Stimuli-responsive siRNA carriers for efficient gene silencing in tumors via systemic delivery
Min Suk
Shim
a,
Seong-Sil
Chang
b and
Young Jik
Kwon
*cdef
aDivision of Bioengineering, Incheon National University, Incheon 406-772, Republic of Korea
bDepartment of Occupational and Environmental Medicine, Eulji University Hospital, Daejeon, Republic of Korea
cDepartment of Pharmaceutical Sciences, University of California, Irvine, CA 92697, USA
dDepartment of Chemical Engineering and Materials Science, University of California, Irvine, CA 92697, USA
eDepartment of Biomedical Engineering, University of California, Irvine, CA 92697, USA
fDepartment of Molecular Biology and Biochemistry, University of California, Irvine, CA 92697, USA. E-mail: kwonyj@uci.edu; Fax: +1-949-824-4023; Tel: +1-949-824-8714
First published on 24th October 2013
Abstract
Development of efficient carriers for small interfering RNA (siRNA) delivery and validation tools for assessing in vivo RNA interference (RNAi) efficiency is crucial to advance RNAi-based therapeutics to the clinic. Here, acid-degradable ketalized linear polyethylenimine (KL-PEI) designed for efficient, stimuli-responsive, and biocompatible siRNA delivery was used to complex with GFP-silencing siRNA (GFP siRNA) for in vivo RNAi. The in vivo gene silencing efficiency of GFP siRNA/KL-PEI polyplexes was evaluated at mRNA, protein, and histological levels using a mouse bearing a GFP-expressing tumor. Intravenously injected GFP siRNA/KL-PEI polyplexes significantly reduced GFP expression in tumors and whole blood of mice, depending on the dosage of GFP siRNA and the time course. Average GFP mRNA levels in the tumors of siRNA/KL-PEI polyplex-injected mice were also reduced. The described siRNA carriers and RNAi validation methodologies in this study may provide insightful clues for the development of RNAi-based therapeutics and preclinical trials.
Introduction
Molecular therapy of diseases (e.g., viral infections, neurodegenerative disorders, and cancers) via RNA interference (RNAi) has been of great interest in medicine.1–3 Effective degradation of target mRNA and subsequent translation can be achieved by exogenously administering double-stranded small interfering RNA (siRNA) to diseased cells and tissues.4–6 However, the poor pharmacokinetic and pharmacodynamic properties of naked siRNA (e.g., low stability against nucleases in serum and poor/nonspecific cellular uptake) necessitate the development of effective and biocompatible delivery systems in clinical implementation of RNAi.7–9Various nonviral siRNA delivery systems have been investigated for efficient RNAi due to their advantages, including ease of synthesis and low immune response.10 Recently, cationic polymers have been intensively developed for siRNA delivery because they efficiently condense siRNA into compact nanoparticles via electrostatic interaction, leading to protection of siRNA from enzymatic digestion and efficient intracellular uptake through endocytosis.11,12 Although cationic polymers can sufficiently deliver siRNA in vitro, several issues need to be addressed in order to achieve successful systemic delivery of siRNA. The critical issues with regard to cationic polymeric carriers for in vivo siRNA delivery include the interaction of cationic siRNA/polymer complexes with serum proteins and rapid clearance by the reticuloendothelial system (RES).13,14In vivo stability and blood circulation of cationic polyplexes can be enhanced by surface modification through coupling with nonionic, hydrophilic polyethylene glycol (PEG).15 PEG provides steric hindrance on the surface of the complexes and reduces adsorption of serum proteins.15,16 As a result, the PEG layer increases the stability of the complexes against serum proteins and thus improves the pharmacokinetic behaviours of the complexes in vivo.17–19
Recently, we have demonstrated efficient in vitro gene silencing with improved biocompatibility using acid-degradable ketalized linear polyethylenimine (KL-PEI).20 KL-PEI greatly facilitated the cytoplasmic release of siRNA upon cleavage of acid-degradable ketal branches in the endosome or lysosome.20 As a result, a significantly enhanced gene silencing efficiency was achieved with almost no cytotoxicity.20 Noticeably, unlike the polyplexes prepared with branched PEI, a superior commercial transfecting agent, the gene silencing efficiency of siRNA/KL-PEI polyplexes was not detrimentally affected by serum, indicating the high promise for gene silencing in vivo.20 In this study, the feasibility of using acid-responsive KL-PEI as an effective and biocompatible siRNA carrier for in vivo RNAi was assessed. To improve the in vivo siRNA delivery efficiency of KL-PEI, the surface of siRNA/KL-PEI polyplexes was further conjugated with PEG (MW = 680 Da).
Assessing gene silencing in vivo is a pivotal step in developing siRNA delivery carriers and advancing them to the clinic,9,10,21 which demands the establishment of an effective in vivo model that allows for comprehensive analysis of in vivo gene silencing. Here, we show an in vivo animal model using a mouse bearing a GFP-expressing tumor and various RNAi validation methodologies to evaluate in vivo RNAi efficiency in a tumor via systemic administration of PEGylated siRNA/KL-PEI polyplexes. Intravenously injected PEGylated KL-PEI polyplexes complexing with GFP-silencing siRNA significantly reduced GFP expression in tumors, which was comprehensively validated at mRNA, protein, and histological levels. This study demonstrates that acid-responsive, PEGylated KL-PEI is an efficient carrier for in vivo siRNA delivery into a tumor.
Experimental
Materials
Linear polyethylenimine (MW = 2.5 kDa) and N-hydroxysuccinimide (NHS) ester-activated PEG (MW = 680 Da) were purchased from Polyscience Inc. (Warrington, PA) and Pierce (Rockford, IL), respectively. Silencer anti-GFP siRNA (sense strand 5′-CAAGCUGACCCUGAAGUUCdTdT-3′ and antisense strand 5′-GAACUUCAGGGUCAGCUUGdCdC-3′) and scrambled siRNA (sense strand 5′-AGUACUGCUUACGAUACGGdTdT-3′ and antisense strand 5′-CCGUAUCGUAAGCAGUACUdTdT-3′) were purchased from Ambion (Austin, TX). All primers for reverse transcription-polymerase chain reaction (RT-PCR) were purchased from Invitrogen (Grand Island, NY). Ovalbumin (OVA)-expressing mouse lymphoma E.G7-OVA cells (ATCC, Manassas, VA) were cultured in RPMI-1640 medium (MediaTech, Herndon, VA) supplemented with 10% FBS (Hyclone, Logan, UT), 50 μM of 2-mercaptoethanol (Invitrogen), 2 mM of L-glutamine (MediaTech), 400 mg L−1 of G418 (MediaTech), 2.5 g L−1 of glucose (MediaTech), 10 mM of HEPES (MediaTech), and 1 mM of sodium pyruvate (MediaTech). Female C57BL/6 mice (8–10 weeks old) purchased from Charles River Laboratories (Wilmington, MA) were used for in vivo study, with the approval of the Institutional Animal Care and Use Committee (IACUC) at University of California, Irvine. The mice were kept anesthetized by isoflurane inhalation during injection in order to minimize pain and discomfort, and they were closely monitored for signs of distress (heart rate, breathing, swelling, itching, redness, etc.) during the study, according to the guidelines on humane care of research animals.Preparation and characterization of polyplexes formed with siRNA and ketalized linear polyethylenimine (KL-PEI)
KL-PEI was synthesized by grafting acid-labile amino ketal branches onto the backbone of linear PEI (MW = 2.5 kDa) (Fig. 1), as previously reported.20 To prepare siRNA-encapsulating polyplexes, siRNA (20 μg in 200 μL of DI water) was mixed with KL-PEI (535 μg in 200 μL of DI water) to obtain the N/P ratio of 150, followed by incubation at room temperature for 30 min. Subsequently, 2.82 μg of NHS ester-activated PEG (MW = 680 Da) in 100 μL of DI water (4.15 nmol; equivalent to 0.5% of KL-PEI's primary amines) was added and reacted for 2 h at room temperature. The resulting PEGylated siRNA/KL-PEI polyplexes were purified by a PD-10 size exclusion column (GE Healthcare, Piscataway, NJ) to remove unreacted NHS ester-activated PEG. The purified PEGylated siRNA/KL-PEI polyplexes were re-suspended in 500 μL of saline containing 0.9% (w/v) NaCl. For the characterization of PEGylated siRNA/KL-PEI polyplexes, an aliquot of a PEGylated siRNA/KL-PEI polyplex solution containing 2 μg of siRNA was diluted with 800 μL of DI water. The mean particle size and zeta potential of the PEGylated siRNA/KL-PEI polyplexes were measured using a ZEN3600 Zetasizer (Malvern Instruments, Worcestershire, UK) at 25 °C.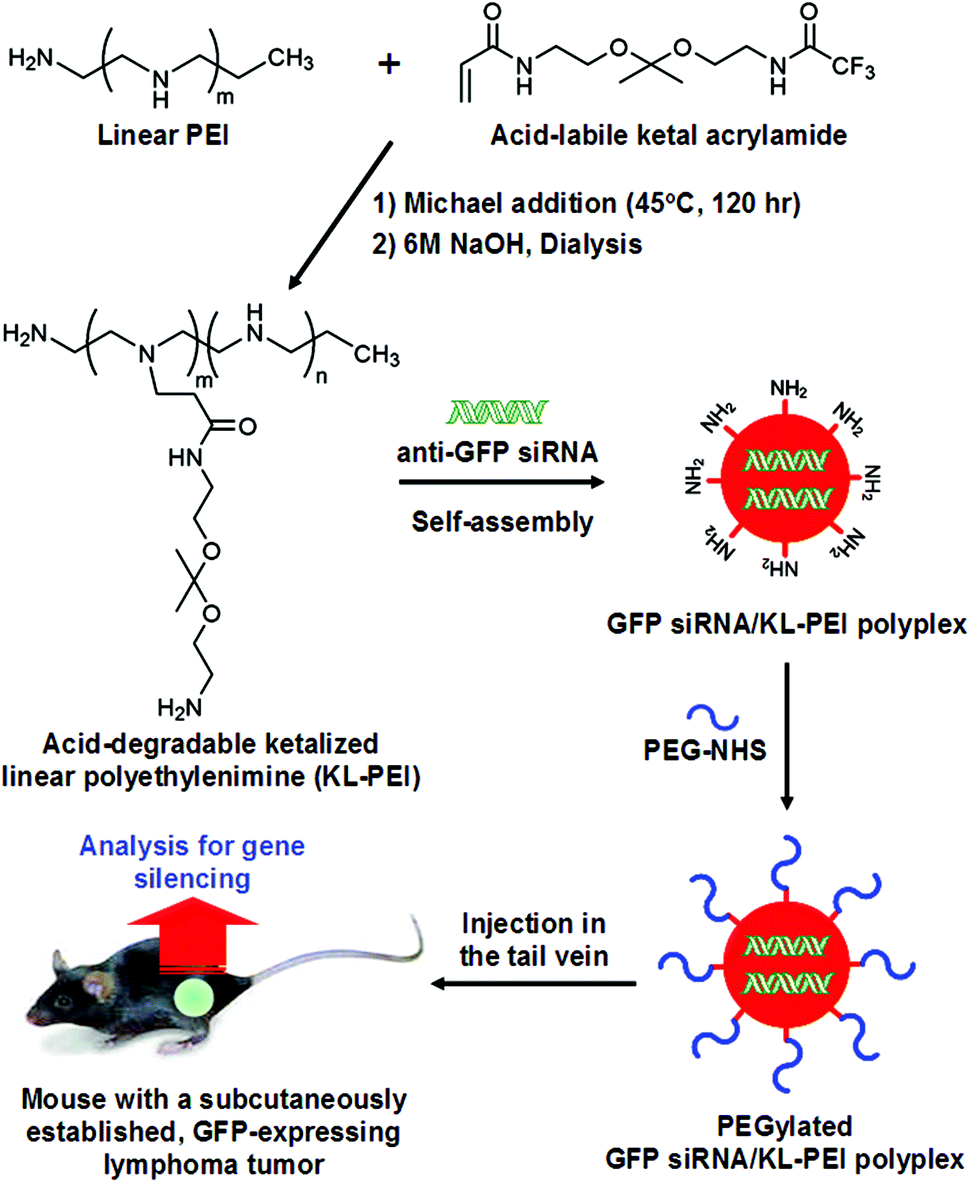 | ||
| Fig. 1 Preparation of PEGylated GFP siRNA/KL-PEI polyplexes and systemic delivery (i.v. injection) for gene silencing in vivo. | ||
Systemic (intravenous) administration of PEGylated siRNA/KL-PEI polyplexes into tumor-bearing mice
E.G7-OVA cells, ovalbumin (OVA)-expressing, C57BL/6-derived T cell lymphoma cells, were transduced to express GFP using VSVG-pseudotyped retroviral vectors.22 The GFP-positive E.G7-OVA cells after transduction were sorted and cloned by a serial dilution technique.23 GFP expression of E.G7-OVA cells was verified by flow cytometry. OVA-expressing tumor cells can be easily identified and separated, and the relative fluorescence of GFP can serve as a quantitative indicator of gene expression levels. One and a half million of the resulting E.G7-OVA/GFP cells per mouse were subcutaneously injected into the right flank of C57BL/6 mice. The mice were kept for 7–10 days until a solid tumor lump with a volume of 160–180 mm3 developed. PEGylated siRNA/KL-PEI polyplexes encapsulating 10 μg or 20 μg of GFP-silencing siRNA (GFP siRNA) or scrambled siRNA (Scr siRNA) in 500 μL of saline were intravenously injected into the tail vein of the mice (4 mice per group) (Fig. 1). All mice received only a single intravenous (i.v.) injection, and a control group of mice were injected with the same volume of saline. After 3 days, blood samples were collected from anesthetized animals and tumors were harvested from euthanized animals. After 5 days, blood and tumor samples were collected from additional groups of mice (3 mice per group) injected with the same doses of PEGylated GFP siRNA/KL-PEI polyplexes in order to evaluate their efficiencies in knockdown of GFP expression after an extended period of time.GFP expression in tumor cells by flow cytometry
To quantify silenced GFP expression, harvested tumors were homogenized using a tissue digestion solution (Sigma-Aldrich, Milwaukee, WI) formulated with collagenase, hyaluronidase, and deoxyribonuclease I. The cell- and tissue-debris in the resulting tumor homogenate were removed three times by centrifugation at 1200 rpm for 3 min, removal of the supernatant, and re-suspension of the cell pellet in PBS. E.G7-OVA/GFP cells were then selectively collected after incubation of the resulting cell-containing solutions with biotinylated anti-OVA antibodies (25.D1–16) (eBioscience, San Diego, CA) at a concentration of 5 μg mL−1 for 40 min at 4 °C. After unbound antibodies were removed by a brief centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 s, the cells were further incubated with anti-biotin magnetic cell sorting (MACS) beads (20 μL per 107 cells) (Miltenyi Biotec, Auburn, CA) for 15 min at 4 °C. The magnetically labeled cells were sorted using an MS column (Miltenyi Biotec) according to the manufacturer's protocol. GFP expression in the collected cells was quantified by flow cytometry using a Guava EasyCyte Plus flow cytometer (Guava Technologies Inc., Hayward, CA).
000 rpm for 15 s, the cells were further incubated with anti-biotin magnetic cell sorting (MACS) beads (20 μL per 107 cells) (Miltenyi Biotec, Auburn, CA) for 15 min at 4 °C. The magnetically labeled cells were sorted using an MS column (Miltenyi Biotec) according to the manufacturer's protocol. GFP expression in the collected cells was quantified by flow cytometry using a Guava EasyCyte Plus flow cytometer (Guava Technologies Inc., Hayward, CA).
GFP quantification in tumors and blood using the enzyme-linked immunosorbent assay (ELISA)
One hundred μL of lysis buffer provided from a GFP fluorometry kit (Cell Biolabs, San Diego, CA) was added to 1 mg of tumor tissue or 10 mg of whole blood collected from the mice. After the resulting tumor lysate was diluted 1000 times, the amount of GFP in 1 μL of the resulting solution was quantified using a GFP ELISA kit (Cell Biolabs) according to the manufacturer's protocols, while the blood lysate was analyzed without dilution. The amounts of GFP in both lysates were normalized by total protein amounts quantified using a BCA protein analysis kit (Pierce).mRNA quantification in tumors and blood using real-time quantitative reverse transcription-polymerase chain reaction (RT-qPCR)
RNA was extracted from tumor and whole blood samples using an RNeasy plus kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. The primer sequences used for 206 bps of GFP were 5′-CTTCTTCAAGTCCGCCATGC-3′ and 5′-AGACGTTGTGGCTGTTGTAGTT-3′. The primers 5′-AGGTCGGTGTGAACGGATTTG-3′ and 5′-ATGACAAGCTTCCCATTCTCG-3′ were used as primers for 193 bps of GAPDH. The primer sequences used for 100 bps of OVA were 5′-AGCCAATCTGTCTGGCATCT-3′ and 5′-TTCGTCCGTCTCTCCACCAT-3′. RT-qPCR was performed using a SYBR Green PCR Master Mix kit (Applied Biosystems, Foster City, CA) with an ABI PRISM 7000 Detection System (Applied Biosystems). Briefly, 2.75 μL of total RNA was mixed with 1 μL of 10 pM primer, 0.2 μL of RNase inhibitor (1 U μL−1) (Applied Biosystems), 0.05 μL of MultiScribe reverse transcriptase (50 U μL−1) (Applied Biosystems), and 5 μL of Master Mix. The thermocycling conditions were 48 °C for 30 min for the synthesis of cDNA and 95 °C for 10 min, 40 cycles of 95 °C for 15 s, and 60 °C for 1 min for the amplification of target cDNA. The final step of RT-qPCR was a dissociation curve analysis of one cycle of 95 °C for 15 s, 60 °C for 15 s, and 95 °C for 15 s to characterize the amplified target product. Analyses of every sample were performed in duplicate for statistical significance. The amount of amplified products due to contaminated DNA was also calculated by carrying out RT-qPCR without the reverse transcriptase under the same conditions. Amplification was confirmed by a specific melting temperature (Tm) peak of a target product along with gel electrophoresis. Only tumor tissues showed a specific Tm peak in the dissociation curves of GFP and OVA obtained from RT-PCR, while a nonspecific or no Tm peak was found in the whole blood samples. This observation is in agreement with previous studies which showed a limited detection of circulatory tumor RNA in the blood.24Histological assessment of tumor tissues
Tumor tissues harvested from the mice were fixed in 4% (w/v) p-formaldehyde solution in PBS overnight at 4 °C, cryo-protected in 10% to 20% graded sucrose solutions in DI water, and stored in the dark at 4 °C. The fixed tissues were then mounted on a Tissue-Tek 4583 optimum cutting temperature (OCT) compound (Sakura Finetek USA Inc., Torrance, CA) and frozen in pentane using liquid nitrogen. The frozen tumor tissues were sectioned at 10 μm thickness from the OCT compound blocks using a cryostat at −20 °C. After staining with DAPI (Vector Laboratories, Burlingame, CA), histological sections of tumor tissues were examined under an inverted fluorescent microscope equipped with fluorescent filter sets for DAPI and GFP (Leica DM4000, Leica Microsystems Inc., Wetzlar, Germany).Results and discussion
KL-PEI was synthesized by grafting acid-cleavable amino ketal branches onto the backbone of linear PEI (Fig. 1), as reported previously.20 Acid-degradable KL-PEI was specifically designed to facilitate the release of siRNA into the cytoplasm through hydrolysis of ketal branches in the mildly acidic endosome or lysosome. To inhibit GFP expression in a GFP-expressing tumor of mice, KL-PEI was complexed with GFP-silencing siRNA (GFP siRNA) and formed nanoparticles. The size of GFP siRNA/KL-PEI polyplexes in the N/P ratio of 150 was around 130 nm, determined by dynamic light scattering (DLS), with a surface charge of +22.3 mV. The surface of the resulting GFP siRNA/KL-PEI polyplexes was further tethered with PEG for shielding the polyplexes from immune response, aggregation in blood, and rapid clearance by the RES (Fig. 1).13,14 The size and the surface charge of the PEGylated GFP siRNA/KL-PEI polyplexes were 140 nm (by DLS analysis) and +2.5 mV, respectively. The small molecular weight of PEG minimally increased the size of the polyplexes, while a significant decrease in the surface charge was obtained. The neutral PEG shell on the surface of the polyplexes could contribute to a dramatic decrease in the surface charge of the polyplexes. This PEGylation could enhance the in vivo gene silencing efficiency of GFP siRNA/KL-PEI polyplexes by stabilizing the polyplexes against aggregation and prolonging circulation in blood.25To establish a syngeneic tumor model that expresses an easily quantifiable protein (i.e., GFP) as well as a distinct marker for initially injected cells (i.e., OVA) for in vivo systemic siRNA delivery, engineered ovalbumin (OVA)- and GFP-expressing murine T lymphoma E.G7-OVA/GFP cells were subcutaneously injected into the flank of C57BL/6 mice (Fig. 1). GFP siRNA/KL-PEI polyplexes complexing with 20 μg of siRNA were intravenously injected into the tail vein of the mice bearing an E.G7-OVA/GFP tumor. In order to confirm sequence-specific silencing of GFP expression, polyplexes complexing with the same amount of siRNA with a scrambled sequence (Scr siRNA/KL-PEI polyplexes) were also injected. The gene silencing efficiency of the polyplexes was evaluated by comparing their GFP suppression in the tumor cells to that of the saline-treated control group.
Tumor tissues were harvested from polyplex- and saline-injected mice 3 days after injection, followed by separating the E.G7-OVA/GFP tumor cells from other cells and tissue components by OVA-specific magnetic cell sorting. The tumor cells were then analyzed by flow cytometry for relative GFP expression (Fig. 2). The result demonstrated that GFP siRNA/KL-PEI polyplexes silenced the GFP expression in the tumor cells more efficiently than saline and Scr siRNA/KL-PEI polyplexes (Fig. 2). The tumor cells harvested from the mice injected with Scr siRNA/KL-PEI polyplexes did not show any meaningful reduction of GFP, indicating sequence-specific inhibition of GFP expression by GFP siRNA (Fig. 2). Dose-dependent gene silencing was also investigated by injecting the GFP siRNA/KL-PEI polyplexes containing 10 or 20 μg of GFP siRNA (Fig. 3). Systemic administration of 20 μg GFP siRNA/KL-PEI polyplexes resulted in a ∼30% reduction in overall GFP expression (i.e., the mean fluorescence intensity), while only statistically insignificant gene silencing was observed when 10 μg of GFP siRNA was injected (Fig. 3). The dose of 20 μg GFP siRNA per mouse used in this study is relatively lower than the doses used in other studies,26,27 demonstrating efficient gene silencing in tumors via systemic siRNA delivery using KL-PEI.
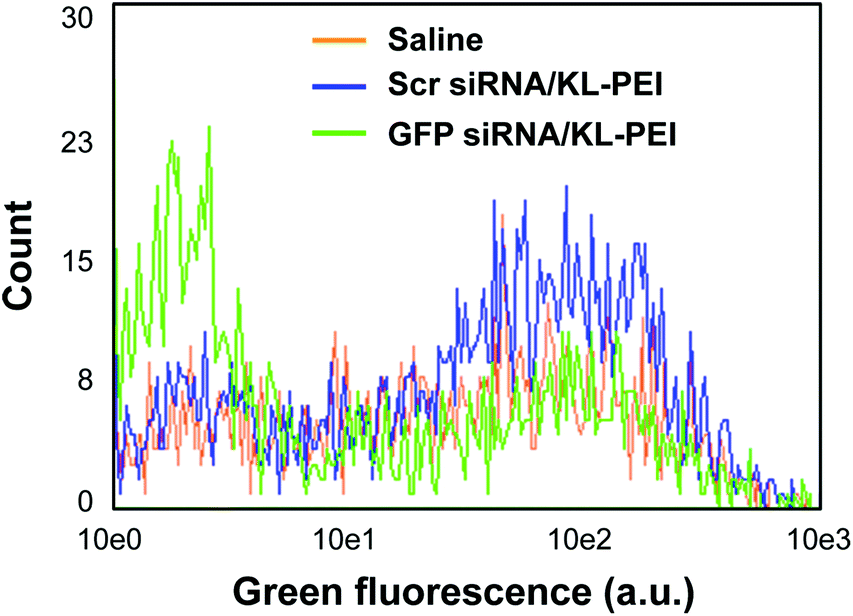 | ||
| Fig. 2 Flow cytometric analysis of silenced GFP expression in the E.G7-OVA/GFP tumor cells harvested 3 days after i.v. injection. Mice were injected with saline, Scr siRNA/KL-PEI polyplexes, and GFP siRNA/KL-PEI polyplexes. | ||
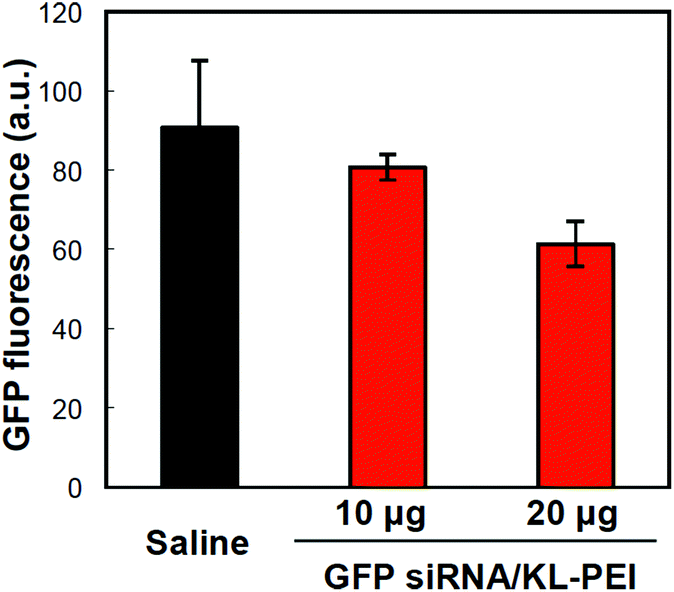 | ||
| Fig. 3 Dose-dependent GFP silencing in the tumor cells harvested 3 days after i.v. injection of polyplexes complexing with 10 or 20 μg of GFP siRNA into E.G7-OVA/GFP tumor-bearing mice. The average GFP expression in the tumor cells was analyzed by flow cytometry. | ||
In addition to the flow cytometric analysis of the harvested tumor cells, silenced GFP expression in the tumor and whole blood was also biochemically quantified. The amount of GFP in the tumors harvested from the GFP siRNA/KL-PEI polyplex-injected mice on the third post-injection day (Group 3 in Fig. 4A) was lower than that in the tumors harvested from the saline- and Scr siRNA/KL-PEI polyplex-injected mice (Groups 1 and 2, respectively, in Fig. 4A). The GFP silencing by GFP siRNA/KL-PEI polyplexes became greater after two more days (Group 4 in Fig. 4A). This observation indicates that gene silencing was sustained for several days by GFP siRNA/KL-PEI polyplexes, although the gene silencing duration needs to be investigated further. The gene silencing efficiency of various KL-PEI polyplexes was also evaluated by quantifying the amount of circulating GFP leaked out of the GFP-expressing tumor. It was reported that tumor cells can detach from the edge of a tumor during proliferation.28,29 These circulating tumor cells can remain in blood and thus be used as a biomarker for cancer diagnosis or treatment.30 Efficient gene silencing by GFP siRNA/KL-PEI polyplexes was confirmed by the reduced amount of GFP in the circulating tumor cells leaked out of the GFP-expressing tumor (Group 3 in Fig. 4B) as compared to saline- or Scr siRNA/KL-PEI polyplex-injected mice (Groups 1 and 2 in Fig. 4B). The amount of circulating GFP was further reduced for an additional two days in the mice injected with GFP siRNA/KL-PEI polyplexes (Group 4 in Fig. 4B), as similarly shown in Fig. 4A. This result implies that a circulating protein leaked out of a tumor can be an effective, predictable, and convenient marker for indirectly validating gene silencing in a tumor. The gene silencing efficiency was also assessed at mRNA levels by quantifying the amount of GFP mRNA in the tumors by RT-qPCR and further normalizing by endogenous GAPDH mRNA (Fig. 4C). A reduced average GFP mRNA level was observed in the tumors harvested from the mice injected with GFP siRNA/KL-PEI polyplexes in a time-dependent manner (Groups 3 and 4 in Fig. 4C), compared to the mice injected with saline or Scr siRNA/KL-PEI polyplexes (Groups 1 and 2 in Fig. 4C). However, the reduction in GFP mRNA levels was not as significant as silenced GFP expression (Fig. 4A and C), possibly due to various post-transcriptional, translational, and protein degradation processes.31 Measuring GFP mRNA in whole blood as another indirect maker for gene silencing in vivo was also attempted, but it was not detectable (data not shown). This might be due to very low mRNA levels in the blood and/or rapid degradation of mRNA by RNases during circulation. Histological evaluation clearly demonstrated significantly reduced GFP expression in the tumor harvested from a GFP siRNA/KL-PEI polyplex-injected mouse on the fifth post-injection day (Fig. 4F), as compared to saline- and Scr siRNA/KL-PEI polyplex-injected groups (Fig. 4D and E). The results shown in Fig. 4 clearly demonstrate efficient, sequence-specific, days-long gene silencing in a tumor via systemic delivery of GFP siRNA/KL-PEI polyplexes.
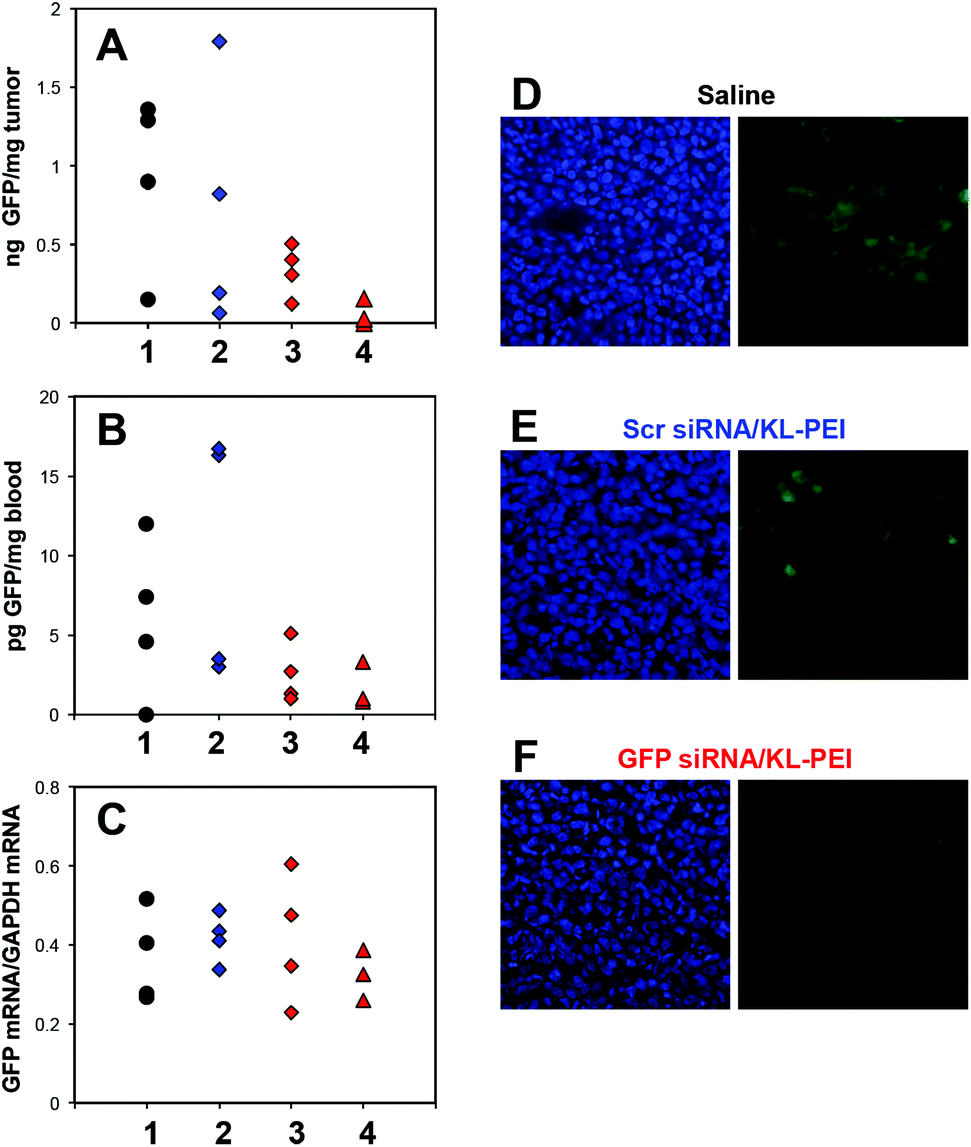 | ||
| Fig. 4 Multi-level analyses of silenced GFP expression in vivo after i.v. injection of GFP siRNA/KL-PEI polyplexes (20 μg of siRNA). The amount of GFP (A) in the tumor and (B) in the blood (ELISA). (C) The amount of GFP mRNA in the tumor (RT-qPCR). The mice were injected with saline (Group 1), Scr siRNA/KL-PEI polyplexes (Group 2), and GFP siRNA/KL-PEI polyplexes (analyzed 3 days after injection, Group 3; analyzed 5 days after injection, Group 4). (D–F) Representative histological images of the tumor tissues harvested from Groups 1, 2, and 4, respectively (left images: DAPI-stained individual cells; right image: GFP). | ||
Conclusions
In summary, acid-degradable, PEGylated KL-PEI polyplexes complexing with siRNA were prepared, and their potential for in vivo RNAi in a tumor was explored. Efficient gene silencing in a tumor by PEGylated GFP siRNA/KL-PEI polyplexes via single i.v. injection was confirmed by mRNA, protein, and histological analyses. Biodistribution of the polyplexes, the effects of a wider range of doses, and long-term immune- and toxicological responses to siRNA and KL-PEI would greatly supplement what was achieved in this study. The in vivo tumor model used in this study is a promising tool for evaluating the efficacy of systemic nonviral siRNA delivery at multiple levels. The described methods to assess in vivo RNAi in this study could provide insights into the design of effective RNAi methodologies in the clinic.Acknowledgements
This work was financially supported by the Incheon National University Research Grant in 2013.Notes and references
- D. H. Kim and J. J. Rossi, Nat. Rev. Genet., 2007, 8, 173 CrossRef CAS PubMed.
- D. Cejka, D. Losert and V. Wacheck, Clin. Sci., 2006, 110, 47 CrossRef CAS PubMed.
- F. Takeshita and T. Ochiya, Cancer Sci., 2006, 97, 689 CrossRef CAS PubMed.
- M. T. McManus and P. A. Sharp, Nat. Rev. Genet., 2002, 3, 737 CrossRef CAS PubMed.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129 CrossRef CAS PubMed.
- F. Czauderna, M. Fechtner, S. Dames, H. Aygün, A. Klippel, G. J. Pronk, K. Giese and J. Kaufmann, Nucleic Acids Res., 2003, 31, 2705 CrossRef CAS PubMed.
- D. Grimm and M. A. Kay, J. Clin. Invest., 2007, 117, 3633 CrossRef CAS PubMed.
- M. S. Shim, S. Wong and Y. J. Kwon, Drug Discovery Today Technol., 2012, 9, e167 CrossRef CAS PubMed.
- M. S. Shim and Y. J. Kwon, FEBS J., 2010, 277, 4814 CrossRef CAS PubMed.
- S. Werth, B. Urban-Klein, L. Dai, S. Höbel, M. Grzelinski, U. Bakowsky, F. Czubayko and A. Aigner, J. Controlled Release, 2006, 112, 257 CrossRef CAS PubMed.
- K. A. Howard, Adv. Drug Delivery Rev., 2009, 61, 710 CrossRef CAS PubMed.
- R. I. Mahato, K. Kawabata, T. Nomura, Y. Takakura and M. Hashida, J. Pharmacol. Sci., 1995, 84, 1267 CrossRef CAS.
- P. R. Dash, M. L. Read, L. B. Barrett, M. A. Wolfert and L. W. Seymour, Gene Ther., 1999, 6, 643 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS PubMed.
- D. E. Owens 3rd and N. A. Peppas, Int. J. Pharm., 2006, 307, 93 CrossRef PubMed.
- M. Ogris, S. Brunner, S. Schüller, R. Kircheis and E. Wagner, Gene Ther., 1999, 6, 595 CrossRef CAS PubMed.
- M. T. Peracchia, S. Harnisch, H. Pinto-Alphandary, A. Gulik, J. C. Dedieu, D. Desmaële, J. d'Angelo, R. H. Müller and P. Couvreur, Biomaterials, 1999, 20, 1269 CrossRef CAS.
- Y. H. Choi, F. Liu, J.-S. Kim, Y. K. Choi, J. S. Park and S. W. Kim, J. Controlled Release, 1998, 54, 39 CrossRef.
- M. S. Shim and Y. J. Kwon, Bioconjugate Chem., 2009, 20, 488 CrossRef CAS PubMed.
- P. Y. Lu, F. Xie and M. C. Woodle, Adv. Genet., 2005, 54, 117 CAS.
- Y. J. Kwon, H. Yu and C.-A. Peng, Biotechnol. Bioeng., 2001, 72, 331 CrossRef CAS.
- R. I. Freshney, Culture of animal cells: a manual of basic technique and specialized applications, ed. N. J. Hoboken, Wiley-Blackwell, 6th edn, 2010, p. 208 Search PubMed.
- M. S. Kopreski, F. A. Benko and C. D. Gocke, Ann. N. Y. Acad. Sci., 2001, 945, 172 CrossRef CAS.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Curr. Opin. Colloid Interface Sci., 2001, 6, 3 CrossRef CAS.
- M. S. Huh, S.-Y. Lee, S. Park, S. Lee, H. Chung, S. Lee, Y. Choi, Y.-K. Oh, J. H. Park, S. Y. Jeong, K. Choi, K. Kim and I. C. Kwon, J. Controlled Release, 2010, 144, 134 CrossRef CAS PubMed.
- Z.-X. Lu, L.-T. Liu and X.-R. Qi, Int. J. Nanomed., 2011, 6, 1661 CrossRef CAS.
- S. C. P. Williams, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4861 CrossRef CAS PubMed.
- S. Riethdorf, H. Fritsche, V. Müller, T. Rau, C. Schindlbeck, B. Rack, W. Janni, C. Coith, K. Beck, F. Jänicke, S. Jackson, T. Gornet, M. Cristofanilli and K. Pantel, Clin. Cancer Res., 2007, 13, 920 CrossRef CAS PubMed.
- J.-P. Eliane, M. Repollet, K. E. Luker, M. Brown, J. M. Rae, G. Dontu, A. F. Schott, M. Wicha, G. V. Doyle, D. F. Hayes and G. D. Luker, Cancer Res., 2008, 68, 5529 CrossRef CAS PubMed.
- C. Vogel and E. M. Marcotte, Nat. Rev. Genet., 2012, 13, 227 CAS.
| This journal is © The Royal Society of Chemistry 2014 |