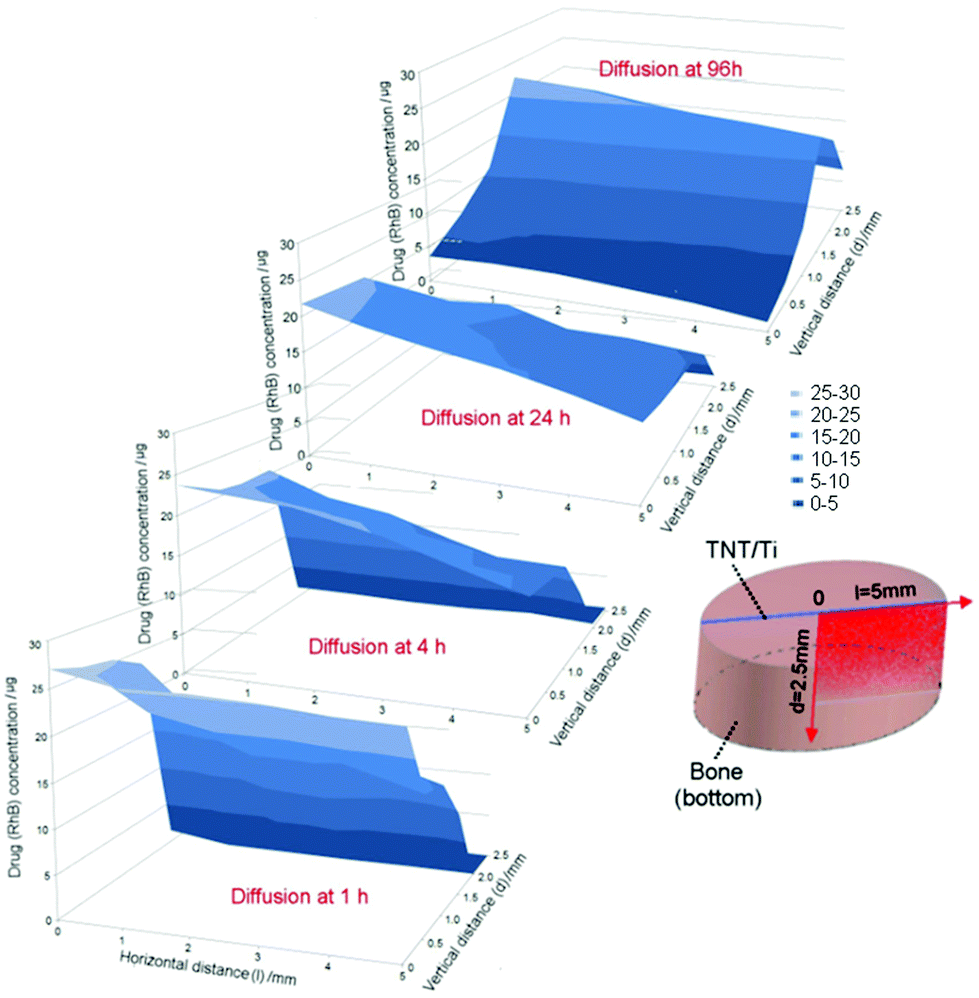 | ||
| Fig. 21 Three-dimensional concentration graphs of the model drug (rhodamine B [RhB]) released from TNT/Ti wires implants. The graphs show spatial distribution in the trabecular bone samples: drug distribution is presented at the vertical plane (x–z) of the bottom part of the bone and the horizontal plane (x–y) from the implant surface at different release times (1, 4, 24, 96, and 120 h). The concentration changes are presented only for a selected area of bone (one quadrant), as illustrated in the bone diagrams (bottom right). Reprinted with permission from ref. 175. Copyright 2012, Dove Medical Press Ltd. | ||
7. Biocompatibility: in vitro and in vivo studies
Biocompatibility is the most pivotal prerequisite for the application of new biomaterials and it is defined in terms of cellular response and tissue integration of implantable biomaterials. Whether or not all of the aforementioned concepts for controlling drug release in NAA and TNT implants are feasible, their clinical applications are mainly dependent on their biocompatibility, which is the ultimate and the most crucial question. In an orthopedic context, the inspiration for developing implants based on electrochemically anodized NAA and TNT originates from the fact that the human bone is, by nature, nanostructured and it comprises of inorganic particles within the same size range. Experiments reported in the past reveal that both nanostructured bone implants made from alumina and titania with nanophase surface topography and nano-fibers promote enhanced calcium deposition and alkaline phosphatase synthesis in osteoblast culture.181–183Aluminium and aluminium oxide are in general recognized as biocompatible materials that are being widely used in orthopedic proteases,184 dental185 and coronary stents,186 bone in-growth187 and blood implants for immuno-isolation.188 The ease of material handling, scalable and low manufacturing cost of NAA substrates are important features for their implant applications. In particular, NAA with pore size measuring between 30 and 80 nm showed favorable osteoblast adhesion, mimicking the natural process.189 Studies on the biocompatibility for the application of anodized alumina biocapsules in immunoisolation reported that the device is not only non-toxic, it also does not elicit significant complement activation of biological responses. Although further in vivo investigation on the implantation of these capsules into the peritoneal cavity of rats revealed a transient inflammatory response, it was found to be minimized by using poly(ethylene glycol) as an organic modifier.190 Surface roughness is one of the key factors that determine the degree of attachment of the implant surface to any biological implant site, as it is the proper cue to induce a positive osteoblast response to implants.191 Erin et al. reported that surface immobilization of RGDC peptides (arginine-glycine-aspartic acid-cysteine) on NAA further enhanced in vitro osteoblast adhesion and bone matrix formation within only two days of cell culture, as compared to unmodified NAA, which despite revealing osteoblast proliferation, did not show any apparent extracellular matrix formation.192 This finding is confirmed by other studies showing the ability of NAA to increase osteoblastic cell growth, and the cell proliferation of normal epithelial cells.193,194 Another important application of NAA was demonstrated as an eluting layer for a type of drug namely, tacrolimus – an immunosuppressive drug, used in coronary stents to suppress neointima proliferation and any possible restenosis. By using a rabbit restenosis model, the biocompatibility of these coatings was proved, showing the effective use of NAA in vivo without any inflammatory responses.195
Compared to titanium (Ti), titania (TiO2) is more highly regarded in the production of medical devices and implants by virtue of its excellent biocompatibility, robustness, mechanical strength and chemical stability. It is broadly applied as implants in orthopedics, cardiovascular surgery and dentistry, consisting about 40% of all medical implants. Most importantly, in regard to the implant application of TNT, it has a high affinity for bone cell adhesion and differentiation. Additionally, it is a favorable template for bone growth, bone support, is a remarkable surface for stem cell proliferation and hydroxyapatite (HAp) formation and is biocompatible due to its biochemical inertness.196,197 This is because TNTs are recognized as novel and promising substrates for developing drug-eluting implants for enhanced bone osseo-integration, interfaces for blood-contacting devices, vascular stents with better control over blood clotting and hemorrhage.198–200
The pioneering work reported by Schmuki's group demonstrated size-dependent interaction between mesenchymal stem cells with TNTs, showing that diameters of 15 nm strongly promote cell adhesion, proliferation and differentiation. However, nanotubes with diameters larger than 100 nm were found to be detrimental.201 Other biocompatibility research determining cellular responses of human osteoblast-like and MC3T3-E1 cells to TNTs in dentistry and bone tissue engineering, was performed by both Kim's and Advincula's groups.202,203 They have observed positive outcomes including enhanced ALP activity, increased amount of mineralized nodules, improved corrosion resistance and cell adhesion on TNT surfaces, re-asserting their biocompatibility properties. To assess if anodized TNTs can be suitable for use as a biomaterial in RMSC cells, similar studies were also carried out by Popat et al. and the outcome is in agreement with the first scenario.204 As shown in Fig. 22, marrow stromal cells (MSC) morphology on Ti and TNT surfaces were shown after 1, 4 and 7 days of culture. After 4 days of culture, despite the fact that the MSCs were still in spherical form on Ti surfaces (Fig. 22(c)), they were more widespread (Fig. 22(d)). Conversely, MSCs on TNT produced a network indicative of cell–cell communication (Fig. 22(f)), proving that they grow more rapidly on the TNT over Ti after 7 days of culture. High resolution SEM images of an MSC extension probing the nanotubes were shown in Fig. 22g,h. These extensions most likely helped the cell anchor itself to the TNTs, due to much greater length of the extension over the cell width; the cells therefore can adhere and propagate on the surface for improved long-term differentiation.
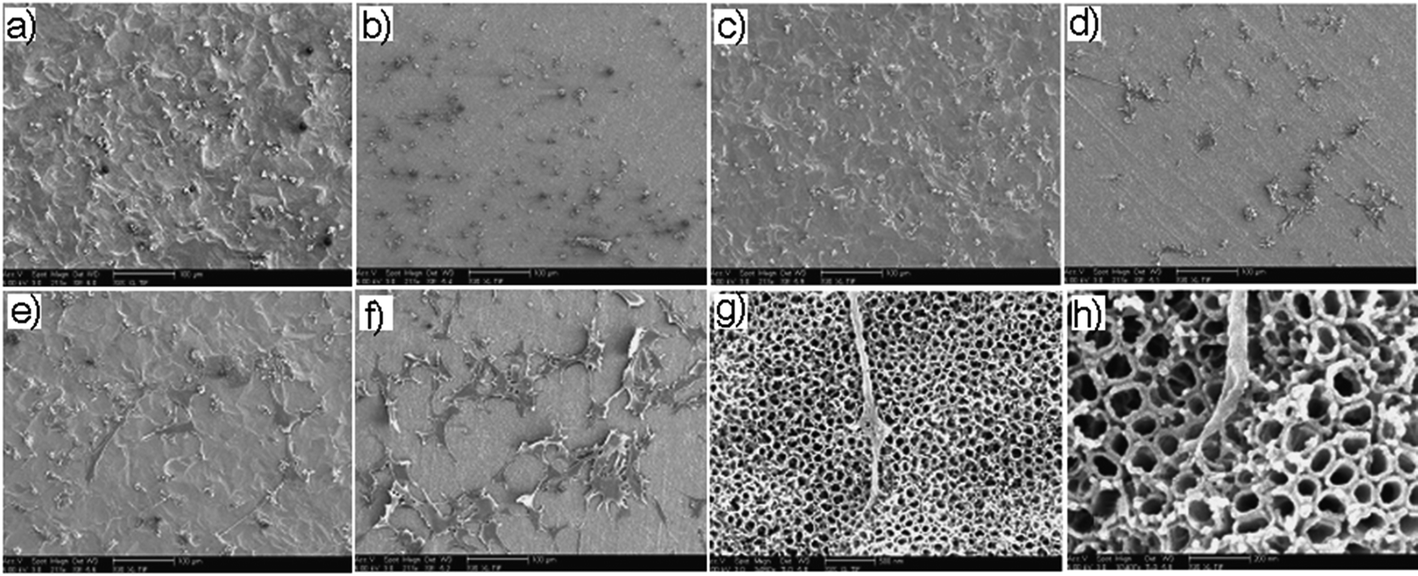 | ||
| Fig. 22 SEM images of marrow stromal cells on titanium and nanotubular surfaces (TNTs) for up to 7 days of cell culture. Cells show spherical morphology on Ti surface (a) compared to spreading morphology on TNT surface (b) after 1 day of culture. After 4 days of culture, cells still show spherical morphology on titanium surface (c) compared to spreading and clustering morphology on TNT surface (d) after 7 days of culture, some of the cells on titanium seem to be spreading (e) however the cells show high degree of spreading and have started communicating on nanotubular surface. (f) High magnification SEM image after 7 days of culture (g) TNT surface shows that cell extensions are protruding into the nanotubular architecture. Reprinted with permission from ref. 204. Copyright 2007, Elsevier, Inc. | ||
An in vivo study of TNT implants on pigs as the animal model was also performed by Schmuki and co-authors to investigate the effects of a TNT layer on bone formation.205Fig. 23 shows a bone-implant that was explored by immuno-histological analysis. This study showed enhanced peri-implant bone formation and confirms the ability of the TNT structure to stimulate bone formation and growth around the implant surface. The study also showed that after 90 days of implant insertion, the nanotubular layer stayed stable without any destruction. These studies clearly indicate the important features of TNTs toward their real in vivo applications. Moreover, another study displays two cross-sectional histology section with hematoxylin and eosin staining, comparing bone growth and bone-to-implant contact area. It showed that after four weeks of experimental implantation in rabbit tibias, pull-out testing indicated that TNTs have significantly improved bone bonding strength to the surrounding tissues, whereas their histological analysis confirmed an excellent bone-to-implant interfacial contact, new bone formation, and satisfactory levels of calcium and phosphorus content on the nanotube surfaces.206 All these case studies ascertain the safety of TNT and NAA as biocompatible implants and thus evidence their safe usage both in the in vivo and ex vivo contexts. More in vivo studies are required in the future to address many still unanswered questions related to biocompatibility, biofilm formation, and integration of a TNT layer with bone. Specifically, these novel concepts and the effect of the surface modification of TNTs on their biocompatibility and osteointegration properties need to be explored and proved by in vivo studies.
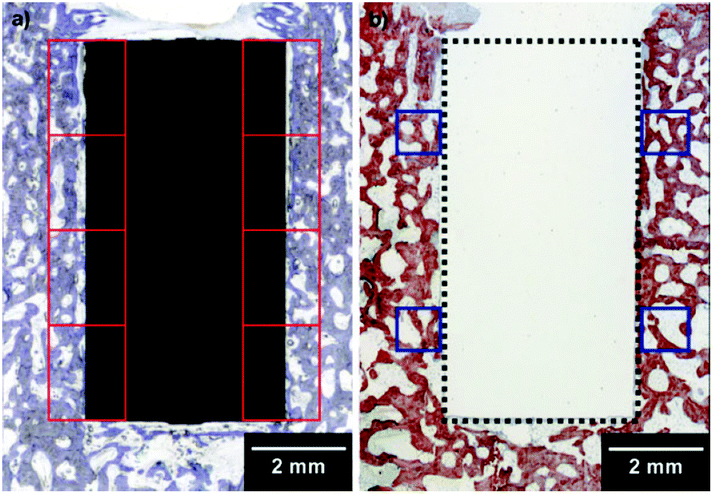 | ||
| Fig. 23 (a) Regions of interests for the histological evaluation of the bone-implant contact for immunohistological analysis. The dashed lines show the dimensions of the TNT implant. Reprinted with permission from ref. 206. Copyright 2008, Willey Periodical. | ||
8. Challenges, conclusions and perspectives
This review article presents a concise account of the concepts for controlling drug release from non-eroding drug-releasing implants with ordered nanoporous and nanotubular structures. In the search for effective, safe, predictable and reproducible DDS, TNT and NAA have proved to be suitable and attractive non-eroding implants for localized DD. From the drug-releasing strategies elucidated in this review, we learnt about the feasibility and the flexibility regarding the delivery of various drugs to meet different therapeutic demands, offering many reasonable and exciting prospects for TNT and NAA in localized DD applications. It was shown that the drug loading and drug release characteristics of non-eroding implants with nanopore and nanotube structures are dependent on the following parameters: the dimensions of nanopore and nanotube structures (pore size and length), their interfacial and chemical properties, molecular size, chemical and diffusion properties of drug molecules, as well as drug and drug–carrier interactions with the nanopores/nanotubes. It was found that a higher aspect ratio of nanotubes/nanopores provides prolonged release. The hydrophilic surface favors a higher drug payload and extend release time, whereby the properties of drug molecules are also an influencing factor. There are challenging problems associated with these materials that need to be addressed including drug incorporating efficiency and the elimination of burst release. This is because drug remnants might still be present on the top surface of these implants as detected in our in vitro studies using our drug loading method, i.e. repeated drop-wise loading of drugs under vacuum conditions. In order to achieve absolute clearance of the drug from the top surface of these implants and complete localization of drug molecules inside the nanotubes and nanopores, alternative drug loading methods may be explored, such as co-solubilization of drugs and polymers in nanoparticles based on compressed or supercritical fluid.207 Furthermore material properties, for instance, mechanical strength, elastic modulus, stress shielding, optimal skeletal load for these implants are not well explored and also need to be assessed by future studies.208,209The application of polymeric micelles as drug nano-carriers for encapsulating drugs was demonstrated to be an elegant approach to achieving extended and sustained drug release with zero-order kinetics. Controlling the pore opening and film thickness of NAA and TNT implants by means of precise deposition using different types of biopolymers (via plasma polymerization or dip-coating) with varied chemical composition and degradation properties was presented as an excellent approach for tuning the properties of implants with desirable characteristics. More studies are required to explore the possibility and limitation of these concepts to provide ultra-long drug delivery of more than 4–6 months. Another challenge here is the need to balance the dosage, drug release rate and quantities of drugs released that are clinically essential for drug administration during this time.
The development of LDDS to design multiple and delayed drug release characteristics using drug nano-carriers shows that by varying the ratio of drug-loaded carriers (i.e. micelles) and their position inside the TNT implants, it is possible to control their release time, the delayed duration, amount of drug released at a given time, release order and release kinetics. The system can be applied for both insoluble and/or water soluble drugs with immediate, delayed, sequential and sustained therapeutic actions. These approaches are generic as they can be applied for other biomaterials with porous structures currently used for local drug delivery (mesoporous silica and porous silicon). Specifically, the more advanced system possesses essential attributes to simultaneously address the complex requirements for bone therapies, wherein multi-drug treatment is required over a long period to prevent bacterial infections, suppress inflammation, improve implant integration and facilitate tissue regeneration. Even though the motivation for this approach is primarily to address challenges for bone implants, it can be applied for other implants and local delivery applications, for instance as coronary stents as well.
Several external stimuli sourced from magnetic field, USW and RF were discussed, showing the influence of triggering protocols to generate time-programmable or on-demand drug release. Non-invasive external sources for stimuli-responsive LDDS such as RF-triggered implantable drug release have the potential to either directly eliminate tumors through irradiation or render them more susceptible to therapeutics. These strategies are also expected to be viable for local chemotherapy in combination with systemic chemotherapy. The formulation of these porous-implant based LDDS with a plethora of stimulating/coating/modifying agents provides comprehensive release characteristics, such as prolonged, zero- or first-order, delayed, multidrug, sequential, stimulated and concurrent multiple drug release. Certain parameters which were not investigated in-depth in these studies remain to be quantified using in vitro and in vivo studies, for example, distance from external field to the implant, energy level and application rate, the size of the implant, its orientation inside the body, the influence of flow rate of physiological solution, drug dosage, size and interfacial properties of drug molecules etc. New mathematical models with advanced theoretical drug release equations would have to be computed to elucidate these conditions in conjunction with practical studies. In conclusion, these external stimulus strategies, even still at initial stage, can be described as promising for developing smart pharmaceutical drug-releasing implants, whereby the loading and release parameters can be customized so that the release characteristics are adjustable and can be regulated with different internal or external sources/approaches, not fixed or rigid such as other previously designed DDS.
The application of TNT implants incorporated into a Zetos™ three-dimensional bone bioreactor was successfully demonstrated as a characterization tool for the ex vivo study of drug release kinetics and drug distribution in trabecular bone core. Successful ex vivo experiments led to the conclusion that the release pattern and molecular disposition of drugs can be readily estimated in the bone core with respect to time and location. The study shows the potential of this technology to predict drug concentration and distribution in a real bone environment, which is not possible with currently existing in vitro models. Regarding the biocompatibility of these two materials, there is a considerable number of studies based on cells, ex vivo and in vivo animal models studies that proved their excellent biocompatibility, especially for TNTs implants. We envisage that these concepts can be realized once long term toxicity assay and tolerability studies are conducted on animals, before proceeding with human clinical trials encompassing plans for evaluating safety, undertaking physical examination to confirm negligible acute side effects pertaining to blank TNT and TNT loaded with drugs before real-life implantation of these materials in patients.210 Hence, more in vivo studies are urgently required before these LDDS can proceed into clinical trials.
In summary, non-eroding TNT drug-releasing implants of different forms (plates, wires, stents etc.) can be considered as a safe, scalable, inexpensive solution for localized drug delivery in a broad range of applications, including bone therapies, infection, inflammation, post-surgical healing, treatment of localized cancer, cardiovascular stents and other therapies in which an implantable device can be introduced. These unprecedented and multi-faceted LDD strategies using these devices to control the delivery of therapeutics locally with optimized concentration for a range of time scales from minutes, days, weeks, to a month, is expected to facilitate their smoother translation into clinical practice in the near future.
Acknowledgements
The authors gratefully acknowledge the financial support provided by the Australian Research Council (ARC) (DP 120101680 and FT 11010071), the University of South Australia, the University of Adelaide for this work. This work is part of M.S.A.'s PhD project. Contributions from the following group members are acknowledged including: S. Simovic, K. Gulati, S. Ramakrishnan, K. Kant, M. Bariana, T. Kumeria, Kamarul Ariffin Khalid, D. A. Findlay and G. Atkins.Notes and references
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. Chan, Expert Opin. Drug Deliv., 2007, 4, 621–633 CrossRef CAS PubMed.
- Series: Fundamental Biomedical Technologies, Multifunctional Pharmaceutical Nanocarriers, ed. V. P. Torchilin, SpringerLink, New York, USA, 2008, 4, 13–41 Search PubMed.
- A. S. Hoffman, J. Controlled Release, 2008, 132, 153–163 CrossRef CAS PubMed.
- Pharmaceutical Product Development-In Vitro-In Vivo Correlation, ed. M. D. Chilukuri, G. Sunkara and D. Young, Informa Healthcare, New York, 2007, 7, pp. 166–186 Search PubMed.
- J. B. Wolinsky, Y. L. Colson and M. W. Grinstaff, J. Controlled Release, 2012, 159, 14–26 CrossRef CAS PubMed.
- R. M. Mainardes and L. P. Silva, Curr. Drug Targets, 2004, 5, 449–455 CrossRef CAS.
- X. S. Zhao, J. Mater. Chem., 2006, 16, 623–625 RSC.
- G. S. Kwon and T. Okano, Adv. Drug Deliv. Rev., 1996, 21, 107–116 CrossRef CAS.
- R. Singh and J. W. Lillard Jr., Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed.
- H. Oonishi, H. Akiyama, M. Takemoto, T. Kawai, K. Yamamoto, T. Yamamuro, H. Oonishi and T. Nakamura, Acta Orthop., 2011, 82, 553–558 CrossRef PubMed.
- H.-H. Lee, U. S. Shin, J. H. Lee and H.-W. Kim, J. Biomed. Mater. Res. Part B: Appl. Biomater., 2011, 98B, 246–254 CrossRef CAS PubMed.
- H. A. Gad, M. A. El-Nabarawi and S. S. Abd El-Hady, AAPS PharmSciTech, 2008, 9, 878–884 CrossRef CAS PubMed.
- T. Arca, J. Proffitt and P. Genever, Biomed. Mater. Eng., 2012, 22, 261–276 CAS.
- M. Morra, C. Cassinelli, G. Cascardo, M. Fini, G. Giavaresi and R. Giardino, J. Orthop. Res., 2009, 27, 657–663 CrossRef CAS PubMed.
- N. Torio-Padron, J. Borges, A. Momeni, M. C. Mueller, F. T. Tegtmeier and G. B. Stark, Minim. Invasive Ther. Allied Technol., 2007, 16, 155–162 CrossRef PubMed.
- E. Luo, J. Hu, C. Bao, Y. Li, Q. Tu, D. Murray and J. Chen, Acta Biomater., 2012, 8, 734–743 CrossRef CAS PubMed.
- M. Kazemzadeh-Narbat, J. Kindrachuk, K. Duan, H. Jenssen, R. E. W. Hancock and R. Wang, Biomaterials, 2010, 31, 9519–9526 CrossRef CAS PubMed.
- J. M. Holzwarth and P. X. Ma, Biomaterials, 2011, 32, 9622–9629 CrossRef CAS PubMed.
- D. H. Betz, R. T. Epperson, B. M. Holt, R. D. Bloebaum and S. Jeyapalina, J. Histotechnol., 2012, 35, 164–170 CrossRef CAS PubMed.
- S. Krol, A. Diaspro, O. Cavalleri, D. Cavanna, P. Ballario, B. Grimaldi, P. Filetici, P. Ornaghi and A. Gliozzi, Nato Sci. Ser. II: Math. Phys. Chem., 2005, 152, 439–446 CrossRef.
- D. K. Kuechle, G. C. Landon, D. M. Musher and P. C. Noble, Clin. Orthop. Rel. Res., 1991, 264, 302–308 Search PubMed.
- N. Arden and M. C. Nevitt, Best Pract. Res. Clin. Rheumatol., 2006, 20, 3–25 CrossRef PubMed.
- L. De Filippis, S. Gulli, A. Caliri, C. Romano, F. Munaò, G. Trimarchi, D. La Torre, C. Fichera, A. Pappalardo, G. Triolo, M. Gallo, G. Valentini, G. Bagnato and Gruppo OASIS, Reumatismo, 2004, 56, 169–184 CAS.
- D. Shi, Biomedical Devices and Their Applications. Biological and Medical Physics, Biomedical Engineering, Springer-Verlag, Berlin, Heidelberg, 2004, XIV, 201, pp. 89–240 Search PubMed.
- O. Bostman, E. Hirvensalo, J. Makinen and P. Rokkanen, J. Bone Joint Surg. Br., 1990, 72-B, 592–596 Search PubMed.
- J. E. Bergsma, W. C. de Bruijn, F. R. Rozema, R. R. M. Bos and G. Boering, Biomaterials, 1995, 16, 25–31 CrossRef CAS.
- D. Losic and S. Simovic, Exp. Opin. Drug Deliv., 2009, 6, 1363–1381 CrossRef CAS PubMed.
- A. J. Thorley and T. D. Tetley, Pharm. Ther., 2013, 140, 176–185 CrossRef CAS PubMed.
- A. Ghicov, S. Aldabergerova, H. Tsuchiya and P. Schmuki, Angew. Chem., Int. Ed., 2006, 45, 6993–6996 CrossRef CAS PubMed.
- K. Shankar, J. I. Basham, N. K. Allam, O. K. Varghese, G. K. Mor, X. Feng, M. Paulose, J. A. Seabold, K.-S. Choi and C. A. Grimes, J. Phys. Chem. C, 2009, 113, 6327–6359 CAS.
- P. Schmuki, in Electrochemistry at the Nanoscale Nanostructure Science and Technology, ed. P. Schmuki and S. Virtanen, Springer, LLC, New York, 2009, pp. 435–466 Search PubMed.
- D. Mao, X. F. Xiao, C. Y. Wang, H. Z. Tang and R. F. Liu, Adv. Mater. Res., 2011, 335–336, 343–346 CrossRef CAS PubMed.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- D. Losic, L. Velleman, K. Kant, T. Kumeria, K. Gulati, J. Shapter, D. A. Beattie and S. Simovic, Aust. J. Chem., 2011, 64, 294–301 CrossRef CAS.
- K. Subramanian, D. Tran and K. T. Nguyen, Emerging Nanotechnologies in Dentistry, Elsevier Inc., 2012, ch. 8, pp. 113–136 Search PubMed.
- H. Jia and L. L. Kerr, J. Pharm. Sci., 2013, 102, 2341–2348 CrossRef CAS PubMed.
- M. Fazil, A. Ali, S. Baboota, J. K. Sahni and J. Ali, Exp. Opin. Drug Deliv., 2013, 10, 1123–1136 CAS.
- S. Bauer, S. Kleber and P. Schmuki, Electrochem. Commun., 2006, 8, 1321–1325 CrossRef CAS PubMed.
- S. P. Albu, D. Kim and P. Schmuki, Angew. Chem., Int. Ed., 2008, 47, 1916–1919 CrossRef CAS PubMed.
- Y. Y. Song, F. Schmidt-Stein, S. Bauer and P. Schmuki, J. Am. Chem. Soc., 2009, 131, 4230–4232 CrossRef CAS PubMed.
- A. M. Md. Jani, D. Losic and N. H. Voelcker, Prog. Mater. Sci., 2013, 58, 636–704 CrossRef CAS PubMed.
- B. S. Smith and K. C. Popat, J. Biomed. Nanotechnol., 2010, 8, 642–658 CrossRef PubMed.
- A. Ghicov and P. Schmuki, Chem. Commun., 2009, 2791–2808 RSC.
- C. A. Grimes, J. Mater. Chem., 2007, 17, 1451–1457 RSC.
- S.-H. Moon, S.-J. Lee, I.-S. Park, M.-H. Lee, Y.-J. Soh, T.-S. Bae and H.-S. Kim, J. Biomed. Mater. Res. Part B: Appl. Biomater., 2012, 100B, 2053–2059 CrossRef CAS PubMed.
- M. Vallet-Regí, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548–7558 CrossRef PubMed.
- S. Simovic, N. Ghouchi-Eskandar, M. S. Aw, D. Losic and C. Prestidge, Curr. Drug Discov. Technol., 2011, 8, 250–268 CrossRef CAS.
- J. L. Xu, F. Liu, J. M. Luo and Z. C. Zhong, Appl. Mech. Mater., 2012, 184–185, 1021–1024 CAS.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed.
- S. Angelos, M. Liong, E. Choi and J. I. Zink, Chem. Eng. J., 2008, 137, 4–13 CrossRef CAS PubMed.
- M. D. Popova, A. Szegedi, I. N. Kolev, J. Mihaly, B. S. Tzankov, G. T. Momekov, N. G. Lambov and K. P. Yoncheva, Int. J. Pharm., 2012, 436, 778–785 CrossRef CAS PubMed.
- E. J. Anglin, L. Cheng, W. R. Freeman and M. J. Sailor, Adv. Drug Deliv. Rev., 2008, 60, 1266–1277 CrossRef CAS PubMed.
- J. Salonen, A. M. Kaukonen, J. Hirvonen and V.-P. Lehto, J. Pharm. Sci., 2008, 97, 632–653 CrossRef CAS PubMed.
- J. M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer and P. Schmuki, Curr. Opin. Solid State Mater. Sci., 2007, 11, 3–18 CrossRef CAS PubMed.
- E.-L. Ticu, D. Vercaigne-Marko, R. Froidevaux, A. Huma, V. Artenie and D. Guillochon, Process Biochem., 2005, 40, 2841–2848 CrossRef CAS PubMed.
- S. Freiberg and X. X. Zhu, Int. J. Pharm., 2004, 282, 1–18 CrossRef CAS PubMed.
- M. Vallet-Regí, J. Int. Med., 2010, 267, 22–43 CrossRef PubMed.
- M. Vallet-Regí and F. Balas, Open Biomed. Eng. J., 2008, 2, 1–9 CrossRef PubMed.
- M. Vallet-Regí, I. Izquierdo-Barba and M. Colilla, Phil. Trans. R. Soc. A, 2012, 370, 1400–1421 CrossRef PubMed.
- K. C. Popat, M. Eltgroth, T. J. LaTempa, C. A. Grimes and T. A. Desai, Small, 2007, 3, 1878–1881 CrossRef CAS PubMed.
- J. A. Puértolas, J. L. Vadillo, S. Sánchez-Salcedo, A. Nieto, E. Gómez-Barrena and M. Vallet-Regí, J. Mater. Sci. Mater. Med., 2012, 23, 229–238 CrossRef PubMed.
- N. Ferraz, M. Karlsson Ott and J. Hong, Microsc. Res. Tech., 2010, 73, 1101–1109 CrossRef CAS PubMed.
- M. Xu, D. Feng, R. Dai, H. Wu, D. Zhao and G. Zheng, Nanoscale, 2011, 8, 3329–3333 RSC.
- M. Manzano and M. Vallet-Regí, Prog. Solid State Chem., 2012, 40, 17–30 CrossRef CAS PubMed.
- D. Molina-Manso, M. Manzano, J. C. Doadrio, G. Del Prado, A. Ortiz-Pérez, M. Vallet-Regí, E. Gómez-Barrena and J. Esteban, Int. J. Antimicrob. Agents, 2012, 40, 252–256 CrossRef CAS PubMed.
- P. Costa and J. M. S. Lobo, J. Pharm. Sci., 2001, 13, 123–133 CrossRef CAS.
- M. Colilla, B. Gonzalez and M. Vallet-Regí, Biomater. Sci., 2013, 1, 114–134 RSC.
- X. Y. Li, K. H. Nan, H. Chen and Y. Xu, Int. J. Pharm., 2011, 420, 371–377 CrossRef CAS PubMed.
- E. Gultepe, D. Nagesha, S. Sridhar and M. Amiji, Adv. Drug Deliv. Rev., 2010, 62, 305–315 CrossRef CAS PubMed.
- F. Martin, R. Walczak, A. Boiarski, M. Cohen, T. West, C. Cosentino and M. Ferrari, J. Controlled Release, 2005, 102, 123–133 CrossRef CAS PubMed.
- I. Izquierdo-Barba, Á. Martinez, A. L. Doadrio, J. Pérez-Pariente and M. Vallet-Regí, Eur. J. Pharm. Sci., 2005, 26, 365–373 CrossRef CAS PubMed.
- A. A. Ayon, M. Cantu, K. Chava, C. M. Agrawal, M. D. Feldman, D. Johnson, D. Patel, D. Marton and E. Shi, Biomed. Mater., 2006, 1, L11–L15 CrossRef CAS PubMed.
- L. Velleman, G. Triani, P. J. Evans, J. G. Shapter and D. Losic, Microporous Mesoporous Mater., 2009, 126, 87–94 CrossRef CAS PubMed.
- J. Andersson, J. Rosenholm and M. Linden, Multifunctional Biomater. Devices, 2008, ch. 6, pp. 1–19 Search PubMed.
- D.-H. Kwak, J.-B. Yoo and D. J. Kim, J. Nanosci. Nanotechnol., 2010, 10, 345–348 CrossRef CAS PubMed.
- D. Gong, V. Yadavalli, M. Paulose, M. Pishko and C. A. Grimes, Biomed. Microdevices, 2003, 5, 75–80 CrossRef CAS.
- P. Horcajada, A. Ramila, J. Perez-Pariente and M. Vallet-Regí, Microporous Mesoporous Mater., 2004, 68, 105–109 CrossRef CAS PubMed.
- M. Manzano, V. Aina, C. O. Arean, F. Balas, V. Cauda, M. Colilla, M. R. Delgado and M. Vallet-Regi, Chem. Eng. J., 2008, 137, 30–37 CrossRef CAS PubMed.
- C. Charnay, S. Begu, C. Tourne-Peteilh, L. Nicole, D. A. Lerner and J. M. Devoisselle, Biopharm., 2004, 57, 533–540 CrossRef CAS PubMed.
- J. Siepmann and F. Siepmann, J. Controlled Release, 2012, 161, 351–362 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2005, 19, 311–330 CrossRef CAS PubMed.
- L.-X. Wen, H.-M. Ding, J.-X. Wang and J.-F. Chen, J. Nanosci. Nanotechnol., 2006, 6, 3139–3144 CrossRef CAS PubMed.
- E. Ajami and K.-F. Aguey-Zinsou, J. Colloid Interface Sci., 2012, 385, 258–267 CrossRef CAS PubMed.
- R. De Palma, S. Peeters, M. J. Van Bael, H. V. den Rul, K. Bonroy, W. Laureyn, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef CAS.
- Y. Jin, S. Lohstreter, D. T. Pierce, J. Parisien, M. Wu, C. Hall III and J. X. Zhao, Chem. Mater., 2008, 20, 4411–4419 CrossRef CAS.
- R. Evans, U. M. B. Marconi and P. Tarazona, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 1763–1787 RSC.
- E. Charlaix and M. Ciccotti, in Capillary Condensation in Confined Media. Handbook of Nanophysics, ed. K. Sattler, CRC. Press, 2010, 1, p. 28 Search PubMed.
- S. Kapoor, R. Hegde and A. J. Bhattacharyya, J. Controlled Release, 2009, 140, 34–39 CrossRef CAS PubMed.
- P. W. Kämmerer, M. Gabriel, B. Al-Nawas, T. Scholz, C. M. Kirchmaier and M. O. Klein, Clin. Oral Impl. Res., 2012, 23, 504–510 CrossRef PubMed.
- E. H. Williams, A. V. Davydov, A. Motayed, S. G. Sundaresan, P. Bocchini, L. J. Richter, G. Stan, K. Steffens, R. Zangmeister, J. A. Schreifels and M. V. Rao, Appl. Surf. Sci., 2012, 258, 6056–6063 CrossRef CAS PubMed.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- B. Munoz, A. Ramila, J. Perez-Pariente, I. Diaz and M. Vallet-Regí, Chem. Mater., 2003, 15, 500–503 CrossRef CAS.
- M. Das, R. P. Singh, S. R. Datir and S. Jain, Bioconjugate Chem., 2013, 24, 626–639 CrossRef CAS PubMed.
- A. M. Md Jani, E. J. Anglin, S. J. P. McInnes, D. Losic, J. G. Shapter and N. H. Voelcker, Chem. Commun., 2009, 3062–3064 RSC.
- D. Losic, M. A. Cole, B. Dollmann, K. Vasilev and H. J. Griesser, Nanotechnology, 2008, 19, 245704 CrossRef PubMed.
- S. K. Jain, M. K. Chourasia, M. Sabitha, R. Jain, A. K. Jain, M. Ashawat and A. K. Jha, Drug Deliv., 2003, 10, 169–177 CrossRef CAS PubMed.
- S. Simovic, D. Losic and K. Vasilev, Chem. Commun., 2010, 46, 1317–1319 RSC.
- L. S. Nair and C. T. Laurencin, Adv. Biochem. Eng./Biotechnol., 2006, 102, 47–90 CrossRef CAS.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nature Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- A. P. Schulz, C. Jürgens and M. Faschingbauer, Internet J. Orthop. Surgery, 2007, 4, 1 Search PubMed.
- S. Y. Yang, J.-A. Yang, E.-S. Kim, G. Jeon, E. J. Oh, K. Y. Choi, S. K. Hahn and J. K. Kim, ACS Nano, 2010, 4, 3817–3822 CrossRef CAS PubMed.
- L. Leoni and T. A. Desai, Adv. Drug Deliv. Rev., 2004, 56, 211–229 CrossRef CAS PubMed.
- T. Desmet, R. Morent, N. De Geyter, C. Leys, E. Schacht and P. Dubruel, Biomacromolecules, 2009, 10, 2351–2378 CrossRef CAS PubMed.
- T. H. Tran, M. Rattu and B. Michael, US Pharm., 2011, 36, 11 Search PubMed , Epub.
- R. Virmani, A. Farb, G. Guagliumi and F. Kolodgie, Coronary Artery Dis., 2004, 15, 313–318 CrossRef PubMed.
- B. D. Ratner and A. S. Hoffman, Physicochemical Surface Modification of Materials Used in Medicine, in Biomaterials Science: An Introduction to Materials in Medicine, ed. B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Elsevier Academic Press, 2012, ch. 1.2, pp. 259–273 Search PubMed.
- B. Aksakal and C. Hanyaloglu, J. Mater. Sci. Mater. Med., 2008, 19, 2097–2104 CrossRef CAS PubMed.
- C. Ketonis, J. Parvizi and L. C. Jones, J. Am. Acad. Orthop. Surg., 2012, 20, 478–480 CrossRef PubMed.
- W. Tan, R. Krishnaraj and T. A. Desai, Tissue Eng., 2001, 7, 203–210 CrossRef CAS PubMed.
- K. Gulati, S. Ramakrishnan, M. S. Aw, G. J. Atkins, D. M. Findlay and D. Losic, Acta Biomater., 2012, 8, 449–456 CrossRef CAS PubMed.
- T. Xi, R. Gao, B. Xu, L. Chen, T. Luo, J. Liu, Y. Wei and S. Zhong, Biomaterials, 2010, 31, 5151–5158 CrossRef CAS PubMed.
- M. S. Aw, S. Simovic, J. Addai-Mensah and D. Losic, J. Mater. Chem., 2011, 21, 7082–7089 RSC.
- Y. Yan, J. Lu, C. Deng and X. Zhang, Talanta, 2013, 107, 30–35 CrossRef CAS PubMed.
- M. Paulose, L. Peng, K. C. Popat, O. K. Varghese, T. J. LaTempa, N. Bao, T. A. Desai and C. A. Grimes, J. Membr. Sci., 2008, 319, 199–205 CrossRef CAS PubMed.
- L. Zhao, L. Liu, Z. Wu, Y. Zhang and P. K. Chu, Biomaterials, 2012, 33, 2629–2641 CrossRef CAS PubMed.
- B. S. Smith, P. Capellato, S. Kelley, M. Gonzalez-Juarrero and K. C. Popat, Biomater. Sci., 2013, 1, 322–332 RSC.
- L. Xia, B. Feng, P. Wang, S. Ding, Z. Liu, J. Zhou and R. Yu, Int. J. Future Med.: Nanomed., 2012, 7, 4873–4881 CAS.
- K. Sternberg, S. Petersen, N. Grabow, V. Senz, H. M. zu Schwabedissen, H. K. Kroemer and K.-P. Schmitz, Biomed. Tech./Biomed. Eng., 2013, 0, 1–11 Search PubMed.
- M. I. Papafaklis, Y. S. Chatzizisis, K. K. Naka, G. D. Giannoglou and L. K. Michalis, Pharmacol. Ther., 2012, 134, 43–53 CrossRef CAS PubMed.
- M. S. Aw, K. Gulati and D. Losic, J. Biomater. Nanobiotech., 2011, 2, 477–484 CrossRef CAS.
- J. Panyam and V. Labhasetwar, Adv. Drug Deliv. Rev., 2012, 64, 61–71 CrossRef PubMed.
- D. L. Clemens, B.-Y. Lee, M. Xue, C. R. Thomas, H. Meng, D. Ferris, A. E. Nel, J. I. Zink and M. A. Horwitz, Antimicrob. Agents Chemother., 2012, 56, 2535–2545 CrossRef CAS PubMed.
- J. Shen and D. J. Burgess, Long Acting Injections and Implants, in Adv. In Deliv. Sci. and Tech., ed. J. Wright and D. J. Burgess, Springer US, Controlled Release Society Press, 2012, ch. 5, pp. 73–92 Search PubMed.
- C. Li, J. Deng and D. Stephens, Adv. Drug Deliv. Rev., 1996, 21, 107–116 CrossRef.
- M. Yokoyama and T. Okano, Adv. Drug Deliv. Rev., 1996, 21, 77–80 CrossRef CAS.
- N. Rapoport, W. G. Pitt, H. Sun and J. L. Nelson, J. Controlled Release, 2003, 91, 85–95 CrossRef CAS.
- A. V. Kabanov, E. V. Batrakova and V. Y. Alakhov, Adv. Drug Deliv. Rev., 2002, 54, 759–779 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Deliv. Rev., 2001, 47, 113–131 CrossRef CAS.
- L. Liu, K.-T. Yong, I. Roy, W.-C. Law, L. Ye, J. Liu, J. Liu, R. Kumar, X. Zhang and P. N. Prasad, Theranostics, 2012, 2, 705–713 CrossRef CAS PubMed.
- M. S. Muthu, S. A. Kulkarni, Y. Liu and S.-S. Feng, Future Med.: Nanomed., 2012, 7, 353–364 CAS.
- E.-M. Collnot, C. Baldes, M. F. Wempe, R. Kappl, J. Hüttermann, J. A. Hyatt, K. J. Edgar, U. F. Schaefer and C. M. Lehr, Mol. Pharm., 2007, 4, 465–474 CrossRef CAS PubMed.
- W. Xu, P. Ling and T. Zhang, J. Drug Deliv., 2013, 340315 CAS.
- A. N. Lukyanov and V. P. Torchilin, Adv. Drug. Deliv. Rev., 2004, 56, 1273–1289 CrossRef CAS PubMed.
- R. M. Sawant, J. P. Hurley, S. Salmaso, A. Kale, E. Tolcheva, T. S. Levchenko and V. P. Torchilin, Bioconjug. Chem., 2006, 17, 943–949 CrossRef CAS PubMed.
- R. Pjanović, N. Bošković-Vragolović, J. Veljković-Giga, R. Garić-Grulović, S. Pejanović and B. Bugarski, J. Chem. Tech. Biotechnol., 2010, 85, 693–698 CrossRef.
- D. Conway and J. A. Cohen, Lancet Neurol., 2010, 9, 299 CrossRef.
- M. Chowers, B.-S. Gottesman, L. Leibovici, J. M. Schapiro and M. Paul, Eur. J. Clin. Microbiol. Infect. Dis., 2010, 29, 779–786 CrossRef CAS PubMed.
- A. M. Jacobine, J. R. Mazzone, R. D. Slack, A. K. Tripathi, D. J. Sullivan and G. H. Posner, J. Med. Chem., 2012, 55, 7892–7899 CrossRef CAS PubMed.
- Y. Yan, C. J. Ochs, G. K. Such, J. K. Heath, E. C. Nice and F. Caruso, Adv. Mater., 2010, 22, 5398–5403 CrossRef CAS PubMed.
- L. E. van Vlerken and M. M. Amiji, Expert Opin. Drug Deliv., 2006, 3, 205–216 CrossRef CAS PubMed.
- S. Harada, S. Ehara, K. Ishii, K. Sera, H. Yamazaki, S. Matsuyama, T. Satoh, S. Oikawa, T. Kamiya and J. Ito, Int. J. Radiat. Oncol., 2008, 72, S710–S711 CrossRef PubMed.
- W. Wu, Q. Zheng, X. Guo, J. Sun and Y. Liu, Biomed. Mater., 2009, 4, 065005 CrossRef PubMed.
- R. Fattori and T. Piva, Lancet, 2003, 361, 247–249 CrossRef.
- M. S. Aw, J. Addai-Mensah and D. Losic, Chem. Commun., 2012, 48, 3348–3350 RSC.
- K. Gulati, M. S. Aw, D. Findlay and D. Losic, Ther. Deliv., 2012, 3, 857–873 CrossRef CAS.
- M. S. Aw, J. Addai-Mensah and D. Losic, Macromol. Biosci., 2012, 12, 1048–1052 CrossRef CAS PubMed.
- A. Schloßbauer, A. M. Sauer, V. Cauda, A. Schmidt, H. Engelke, U. Rothbauer, K. Zolghadr, H. Leonhardt, C. Bräuchle and T. Bein, Adv. Healthcare Mater., 2012, 1, 316–320 CrossRef PubMed.
- S. Savonitto, M. D'Urbano, M. Caracciolo, F. Barlocco, G. Mariani, M. Nichelatti, S. Klugmann and S. De Servi, Br. J. Anaesth., 2010, 104, 285–291 CrossRef CAS PubMed.
- H. H. Ho, T. W. Lau, F. Leung, H.-F. Tse and C.-W. Siu, Osteoporosis Int., 2010, 21, 573–577 CrossRef PubMed.
- M. K. Kang and R. Huang, J. Mech. Phys. Solids, 2010, 58, 1582–1598 CrossRef CAS PubMed.
- K. Jain, P. Kesharwani, U. Gupta and N. K. Jain, Int. J. Pharm., 2010, 394, 122–142 CrossRef CAS PubMed.
- R. Challa, A. Ahuja, J. Ali and R. K. Khar, AAPS PharmSciTech., 2005, 6, E329–E357 CrossRef PubMed.
- H. M. E. Azzazy, M. M. H. Mansour and S. C. Kazmierczak, Clin. Biochem., 2007, 40, 917–927 CrossRef CAS PubMed.
- M. S. Aw, J. Addai-Mensah and D. Losic, J. Mater. Chem., 2012, 22, 6561–6563 RSC.
- B. Kozissnik, L. A. W. Green, K. A. Chester and T. K. Nguyen Thanh, in Magnetic Nanoparticles: From Fabrication to Clinical Applications, ed. T. K. Nguyen Thanh, CRC Press, Taylor and Francis Group, LLC, 2012, Part III, ch. 5, pp. 129–150 Search PubMed.
- R. F. Donnelly, T. R. Raj Singh and A. D. Woolfson, Drug Deliv., 2010, 17, 187–207 CrossRef CAS PubMed.
- C. R. Thomas, D. P. Ferris, J.-H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J.-S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS PubMed.
- G. Valdés-Ramírez, J. R. Windmiller, J. C. Claussen, A. G. Martinez, F. Kuralay, M. Zhou, N. Zhou, R. Polsky, P. R. Miller, R. Narayan and J. Wang, Sensors Actuators B: Chem., 2012, 161, 1018–1024 CrossRef PubMed.
- P. Lokwani, Int. J. Res. Pharm. Biomed. Sci., 2011, 2, 465–473 Search PubMed.
- J. Ophir, Ultrasound Med. Biol., 1989, 15, 319–333 CrossRef CAS.
- S.-T. Kang and C.-K. Yeh, Chang Gung Med. J., 2012, 35, 125–139 Search PubMed.
- M. S. Aw and D. Losic, Int. J. Pharm., 2013, 443, 154–162 CrossRef CAS PubMed.
- G. A. Husseini and W. G. Pitt, Adv. Drug Deliv. Rev., 2008, 60, 1137–1152 CrossRef CAS PubMed.
- W. G. Pitt, G. A. Husseini and B. J. Staples, Exp. Opin. Drug Deliv., 2004, 1, 37–56 CrossRef CAS PubMed.
- G. A. Husseini and W. G. Pitt, J. Pharm. Sci., 2009, 98, 795–811 CrossRef CAS PubMed.
- M.-S. Martina, J.-P. Fortin, C. Menager, O. Clement, G. Barratt, C. Grabielle-Madelmont, F. Gazeau, V. Cabuil and S. Lesieur, J. Am. Chem. Soc., 2005, 127, 10676–10685 CrossRef CAS PubMed.
- C. Lorenzato, A. Cernicanu, M.-E. Meyre, M. Germain, A. Pottier, L. Levy, B. D. de Senneville, C. Bos, C. Moonen and P. Smirnov, Contrast Media Mol. Imaging, 2013, 8, 185–192 CrossRef CAS PubMed.
- M. Bariana, M. S. Aw, E. Moore, N. H. Voelcker and D. Losic, Future Med.: Nanomed., 2013 DOI:10.2217/NNM13.93 , in press.
- M. Raoof and S. A. Curley, Int. J. Hepatol., 2011, 6, 676957 Search PubMed.
- J. Cardinal, J. R. Klune, E. Chory, G. Jeyabalan, J. S. Kanzius, M. Nalesnik and D. A. Geller, Surgery, 2008, 144, 125–132 CrossRef PubMed.
- G. A. Rodan and T. J. Martin, Science, 2000, 289, 1508–1514 CrossRef CAS.
- D. A. LaVan, T. McGuire and R. Langer, Nat. Biotechnol., 2003, 21, 1184–1191 CrossRef CAS PubMed.
- L. L. Peng, A. D. Mendelsohn, T. J. LaTempa, S. Yoriya, C. A. Grimes and T. A. Desai, Nano Lett., 2009, 9, 1932–1936 CrossRef CAS PubMed.
- M. S. Aw, K. Khalid, K. Gulati, G. J. Atkins, P. Pivonka, D. M. Findlay and D. Losic, Int. J. Nanomed., 2012, 7, 4883–4892 CAS.
- C. M. Davies, D. B. Jones, M. J. Stoddart, K. Koller, E. Smith, C. W. Archer and R. G. Richards, Eur. Cell Mater., 2006, 11, 57–75 CAS.
- V. David, A. Guignandon, A. Martin, L. Malaval, M. H. Lafage-Proust, A. Rattner, V. Mann, B. Noble, D. B. Jones and L. Vico, Tissue Eng. Part A., 2008, 14, 117–126 CrossRef CAS PubMed.
- S. Endres, M. Kratz, S. Wunsch and D. B. Jones, J. Musculoskelet. Neuronal Interact., 2009, 9, 173–183 CAS.
- V. David, A. Martin, M. H. Lafage-Proust, L. Malaval, S. Peyroche, D. B. Jones, L. Vico and A. Guignandon, Endocrinology, 2007, 148, 2553–2562 CrossRef CAS PubMed.
- D. P. Pioletti, O. Gauthier, V. A. Stadelmann, B. Bujoli, J. Guicheux, P.-Y. Zambelli and J.-M. Bouler, Curr. Drug Deliv., 2008, 5, 59–63 CrossRef CAS.
- M. Vandrovcova and L. Bacakova, Physiol. Res., 2011, 60, 403–417 CAS.
- L. Leoni and T. A. Desai, IEEE Trans. Biomed. Eng., 2001, 48, 1335–1341 CrossRef CAS PubMed.
- B. S. Smith, S. Yoriya, L. Grissom, C. A. Grimes and K. C. Popat, J. Biomed. Mater. Res. A., 2010, 95, 350–360 CrossRef PubMed.
- L. Hu, Y. Zhang, S. Dong, S. Zhang and B. Li, Ceramics Int., 2013, 39, 6287–6291 CrossRef CAS PubMed.
- S. P. Adiga, C. Jin, L. A. Curtiss, N. A. Monteiro-Riviere and R. J. Narayan, Nanomed. Nanobiotechnol., 2009, 1, 568–581 CrossRef CAS PubMed.
- D. Rai, J. Deng and C.-S. Toh, Talanta, 2012, 98, 112–117 CrossRef PubMed.
- A. Zelikin and B. Städler, in Intelligent Surfaces in Biotechnology. Scientific and Engineering Concepts, Enabling Technologies, and Translation to Bio-oriented Applications, ed. H. M. Grandin and M. Textor, John Wiley and Sons, Inc., New Jersey, 2012, 7, pp. 228–270 Search PubMed.
- K. C. Popat, E. E. Leary Swan, V. Mukhatyar, K.-I. Chatvanichkul, G. K. Mor, C. A. Grimes and T. A. Desai, Biomaterials, 2005, 26, 4516–4522 CrossRef CAS PubMed.
- T. J. Webster, R. W. Siegel and R. Bizios, Biomaterials, 1999, 20, 1221–1227 CrossRef CAS.
- K. E. La Flamme, K. C. Popat, L. Leoni, E. Markiewicz, T. J. La Tempa, B. B. Roman, C. A. Grimes and T. A. Desai, Biomaterials, 2007, 28, 2638–2645 CrossRef CAS PubMed.
- M. Vallet-Regí, A. Rámila, R. P. del Real and J. Pérez-Pariente, Chem. Mater., 2000, 13, 308–311 CrossRef.
- E. E. Leary Swan, K. C. Popat and T. A. Desai, Biomaterials, 2005, 26, 1969–1976 CrossRef CAS PubMed.
- M. Karlsson, E. Pålsgård, P. R. Wilshaw and L. Di Silvio, Biomaterials, 2003, 24, 3039–3046 CrossRef CAS.
- S. Chung, S. Son and J. Min, Nanotechnology, 2010, 21, 125104 CrossRef CAS PubMed.
- I. Karoussos, H. Wieneke, T. Sawitowski, S. Wnendt, A. Fischer, O. Dirsch, U. Dahmen and R. Erbell, Materialwissenschaft Werkstofftechnik, 2002, 33, 738–746 CrossRef CAS.
- Z. Li, X. Yang, H. Guo, X. Yang, L. Sun and S. Dong, J. Nanopart. Res., 2012, 14, 1145–1153 CrossRef CAS.
- W. Shi, M. S. Mozumder, H. Zhang, J. Zhu and H. Perinpanayagam, Biomed. Mater., 2012, 7, 055006 CrossRef CAS PubMed.
- X. Fan, B. Feng, J. Weng, J. Wang and X. Lu, Mater. Lett., 2011, 65, 2899–2901 CrossRef CAS PubMed.
- C. C. Mohan, K. P. Chennazhi and D. Menon, Acta Biomater., 2013, 9, 9568–9577 CrossRef CAS PubMed.
- S. L. Tao and T. A. Desai, Adv. Drug Deliv. Rev., 2003, 55, 315–328 CrossRef CAS.
- J. Park, S. Bauer, K. von der Mark and P. Schmuki, Nano Lett., 2007, 7, 1686–1691 CrossRef CAS PubMed.
- H. W. Kim, Y. H. Koh, L. H. Li, S. Lee and H. E. Kim, Biomaterials, 2004, 25, 2533–2538 CrossRef CAS PubMed.
- M. C. Advincula, F. G. Rahemtulla, R. C. Advincula, E. T. Ada, J. E. Lemons and S. L. Bellis, Biomaterials, 2006, 27, 2201–2212 CrossRef CAS PubMed.
- K. C. Popat, L. Leoni, C. A. Grimes and T. A. Desai, Biomaterials, 2007, 28, 3188–3197 CrossRef CAS PubMed.
- C. von Wilmowsky, S. Bauer, R. Lutz, M. Meisel, F. W. Neukam, T. Toyoshima, P. Schmuki, E. Nkenke and K. A. Schlegel, J. Biomed. Mater. Res. Part B: Appl. Biomater., 2009, 89, 165–171 CrossRef PubMed.
- L. M. Bjursten, L. Rasmusson, S. Oh, G. C. Smith, K. S. Brammer and S. Jin, J. Biomed. Mater. Res. Part A., 2010, 92, 1218–1224 Search PubMed.
- C. Pinto Reis, R. J. Neufeld, A. J. Ribeiro and F. Veig, Nanomed. Nanotech., Biol. Med., 2006, 2, 8–21 CrossRef PubMed.
- M. Geetha, A. K. Singh, R. Asokamani and A. K. Gogia, Prog. Mater. Sci., 2009, 54, 397–425 CrossRef CAS PubMed.
- S. Nag and R. Banerjee, ASM Handbook:Mater. for Med. Device, 2012, vol. 23, pp. 6–17 Search PubMed.
- D. Manickavasagam and M. O. Oyewumi, J. Drug Deliv., 2013, 895013 CAS.
| This journal is © The Royal Society of Chemistry 2014 |