Translocation of flexible polymersomes across pores at the nanoscale†
Carla
Pegoraro
abc,
Denis
Cecchin
efg,
Jeppe
Madsen
ad,
Nicholas
Warren
d,
Steven P.
Armes
d,
Sheila
MacNeil
bc,
Andrew
Lewis
g and
Giuseppe
Battaglia
*def
aDepartment of Biomedical Sciences, University of Sheffield, Sheffield, UK
bThe Kroto Research Institute, University of Sheffield, Sheffield, UK
cDepartment of Material Science and Engineering, University of Sheffield, Sheffield, UK
dDepartment of Chemistry, University of Sheffield, Sheffield, UK
eDepartment of Chemistry, University College London, London, UK
fThe MRC/UCL Centre for Medical Molecular Virology, University College London, London, UK. E-mail: g.battaglia@ucl.ac.uk; Tel: +44 (0)2076794688
gBiocompatibles UK Ltd, Farnham, UK
First published on 25th March 2014
Abstract
Hierarchical biological systems such as tissues and organs are often characterised by highly crowded and packed environments with nanoscopic interconnections between them. Engineering nanovectors that can penetrate and diffuse across these is critical to ensure enhanced delivery and targeting. Here we demonstrate that flexible polymeric vesicles, known as polymersomes, enable the translocation of large macromolecules across both synthetic and biological porous systems. We compare the translocation across narrow pores of different polymersome formulations. We demonstrate that effective translocation depends on the right combination of mechanical properties and surface lubrication. We prove that with the effect of external gradients (e.g. osmotic pressure, capillarity, hydration, etc.) polymersomes can translocate across pores with diameters one order of magnitude smaller without breaking. We demonstrate that these properties are essential to develop effective tissue penetration and show polymersome mediated transdermal delivery of large macromolecules such as dextran and antibodies using human ex vivo skin.
The translocation of large molecules or cells across narrow pores is a ubiquitous and fundamental phenomenon in biology. It is a pre-requisite for cell and tissue dynamics in both healthy and diseased states, as well as being at the heart of most drug delivery strategies. Immune cells are able to perform their fundamental role of immunosurveillance by trafficking through endothelial barriers, basement membranes and vascular networks without loss to themselves or to the surrounding tissue structure.1,2 Similarly, bacteria cross biological barriers to initiate infections and metastatic cancerous cells spread by entering the vasculature and lymphatic system to establish secondary tumours.3,4 At a smaller scale lipid vesicles extravasate out of the circulatory system and large nucleic acids can translocate across small pores when injected from viral capsids.5,6 Proteins cross cell bilayers either via membrane pores or via the active transportation of other protein complexes, whilst RNA traverses nuclear pores during transcription.7–9 All such biological systems share the necessity to cross these narrow gaps as a single unit and must therefore undergo significant deformation prior to translocation.
Gene therapy and drug delivery strategies all depend on the ability to transport therapeutic molecules to the appropriate site with minimal (bio)chemical degradation. To achieve this goal, vectors must traverse biological barriers via their narrow pores without damaging the healthy tissues. For optimal delivery strategies it is still unclear whether vectors must be no larger than the actual pores or can be significantly larger, mechanically flexible and guided across under the effects of active transportation. Lipid based nanometer sized vesicular systems, liposomes, have been widely used due to their ability to efficiently encapsulate and deliver therapeutic molecules. When liposome delivery is dependent on their ability to cross much smaller pores (e.g. transdermal drug delivery applications) they are not able to deform to the same extent as that demonstrated by cells or biopolymers without fragmentation.10–13 Polymeric vesicles made from amphiphilic block copolymers, polymersomes, have a similar drug delivery potential compared to liposomes as they also comprise an enclosed aqueous lumen that can be loaded with hydrophilic molecules and macromolecules. However, polymersomes can be easily designed with specific mechanical and chemical properties that render them more suitable for narrow pore translocations than liposomes. Their much higher molecular weight allows polymersomes to form highly entangled polymeric membranes that self-assemble into vesicles. Because of this they exhibit significantly greater mechanical stability and flexibility, which allows them to withstand much higher stresses prior to rupture compared to conventional liposomes.14–19 Previous research has demonstrated the influence on vesicle translocation of (i) the presence or absence of an active transportation mechanism (e.g. hydration or osmotically driven transport), (ii) of the interaction between vesicle and pore channel and (iii) of the adhesion properties of the barrier, which can initiate a substrate-induced transport, etc.6,20–22 In this paper we present an experimental set-up for modelling the physical problem of polymersome translocation and drug delivery under the influence of active/passive transportation. We propose that polymersomes are an ideal model system for studying the permeation of large objects across narrow pores that possess the appropriate mechanical properties and surface chemistry. We hypothesise that successful translocation will require flexible and mechanically stable vesicles that combine the appropriate surface interaction with the barrier surface together with inherent sensitivity to external forces such as hydration, concentration gradient, capillary forces and osmotic pressure.
For large vesicles to deform across smaller pores, they must overcome the energetic penalty associated with the spatial restrictions, the increase in elastic energy and reduction in entropy. The energy required to deform a large spherical vesicle into a smaller pore is proportional to the bending rigidity, k, and the square of the ratio of the vesicle and pore radii.20 Typically, k is approximately 10–20kBT for most lipids. Depending on the hydrophobic block length and chemistry, k can be an order of magnitude larger for polymersome membranes.23 Thermal fluctuations are not enough to overcome the energetic barrier and an external driving force is therefore required to push the vesicle into the pore. Linke et al. have modelled osmotic pressure as the main driving force but a more complete model is likely to be a combination of this parameter, the hydration gradient, the polymersome concentration gradient and other external forces, depending on the specific biological environment.21 If these forces are sufficient to overcome the energetic barrier and deform the vesicles into pores, then the vesicle will spontaneously move through the pore in order to lower its potential energy by minimising the spatial constriction. Whether or not the vesicle can do this without membrane rupture depends largely on its mechanical properties, which are dictated by its chemical composition.
Polymersome translocation across narrow pores was modelled using an adapted Franz diffusion cell in which the vesicle solution was placed on top of a polycarbonate porous barrier between a donor chamber and an acceptor chamber (Fig. 1).24 SEM analysis of the surface of the polycarbonate membranes showed a uniformly porous structure with pores crossing the whole section of the 10 μm thick barrier. This model system was constructed in-house and allowed us to alter the average pore diameter (from 50 to 400 nm) and barrier porosity (from 5 to 70%). This enabled us to determine (i) the optimal polymersome/pore diameter ratios required for translocation; (ii) the polymersome concentration (in order to study the influence of a concentration gradient); (iii) the type of polymersome chemistry (so as to determine whether an attractive interaction with the barrier surface leads to more effective translocation); (iv) the hydration pull across the barrier by connecting the acceptor chamber to a peristaltic pump (so as to assess the effects of capillarity, osmotic and hydration forces).
 | ||
| Fig. 1 Translocation chamber. (A) Side view schematic of the permeation system showing the donor and acceptor chambers separated by the porous membrane (shown in pink) and the polymersome solution placed on top of it. Flow in the acceptor chamber (shown with the yellow arrow) is imposed by a peristaltic pump connected to the acceptor chamber. (B) Three dimensional schematic of the perfusion system. (C) Surface and cross-section SEM images of 50 and 400 nm pore sized polycarbonate membranes. (D) Scanning Ion Conductance Microscope image of 200 nm sized PMPC25-PDPA70 polymersomes placed on top of a 50 nm pore sized membrane. | ||
Fig. 2 shows the chemical structures and corresponding TEM images of the diblock copolymers used to prepare polymersome formulations and their rates of permeation across a 50 nm pore diameter polycarbonate membrane: PMPC25-PDPA70, PMPC25-PDPA500, poly(2-(methacryloyloxy)ethyl phosphorylcholine)-co-poly(2-(diisopropylamino)ethyl methacrylate) and PEO113-PDPA70, poly(ethylene oxide)-co-poly(2-(diisopropylamino)ethyl methacrylate). It is apparent that PMPC25-PDPA70 permeates across 50 nm pores more rapidly than the other two formulations (Fig. 2D). This formulation was characterised by a permeation profile with a fast initial slope followed by a plateau with a maximum relative extent of permeation of around 94%. PMPC25-PDPA500 and PEO113-PDPA70 had much slower and nearly linear permeation profiles (maximum permeation of 28% and 20% respectively). Only PMPC25-PDPA500 micelles had an average diameter comparable with the pore size (85 nm). The other formulations each had mean polymersome diameters ranging between 200 and 400 nm (see Fig. 3).
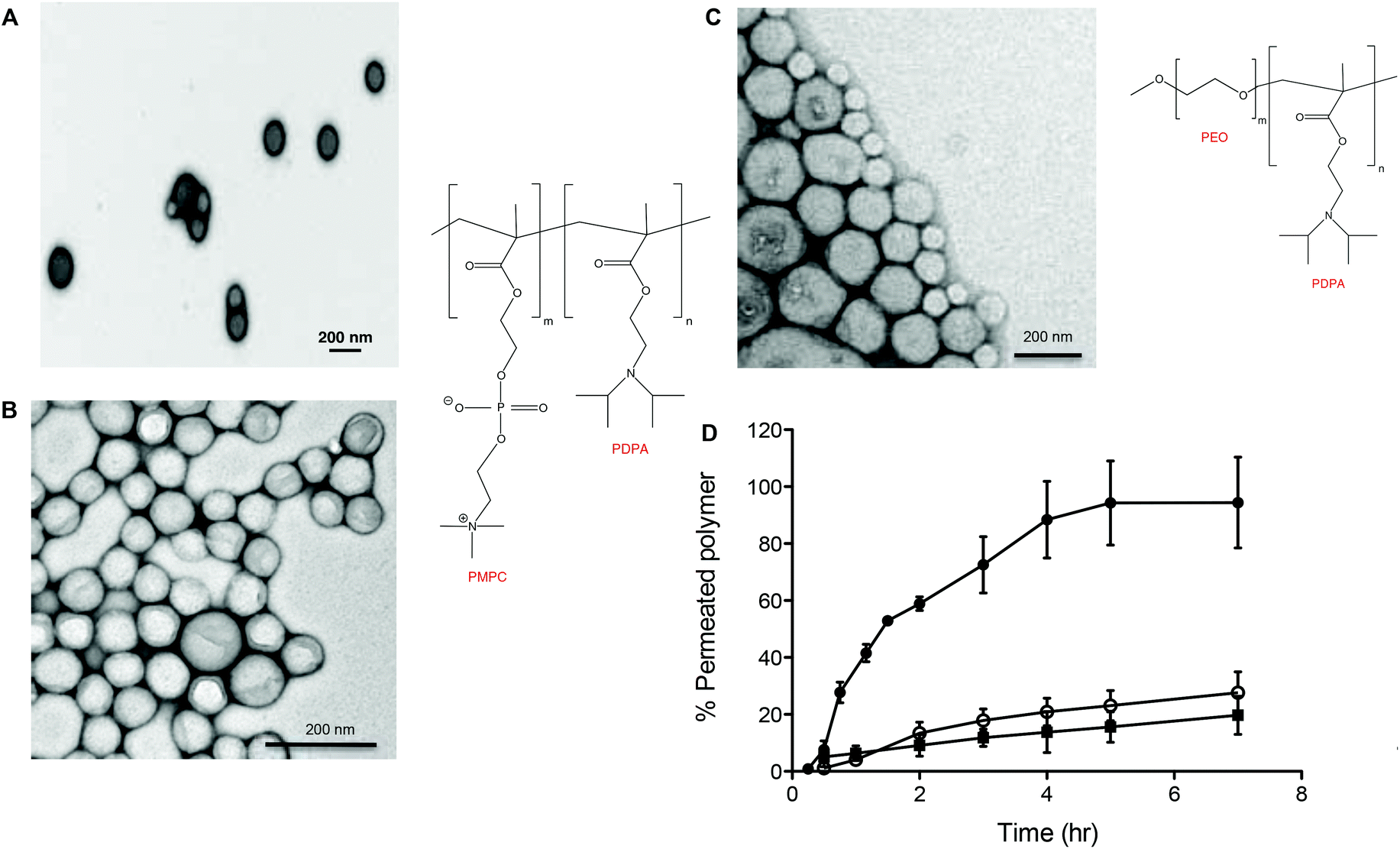 | ||
| Fig. 2 Polymersome formulations and relative permeations across a 50 nm pore sized membrane. (A) TEM micrograph and chemical structure of PMPC25-PDPA70. (B) TEM micrograph and chemical structure of PMPC25-PDPA500. (C) TEM micrograph and chemical structure of PEO113-PDPA70. (D) Permeation profiles as a function of time of the polymersome and polymersomes/micelles formulations expressed as a function of the initial amount placed in the donor chamber: (●) PMPC25-PDPA70 (400 nm), (○) PMPC25-PDPA500 (85 nm) and (■) PEO113-PDPA70 (200 nm). | ||
 | ||
| Fig. 3 DLS analysis of polymersome dimensions before (–) and after permeation (---) across a 50 nm pore membrane. (A) Intensity-average diameter distributions of PMPC25-PDPA500 micelles and vesicles. (B) Intensity-average diameter distributions of PMPC25-PDPA70. (C) Intensity-average diameter distributions of PEO113-PDPA70. The difference in the scattered light intensity between PMPC25-PDPA70 and the other polymersomes is due to the use of a Brookhaven DLS rather than a Malvern instrument. | ||
DLS analysis of the polymersome dimensions before and after permeation confirms that only the PMPC-PDPA formulations have almost identical size distributions, thereby achieving translocation across narrow pores without loss of their mechanical integrity. PEO113-PDPA70 appears to undergo fragmentation and then reassemble into various size populations on the acceptor side of the membrane. Intact translocation without barrier disruption is one of the major challenges in the field of drug delivery and therapy. If the delivery vehicles do not remain intact while crossing the barrier, then the cargo will not be delivered according to the specific requirements of the application. Consequently the fact that the PMPC-PDPA formulations remain intact while crossing pores up to 8 times smaller is a major result. Translocation across the barrier via dissociation of PMPC-PDPA polymersomes and their subsequent reassembly on the other side to form polymersomes with the same mean diameter is highly unlikely given that such block copolymer assemblies have unimer concentrations close to zero and that several studies have demonstrated that once formed, polymersomes do not exchange material amongst themselves or with the surrounding solution, in other words they are non-ergodic.25–30
The main differences between the polymersome formulations examined in this study are the degree of hydration of the external polymer brush (i.e. PMPC or PEO) and the length of the hydrophobic block (i.e. PDPA) in the polymersome membrane. On average each MPC monomer in the block copolymer is solvated by up to 24 water molecules, while EO is significantly less hydrophilic, with just 3 water molecules per monomer.31,32 This leads to the expression of a highly hydrophilic polymer brush on the outer surface of PMPC-PDPA polymersomes with a reported contact angle of around 0°.33 The water-rich PMPC brush has also been reported to have a very strong lubrification effect, which drastically reduces the corresponding friction coefficient and hence enhances the rate of permeation.34–36 As previously demonstrated by Cevc et al., if the water activity around the transportant diminishes, it will partly dehydrate (thereby reducing the flexibility of the vesicles and allowing them to collapse). In contrast, if the water activity increases then hydration-driven transport becomes more efficient.37 In other words, by maintaining the flow in the perfusion system, the polymer membranes remain hydrated, thus transport across the membrane of PMPC-based polymersome formulations with higher water contents is favoured. A strongly attractive barrier surface and pore wall can effectively enhance translocation.6 Contact angle measurements obtained using a contact angle goniometer have shown that polycarbonate membranes are highly hydrophilic. Thus a layer of water will coat the outer surface of the barrier as well as that of the pore walls when such membranes are immersed in an aqueous environment. The most hydrophilic polymer brushes will therefore interact more strongly with the membrane surface than those with lower water contents. Indeed Scanning Ion Conductance Microscopy (SICM) studies indicate that when PMPC-PDPA polymersomes were placed on a 50 nm pore diameter membrane they were closely associated with the polycarbonate surface (Fig. 1D). Membrane rupture is caused by both the external pressure, given by the deformation across narrow pores, and the internal pressure, given by the excess volume of water inside the vesicle, which is controlled by the membrane permeability. The comparison between PMPC25-PDPA70 and PEO113-PDPA70 polymersomes with the same molecular weight hydrophobic PDPA block shows that water permeability does not affect the ability of these vesicles to deform and cross narrow pores. Considering the identical membrane chemistry, the enhanced translocation of PMPC25-PDPA70 must be due to the different surface chemistry and lubrification effect.
The mean degree of polymerisation of the hydrophobic membrane-forming block strongly affects polymersome flexibility. Previous studies have demonstrated that membrane thickness scales with block copolymer molecular weight and that thicker membranes lead to greater bending rigidities.23,38 Due to the relatively long PDPA block, PMPC25-PDPA500 micelles are characterised by a relatively thick hydrophobic membrane surrounded by a much shorter hydrophilic polymer brush. Compared to PMPC25-PDPA70 and PEO113-PDPA70 polymersomes, this self-assembled micelle has a larger bending rigidity (due to its thicker membrane and reduced surface area). Thus, although the average dimensions of PMPC25-PDPA500 micelles were more comparable with the 50 nm diameter pores and no fragmentation occurred during translocation, permeation across the pores was still three-fold slower due to the time required to deform such relatively rigid micelles (Fig. 2E).
Both 400 nm sized PMPC25-PDPA70 polymersomes and 85 nm sized PMPC25-PDPA500 micelles were able to traverse 50 nm pores intact. Due to their greater flexibility the former polymersomes exhibited a faster rate of permeation compared to micelles. To examine the optimal vesicle-pore size ratio we compared the permeation of 100, 200 and 400 nm sized PMPC25-PDPA70 polymersomes across both 50 and 400 nm pores (Fig. 4A). Translocation through 50 nm pores was strongly enhanced: 98% of 400 nm, 69% of 200 nm and 61% of 100 nm sized polymersomes permeated intact. For the larger pores, translocation was reduced by a factor 3–4: 30% of 400 nm, 22% of 200 nm and 13% of 100 nm sized polymersomes permeated intact. Considering the large surface area compared to polymersome membrane thickness and its effect on surface tension, it is reasonable to assume that larger polymersomes will be relatively more flexible than smaller ones and will therefore be able to deform more quickly across the pore. The 400 nm pore membrane has an average porosity of 70%, while for the 50 nm pore membrane it is approximately 5%. Consequently the attractive interaction between a single vesicle and the membrane surface is drastically reduced for the former and hence such polymersomes are more likely to remain dispersed in solution rather than move towards the membrane surface. Moreover, the sensitivity to the external gradients increases with the aggregation number for a given polymersome and the total change in osmotic pressure, to which a permeating polymersome is subjected, is proportional to the difference in water activity and the square of the vesicle radius.21,37 These two factors explain why larger polymersomes cross the porous barrier intact more efficiently than smaller ones. Further proof of the attractive interaction between membrane and polymersome surfaces was found when comparing the permeation profiles between the molecularly dissolved block copolymers and homopolymers (Fig. 4B and C). PMPC-PDPA copolymers facilitate such a comparison as they are pH sensitive and can be tested as vesicles or as unimers depending on solution pH (see Materials and methods for further details).39,40 Molecularly dissolved PMPC25-PDPA70 polymer chains translocated across 50 nm pores with a final permeation of 96%. The kinetics in this case was only slightly faster than those obtained for 200 nm sized polymersomes and much slower than those observed for the 400 nm polymersomes. Molecularly dissolved PEO113-PDPA70 chains exhibited similar permeation behaviour with a much faster initial linear region followed by a plateau. Around 81% of the molecularly dissolved PEO113-PDPA70 chains crossed the membrane after 7 h compared to just 20% of the corresponding polymersome formulation. As with RNA traversing nuclear pores, copolymer chains must become disentangled and stretch before they can be threaded across narrow pores, with an average residence time of a copolymer passing through a narrow pore scaling with its length.8,41,42 In this case the pore diameter is 50 nm, so we would expect the individual copolymer chains to permeate faster compared to polymersomes. However, the PMPC100 homopolymer had similar permeation kinetics to that of PMPC25-PDPA70 polymersomes and the longer PMPC400 homopolymer exhibited significantly slower kinetics (Fig. 4C). The radius of gyration (Rg) for such homopolymers was estimated by using Rg = bN0.5, where b is the C–C length and N is the degree of polymerisation, which resulted in 2.4 and 5.6 nm, respectively. Furthermore, the influence of external gradients and the membrane surface interaction for both homopolymers are drastically reduced due to the much smaller aggregation number (unity). Our premise is that there must be an active transportation mechanism (governed by the sum of the strong interaction with the membrane, capillarity, flow and concentration gradient across the membrane, osmotic pressure, etc.), which favours the deformation and translocation of large, flexible and highly hydrated polymersomes.
 | ||
| Fig. 4 Translocation across polycarbonate membranes. (A) Permeation profiles of PMPC25-PDPA70 polymersomes as a function of vesicle and pore size: (-●-) 400 nm, (-▲-) 200 nm, (-■-) 100 nm PMPC25-PDPA70 vesicles traversing 50 nm pores, (--○--) 400 nm, (--△--) 200 nm, (--□--) 100 nm sized PMPC25-PDPA70 vesicles traversing 400 nm pores. (B) Comparison between the molecularly dissolved and the vesicular form of PMPC25-PDPA70 and PEO113-PDPA70 traversing 50 nm pores: (-●-) 400 nm PMPC25-PDPA70 polymersomes, (-■-) 200 nm PMPC25-PDPA70 polymersomes, (-▲-) 200 nm PEO113-PDPA70 polymersomes, (--○--) molecularly dissolved PMPC25-PDPA70, (--△--) molecularly dissolved PEO113-PDPA70. (C) Comparison between the PMPC homopolymer and the vesicular form: (-●-) 400 nm PMPC25-PDPA70 polymersomes, (-□-)PMPC100 homopolymer, (-△-) PMPC400 homopolymer. | ||
Fig. 5 shows a schematic of the proposed translocation mechanism across narrow pores. Once polymersomes are placed on the surface of the polycarbonate membrane they will feel the effects of external driving forces depending on their size and surface chemistry and begin to move towards and across the barrier. This movement will be favoured by the existence of concentration and hydration gradients across the membrane (see ESI† regarding the influence of flow and concentration on translocation). These external gradients will gradually push the vesicles towards the pore openings and begin to deform them and drag them inside (Fig. 5B). Once the vesicle has been dragged into the pore a distance equal to at least the pore radius, this delicate equilibrium will be suddenly broken due to the sudden increase in osmotic pressure (which results from the vesicles blocking the pore opening), which together with the previously mentioned gradients will push the vesicle to the other side of the pore to reach a new stable equilibrium at a lower energetic state than the one inside the pore. Capillarity also plays a role in the sucking of the vesicle across the pore. Whilst the vesicles are moving to the opposite side of the membrane, water will move along the inside wall of the pore (indicated by the white arrows in Fig. 5C) in the opposite direction. In the case of PMPC25-PDPA70 based polymersomes, the highly hydrated polymeric brush allows for the movement of water along the pore wall without forcing the polymeric membrane farther away from the pore wall and increasing the mechanical stresses placed upon it. Since PEO113-PDPA70 polymersomes have a more hydrophobic polymer brush, the interaction with the layer of water on the inside wall of the pore will force the vesicle into more stringent volume constraints (pushing the vesicle membrane farther away from the wall). This is another factor that determines PEO113-PDPA70 fragmentation during permeation. PMPC25-PDPA500 polymersomes–micelles mixture do not present this problem but, as can be seen from Fig. 5, the volume constraints due to the hydrophobic block are higher, and this slows down the translocation kinetics, even if they do not cause fragmentation.
 | ||
| Fig. 5 Schematic of the proposed method of polymersome translocation across narrow pores. (A) Polycarbonate membrane with 50 nm pores and average thickness of 10 μm with 200 nm vesicles translocating across it. (B) Enlarged sections of vesicles deforming and permeating across the narrow pore. The red line indicates the vesicle polymer membrane, the dark blue contour the hydrophilic polymer brush, the lighter blue contour the water layer surrounding the membrane surface, the black and white arrows indicate respectively the direction of vesicle translocation and the direction in which water moves due to capillarity. (C) Interaction between polymersomes, micelles and pore wall. PMPC25-PDPA70 is characterised by the smallest distance between polymer-brush and pore wall due to the high level of hydration and lubrification (the internal radius, r1, is the largest compared to other formulations). PMPC25-PDPA500 has the same hydrophilic outer brush but r1 > r2 because of the increased hydrophobic block length. This does not cause the polymersomes and micelles present on the sample to fragment since they are similar in size to the pore dimensions but it drastically reduces translocation kinetics. PEO113-PDPA70 potentially fragments during permeation across the pore because of the added space constriction imposed by the relatively more hydrophobic brush that pushes the membrane farther away (r1 > r3). Molecularly dissolved block copolymers will stretch and translocate across the pore with kinetics depending on the block length (residence time within the pore scales with length). | ||
This shows that between the formulations tested, PMPC25-PDPA70 is the most efficient at crossing the porous barrier intact. For this reason a high molecular weight biopolymer was encapsulated within PMPC25-PDPA70 polymersomes to demonstrate their ability not only of translocating across narrow pores but of being able to deliver potentially therapeutic molecules across a biological barrier for drug delivery applications. Fig. 6 shows the translocation profiles of free FITC-labelled dextran (40 kDa), Rh 3.5 nm,51 empty 200 nm PMPC25-PDPA70 polymersomes and encapsulated FITC-labelled dextran within these polymersomes across a 50 nm pore diameter membrane. Free dextran was characterised by a slow initial permeation rate, which reached a plateau after 2–3 hours when the molecule saturated the pores or aggregated around them (final relative extent of permeation of 33%). In contrast, the encapsulated dextran exhibited a permeation profile that followed very closely that of empty polymersomes with a final translocation of 60%. Overall dextran delivery was enhanced by a factor 2 and, more importantly, the permeation profile closely matched that of the empty polymersomes. This proof-of-concept experiment clearly demonstrated the ability of polymersomes to deliver large biomolecules across porous barriers and it is a further demonstration that they do this intact. If polymersome had ruptured during translocation, then the encapsulated dextran would have been released into solution and its trans-barrier flow would not have resembled that of empty polymersomes but that of the free non-encapsulated dextran or a combination thereof.
 | ||
| Fig. 6 Polymersome delivery across the barrier. Permeation profiles across 50 nm pores of free (-●-), encapsulated dextran (-○-) and 200 nm sized PMPC25-PDPA70 polymersomes (-□-). | ||
Permeation across skin depends on the ability of the delivery agent to deform and squeeze across the tight gaps between the tightly packed dead cells of the top most layer of skin (the stratum corneum, SC). Cells in this layer of skin can be seen as protein envelops surrounded by a lipid rich matrix. The average gaps between cells are around 50 nm or less so the polycarbonate membrane model is a good approximation of the biological reality.43 Both 200 and 400 nm rhodamine labelled PMPC25-PDPA70 polymersomes cross human ex vivo skin (Fig. 7A) and as with the membrane model, larger vesicles have faster kinetics. Importantly, when a large macromolecule like far-red labelled dextran (10 kDa) is encapsulated within 200 nm PMPC25-PDPA70 polymersomes, permeation compared to free dextran is nearly 7 fold higher (Fig. 7B). Confocal Laser Scanning Microscopy (CLSM) analysis clearly shows that rhodamine-labelled PMPC25-PDPA70 polymersomes have crossed the impermeable SC layer and reached the viable dermal layer (Fig. 7C and D). 3D reconstructed confocal images of rhodamine-labelled PMPC25-PDPA70 and PEO113-PDPA70 polymersomes crossing the biological barrier show that, as with the membrane model, they first formulation is more efficient, while the second does not penetrate as far into the skin layers after 24 h of topical application. Similarly, encapsulated dextran was evenly distributed across the epidermis and dermis when delivered by PMPC25-PDPA70 polymersomes. In contrast, free dextran showed little or no penetration and was mostly found on the top skin surface or in the skin appendages.
 | ||
| Fig. 7 Polymersome translocation and delivery across biological barriers. (A) Permeation profiles of 200 (-■-) and 400 nm (-▲-) PMPC25-PDPA70 polymersomes across ex vivo human skin. (B) Permeation profiles across ex vivo human skin of free (-●-), encapsulated dextran (-○-) and 400 nm sized PMPC25-PDPA70 polymersomes (-■-). (C) 3D reconstructed CLSM images of rhodamine-labelled PMPC25-PDPA70 and PEO113-PDPA70 polymersomes across ex vivo human skin after 24 h, together with an H&E section of skin showing that there has been no structural damage to the tissue after polymersome penetration. (D) CLSM cross-sections of skin after permeation of free and PMPC25-PDPA70 encapsulated dextran after 48 h. | ||
As further proof of transdermal delivery and as a way to determine the pathway across skin, 1.4 nm gold nanoparticle labelled antibodies (IgG, 160 kDa, Rh 5.22 nm52) were encapsulated in PMPC25-PDPA70 polymersomes, topically applied to human ex vivo skin and analysed via TEM (Fig. 8, for the control images see ESI†). As with the high molecular dextran, IgGs are not able to permeate across skin because of their size. Fig. 8 however clearly shows that encapsulated gold-labelled antibodies are distributed all the way from the top most layers of the epidermis to the top layers of the dermis (see white arrows). These observations further confirm the ability of PMPC25-PDPA70 polymersomes to translocate across skin and carry the desired encapsulated molecule for drug delivery applications (Fig. 8A–F). The enlarged images of the SC indicate that the particles diffused across skin by translocating across the narrow gaps between cells. TEM analysis therefore supports our suggested translocation pathway across narrow pores in a biological barrier such as skin. Polymersomes were indeed able to deform and cross the narrow pores as a function of the previously discussed driving forces, and reach the deeper epidermal and dermal skin layers (Fig. 8G–K). Moreover PMPC25-PDPA70 polymersome were able to target epidermal cells, which compared to the SC cells are still capable of intracellular uptake, and deliver the gold-labelled antibody within the cell interior (Fig. 8L). Co-localisation studies of the CLSM skin sections are a further confirmation of this intracellular delivery (see ESI†).
 | ||
| Fig. 8 Polymersome delivery across the barrier. TEM micrographs of ex vivo human skin after 48 h translocation of gold nanoparticle labelled antibodies loaded PMPC25-PDPA70 polymersomes. The white arrows indicate where the gold nanoparticles are in the tissue, SC stands for stratum corneum, EP for epidermis, K for corneocytes, the cells of the SC, L for the lipid matrix surrounding the cells, GC for the granular cells of the epidermal layer immediately below the SC. For TEM skin controls see ESI.† | ||
Both CLSM and TEM analysis clearly demonstrate the potential for using large flexible polymersomes as delivery vehicles compared to the free diffusion of high molecular weight biomolecules. In the past the delivery of small molecules without carriers or skin solubilisers has been relatively successful. For example, Zheng and coworkers have topically delivered 13 nm gold nanoparticles conjugated with siRNA.44 The models used however were rat and human skin substitutes, which are generally less impermeable compared to natural human skin, and the size of the nanoparticles is such that even with human models they can easily cross the skin barrier by moving along the gaps between cells.
Compared to previous lipid- or polymer-based delivery vehicles, the present study represents a marked improvement, both from the point of view of the amount delivered across skin (Fig. 9 compares the amount of topically delivered dextran by our polymersome formulation and some of the most commonly used transdermal delivery enhancers) and from the point of view of the delivery pathway (generally other delivery vehicles are localized in the hair follicles or other larger openings in skin (>1 μm) after cutaneous application). Such openings (e.g. wrinkles, etc.) constitute only 0.1% of the total skin surface and are therefore a less efficient route of administration.45,46
 | ||
| Fig. 9 Comparison between polymersomes and other delivery systems of dextran across skin (the amount of transportation was calculated as a percentage of the original solution placed on the skin as reported by the authors).47–50 | ||
We have demonstrated that active pore translocation can be achieved by the right combination of surface chemistry and mechanical properties. We propose that nanoscopic flexible vectors can diffuse more effectively across narrow pores than molecularly dissolved polymers under the influence of external driving forces such as capillarity, osmotic pressure, hydration, etc. Importantly, we have demonstrated the increased ability of specific polymersome formulations compared to more traditional delivery approaches to transport clinically relevant large molecular weight biopolymers across both synthetic and biological barriers with potential applications in the field of transdermal drug delivery (e.g. needle free vaccinations, melanoma treatments, etc.).
Materials and methods
Perfusion system
The perfusion system was built by cutting the tops from 5 ml bijous tubes at the base of the thread and attaching them to the base of a Petri dish using chloroform. Holes were drilled on opposite sides of the bijou head and lid to create the input and output for a continuous laminar flow of media or PBS in the perfusion device. A hole was drilled in the top of the lid to create the donor and acceptor chambers and a porous barrier (polycarbonate membranes or ex vivo skin) was placed between them creating an air–liquid interface. The acceptor chamber was connected to a 205s Watson Marlow peristaltic pump with Watson Marlow marprene tubes (inner diameter 0.25 mm) set to 0.5 rpm, so as to create a controllable laminar flow at the air–liquid-interface.Polymersome preparation
The pH sensitive PMPC25-PDP A70, PEO113-PDPA70 and PMPC25-PDPA500 copolymers as well as the rhodamine labelled copolymers (excitation = 540 nm, emission = 600 nm) were synthesised by atom transfer radical polymerization (ATRP) as reported by Du and coworkers.39 The former polymers form stable vesicles while the longer PDPA chain on PMPC25-PDPA500 allow the formation of polymersomes as well as micelles. This is due to the protonation and deprotonation of the tertiary amine groups on the PDPA chains, which switch from being hydrophilic at acid pH to hydrophobic at physiological pH. These pH sensitive copolymers were used for their previously reported low toxicity, their ability to enter cells via the endocytotic pathway and to escape the endosome (the endosomal internal environment is slightly acidic, which disassembles the vesicles creating an osmotic pressure shock inside the endosome that causes its rupture and release of the block copolymer chains into the cell cytosol40). Empty polymersomes were either prepared via pH switch or by dissolving directly the polymer in phosphate-buffered saline solution (PBS, prepared from tablets purchased from Oxoid Ltd following the manufacturer's instructions) at pH 7. In both cases the first step was the formation of a block copolymer film in a glass vial. The glass vial was autoclaved before use for sterility purposes. The block copolymers with 10% w/w% of Rhodamine labelled block copolymer were dissolved in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chloroform–methanol solution with a final concentration of 2 mg ml−1 (Chloroform and Methanol purchased from Fisher Scientific and Sigma-Aldrich UK respectively). This solution was left to evaporate overnight in a vacuum oven or in a desiccator so as to obtain a uniform film on the inside of the glass vial. The glass vial was sealed with a solvent resistant nylon membrane that allowed evaporation and maintained sterility inside the vial (membranes purchased from Millipore UK Limited). For the first technique, once all the solvent has been removed the film was rehydrated in sterile PBS at pH 2 to produce a 10 mg ml−1 polymer solution. This solution was filter-sterilised and the pH slowly raised to 7 to allow the formation of polymersomes. The polymersome solution was sonicated for 30 minutes and then extruded across polycarbonate porous membranes of known pore size to obtain polymersomes with a mean diameter of either 100, 200 or 400 nm (extrusion system purchased from Avanti Polar Lipids Inc. and membranes from AVESTIN Inc.). The second technique involved dissolving the block copolymer film directly in 2 ml of PBS at pH 7 (again obtaining a 10 mg ml−1 solution). The polymer solution was left to stir for at least two weeks at ambient temperature to allow the formation of polymersomes under the effect of continuous shear and hydration. The average polymersome size in this case was 200 nm. The final solution, obtained with either technique, was then purified by preparative gel permeation chromatography using a Sepharose 4B size exclusion column (Sepharose 4B purchased from Sigma-Aldrich UK).
:1 chloroform–methanol solution with a final concentration of 2 mg ml−1 (Chloroform and Methanol purchased from Fisher Scientific and Sigma-Aldrich UK respectively). This solution was left to evaporate overnight in a vacuum oven or in a desiccator so as to obtain a uniform film on the inside of the glass vial. The glass vial was sealed with a solvent resistant nylon membrane that allowed evaporation and maintained sterility inside the vial (membranes purchased from Millipore UK Limited). For the first technique, once all the solvent has been removed the film was rehydrated in sterile PBS at pH 2 to produce a 10 mg ml−1 polymer solution. This solution was filter-sterilised and the pH slowly raised to 7 to allow the formation of polymersomes. The polymersome solution was sonicated for 30 minutes and then extruded across polycarbonate porous membranes of known pore size to obtain polymersomes with a mean diameter of either 100, 200 or 400 nm (extrusion system purchased from Avanti Polar Lipids Inc. and membranes from AVESTIN Inc.). The second technique involved dissolving the block copolymer film directly in 2 ml of PBS at pH 7 (again obtaining a 10 mg ml−1 solution). The polymer solution was left to stir for at least two weeks at ambient temperature to allow the formation of polymersomes under the effect of continuous shear and hydration. The average polymersome size in this case was 200 nm. The final solution, obtained with either technique, was then purified by preparative gel permeation chromatography using a Sepharose 4B size exclusion column (Sepharose 4B purchased from Sigma-Aldrich UK).
Loaded polymersome preparation
Fluorescein isothiocyanate-dextran (FITC-labelled dextran) loaded polymersomes were obtained by directly dissolving the PMPC25-PDPA70 copolymer chains with dextran in a pH 7 solution and stirred for two weeks at ambient temperature for high encapsulation. The average polymersome size in this case was 200 nm (FITC-labelled dextran was purchased from Sigma-Aldrich UK, Mw 40 kDa, excitation = 488 nm, emission = 520 nm). Far-red labelled dextran (Alexa Fluor® dextran, Mw 10 kDa, excitation = 647 nm, emission = 668 nm, purchased from Invitrogen UK) and gold labelled antibodies (rabbit polyclonal secondary antibody to goat IgG, 1.4 nm gold, was purchased from Abcam) were encapsulated in PMPC25-PDPA70 polymersomes via the pH switch technique. A solution of known concentration was added to the rehydrated polymer film at pH 6. The pH was then increased to 7.3 to form polymersomes with gold nanoparticles trapped inside the hydrophilic core. The resulting polymersome solution was then sonicated and purified as previously discussed (average size of dextran and gold nanoparticles encapsulated polymersomes 400 and 200 nm respectively).Separation of polymersome from micelles
All the polymersome preparations were purified by centrifugation to decrease as much as possible the micelles and aggregates that inevitably form during both pH-switch and film re-hydration. The first step was the centrifugation at 15000 rpm for 15 minutes to eliminate aggregates. The supernatant was collected and centrifuged at 20000 rpm for 20 minutes. This allowed the separation between micelles (supernatant) and polymersomes (pellet).
Polymersome transport
Permeation of the polymersome formulations across porous barriers was performed by placing 75–100 μl of the solution, at a known concentration, in the donor chamber of the perfusion system on top of the porous barrier (polycarbonate membranes or ex vivo skin under sterile conditions in an incubator). The aqueous solution in the acceptor chamber was collected and analysed as a function of time via fluorescence using a Varian CaryEclipse fluorescence spectrophotometer or HPLC. All the results were analysed using Excel or Matlab. Intact imaging of penetration and delivery across ex vivo skin was analysed via non-invasive CLSM (Zeiss LSM 510 META). The z-stack images were obtained by placing the tissue samples in PBS and using a 40 times magnification immersion objective lens and were later 3D rendered using ImageJ software. Skin sections were also analysed via CLSM.Synthetic porous membranes
Polycarbonate porous membranes were used to model translocation across narrow pores. They were purchased from AVESTIN Inc. and were characterised by an average diameter of 19 mm and width of 10 μm. They were produced via chemical etching and were guaranteed by the company to be uniformly porous.Ex vivo skin
All ex vivo skin was obtained from abdominoplasties or breast reduction operations from patients who gave informed consent for tissue not required for their treatment to be used for research. This tissue was used under a Human Tissue Authority (HTA) Research Tissue Bank licence number 12179. Skin was stored at 4 °C in culture medium until use (either for cell isolation of for permeation experiments) for a maximum time of 1 week. Cryo-sections of skin samples were obtained using a Cryo-Star HM 560 Cryostat (MICROM International). Tissue samples were fixed in formaldehyde and subsequently placed on the sample holders using OCT mounting medium and frozen at −40 °C. Sections were cut, placed on slides and stained with DAPI (4′,6-diamidino-2-phenylindole, Invitrogen UK). Confocal images were obtained using a Carl Zeiss LSM 510 META microscope with 10, 20, 40 and 63 times magnification oil lenses.Concentration analysis: fluorescence spectrophotometer and HPLC
The final concentration of every polymersome formulation and of the encapsulated molecules was determined either by fluorescence analysis using a Varian CaryEclipse fluorescence spectrophotometer or high-performance liquid chromatography system (HPLC). For the former technique, the rhodamine labelled polymers was excited at 540 nm and the emission was detected at 562 nm. The FITC labelled dextran was excited at 488 nm and detected at 520 nm. The HPLC set-up used was an UltiMate® 3000 HPLC series provided with a UV-VIS and fluorescence detector and a Chromeleon Data system software purchased from Dionex. A Jupiter C18 300 A column (size 150 × 4.60 mm, purchased from Phenomenex Inc. UK) was used in this work. For the PDPA-based copolymers, the UV-VIS detector was set at 524 nm and the fluorescence detector set to excite at 540 and emit at 565 nm.Dynamic light scattering analysis
The average size of the vesicles was determined by Dynamic Light Scattering analysis (DLS). A Brookhaven Instruments 200SM laser light scattering goniometer and a Zetaseizer Nano ZS (Malvern Instruments) with a 633 nm HeNe laser were used. For the Brookhaven system polymersome dispersions were placed into glass vials. Single scans of 10 min exposures were performed and particle sizes were estimated using the CONTIN multiple-pass method of data analysis at 90°. For the Zetaseizer system samples with a minimum volume of 800 μl were placed in polystyrene cuvettes and scanned for 10 s at 173° and averaged over thirteen runs. This measurement was repeated three times and then averaged. Particle size and polydispersity was estimated using the CONTIN multiple-pass method of data analysis.Electron microscopy
SEM imaging was performed using a FEI Inspect™ F Scanning Electron Microscope. Membranes were cut into small pieces of less than 0.5 cm2 and stuck onto a carbon sticky pad. They were placed on the sample holder and gold coated using a gold coating EMITech K950 Emscope. Images were obtained under high vacuum with a 5–7 kV beam. TEM imaging was performed using a FEI TECNAI G2 Spirit transmission electron microscope. Polymersomes samples were placed on carbon-coated grids and negatively stained with PTA. Skin samples were fixed in glutaraldehyde for at least 24 h, sectioned and then transferred to the carbon-coated copper grids.Author contributions
Conceived and designed the experiments: CP, GB, SM. Performed the experiments: CP. Analysed the data: CP, GB. Contributed reagents/materials/analysis tools: SM, SPA, JM, NW, AB, DC, GB. Wrote the paper: CP, DC, GB, SM, SPA.Acknowledgements
The authors thank Russell Pearson for supplying the TEM images of PMPC-PDPA and PEO-PDPA polymersomes, Dr Robert Field, Ph.D., Ionscope Ltd for the use of the Scanning Ion Conductance Microscope, Biocompatibles UK for support of the project and the EPSRC for funding the White Rose Doctoral Training Programme of Leeds and Sheffield Universities, which supported Carla Pegoraro.References
- P. Friedl and K. Wolf, Proteolytic interstitial cell migration: a five-step process, Cancer Metast. Rev., 2009, 28, 129–135 CrossRef PubMed.
- C. D. Madsen and E. Sahai, Cancer Dissemination-Lessons from Leukocytes, Dev. Cell, 2010, 19, 13–26 CrossRef CAS PubMed.
- M. F. Stins, J. Badger and K. S. Kim, Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells, Microb. Pathog., 2001, 30, 19–28 CrossRef CAS PubMed.
- K. S. Kim, Microbial translocation of the blood-brain barrier, Int. J. Parasitol., 2006, 36, 607–614 CrossRef CAS PubMed.
- R. Ledesma-Aguilar, T. Sakaue and J. M. Yeomans, Length-dependent translocation of polymers through nanochannels, Soft Matter, 2012, 8, 1884–1892 RSC.
- C. Tordeux and J. B. Fournier, Extravasation of adhering vesicles, Europhys. Lett., 2002, 60, 875–881 CrossRef CAS.
- D. Panja, G. T. Barkema and R. C. Ball, Polymer translocation out of planar confinements, J. Phys.: Condens. Matter, 2008, 20, 075101 CrossRef.
- J. K. Wolterink, G. T. Barkema and D. Panja, Passage times for unbiased polymer translocation through a narrow pore, Phys. Rev. Lett., 2006, 96, 208301 CrossRef PubMed.
- D. K. Lubensky and D. R. Nelson, Driven polymer translocation through a narrow pore, Biophys. J., 1999, 77, 1824–1838 CrossRef CAS PubMed.
- M. Mezei and V. Gulasekharam, Liposomes–a selective drug delivery system for the topical route of administration. Lotion dosage form, Life Sci., 1980, 26, 1473–1477 CrossRef CAS PubMed.
- A. Gillet, B. Evrard and G. Piel, Liposomes and parameters affecting their skin penetration behaviour, J. Drug Delivery Sci. Technol., 2011, 21, 35–42 CrossRef CAS.
- G. M. Maghraby, B. W. Barry and A. C. Williams, Liposomes and skin: From drug delivery to model membranes, Eur. J. Pharm. Sci., 2008, 34, 203–222 CrossRef PubMed.
- C. Pegoraro, S. Macneil and G. Battaglia, Transdermal drug delivery: from micro to nano, Nanoscale, 2012, 4, 1881–1894 RSC.
- B. M. Discher, et al., Polymersomes: tough vesicles made from diblock copolymers, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- D. E. Discher, H. Bermudez, A. K. Brannan, D. A. Hammer and F. S. Bates, Molecular weight dependence of polymersome membrane structure, elasticity, and stability, Macromolecules, 2002, 35, 8203–8208 CrossRef.
- T. Smart, et al., Block copolymer nanostructures, Nano Today, 2008, 3, 38–46 CrossRef CAS.
- H. Bermudez, D. A. Hammer and D. E. Discher, Effect of bilayer thickness on membrane bending rigidity, Langmuir, 2004, 20, 540–543 CrossRef CAS PubMed.
- G. Battaglia, C. LoPresti, H. Lomas, M. Massignani and T. Smart, Polymersomes: nature inspired nanometer sized compartments, J. Mater. Chem., 2009, 19, 3576–3590 RSC.
- G. Battaglia and A. J. Ryan, Bilayers and interdigitation in block copolymer vesicles, J. Am. Chem. Soc., 2005, 127, 8757–8764 CrossRef CAS PubMed.
- G. Gompper and D. M. Kroll, Driven Transport of Fluid Vesicles through Narrow Pores, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1995, 52, 4198–4208 CrossRef CAS.
- G. T. Linke, R. Lipowsky and T. Gruhn, Osmotically induced passage of vesicles through narrow pores, Europhys. Lett., 2006, 74, 916–922 CrossRef CAS.
- J. A. Cohen, A. Chaudhuri and R. Golestanian, Active polymer translocation through flickering pores, Phys. Rev. Lett., 2011, 107, 238102 CrossRef PubMed.
- H. Bermudez, D. A. Hammer and D. E. Discher, Effect of bilayer thickness on membrane bending rigidity, Langmuir, 2004, 20, 540–543 CrossRef CAS PubMed.
- T. J. Franz, Percutaneous absorption on the relevance of in vitro data, J. Invest. Dermatol., 1975, 64, 190–195 CAS.
- F. S. Bates and S. Jain, On the origins of morphological complexity in block copolymer surfactants, Science, 2003, 300, 460–464 CrossRef PubMed.
- F. S. Bates, Y. Y. Won and H. T. Davis, Molecular exchange in PEO-PB micelles in water, Macromolecules, 2003, 36, 953–955 CrossRef.
- F. S. Bates and S. Jain, Consequences of nonergodicity in aqueous binary PEO-PB micellar dispersions, Macromolecules, 2004, 37, 1511–1523 CrossRef.
- G. Battaglia and A. J. Ryan, Neuron-like tubular membranes made of diblock copolymer amphiphiles, Angew. Chem., Int. Ed., 2006, 45, 2052–2056 CrossRef CAS PubMed.
- G. Battaglia and A. J. Ryan, Pathways of polymeric vesicle formation, J. Phys. Chem. B, 2006, 110, 10272–10279 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and surface forces, Academic Press, 3rd edn, 2010 Search PubMed.
- T. P. Smart, O. O. Mykhaylyk, A. J. Ryan and G. Battaglia, Polymersomes hydrophilic brush scaling relations, Soft Matter, 2009, 5, 3607–3610 RSC.
- S. P. Armes, A. Blanazs, N. J. Warren, A. L. Lewis and A. J. Ryan, Self-assembly of double hydrophilic block copolymers in concentrated aqueous solution, Soft Matter, 2011, 7, 6399–6403 RSC.
- M. Kobayashi, et al., Structure and Surface Properties of High-density Polyelectrolyte Brushes at the Interface of Aqueous Solution, Macromol. Symp., 2009, 279, 79–87 CrossRef CAS.
- M. Chen, W. H. Briscoe, S. P. Armes and J. Klein, Lubrication at Physiological Pressures by Polyzwitterionic Brushes, Science, 2009, 323, 1698–1701 CrossRef CAS PubMed.
- M. Chen, W. H. Briscoe, S. P. Armes, H. Cohen and J. Klein, Polyzwitterionic brushes: Extreme lubrication by design, Eur. Polym. J., 2011, 47, 511–523 CrossRef CAS.
- M. Kobayashi and A. Takahara, Tribological Properties of Hydrophilic Polymer Brushes Under Wet Conditions, Chem. Rec., 2010, 10, 208–216 CrossRef CAS PubMed.
- G. Cevc and D. Gebauer, Hydration-driven transport of deformable lipid vesicles through fine pores and the skin barrier, Biophys. J., 2003, 84, 1010–1024 CrossRef CAS PubMed.
- H. Bermudez, A. K. Brannan, D. A. Hammer, F. S. Bates and D. E. Discher, Molecular weight dependence of polymersome membrane structure, elasticity, and stability, Macromolecules, 2002, 35, 8203–8208 CrossRef CAS.
- J. Du, Y. Tang, A. L. Lewis and S. P. Armes, pH-sensitive vesicles based on a biocompatible zwitterionic diblock copolymer, J. Am. Chem. Soc., 2005, 127, 17982–17983 CrossRef CAS PubMed.
- H. Lomas, et al., Non-cytotoxic polymer vesicles for rapid and efficient intracellular delivery, Faraday Discuss., 2008, 139, 143–159 RSC.
- M. Muthukumar, Polymer translocation through a hole, J. Chem. Phys., 1999, 111, 10371–10374 CrossRef CAS.
- E. Slonkina and A. B. Kolomeisky, Polymer translocation through a long nanopore, J. Chem. Phys., 2003, 118, 7112–7118 CrossRef CAS.
- G. Cevc, A. G. Schaetzlein, H. Richardsen and U. Vierl, Overcoming semipermeable barriers, such as the skin, with ultradeformable mixed lipid vesicles, transfersomes, liposomes, or mixed lipid micelles, Langmuir, 2003, 19, 10753–10763 CrossRef CAS.
- D. Zheng, et al., Topical delivery of siRNA-based spherical nucleic acid nanoparticle conjugates for gene regulation, Proc. Natl. Acad. Sci. U. S. A., 2012, 11975–11980 CrossRef CAS PubMed.
- A. Gillet, et al., Skin penetration behaviour of liposomes as a function of their composition, Eur. J. Pharm. Biopharm., 2011, 43–53 CrossRef CAS PubMed.
- A. Schatzlein and G. Cevc, Non-uniform cellular packing of the stratum corneum and permeability barrier function of intact skin: a high-resolution confocal laser scanning microscopy study using highly deformable vesicles (Transfersomes), Br. J. Dermatol., 1998, 138, 583–592 CrossRef CAS PubMed.
- G. Cevc, Transfersomes, liposomes and other lipid suspensions on the skin: Permeation enhancement, vesicle penetration, and transdermal drug delivery, Crit. Rev. Ther. Drug Carrier Syst., 1996, 13, 257–388 CrossRef CAS PubMed.
- M. Mezei and V. Gulasekharam, Liposomes - a Selective Drug Delivery System for the Topical Route of Administration .1. Lotion Dosage Form, Life Sci., 1980, 26, 1473–1477 CrossRef CAS PubMed.
- J. A. Bouwstra, et al., Assembled microneedle arrays enhance the transport of compounds varying over a large range of molecular weight across human dermatomed skin, J. Controlled. Release, 2007, 117, 238–245 CrossRef PubMed.
- A. Yamamoto, et al., Trypsin as a novel potential absorption enhancer for improving the transdermal delivery of macromolecules, J. Pharm. Pharmacol., 2009, 61, 1005–1012 CrossRef.
- M. Weiss, et al., Anomalous subdiffusion is a measure for cytoplasmic crowding in living cells, Biophys. J., 2004, 87, 3518–3524 CrossRef CAS PubMed.
- R. H. Huang, Y. Wang, R. Roth, X. Yu, A. R. Purvis, J. E. Heuser, E. H. Egelman and J. E. Sadler, et al., Assembly of Weibel–Palade body-like tubules from N-terminal domains of von Willebrand factor, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(2), 482–487 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60294j |
| This journal is © The Royal Society of Chemistry 2014 |