Incorporation of sulfated hyaluronic acid macromers into degradable hydrogel scaffolds for sustained molecule delivery
Brendan P.
Purcell
,
Iris L.
Kim
,
Vanessa
Chuo
,
Theodore
Guenin
,
Shauna M.
Dorsey
and
Jason A.
Burdick
*
Department of Bioengineering, University of Pennsylvania, 240 Skirkanich Hall, 210 S. 33rd Street, Philadelphia, PA 19104, USA. E-mail: burdick2@seas.upenn.edu; Fax: +1 215-573-2071; Tel: +1 215-898-8537
First published on 8th November 2013
Abstract
Synthetically sulfated hyaluronic acid (HA) has been shown to bind proteins with high affinity through electrostatic interactions. While HA-based hydrogels have been used widely in recent years for drug delivery and tissue engineering applications, incorporation of sulfated HA into these networks to attenuate the release of proteins is yet to be explored. Here, we developed sulfated and methacrylate-modified HA macromers and incorporated them into HA hydrogels through free radical-initiated crosslinking. The sulfated HA macromers bound to a heparin-binding protein (i.e., stromal cell-derived factor-1 alpha, SDF-1α) with an affinity comparable to heparin and did not alter the gelation behavior or network mechanics when copolymerized into hydrogels at low concentrations. Further, these macromers were incorporated into electrospun nanofibrous hydrogels to introduce sulfate groups into macroporous scaffolds. Once incorporated into either uniform or fibrous HA hydrogels, the sulfated HA macromers significantly slowed encapsulated SDF-1α release over 12 days. Thus, these macromers provide a useful way to introduce heparin-binding features into radical-crosslinked hydrogels to alter protein interactions for a range of applications.
1. Introduction
Hyaluronic acid (HA)-based hydrogels have been used widely in recent years for applications in drug delivery and tissue engineering.1 HA is a ubiquitous biological polymer composed of repeating D-glucuronic acid [β-1–3] and N-acetyl-D-glucosamine [β-1–4] disaccharides and is found abundantly in the extracellular matrix (ECM) of many tissues. During endogenous tissue remodeling, HA plays a critical role in regulating cell motility through CD44 receptor interactions and provides a hydrated microenvironment for growth factor and cytokine stability and diffusion.2HA is unique among glycosaminoglycans (GAGs) in that it is produced and secreted from cells as a linear polymer unattached to a polypeptide. This feature makes HA production amendable to typical genetic engineering approaches using microbial fermentation.3 These recombinant HAs are non-immunogenic, available in a wide range of well-defined molecular weights, and have been used in numerous biomedical applications. Further, due to the abundance of hydroxyl and carboxylic acid groups, HA is readily modified with reactive groups to form hydrogels.1 In particular, modification of HA with methacrylate groups permits hydrogel formation through free-radical initiated crosslinking. This approach has allowed spatiotemporal control of network architectures to direct stem cell differentiation in vitro,4,5in situ gelation to localize therapeutic protein delivery in vivo,6 and construction of nanofibers to mimic fibrous tissue architectures.7
While protein binding to methacrylate-modified HA macromers has been observed,6 HA lacks the sulfate groups typically associated with high protein-ECM binding affinities. For example, sulfated glycosaminoglycans have been shown to bind, stabilize, and control the activity of hundreds of proteins through electrostatic interaction between sulfates and basic amino acid residues.8 These interactions are critical for regulating tissue remodeling processes by localizing protein activity in a temporally controlled fashion (i.e., protein presentation).9,10 In order to control protein presentation and therefore engineer tissue remodeling, sulfated polymers have been incorporated into biomaterial scaffolds. Heparin isolated from animal tissues is most commonly incorporated into these scaffolds due to its high degree of sulfation;11–13 however, synthetically produced heparin mimetics have recently been developed as an alternative source of protein-binding polymers that provide greater chemical versatility for incorporation into scaffolds.
Heparin mimetics are highly sulfated polymers that are structurally distinct from naturally occurring GAGs, but perform some of the same functions as heparin (e.g., protein presentation).8,14 These sulfated polymers have been shown to bind proteins with high affinity and when delivered in vivo significantly enhance tissue repair, presumably by replacing degraded heparin and heparan sulfate and increasing the bioavailability of proteins.15 While HA is unique among GAGs in that it lacks sulfate groups, researchers have sulfated HA through nucleophilic substitution of primary hydroxyl hydrogens on HA with SO3 by simply reacting SO3 complexes with HA in an organic solvent.16–18 Reaction of an SO3/DMF complex with HA results in a specific sulfation pattern that binds heparin-binding proteins (HBPs) with a high affinity.17,19 Further, sulfated HA polymers exhibit good cytocompatibility and enhance HBP presentation to cells in vitro.20 Also, sulfated HA polymers themselves have been shown to influence cell activity in vitro, including stimulating Wnt and Notch signaling16 and enhancing cell–cell junctions.18 While sulfated HA has been covalently modified to functionalize biomaterial surfaces,21,22 it has not been previously modified for covalent incorporation into hydrogels to alter features such as protein interactions and diffusion within three-dimensional networks.
Here, we synthesized sulfated and methacrylate-modified HA macromers to incorporate protein-binding sulfate groups into radical-crosslinked HA hydrogels. Further, hydrolytically unstable ester groups were included between the reactive methacrylate group and the sulfated HA backbone to liberate bound proteins in a controlled fashion as the hydrogel degrades. This technology was investigated in bulk hydrogels, as well as with electrospun HA nanofibers to mimic heparin binding in a complex three-dimensional scaffold.
2. Materials and methods
2.1 Materials
Sodium hyaluronate (NaHy) was purchased from Lifecore (100 and 440 kDa, as measured with GPC). Recombinant stromal cell-derived factor-1α (rSDF-1α), ELISA kits, and ELISA reagents were purchased from R&D Systems. Heparin binding plates were purchased from BD Biosciences. Polyethylene glycol standards were purchased from Waters. All other materials and chemicals were purchased from Sigma Aldrich.2.2 Macromer synthesis
NaHy (100 kDa) was chemically modified with a hydroxyethyl methacrylate (HEMA) group to incorporate methacrylate reactivity for free-radical initiated crosslinking, as well as hydrolytic degradation through ester group hydrolysis as previously described.6 Briefly, HEMA was reacted with succinic anhydride via a ring opening polymerization in the presence of N-methylimidazole to obtain HEMA–COOH, which was then coupled to a tetrabutylammonium (TBA) salt of HA in the presence of 4-dimethylaminopyridine (DMAP) and di-tert-butyl dicarbonate (BOC2O). The resulting HA macromer with HEMA group modification (HEMA-HA) was purified with dialysis and characterized by 1H NMR.For HEMA-SHA synthesis, a 440 kDa NaHy was first synthesized to HEMA-HA as described above and then converted to a TBA salt through resin exchange (Dowex 50 × 8w hydrogen form) and TBA–OH titration to pH 7.02 to permit polymer solubility in DMF for the sulfation reaction. HEMA-HA TBA was then frozen and lyophilized. Dry HEMA-HA TBA was dissolved in N,N-dimethylformamide (DMF) at 0.25 wt% under N2 at room temperature (RT). The SO3/DMF complex in a DMF solution (10 wt%) was added to the reaction mixture at a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (mol/mol) SO3/DMF:HA repeat unit ratio and reacted for 1 h at RT under N2. The reaction solution was then dialyzed against DI H2O for 14 days at 4 °C to remove the solvent and TBA salts, and was frozen and lyophilized.
:1 (mol/mol) SO3/DMF:HA repeat unit ratio and reacted for 1 h at RT under N2. The reaction solution was then dialyzed against DI H2O for 14 days at 4 °C to remove the solvent and TBA salts, and was frozen and lyophilized.
2.3 Gel permeation chromatography
Molecular weights were determined with gel permeation chromatography (GPC) analysis using a Waters 1525 Binary HPLC Pump, Waters 2414 Refractive Index (RI) Detector, and Waters Ultrahydrogel 1000 columns. The eluent was PBS buffer and the flow rate was 0.5 mL min−1. Size calibrations for molecular weight determination were made with polyethylene glycol standards.2.4 Zeta potential measurements
Polymers were dissolved in MilliQ H2O at 3.5 wt% for HEMA-HA and 4 wt% for HEMA-SHA and loaded into folded capillary cells (Malvern). Zeta potential measurements were made on a ZetaSizer Nano ZS (Malvern).2.5 Dimethylmethylene blue assay
Serial dilutions of each polymer were prepared in MilliQ H2O (50 μg mL−1→0 μg mL−1) and assayed for the presence of sulfate groups using a previously developed dimethylmethylene blue (DMMB) assay.23 Absorbance at 520 nm was measured using a cuvette reader (Tecan). To visualize sulfate groups, 50 μL hydrogels were formed (see the subsequent methods), washed for 2 days in PBS, incubated in a DMMB solution overnight at 37 °C, rinsed and photographed.2.6 Solid-phase binding assay
HEMA-HA, HEMA-SHA and heparin at 2.5 μM in phosphate buffered saline (PBS) were coated overnight at 25 °C onto heparin binding plates.24 Wells were washed in TNC buffer (50 mM Tris/HCl, 150 mM NaCl, 10 mM CaCl2, 0.05% Brij-35 and 0.02% sodium azide) containing 0.1% Tween 20 between each subsequent incubation. Wells were blocked with 0.2% gelatin in TNC buffer and then incubated with rSDF-1α (300–5 ng mL−1 in a blocking solution for 2 h at 37 °C). Bound rSDF-1α was detected using a biotin labeled antibody for rSDF-1α (R&D Systems, part no. 840932, 2 h incubation at 37 °C), followed by streptavidin coupled to horseradish peroxidase (R&D Systems) for 20 min at 37 °C. Hydrolysis of a 1:1 mixture of H2O2 and tetramethylbenzidine (R&D Systems) was measured at 450 nm using a microplate reader (Tecan infinite m200).
2.7 Hydrogel formation
For crosslinking, a redox initiator system consisting of ammonium persulfate (APS) and N,N,N′,N′-tetramethylethylenediamine (TEMED) was utilized. APS and TEMED were mixed with polymers dissolved in PBS to initiate crosslinking at final concentrations of 10 mM APS, 10 mM TEMED, and 4 wt% macromer. HEMA-HA/HEMA-SHA blends were formed by mixing different volumetric ratios of the respective polymers dissolved at the same concentrations in PBS. Kinetics of the crosslinking reaction were characterized with rheometry by monitoring the storage (G′) and loss (G′′) moduli with time (20 mm 1° cone geometry, 1% strain, 1 Hz, TA Instruments AR 2000ex).2.8 Hydrogel swelling
50 μL hydrogels (4 wt% polymer, 10 mM APS/TEMED) were formed in cylindrical molds at 37 °C for 30 min after mixing APS/TEMED initiators. Hydrogels were then incubated in PBS for 2 days at 37 °C. Hydrogel diameters were measured after 30 min of crosslinking and again after 2 days in PBS. Swelling changes were reported as percent change in diameter after 2 days in PBS.2.9 Protein release studies
rSDF-1α or FITC-BSA were dissolved in the polymer solutions at 200 ng and 10 μg of protein per 50 μL hydrogels, respectively. 50 μL hydrogels (4 wt% polymer, 10 mM APS/TEMED) were formed in cylindrical molds at 37 °C for 30 min after mixing APS/TEMED initiators. After a wash in PBS, hydrogels were incubated in PBS supplemented with 1 wt% BSA (R&D Systems) at 37 °C. Hydrogels were moved to a fresh buffer every 2 days and samples were stored at −20 °C until the end of the study. After the final time point, hydrogels were degraded in 0.3 mg mL−1 hyaluronidase to release the remaining protein. FITC-BSA was quantified using fluorescence while rSDF-1α was quantified using ELISA.2.10 Electrospinning
Solutions for electrospinning were prepared as follows: 4 wt% HEMA-HA/HEMA-SHA blends, 2 wt% poly(ethylene oxide) (900 kDa), and 0.5 wt% Irgacure 2959 in deionized water. Electrospinning was performed as previously described.7 Briefly, a syringe was connected to a 12′′ long 18G blunt-ended needle positioned 16 cm away from a grounded aluminum mandrel. Using a high voltage power source (Gamma High Voltage Research) and a syringe pump (KD Scientific), the potential difference between the needle and the mandrel was adjusted to +21 kV, and the solution flow rate was set to 1.4 mL hour−1. After collection, electrospun fibers were purged under nitrogen and then crosslinked for 10 min with 12 mW cm−2 UV light (320–390 nm collimated, Omnicure S1000 UV Spot Cure Systems).2.11 Nanofiber characterization
Average diameters of dry and swollen HA fibers were measured as described previously.7 Briefly, samples were imaged in the dry state with scanning electron microscopy (SEM, JEOL 7500F HRSEM, Penn Regional Nanotechnology Facility) or after swelling in PBS for 48 hours at 37 °C with confocal microscopy (Zeiss AxioObserver inverted microscope, Penn CDB Microscopy Core). For confocal images, methacrylated rhodamine was added to the electrospinning solution at 25 μM to visualize fibers. Fiber diameters were then quantified using ImageJ (NIH, n = 80–90 fibers per group).2.12 SDF-1α release from fibrous HA hydrogels
rSDF-1α was added to each polymer solution at a concentration of 1.67 μg mL−1 prior to electrospinning. After electrospinning and crosslinking as described in the section above, 1.5 × 1.5 cm square samples were cut from the electrospun mats and incubated in PBS supplemented with 1% BSA (R&D Systems) at 37 °C. Samples were moved to fresh buffer every 2 days and then stored at −20 °C. After the final time point, hydrogels were degraded in 2 mg mL−1 hyaluronidase to release the remaining protein. rSDF-1α was quantified using ELISA.2.13 Statistical analysis
Statistical comparisons between groups were performed with a two-tailed Student's t-test; p < 0.05 was considered statistically significant for comparison between two groups. The results are reported as mean values ± standard deviations.3. Results and discussion
3.1 Sulfation of HEMA modified HA
HA has been previously modified with sulfate groups through nucleophilic substitution of primary hydroxyls along the HA backbone.17 When the reaction was performed with the SO3/DMF complex at a 20:1 SO3/DMF:HA ratio, a high degree of sulfation was observed, estimated at 2.8 sulfate groups per HA disaccharide, leading to high protein binding affinities. We applied this sulfation reaction to our previously developed hydroxyethylmethacrylate (HEMA)-modified HA (HEMA-HA) in order to incorporate protein-binding features into degradable, radically crosslinked hydrogels. Sulfated HEMA-HA macromers (HEMA-SHA) could be incorporated into HEMA-HA hydrogels by simply blending the HEMA-SHA and HEMA-HA macromers at a desired ratio and initiating crosslinking with one of many free-radical initiator systems (Scheme 1). Using this strategy, sulfate groups were incorporated into the hydrogels without introducing additional chemistries and crosslinking steps often required for heparin incorporation into hydrogels. In addition, fundamental features of the networks are maintained (wt%, crosslinking density, and chain length), allowing independent variation of sulfate group content for controlled biological studies. Further, degradation kinetics of the system can be controlled through HEMA group modification to release bound proteins in a temporally controlled fashion.
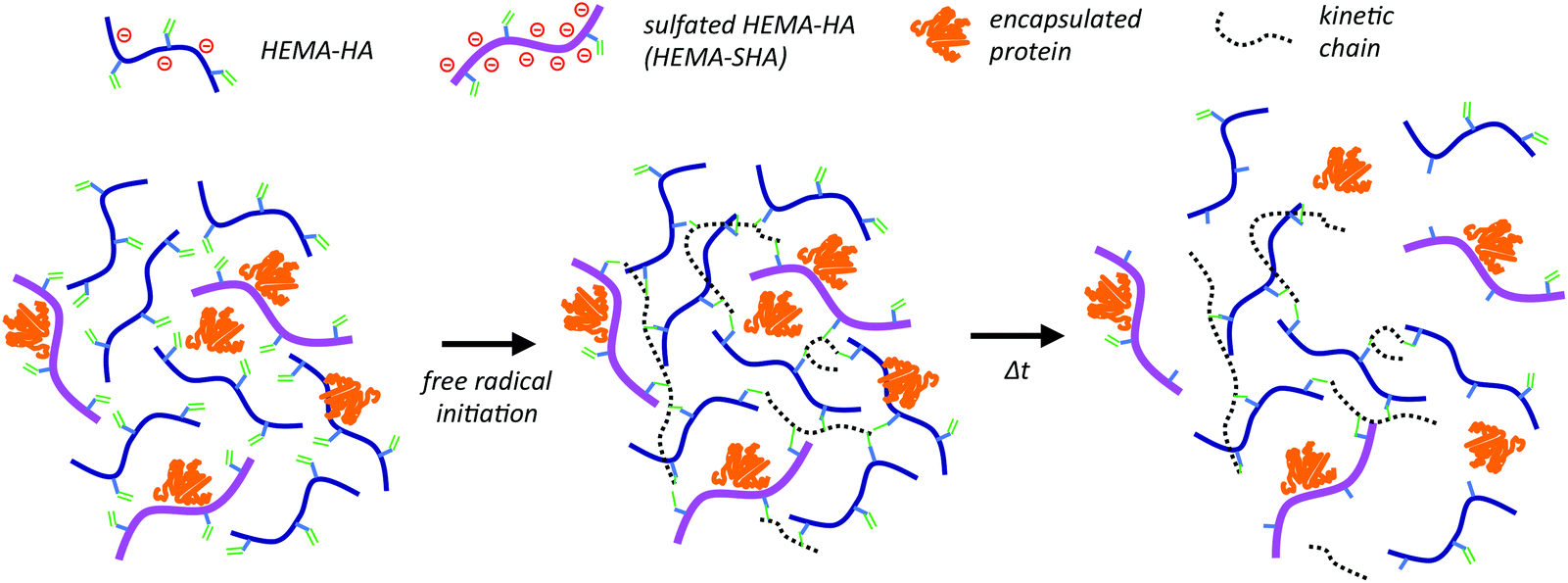 | ||
| Scheme 1 Incorporation of sulfated hyaluronic acid (HA) macromers into hydrogels through free-radical initiated crosslinking. Hydroxyethylmethacrylate (HEMA) modified HA macromers were synthesized with or without sulfate groups to control sulfated polymer concentration in hydrogels by simply blending the two macromers together and crosslinking with free-radical initiators. Release of encapsulated proteins in these networks is controlled by electrostatic interactions with the negatively charged polymers and by gel degradation through ester group hydrolysis of the HEMA crosslinks. | ||
HEMA-HA was synthesized as previously reported by first converting sodium hyaluronate (NaHy, 100 kDa) to a tetrabutylammonium (TBA) salt and then coupling HEMA succinate to primary hydroxyls on HA through an esterification reaction6 (Fig. 1a). The modification process decreased the number average molecular weight (Mn) of the HA polymer by 20% and slightly increased the HA polydispersity, as determined by GPC (Fig. 1b). The decrease in Mn is likely due to acid hydrolysis of the polysaccharide backbone during HA-TBA synthesis as a proton-rich exchange resin was used for protonation of NaHy. The final HEMA-HA had a Mn of ∼80 kDa and a HEMA modification of ∼16%.
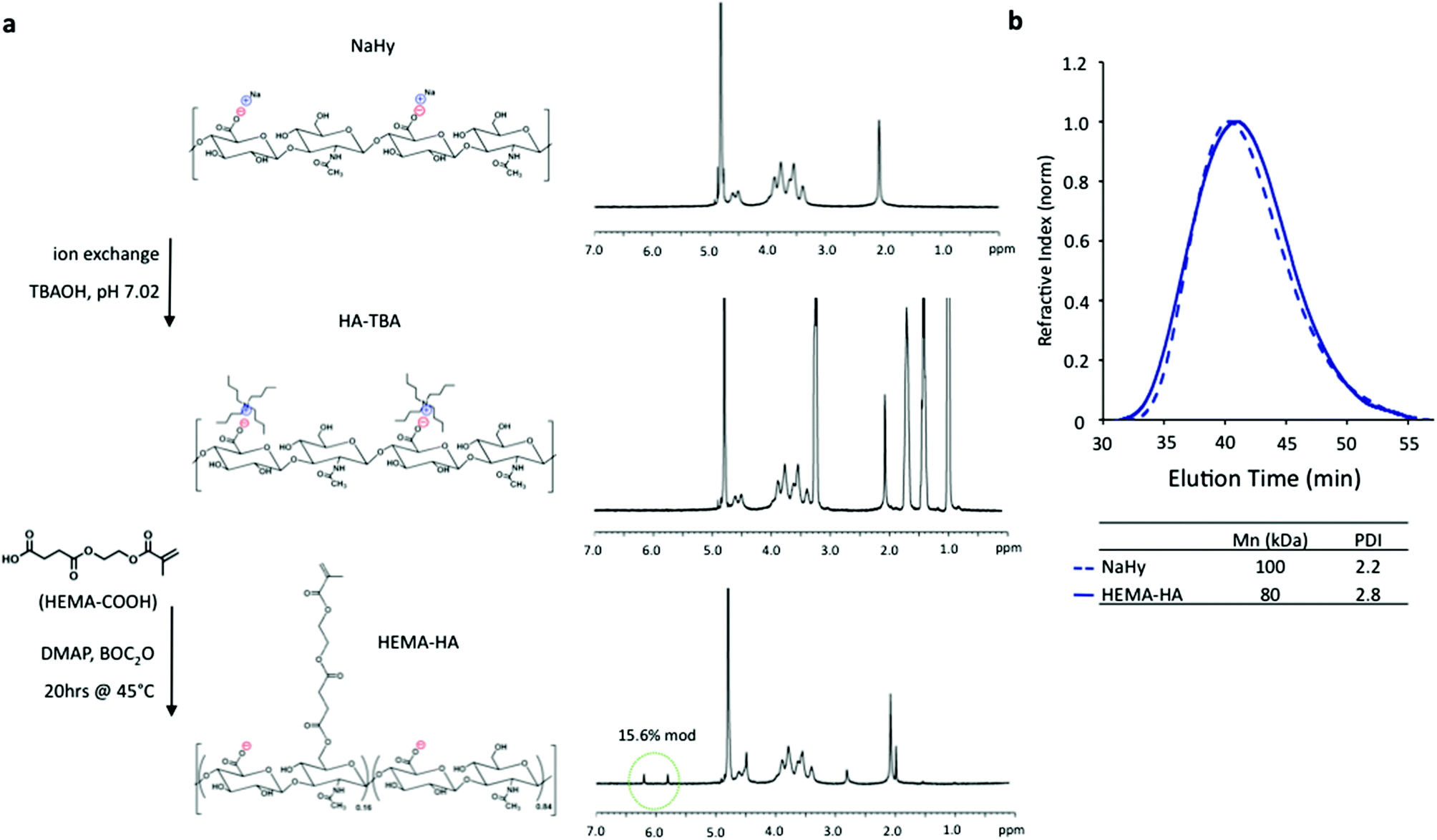 | ||
| Fig. 1 Synthesis of HEMA modified HA. (a) Sodium hyaluronate (NaHy) was converted to a tert-butylammonium (TBA) salt and reacted with HEMA-succinate in dimethyl sulfoxide using dimethylaminopyridine (DMAP)/di-tert-butyl dicarbonate (BOC2O) catalyzed esterification. HEMA modification was quantified by methacrylate group 1H NMR peaks (green circle). (b) This modification slightly reduced the number average molecular weight (Mn) and increased the polydispersity index (PDI) as measured with GPC when compared to unmodified NaHy. | ||
To synthesize HEMA-SHA, HEMA-HA was synthesized starting with NaHy of 440 kDa and converted to a TBA salt through a protonated resin exchange. This step did not affect the HEMA modification as indicated by peak integration of methacrylate protons on the methacrylate group (approximately 5.8 and 6.2 ppm) via1H NMR (Fig. 2a). Next, the TBA salt of HEMA-HA was reacted with SO3 in DMF, which also did not affect HEMA modification as indicated by the methacrylate proton integrations before and after the reaction (Fig. 2a). Also, there was an obvious shift in hydroxyl proton peaks along the HA backbone (approximately 3 to 4 ppm) following the sulfation reaction as expected due to substitution of these protons with sulfate groups (Fig. 2a). Although there was no change in HEMA modification (∼16%) there was an expected and significant reduction of the macromer Mn following the sulfation reaction, which is likely due to acid hydrolysis of HA from H2SO4 formed from a reaction of SO3/DMF and water vapor or from non-specific nucleophilic substitution (Fig. 2b). By starting with a 440 kDa NaHy, the final Mn of HEMA-SHA was maintained at ∼80 kDa so that HEMA-HA and HEMA-SHA could be blended at approximately the same molecular weight and modification. Naturally sulfated GAGs are typically much smaller than 80 kDa; therefore, our approach allows generation of sulfated and crosslinkable polymers with larger molecular weights for covalent incorporation into hydrogels. In addition, the wide range of HA molecular weights available commercially provides control over the molecular weight of the final sulfated HA macromer. Using this approach, the sulfate group content within hydrogels can be easily controlled by blending sulfated and non-sulfated polymers at the desired ratios without altering network crosslink densities.
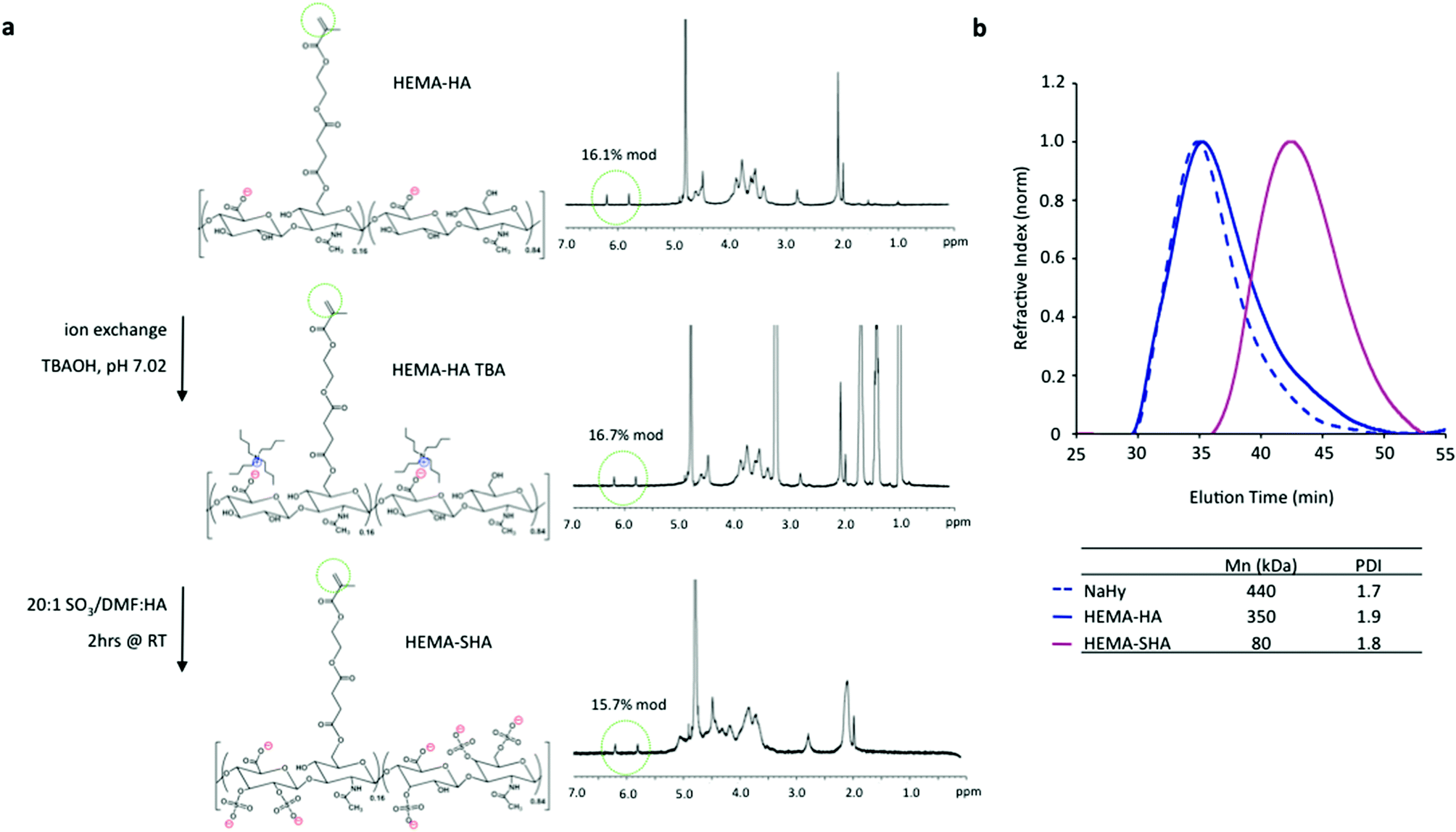 | ||
| Fig. 2 Synthesis of sulfated HEMA-HA. (a) HEMA-HA was converted to a TBA salt and reacted with the SO3/dimethylformamide (DMF) complex in DMF. This reaction altered hydroxyl group 1H NMR peaks as expected, but did not affect methacrylate 1H NMR peaks (green circles). (b) Sulfation reduced the Mn of HEMA-HA by almost 80% as measured by GPC, so a larger 440 kDa NaHy was used to generate HEMA-SHA macromers with comparable Mn to HEMA-HA macromers (∼80 kDa). | ||
3.2 Sulfating HEMA-HA alters polymer charge and increases HBP interactions
Heparin has been shown to bind hundreds of proteins through electrostatic interaction between negatively charged sulfate groups on heparin and positively charged amino acid residues on proteins.8 While intermolecular forces such as hydrogen bonding and van der Waals interactions involving other chemistries on heparin have been shown to contribute to some highly specific binding events,25 charge interactions from sulfate groups are the major force driving heparin-protein binding.26 Similarly, the addition of sulfate groups to HA has been shown to dramatically increase binding of proteins that are rich in basic amino acid residues.17,19 While the degree of sulfation of HA was shown to directly correlate with protein binding affinity, oversulfation of polysaccharides can inhibit protein binding27 and the distribution of sulfate groups along the polysaccharide is important in protein binding affinity.17,19The sulfate content on HEMA-SHA macromers was compared to HEMA-HA and heparin using a modified dimethylmethylene blue (DMMB) assay.23 Using this assay, a significant increase in sulfate content was observed on HEMA-SHA compared to HEMA-HA, confirming success of the sulfation reaction (Fig. 3a). Further, the sulfate content on HEMA-SHA was comparable to that of heparin. To quantify the effect of the sulfate groups on polymer charge, an important contributor to protein binding affinity, zeta potentials of the polymers were measured from pH 2 to 10. HEMA-SHA polymers possessed a more negative charge when compared to HEMA-HA polymers across the entire pH range (Fig. 3b). Further, a clear difference was observed in the protonation states characteristic of sulfate groups on HEMA-SHA polymers compared to carboxylate groups on HEMA-HA polymers. The sulfate groups remained deprotonated across all pHs, while the carboxylate groups were protonated at lower pH.
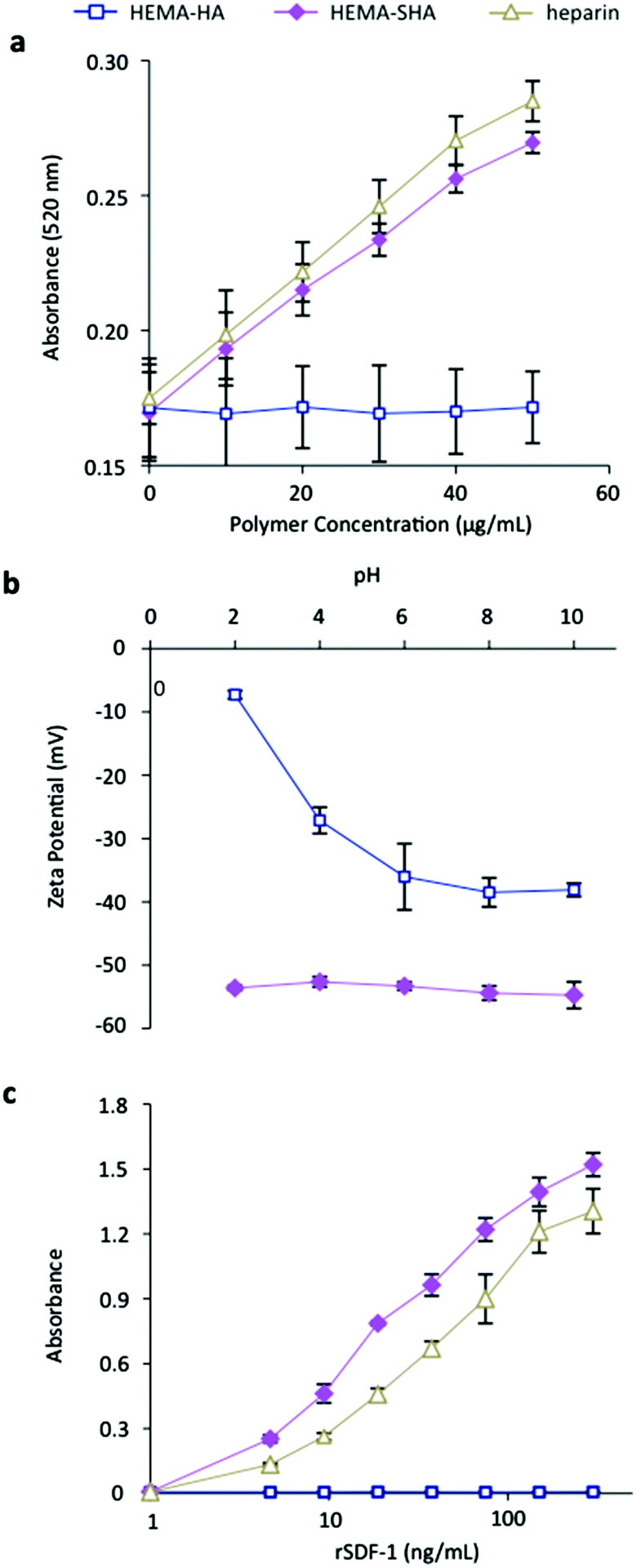 | ||
| Fig. 3 Characterization of HEMA-SHA macromers. (a) Sulfate content was increased in HEMA-SHA macromers when compared to HEMA-HA macromers, as determined by dimethylmethylene blue assay. Sulfate content of HEMA-SHA macromers was comparable to that of heparin. (b) Addition of sulfate groups enhanced the negative charge of carboxylic acid-containing HEMA-HA macromers via zeta potential measurements and (c) enhanced binding to the heparin binding protein SDF-1α. rSDF-1α binding of HEMA-SHA macromers was comparable to that of heparin (n = 3 replicates per group, mean ± SD). | ||
To assess the protein-binding characteristics of the polymers, recombinant stromal cell-derived factor-1 alpha (rSDF-1α) was used as a model HBP. SDF-1α is a chemoattracting cytokine that plays a critical role in regulating progenitor cell trafficking between the bone marrow and remodeling tissues.28,29 Studies have shown that SDF-1α binds to sulfated GAGs with a high affinity through an abundance of basic amino acids on the surface of SDF-1α.30 To assess SDF-1α binding to HEMA-SHA, rSDF-1α was incubated in wells with GAG polymers adsorbed on the surface, and rSDF-1α bound to the polymer-coated wells was quantified using ELISA. Using this technique, a significant increase in rSDF-1α binding was observed with HEMA-SHA polymers compared to non-sulfated HEMA-HA polymers (Fig. 3c). Further, HEMA-SHA polymers had comparable rSDF-1α binding to heparin, although more rSDF-1α was bound to HEMA-SHA. This observation could be explained by the greater Mn of HEMA-SHA compared to heparin (80 kDa vs. 20 kDa by GPC) as both polymers were adsorbed to the wells at the same molar concentration. In addition, differences in the sulfation pattern could affect HBP binding. For example, Hintze and coworkers showed that sulfated HA binds human transforming growth factor beta-1 (TGF-β1) with a stronger affinity than synthetically sulfated chondroitin sulfate, despite having the same degree of sulfation.19 Further, sulfated HA was shown to have different binding specificities for human plasma proteins than heparin.31 Therefore, while HEMA-SHA was shown to bind a HBP (rSDF-1α) in this study, further investigation into protein binding specificity of HEMA-SHA polymers is warranted for specific HBPs.
3.3 HEMA-SHA is incorporated into radically crosslinked hydrogels and alters HBP presentation
While a number of heparin mimetics have been developed as antithrombotic therapeutics, very few have been crosslinked into hydrogels for application in drug delivery or tissue engineering.32–34 After demonstrating successful synthesis of a methacrylate-modified and sulfated HA macromer and its ability to bind a HBP, we crosslinked these macromers into hydrogels to encapsulate and localize HBPs. HEMA-SHA polymers were blended with HEMA-HA polymers at 10% (w/w) HEMA-SHA and 90% (w/w) HEMA-HA with a total polymer concentration of 4 wt% and crosslinking was initiated with APS and TEMED redox initiators. Rheometry was used to quantify gelation, and HEMA-SHA blended hydrogels were compared to 100% HEMA-HA hydrogels. Incorporation of 10% HEMA-SHA did not significantly alter gelation, as both groups had similar storage modulus (G′) profiles over time and crosslinking reached the same G′ plateau after 30 min (Fig. 4a and b). Other ratios were also investigated (higher HEMA-SHA content); however, they led to hydrogels with statistically different mechanical properties, likely due to extensive sulfate charge repulsion and were not pursued further (results not shown).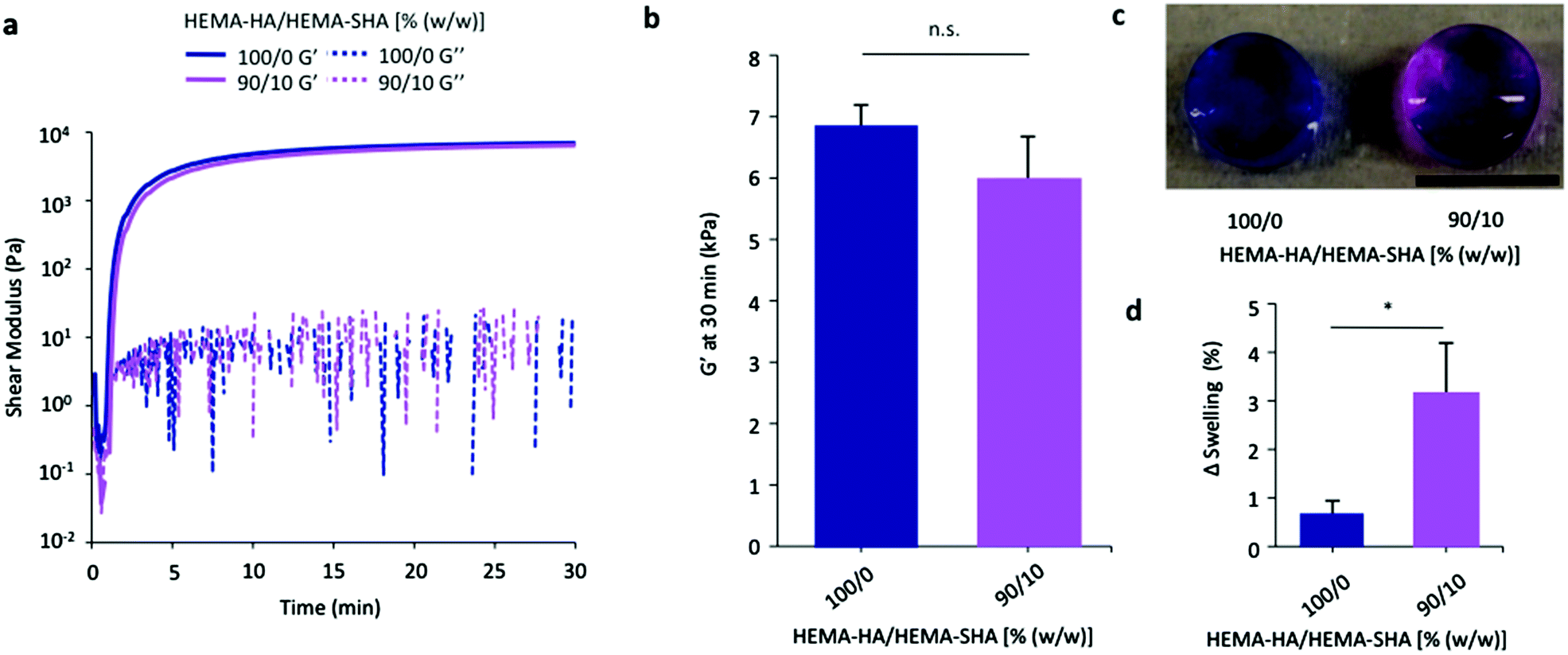 | ||
| Fig. 4 Crosslinking of HEMA-HA/HEMA-SHA hydrogels. HEMA-HA/HEMA-SHA hydrogels were crosslinked using APS/TEMED free-radical initiators and 4 wt% macromers in PBS at 100/0 or 90/10 HEMA-HA/HEMA-SHA compositions (wt/wt). (a) Representative storage (G′) and loss (G′′) moduli profiles over time with and without 10% HEMA-SHA incorporation. (b) Incorporation of 10% HEMA-SHA did not significantly affect G′ after crosslinking had reached a plateau at 30 min. (c) Incorporation of sulfate groups in the hydrogels was visualized using dimethylmethylene blue with purple indicative of sulfate groups (scale bar = 5 mm). (d) Incorporation of sulfate groups slightly increased the swelling of the hydrogels after 2 days in PBS (n = 3 replicates per group, mean ± SD, *p < 0.05 between groups). | ||
To visualize the incorporation of sulfate groups into the HEMA-SHA blended hydrogels, they were incubated with DMMB after 2 days of swelling in PBS. HEMA-SHA blended hydrogels exhibited a purple color compared to blue HEMA-HA hydrogels, indicating the presence of sulfate groups (Fig. 4c). Further, HEMA-SHA blended hydrogels swelled slightly more than HEMA-HA hydrogels, likely due to charge repulsion within the sulfate rich hydrogels or increased water content due to sulfate hydrophilicity (Fig. 4d).
Finally, to illustrate the influence of sulfate group incorporation on HBP presentation within the hydrogels, networks were formed that encapsulated either a model HBP, rSDF-1α, or bovine serum albumin (BSA) and protein release and hydrogel degradation were monitored over 12 days. HEMA-SHA blended hydrogels degraded in a slightly more rapid fashion compared to HEMA-HA hydrogels (Fig. 5a). This observation is likely due to the increased water content of the hydrogels, which enhances ester group hydrolysis of the HEMA groups. BSA release from HEMA-SHA blended hydrogels was similar to HEMA-HA hydrogel release, indicating little influence of sulfate groups on BSA presentation and release (Fig. 5b). This is not surprising as previous studies also found that albumin does not bind to sulfated HA with a high affinity.31 However, rSDF-1α release was drastically reduced with the incorporation of HEMA-SHA polymers into the network (Fig. 5c). This reduced release of rSDF-1α with the incorporation of HEMA-SHA polymers demonstrates our ability to control HBP presentation within covalently crosslinked and degradable hydrogels, simply through chemical modification of the hydrogel precursors.
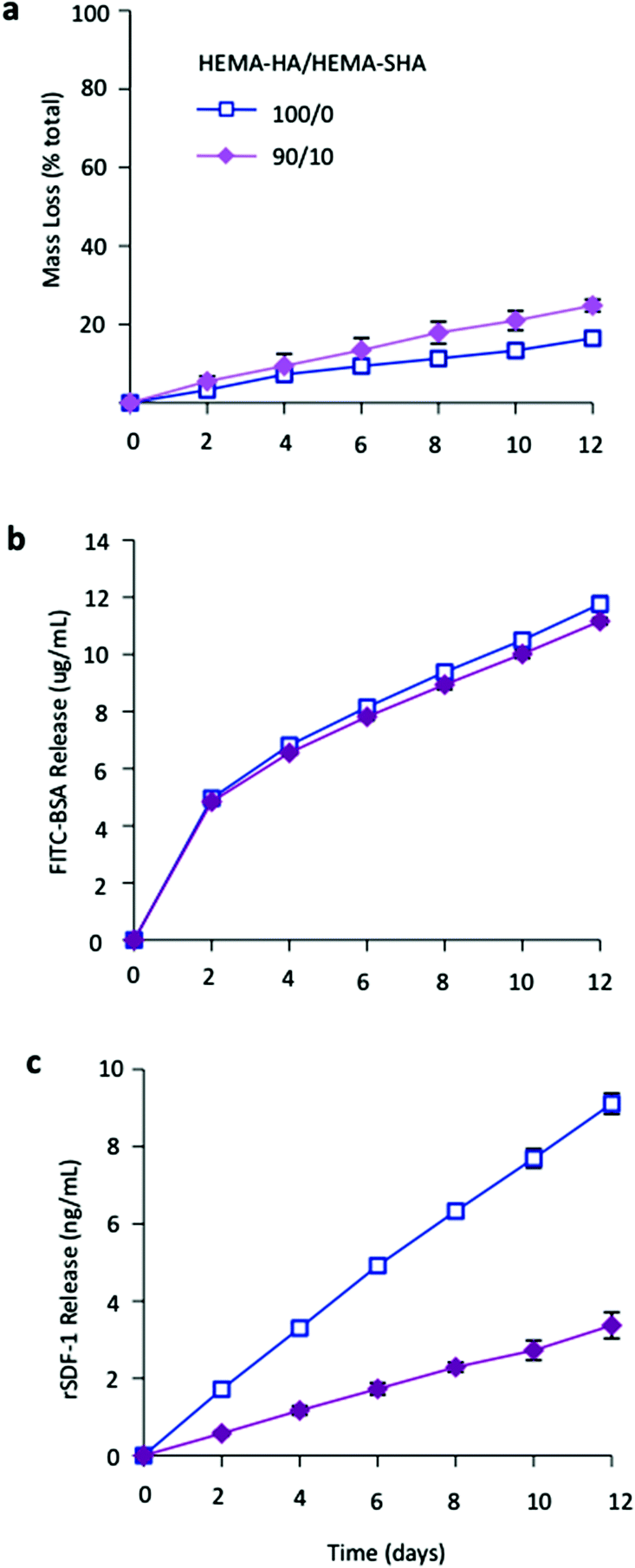 | ||
| Fig. 5 Hydrogel degradation and encapsulated protein release. (a) Hydrogel mass loss as determined by uronic acid content. Incorporation of HEMA-SHA macromers enhanced hydrolytic degradation of the hydrogels over 2 weeks. (b) Encapsulated FITC-BSA and (c) heparin-binding SDF-1α release from hydrogels of HEMA-HA alone or incorporating HEMA-SHA macromers. Encapsulated SDF-1α release was reduced by over 50% with the incorporation of HEMA-SHA macromers (n = 3 hydrogels per group, mean ± SD). | ||
To demonstrate versatility and other possible applications, HEMA-SHA macromers were also incorporated into crosslinked and swollen electrospun nanofibers (Fig. 6). Electrospun scaffolds have been widely explored for their use in tissue engineering applications due to their nanoscale fibrous structure that mimics tissue architectures found in the body.35 In particular, photoinitiated crosslinking of electrospun fibers has permitted tuning of the nanoscale structure to construct scaffolds with unique properties for control of cell alignment,36 cell motility,37 and cell invasion.38 To incorporate protein-binding sulfate groups into these systems, HEMA-SHA macromers were blended with non-sulfated HEMA-HA macromers, electrospun, and crosslinked with UV light exposure. Fiber diameters were visualized and quantified in the dry, pre-swelled state and after swelling in PBS to assess the effects of HEMA-SHA incorporation on the nanoscale structure of the fibers (Fig. 6a and b). There was no significant difference in fiber diameter in the dry state or swollen state with incorporation of HEMA-SHA macromers into HEMA-HA fibers.
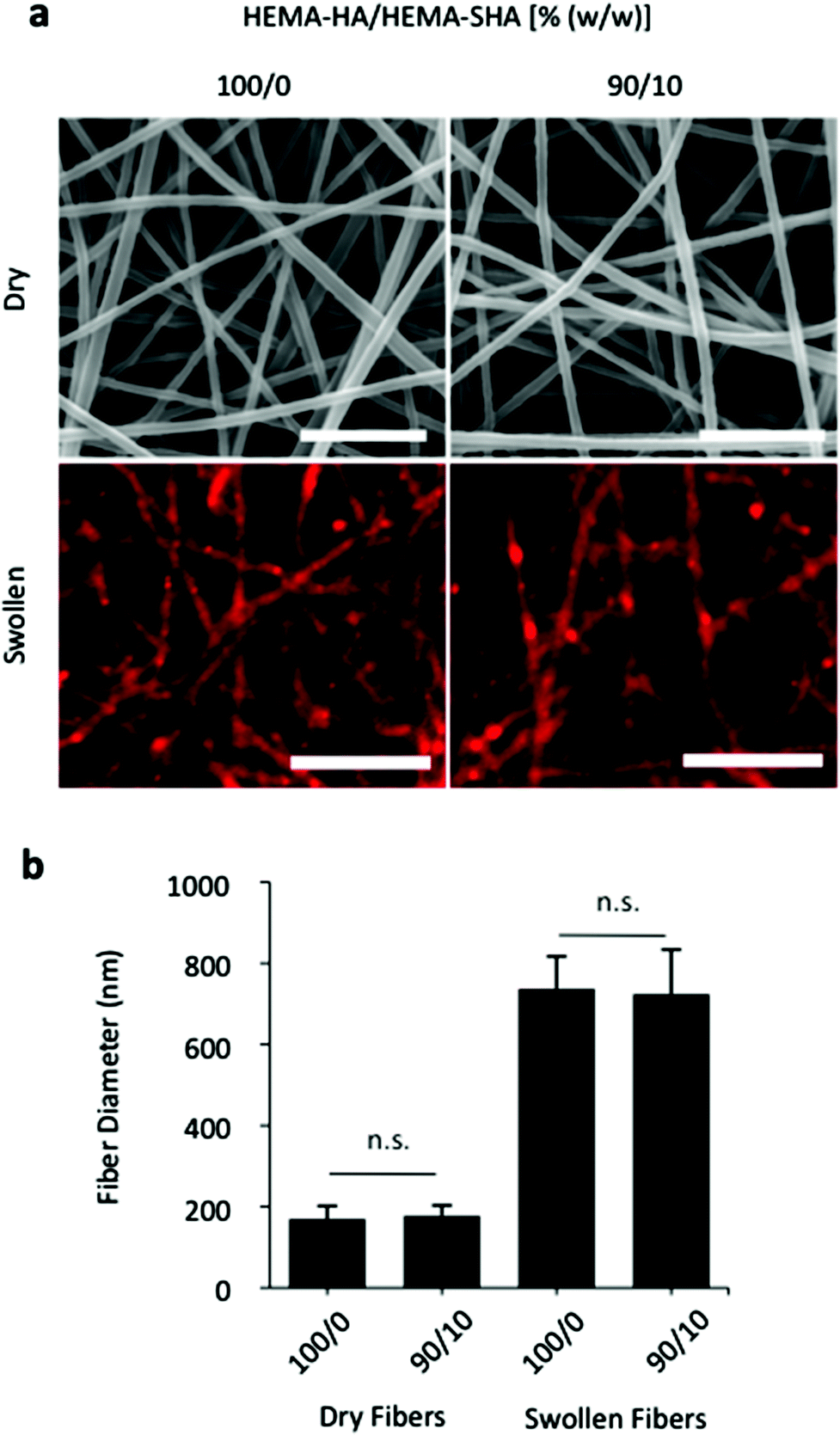 | ||
| Fig. 6 Electrospun HEMA-HA/HEMA-SHA nanofibers. Hydrogels were processed into nanoscale fibers using an electrospinning technique. (a) Fibers visualized in the dry state using scanning electron microscopy (top, scale bars = 2 μm) and after swelling in PBS using fluorescence of incorporated methacrylated rhodamine and optical microscopy (bottom, scale bars = 10 μm). (b) Fiber diameter quantification showed no significant difference between 100/0 and 90/10 HEMA-HA/HEMA-SHA ratios (w/w) in either dry or swollen states (n = 80–90 fibers, mean ± SD, p > 0.05). | ||
To visualize the charge of the crosslinked fibers, scaffolds were incubated with DMMB dye. As with the 50 μL hydrogels, electrospun nanofibers were purple with HEMA-SHA incorporation compared to blue HEMA-HA fibers, confirming incorporation of sulfate groups into the fibers (Fig. 7a). Finally, encapsulated rSDF-1α release was significantly reduced with HEMA-SHA incorporation, indicating a higher protein-binding capacity of the chemically incorporated sulfate groups within the fibers (Fig. 7b). To incorporate heparin-binding features into electrospun nanofibers, heparin is often physically encapsulated into electrospun fibers in its low molecular weight form or conjugated to larger polymers,39–41 and recent studies have chemically conjugated heparin to crosslinked fibers after electrospinning, decorating the outside of the fibers with sulfate groups.42,43 In the HEMA-HA/HEMA-SHA fibers, sulfate groups are chemically crosslinked throughout the fibers in a process amendable to protein encapsulation, allowing further control of encapsulated protein release with hydrolytic degradation of the fibers. This approach can be used with a variety of radically crosslinked scaffolds for drug delivery and tissue engineering applications, either for the controlled release of proteins or to alter the accumulation of proteins surrounding cells.
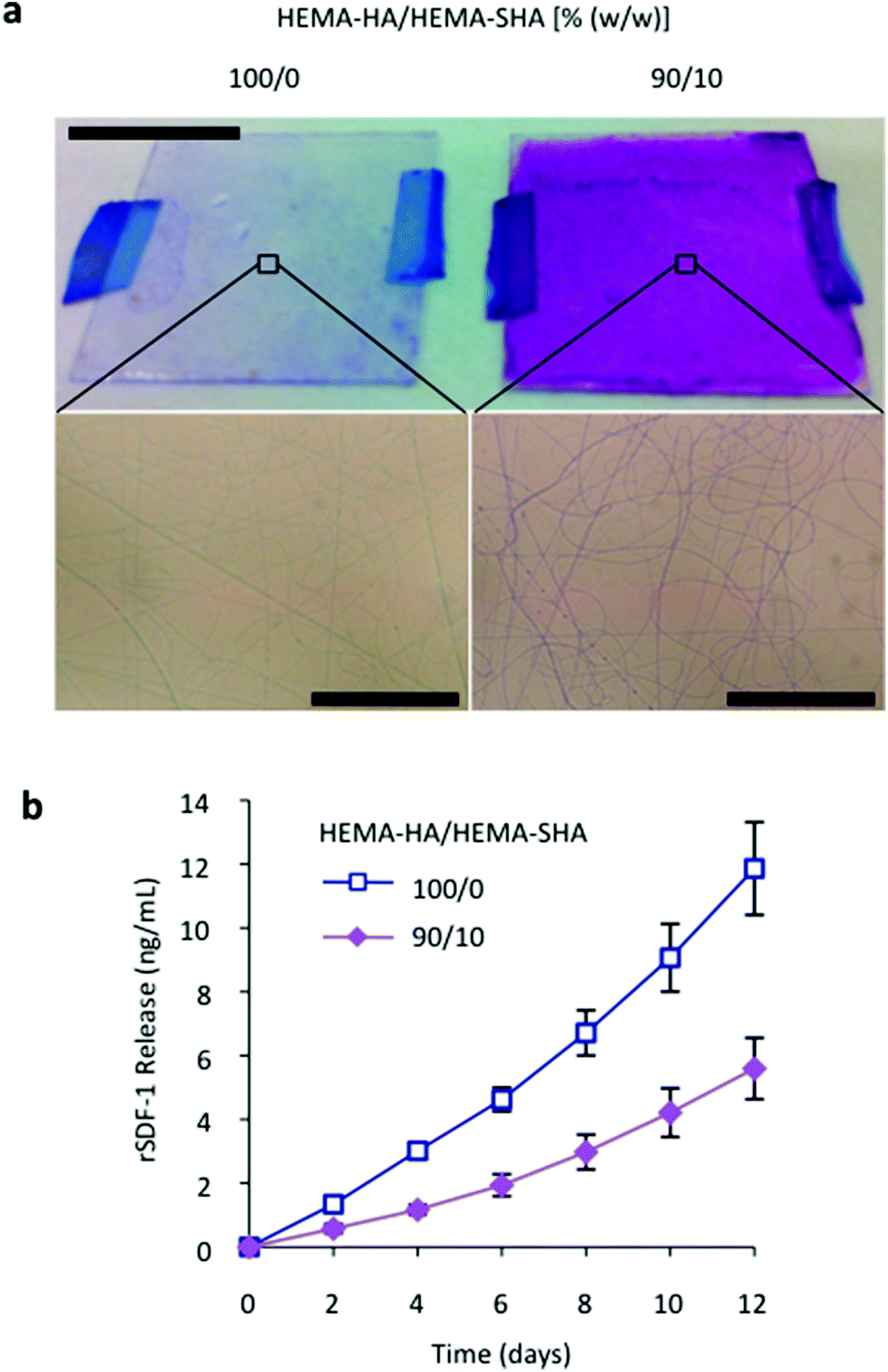 | ||
| Fig. 7 Electrostatic interactions in nanofibers. (a) Sulfation of electrospun nanofibers was visualized with dimethylmethylene blue staining. Incorporation of sulfated HEMA-SHA macromers gave the fibers a more negative charge as indicated by the uniform purple stain of the electrospun mat compared to the blue unsulfated HEMA-HA mat (top, scale bar = 10 mm). DMMB staining was localized to individual nanofibers (bottom, scale bars = 50 μm). (b) Incorporation of sulfated HEMA-SHA macromers reduced the release of encapsulated rSDF-1α from the nanofibers by over 50%, indicating stronger electrostatic interactions between the heparin-binding rSDF-1α and sulfated HA (n = 3 hydrogels per group, mean ± SD). | ||
4. Conclusions
In summary, sulfated and HEMA modified HA macromers were synthesized and incorporated into covalently crosslinked hydrogels. The sulfated HA macromers had a similar sulfate content and HBP binding capacity as heparin and could be blended with non-sulfated HA macromers to form hydrogels that are also degradable. In addition, the sulfated HA macromers could be incorporated into electrospinning processes to control sulfate content in nanofibrous architectures. Therefore, these macromers provide a useful system to introduce heparin mimetic features into covalently-crosslinked hydrogels to control HBP presentation for a variety of drug delivery and tissue engineering applications.Acknowledgements
The authors would like to acknowledge funding from the National Institutes of Health (R01 HL107938, R01 HL111090, and T32 HL007954) and a National Science Foundation Graduate Research Fellowship (ILK).References
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- T. C. Laurent and J. R. Fraser, FASEB J., 1992, 6, 2397–2404 CAS.
- T. Yamada and T. Kawasaki, J. Biosci. Bioeng., 2005, 99, 521–528 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 792 CrossRef PubMed.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS PubMed.
- B. P. Purcell, J. A. Elser, A. Mu, K. B. Margulies and J. A. Burdick, Biomaterials, 2012, 33, 7849–7857 CrossRef CAS PubMed.
- I. L. Kim, S. Khetan, B. M. Baker, C. S. Chen and J. A. Burdick, Biomaterials, 2013, 34, 5571–5580 CrossRef CAS PubMed.
- R. Lever, B. Mulloy and C. P. Page, Heparin - a century of progress, Springer, New York, 2012 Search PubMed.
- R. Raghow, FASEB J., 1994, 8, 823–831 CAS.
- N. Ilan, M. Elkin and I. Vlodavsky, Int. J. Biochem. Cell Biol., 2006, 38, 2018–2039 CrossRef CAS PubMed.
- S. E. Sakiyama-Elbert and J. A. Hubbell, J. Controlled Release, 2000, 65, 389–402 CrossRef CAS.
- T. Nie, A. Baldwin, N. Yamaguchi and K. L. Kiick, J. Controlled Release, 2007, 122, 287–296 CrossRef CAS PubMed.
- S. Nakamura, M. Ishihara, K. Obara, K. Masuoka, T. Ishizuka, Y. Kanatani, B. Takase, T. Matsui, H. Hattori, T. Sato, Y. Kariya and T. Maehara, J. Biomed. Mater. Res., Part A, 2006, 78, 364–371 CrossRef PubMed.
- N. S. Gandhi and R. L. Mancera, Drug Discovery Today, 2010, 15, 1058–1069 CrossRef CAS PubMed.
- E. Petit, D. Papy-Garcia, G. Muller, B. Courtois, J. P. Caruelle and J. Courtois, Biomacromolecules, 2004, 5, 445–452 CrossRef CAS PubMed.
- T. Nagira, M. Nagahata-Ishiguro and T. Tsuchiya, Biomaterials, 2007, 28, 844–850 CrossRef CAS PubMed.
- V. Hintze, S. Moeller, M. Schnabelrauch, S. Bierbaum, M. Viola, H. Worch and D. Scharnweber, Biomacromolecules, 2009, 10, 3290–3297 CrossRef CAS PubMed.
- T. Yamada, R. Sawada and T. Tsuchiya, Biomaterials, 2008, 29, 3503–3513 CrossRef CAS PubMed.
- V. Hintze, A. Miron, S. Moeller, M. Schnabelrauch, H. P. Wiesmann, H. Worch and D. Scharnweber, Acta Biomater., 2012, 8, 2144–2152 CrossRef CAS PubMed.
- U. Hempel, V. Hintze, S. Moller, M. Schnabelrauch, D. Scharnweber and P. Dieter, Acta Biomater., 2012, 8, 659–666 CrossRef CAS PubMed.
- G. Chen, Y. Ito, Y. Imanishi, A. Magnani, S. Lamponi and R. Barbucci, Bioconjugate Chem., 1997, 8, 730–734 CrossRef CAS PubMed.
- L. Cen, K. G. Neoh, Y. Li and E. T. Kang, Biomacromolecules, 2004, 5, 2238–2246 CrossRef CAS PubMed.
- R. W. Farndale, D. J. Buttle and A. J. Barrett, Biochim. Biophys. Acta, 1986, 883, 173–177 CrossRef CAS.
- D. J. Mahoney, J. D. Whittle, C. M. Milner, S. J. Clark, B. Mulloy, D. J. Buttle, G. C. Jones, A. J. Day and R. D. Short, Anal. Biochem., 2004, 330, 123–129 CrossRef CAS PubMed.
- M. C. Bourin and U. Lindahl, Biochem. J., 1993, 289(Pt 2), 313–330 CAS.
- M. Maccarana, B. Casu and U. Lindahl, J. Biol. Chem., 1993, 268, 23898–23905 CAS.
- A. K. Powell, D. G. Fernig and J. E. Turnbull, J. Biol. Chem., 2002, 277, 28554–28563 CrossRef CAS PubMed.
- A. T. Askari, S. Unzek, Z. B. Popovic, C. K. Goldman, F. Forudi, M. Kiedrowski, A. Rovner, S. G. Ellis, J. D. Thomas, P. E. DiCorleto, E. J. Topol and M. S. Penn, Lancet, 2003, 362, 697–703 CrossRef CAS.
- D. J. Ceradini, A. R. Kulkarni, M. J. Callaghan, O. M. Tepper, N. Bastidas, M. E. Kleinman, J. M. Capla, R. D. Galiano, J. P. Levine and G. C. Gurtner, Nat. Med., 2004, 10, 858–864 CrossRef CAS PubMed.
- A. Amara, O. Lorthioir, A. Valenzuela, A. Magerus, M. Thelen, M. Montes, J. L. Virelizier, M. Delepierre, F. Baleux, H. Lortat-Jacob and F. Arenzana-Seisdedos, J. Biol. Chem., 1999, 274, 23916–23925 CrossRef CAS PubMed.
- T. Satoh, K. Nishiyama, M. Nagahata, A. Teramoto and K. Abe, Polym. Adv. Technol., 2004, 15, 720–725 CrossRef CAS.
- S. H. Kim and K. L. Kiick, Peptides, 2007, 28, 2125–2136 CrossRef CAS PubMed.
- I. Freeman, A. Kedem and S. Cohen, Biomaterials, 2008, 29, 3260–3268 CrossRef CAS PubMed.
- C. W. Chang, Y. Hwang, D. Brafman, T. Hagan, C. Phung and S. Varghese, Biomaterials, 2013, 34, 912–921 CrossRef CAS PubMed.
- R. J. Wade and J. A. Burdick, Mater. Today, 2012, 15, 454–459 CrossRef CAS.
- I. L. Kim, R. L. Mauck and J. A. Burdick, Biomaterials, 2011, 32, 8771–8782 CrossRef CAS PubMed.
- H. G. Sundararaghavan, R. L. Saunders, D. A. Hammer and J. A. Burdick, Biotechnol. Bioeng., 2013, 110, 1249–1254 CrossRef CAS PubMed.
- H. G. Sundararaghavan, R. B. Metter and J. A. Burdick, Macromol. Biosci., 2010, 10, 265–270 CrossRef CAS PubMed.
- E. Luong-Van, L. Grondahl, K. N. Chua, K. W. Leong, V. Nurcombe and S. M. Cool, Biomaterials, 2006, 27, 2042–2050 CrossRef CAS PubMed.
- G. Viswanathan, S. Murugesan, V. Pushparaj, O. Nalamasu, P. M. Ajayan and R. J. Linhardt, Biomacromolecules, 2006, 7, 415–418 CrossRef CAS PubMed.
- C. L. Casper, N. Yamaguchi, K. L. Kiick and J. F. Rabolt, Biomacromolecules, 2005, 6, 1998–2007 CrossRef CAS PubMed.
- C. L. Casper, W. D. Yang, M. C. Farach-Carson and J. F. Rabolt, Biomacromolecules, 2007, 8, 1116–1123 CrossRef CAS PubMed.
- K. T. Kurpinski, J. T. Stephenson, R. R. R. Janairo, H. M. Lee and S. Li, Biomaterials, 2010, 31, 3536–3542 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |