Small molecule phosphorescent probes for O2 imaging in 3D tissue models†
Ruslan I.
Dmitriev
a,
Alina V.
Kondrashina
a,
Klaus
Koren
b,
Ingo
Klimant
b,
Alexander V.
Zhdanov
a,
Janelle M. P.
Pakan
c,
Kieran W.
McDermott
c and
Dmitri B.
Papkovsky
*a
aBiochemistry Department, University College Cork, Cork, Ireland. E-mail: d.papkovsky@ucc.ie; Fax: +353-21-4901698; Tel: +353-21-4901699
bInstitute of Analytical Chemistry and Food Chemistry, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria
cDepartment of Anatomy and Neuroscience, University College Cork, Cork, Ireland
First published on 28th January 2014
Abstract
Monitoring of oxygenation is important for physiological experiments investigating the growth, differentiation and function of individual cells in 3D tissue models. Phosphorescence based O2 sensing and imaging potentially allow this task; however, current probes do not provide the desired bio-distribution and analytical performance. We present several new cell-penetrating phosphorescent conjugates of a Pt(II)-tetrakis(pentafluorophenyl)porphine (PtPFPP) dye produced by click-modification with thiols, and perform their evaluation as O2 imaging probes for 3D tissue models. The hydrophilic glucose (Pt-Glc) and galactose (Pt-Gal) conjugates demonstrated minimal aggregation and self-quenching in aqueous media, and efficient in-depth staining of different cell types and multi-cellular aggregates at working concentrations ≤10 μM. The Pt-Glc probe was applied in high-resolution phosphorescence lifetime based O2 imaging (PLIM) in multi-cellular spheroids of cancer cells (PC12), primary neural cells (neurospheres) and slices of brain tissue, where it showed good analytical performance, minimal effects on cell viability and appropriate responses to O2 with phosphorescence lifetimes changing from 20 μs in air-saturated to 57 μs under deoxygenated conditions. In contrast, mono- and tetra-substituted oligoarginine conjugates of PtPFPP showed marked aggregation and unstable photophysical properties precluding their use as O2 sensing probes.
1. Introduction
Two-dimensional cultures of adherent cells are routinely used in many areas of biomedical and life sciences; however, more complex three-dimensional (3D) models are more relevant for mimicking the physiological conditions occurring in vivo and replacing less accessible, more expensive and ethically problematic animal and human models.1–4 3D tissue models, such as multi-cellular spheroids, tissue explants, perfused organs and bioengineered tissue, can resemble more closely the natural microenvironment of the cells, cell–cell interactions, tissue heterogeneity and the diffusion-limited supply of metabolites and drugs occurring in vivo. They are actively used in regenerative medicine, cancer biology, stem cell research and tissue engineering.1,4–7 At the same time such models require careful control of the key physical–chemical, biological and physiological parameters during their preparation and use.Bio-imaging has made it possible to probe many vital biochemical characteristics and processes involved in the normal and abnormal functioning of individual cells and tissue samples. For instance, scientists can examine the heterogeneity of cell populations, polarised cell–cell interactions, differential responses of individual cells to manipulated parameters, including signalling cascades, gradients of metabolites and biomolecules.5 Optical and particularly photoluminescence-based imaging techniques provide high temporal and spatial resolution at subcellular level.8–12 However these methods also face technical challenges, particularly limited light penetration depth (<500 μm) and delivery of synthetic reporter materials to large biological specimens. The latter is particularly complicated for large size biosensor or probe structures such as photo-luminescent plasmid-encoded constructs and nanoparticle structures. In this context, there is a demand for new probes which provide efficient tissue penetration, optimal spectral characteristics and characterized structure–activity relationships.13,14
Molecular oxygen (O2) is one of the main components of the atmosphere that fulfils many vital functions in animal cells and tissues. Cellular O2 levels regulate metabolism, gene expression, formation of free radicals and many other processes.15–17 Importantly, abnormal supply of O2 to the tissue is associated with many common pathological states and diseases. In cultures of cancer and stem cells, spatial and temporal gradients of O2 play a role in intracellular signalling as do H+, Ca2+ and other analytes.18–20 Cells and tissues constantly consume O2 and deoxygenate themselves, and this is highly dependent on the metabolic state of the cells and their micro-environment.15 Therefore, knowledge of the actual levels of O2 in respiring cells and tissues is very important, and analytical systems that can provide accurate, real-time monitoring of oxygenation are in high demand. Conventional methods, such as O2 micro-electrodes,21 pimonidazole labelling, staining of hypoxia markers22 or the use of luciferase reporter systems,23 are mostly invasive, indirect, and lack quantitative and/or live 3D imaging readout. In contrast, the phosphorescence quenching method and particularly phosphorescence lifetime imaging microscopy, PLIM,24–27 which operates by means of dedicated probes, can provide high-resolution and quantitative, real-time mapping and monitoring of O2.
Over the years a number of phosphorescent probes have been developed for measurement of O2 in vasculature of animal models.25,27–29 More recently, cell-penetrating O2 probes have been introduced for in vitro applications, to analyse localised O2 gradients in cell cultures30–33 and more complex 3D tissue models.34,35 However these probes still have drawbacks which limit their use. An “ideal” phosphorescent probe is expected to possess high brightness and photostability, convenient spectral characteristics, ease of loading, optimal in-depth distribution in live tissue, appropriate response to O2, and minimal cytotoxicity and photodamage.
One strategy to achieve these probe specifications is to couple a suitable O2-sensitive reporting moiety, such as Pt-porphyrin, to a specific delivery and cell-targeting vector. Introduction of chemical groups,36,37 conjugation with cell-penetrating peptides30,38–40 or incorporation in polymeric nanoparticle structures31,41,42 have been employed for that. The nanoparticle-based O2 probes provide high specific brightness, the possibility of using highly photostable and hydrophobic dyes such as PtPFPP, and simple light-harvesting antennae systems.43 However, these structures are relatively large (>35 nm), heterogeneous and have limited flexibility for tuning their cell-penetrating properties (determined mainly by the polymeric material and characteristics of the particles41–43). Small molecule O2 probes are more attractive in this regard, but to date, only the conjugates of moderately photostable phosphorescent dyes, such as Pt(II)-coproporphyrin PtCP, Ir(III)-octaethylporphyrin IrOEP38,40 and Pd-meso-tetra(4-carboxyphenyl)tetrabenzoporphyrin),28,44 have been explored. Also very few probes have been evaluated in detail with 3D tissue models and other biomaterials which require rapid penetration through multiple layers of cells and efficient in-depth staining.34,35,43,45
The recently developed click modification of perfluorinated porphyrins, which enables substitution of up to four p-fluorine atoms with thiol or other nucleophilic groups,46–50 has paved the way for synthesising new cell-penetrating small-molecule phosphorescent structures, for example based on the highly photostable and bright PtPFPP dye. However prominent hydrophobicity and aggregation in aqueous solutions have been noted for such compounds.47 In this study, we applied this chemistry to prepare several new conjugates of PtPFPP with monosaccharide and peptide delivery vectors that were expected to facilitate cell staining and penetration. In the experiments with adherent cell cultures, multi-cellular spheroids and tissue slices we showed that PtPFPP derivatives tetra-substituted with glucose and galactose moieties display efficient staining and in-depth penetration, moderate brightness and general suitability for high-resolution 3D mapping of tissue O2 by the PLIM method. Conversely, mono- and tetra-substituted oligoarginine conjugates of PtPFPP demonstrated marked aggregation and unstable photophysical properties in biological samples, which preclude their use as O2 probes.
2. Materials and methods
2.1. Materials
PtPFPP dye was from Frontier Scientific (Inochem Ltd, Lancashire, UK), and β-D-thioglucose and thiogalactose were from Carbosynth Ltd (Berkshire, UK). Synthetic G2R8-amide and R2-amide peptides (purity >90% by HPLC; structures confirmed by mass spectrometry) were from Genscript (Piscataway, NJ, USA). Secondary antibodies labelled with Alexa Fluor 488 and 555, recombinant cholera toxin, subunit B-Alexa Fluor 488 conjugate (CTX), B27 serum-free supplement, Calcein Green AM, tetramethylrhodamine methyl ester (TMRM) and Pro-long Gold Anti-Fade mounting medium were from Invitrogen (BioSciences, Dublin, Ireland). Epidermal growth (EGF) and fibroblast growth (FGF) factors, and monoclonal anti-BrdU antibody BU-1 were from Millipore (Cork, Ireland). Goat anti-nestin antibodies were from Santa Cruz Biotechnology (Heidelberg, Germany). Pimonidazole and anti-pimonidazole antibody (MAB1) were from hpi (Burlington, MA). Anti-GFAP (G-A-5) antibody, bis-Benzimide Hoechst 33342 and all the other reagents were from Sigma-Aldrich (Dublin, Ireland). pH-Xtra, MitoXpress-Xtra and MitoImage-NanO2 probes and high density mineral oil were from Luxcel Biosciences (Cork, Ireland). Standard cell culture grade plasticware was from Sarstedt (Wexford, Ireland) and Corning (VWR, Ireland), glass bottom mini-dishes were from MatTek (Ashland, USA), glass bottom μ-dishes Grid-500 and multiwell slides were from Ibidi (Martinsried, Germany).2.2. Synthesis and characterisation of conjugates
Click modification of PtPFPP was performed according to the modified method.51 Direct tetra-substitution with hexoses was achieved by incubation of PtPFPP with 5 molar equivalents of unprotected β-D-thioglucose or thiogalactose in DMF in the presence of triethylamine (16 h at 40 °C), followed by RP-HPLC purification (gradient of methanol in 0.1% TFA in water). For the conjugation with L-Arg-O-methyl ester or peptides, PtPFPP was first modified at one or four p-fluorine atoms with two or ten equivalents of 3-mercaptopropionic acid in DMF in the presence of triethylamine (3–16 h at 40–70 °C), followed by RP-HPLC purification. The derivatives of PtPFPP were coupled with the peptides by the carbodiimide method, using EDC, DMSO as the solvent, twofold excess of the peptide per carboxylic group and 16 h incubation at room temperature, followed by RP-HPLC purification.38 Purified conjugates were reconstituted in DMSO and stored at 4 °C. Structures were confirmed by MALDI-TOF with spectra recorded on a MicromassTof Spec 2E (reflectron mode at an accelerating voltage of +20 kV).Absorption spectra of the conjugates were measured on an 8453 diode array spectrophotometer (Agilent) in phosphate buffered saline (PBS) containing 0.25% Triton X-100 or 10% fetal bovine serum (FBS). Phosphorescence lifetimes under air-saturated and deoxygenated (5 mg ml−1 KH2PO4, 5 mg ml−1 Na2SO3) conditions were measured on a TR-F reader Victor2 (Perkin Elmer), as described in ref. 40.
2.3. Cell and tissue culture
Murine embryonic fibroblast (MEF), human hepatocellular liver carcinoma (HepG2), human neuroblastoma (SH-SY5Y), human colon carcinoma (HCT116), African green monkey kidney (COS-7), rat pheochromocytoma (PC12) and human colorectal adenocarcinoma (Caco-2) cells from ATCC (Manassas, VA, USA) were handled as described before.38,40 Primary neurons from the embryonic mouse brain (CN E16) were kindly supplied by Dr U. Anilkumar (RCSI, Dublin).PC12 cell aggregates were grown in RPMI-1640 medium supplemented with 10% horse serum (HS), 5% FBS, 10 mM HEPES-Na, pH 7.2, and penicillin–streptomycin. For the assessment of cell staining kinetics, leakage and toxicity in ATP and extracellular acidification (ECA) assays, MEF cells were seeded on collagen-coated 96-well plates and cultured for 48 h to reach 75–100% confluence. Metabolic stimulation of PC12 cell aggregates was performed on 35 mm Cell+ mini-dishes (Sarstedt) in 2 ml of Phenol Red-free DMEM supplemented with NGF (100 ng ml−1) at 37 °C and 20% O2. Drugs were applied by adding 10× stock solutions to the sample (1/10 of the volume).
All procedures with animals were performed under a licence issued by the Irish Government Department of Health and Children (Ireland) and in accordance with the Directive 2010/63/EU adopted by the European Parliament and the Council of the European Union. Briefly, Sprague-Dawley rats (pregnant at embryonic day (E)16 and E18 or postnatal (P) ages P7 and P21) were anesthetized by inhalation of isoflurane (Abbeyville Veterinary, Ireland), decapitated and embryos/brains were dissected out into ice-cold HBSS. Tissue was embedded in 4% low melting agarose (prepared using PBS supplemented with 5 mg ml−1 glucose) and placed on ice. The agarose block was then mounted on a vibratome stage (VT1200, Leica Microsystems) and 300–400 μm thick horizontal sections were sliced in ice cold L15 (Leibovitz) medium. Brain sections were placed onto pre-wet polystyrene scaffold membranes Alvetex™ (Reinnervate, Amsbio, UK) in phenol red-free DMEM supplemented with 25% HBSS, 10% FBS, 1% penicillin–streptomycin, 10 mM Glc, and 20 mM HEPES-Na, pH 7.2 (“Slice medium”) under ambient O2 (20%) and stained for 3 h with Pt-Glc (10 μM) and Cholera toxin, subunit B-Alexa Fluor 488 (1 ng ml−1). After incubation the tissue was transferred to fresh media and imaged as described below.
Neurospheres from cortices of embryonic (E18) rat brain were prepared as described before34,43 and cultured in DMEM/F12 Ham medium supplemented with FGF (20 ng ml−1), EGF (20 ng ml−1), B27 (2%) and penicillin–streptomycin. Staining of the neurospheres (grown for 5–7 days in vitro) was achieved by incubation with 1–5 μM Pt-Glc in the presence or absence of TMRM (20 nM), BrdU (0.2 μM, 18 h) or pimonidazole (200 μM, 18 h) stains. Alternatively, neurospheres were stained by continuous culturing them in the presence of 5 μg ml−1 of the NanO2 probe, adding fresh medium to the probe on days 1, 3, and 5.
2.4. Microscopy imaging experiments
Cells stained with Pt-Glc, Pt-Gal, PEPP338 (2.5 μM, 16 h) or NanO231 (5 μg ml−1, 16 h) probes were washed three times with the medium and counter-stained with Calcein Green (1 μM, 30 min). Cells were then placed in fresh media and imaged.Confocal PLIM imaging was performed on a system based on an upright fluorescent microscope AxioExaminer Z1 (Carl Zeiss) with 20×/1.0 W-Plan-Apochromat objective, heated stage with motorized Z-axis control, DCS-120 confocal scanner (Becker & Hickl) with an R10467U-40 photon counting detector (Hamamatsu Photonics K.K.) and TCSPC hardware (Becker & Hickl).34 Pt-Glc- and Pt-Gal-stained cells were excited with a 405 nm picosecond diode laser BDL-SMC (Becker & Hickl) and emission was collected at 635–675 nm. Calcein Green probe was excited with picosecond supercontinuum laser SC400-4 (Fianium, UK) at 488 nm and emission was collected at 512–536 nm with a bandpass filter. DIC images were recorded with a D3100 digital camera (Nikon) connected to the microscope. Phosphorescence lifetimes were calculated from mono-exponential decay fits in SPCImage software (Becker & Hickl). 2D matrices of lifetime data for each measurement were then correlated with intensity and DIC images, and selected ROIs were converted into O2 concentration using the following calibration function (produced with stained PC12 cells and neurospheres): O2 [μM] = 1768.574 exp(−τ/9.72584).
Comparison of relative brightness and photostability of different probes and O2 calibration for Pt-Glc was performed on a wide-field inverted microscope Axiovert 200 (Carl Zeiss) equipped with 40×/1.3 EC Plan Neofluar objective, excitation module (390 nm, 470 nm and 590 nm LEDs pulsed in PLIM mode), gated CCD camera, ImSpector software (LaVision BioTec, Germany), and integrated CO2/O2 climate control chamber (PeCon) as described before.34
Immunofluorescence was performed as described in ref. 34 Briefly, Pt-Glc-stained neurospheres were attached to PDL-coated glass or plastic mini-dishes, washed with HBSS, fixed with paraformaldehyde (4%, 20 min), permeabilised with Triton X-100 (0.25%, 10 min), blocked with FBS (5% in TBST, 1 h), then immunostained with primary anti-nestin (Santa Cruz Biotechnology) and/or anti-GFAP (G-A-5, Sigma-Aldrich), anti-BrdU (BU-1, Millipore), anti-pimonidazole (MAB1, hpi) and secondary Alexa Fluor 488 and 555-conjugated antibodies, counter-stained with DAPI (300 nM, 10 min), mounted in Pro-Long Gold anti-fade medium and analysed on the confocal PLIM microscope.
2.5. Cell viability and probe toxicity assessment
Viability of cells was assessed by measuring changes in total ATP with a CellTiter-Glo kit, lactate-mediated (L-ECA) and total (T-ECA) extracellular acidification with the pH-Xtra™ probe:52 MEF cells grown on 96 well plates were stained with 0–20 μM of Pt-Glc or Pt-Gal for 16 h, then washed with fresh medium and analysed. In the ECA assays, the samples were pre-incubated for 2 h under CO2-free conditions in phenol-red free DMEM, 10 mM HEPES-Na, pH 7.2, at 37 °C. Then the medium was replaced with unbuffered DMEM, pH 7.4, containing 1 μg ml−1 of pH-Xtra, 1 μM FCCP and 10 μM oligomycin (maximal uncoupling of mitochondria) or DMSO (mock). For L-ECA the samples remained uncovered and for T-ECA they were covered with high density mineral oil. The plate was then measured kinetically for 1 h at 37 °C on a multi-label reader Victor2 (PerkinElmer) using time-resolved fluorescence (TR-F) mode and 340 ± 50 nm excitation, 615 ± 8.5 nm emission filters. In each measurement point two TR-F signals (F1, F2) were collected at delay times t1 = 100 μs and t2 = 300 μs, and phosphorescence lifetimes were calculated as follows: τ [μs] = (t2 − t1)/ln(F1/F2). Lifetime values were then converted into pH according to the calibration function (37 °C): pH = (1893.4 − τ)/227.54. After L-ECA measurement the cells were lysed with a buffer containing 150 mM NaCl, 1 mM EDTA, 1% Igepal CA-630, 50 mM HEPES-Na, pH 7.6, protease inhibitor cocktail (Sigma P2714), centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 15 min and analysed for total protein content with BCA protein assay kit (Pierce, MSC, Ireland). T-ECA and L-ECA results were normalised for total protein concentration.53
000g for 15 min and analysed for total protein content with BCA protein assay kit (Pierce, MSC, Ireland). T-ECA and L-ECA results were normalised for total protein concentration.53
2.6. Data assessment
Fitting of phosphorescence decays was performed either in ImSpector software (wide-field PLIM) or SPCImage software (Becker & Hickl) using single-exponential decay function and pixel binning as appropriate. The resulting 2D matrices with lifetime data were converted to ASCII format and processed in Microsoft Excel to produce O2 concentration values using calibration function. 3D projections of neurosphere staining were produced from intensity images representing individual optical sections in Fiji software (http://fiji.sc/Fiji), using Volume Viewer 2.01 plugin. Plate reader data are presented as mean values with standard deviation (error bars) for 4–6 independent measurement points. To ensure consistency, all the experiments were repeated 2–3 times, as indicated in figure descriptions.3. Results
3.1. Rational design and synthesis of the O2-sensitive PtPFPP conjugates
Recently we studied the structure–activity relationships of PtCP derivatives substituted via peripheral carboxylic groups with peptides and of IrOEP complexes coordinated with histidine-containing axial ligands,38,40 particularly cell-permeability and intracellular distribution of such structures which unfortunately had modest photostability. The conjugates of other porphyrin dyes with saccharide moieties were also reported.46,48,54 Although showing efficient intracellular accumulation, these structures were designed for use as photosensitizers, i.e. to kill the cells upon exposure to light.We applied this knowledge to prepare several new conjugates of the PtPFPP dye with cell-penetrating peptide and saccharide vectors using click modification.51 These structures were expected to have high brightness, photostability and convenient phosphorescence spectral and lifetime (τ0 = 70 μs) characteristics.31,55 Tetra-substituted conjugates were deemed more promising, therefore we functionalised PtPFPP with four arginine or diarginine residues (conjugates 1, 3), and four glucose (Glc) or galactose (Gal) residues (conjugates 4, 5) (Fig. 1, Table 1, ESI†). The mono-substituted conjugate with linear octaarginine peptide (conjugate 2) was also synthesized for comparison. Coupling with the peptides was performed in two steps: first introducing a 3-mercaptopropionic acid linker and then coupling it with a free amino group of the peptide by a carbodiimide method.56 The Glc/Gal conjugates were produced in one-step using thio-hexoses (see Materials and methods, ESI†). In agreement with strong hydrophobicity of bulky pentafluorophenyl groups of PtPFPP, the analysis of retention times in RP-HPLC showed that the tetra-substituted R8 conjugate was more hydrophilic than its mono-substituted analogue (Table 1, conjugates 2, 3). On the other hand, the hydrophilicity of tetra-substituted R4 and R8 conjugates was rather similar (Table 1, conjugates 1, 3). The hydrophobic conjugates 1 and 2 showed largely reduced phosphorescence lifetimes (Table 1), whereas conjugates 3 and 4 were similar to unmodified PtPFPP in solubilised state (0.25% SDS). Aggregation of conjugates 1 and 2 in aqueous media was also evident from their blue-shifted absorption spectra (not shown).
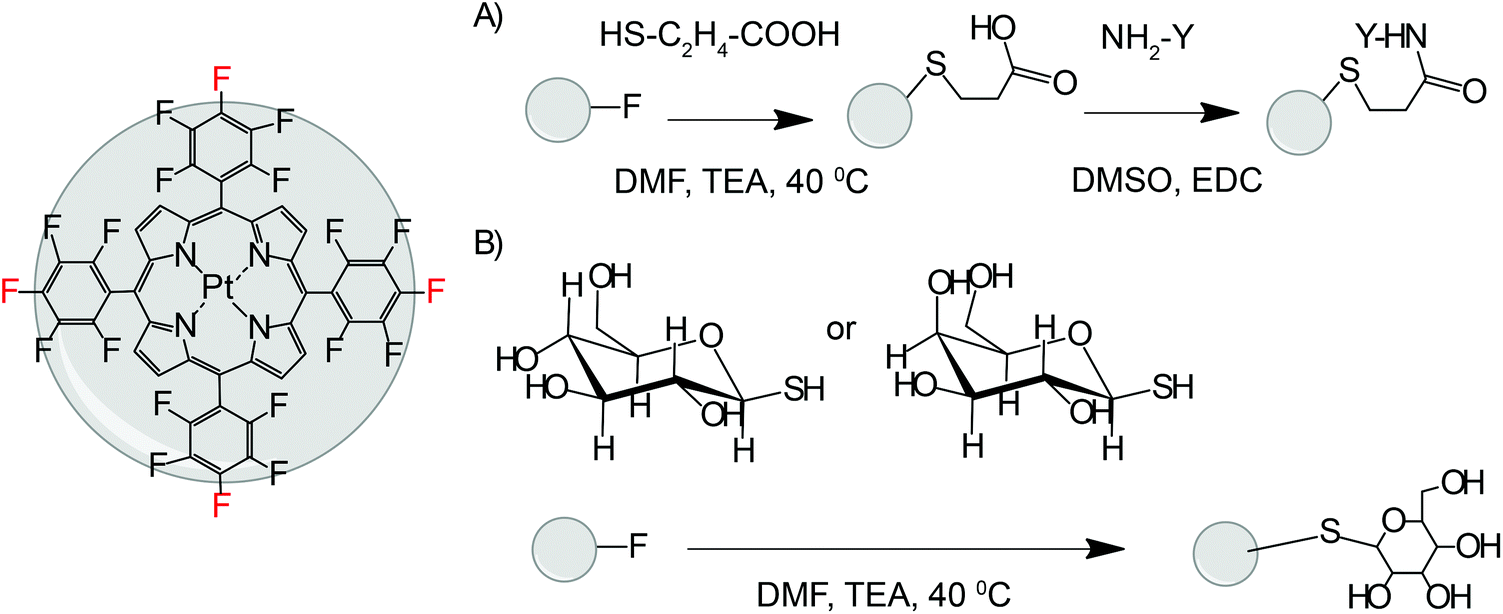 | ||
| Fig. 1 Scheme of synthesis of PtPFPP conjugates by click modification. Parent PtPFPP structure with p-fluorine atoms indicated in red. (A) Synthesis of peptide conjugates 1–3, Y denotes the arginine-containing group. (B) Synthesis of Gal and Glc conjugates 4 and 5. | ||
| Conjugate | Modification | M w [Da] | Charge | Hydrophobicity as solubility in MeOHa [%] |
τ*; τ0b [μs] |
|---|---|---|---|---|---|
| Notes:a Solubility data are based on RP-HPLC retention time of H2O–MeOH/0.1% TFA gradient (relative to unconjugated PtPFPP taken as 100%).b Phosphorescence lifetimes were measured on a Victor2 microplate TR-F reader in PBS containing 0.25% Triton X-100, with (τ0) or without (τ*) deoxygenation (5 mg ml−1 KH2PO4, 5 mg ml−1 Na2SO3). | |||||
| 1 | 4 × Arg (branched) | 2193 | +4 | 64.5% | 23.1; 37.3 |
| 2 | 8 × Arg (linear) | 2616 | +8 | 94.9% | 20.9; 43.0 |
| 3 | 8 × Arg (branched) | 2758 | +8 | 65.8% | 32.4; 51.8 |
| 4 | 4 × Glc (branched) | 1872 | 0 | 64.2% | 20.3; 57 |
| 5 | 4 × Gal (branched) | 1872 | 0 | 64.2% | ND |
Based on this initial assessment the conjugates with four hexose moieties (Pt-Glc and Pt-Gal) were selected for quantitative sensing and imaging of cellular O2, as they possessed small molecular size, easy synthesis procedure, optimal photophysical characteristics and efficient cell staining (similar to conjugates 2 and 3, data not shown). So far, such structures have not been studied as O2 probes.
3.2. Cellular uptake and toxicity of Pt-Glc and Pt-Gal conjugates
Internalisation of porphyrin conjugates with carbohydrates is thought to depend on intracellular transport of glucose, cell glycolytic activity,48 and therefore should show cell-specificity. We analysed the cell-penetrating properties of Pt-Glc and Pt-Gal conjugates with the MEF cell line which was previously used for evaluation of other iO2 probes.31,32 Both conjugates showed similar kinetics of intracellular staining (4–6 h, Fig. 2A), which was 3–4 times faster than that for PtCP-peptide conjugates such as PEPP0 or the nanoparticle probes such as NanO2.25 We observed no saturation of probe internalisation at concentrations up to 20 μM (Fig. 2B). Pt-Glc and Pt-Gal displayed similar patterns of perinuclear distribution in MEF cells (resembling endoplasmic reticulum), and they were sufficiently bright and photostable for high-resolution PLIM measurements (Fig. 2C, S1†). They also showed somewhat shorter retention in cells with a half-clearance rate of less than 24 h (Fig. 2D).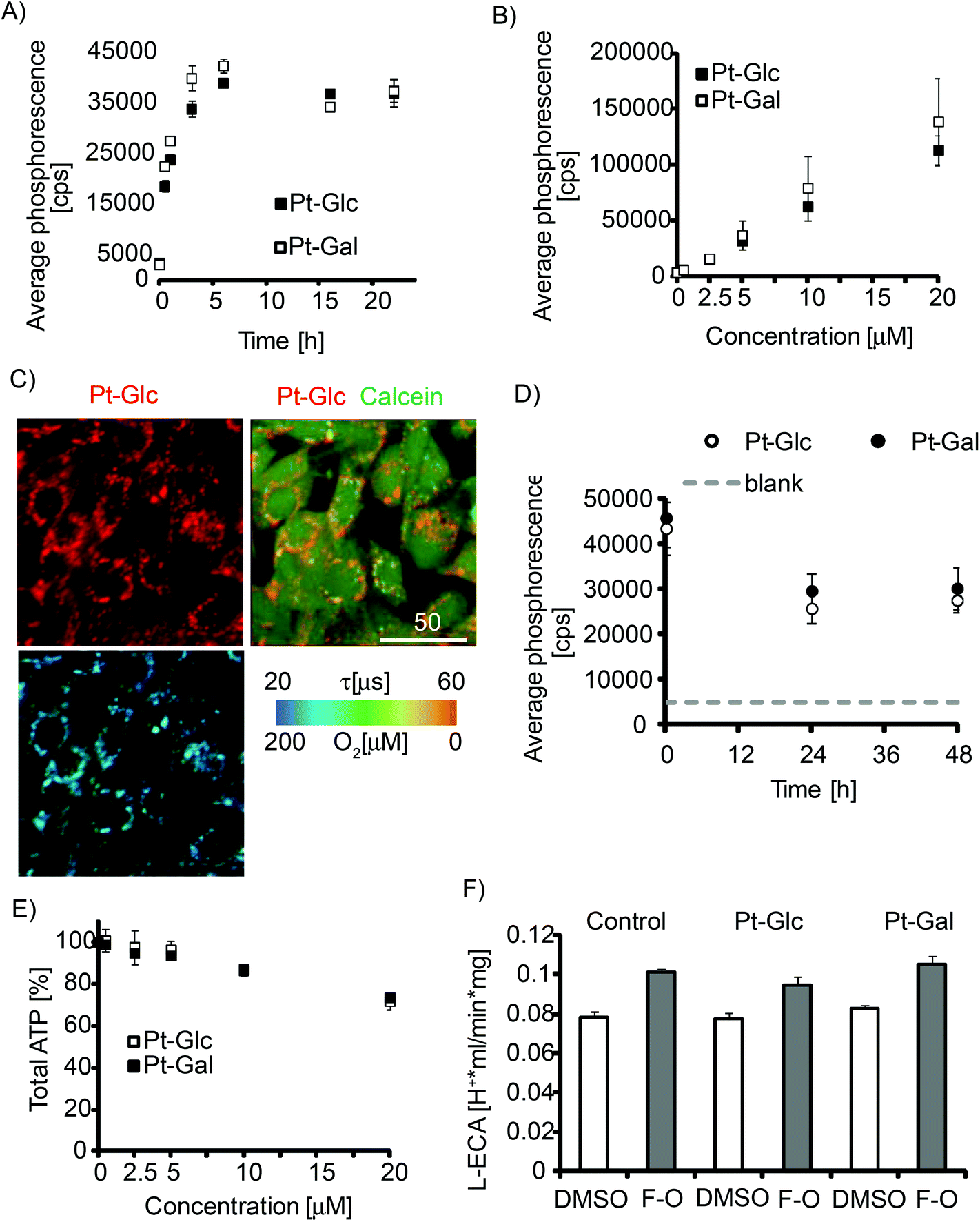 | ||
| Fig. 2 Staining of MEF cells with Pt-Glc and Pt-Gal conjugates and their effects on cell viability. Time (A) and concentration (B) dependence of cell internalisation, measured on a TR-F reader. (A) Concentration of the conjugates – 5 μM; (B) staining time – 16 h. (C) Intensity images of localisation of Pt-Glc (2.5 μM, 16 h) and cytosolic stain Calcein Green (1 μM, 0.5 h); lifetime image of Pt-Glc (405 nm exc./650 nm em.) is shown below. Scale bar is in μm. (D) Time-dependence of retention of the conjugates inside the cells (pre-stained with 10 μM, 16 h) measured on a TR-F reader. (E) Effects of the conjugate on cell viability (total ATP). (F) Effects of the conjugate (5 μM, 16 h staining) on L-ECA under resting (DMSO) and uncoupled (FCCP-Oligomycin treatment) conditions. | ||
Next, we tested the conjugates with extended panel of cell lines including PC12, COS-7, SH-SY5Y, HepG2, HCT116, Caco-2 and primary neurons and found efficient intracellular accumulation in all of them (Fig. S1†). In MEF, HepG2 and SH-SY5Y cells internalisation efficiency of Pt-Glc and Pt-Gal was identical. In other cell lines differences were observed (Pt-Gal > Pt-Glc in Caco-2, Pt-Glc > Pt-Gal in PC12), which we attribute to the differences in glucose uptake by the cells. In PC12 cells and primary neurons, slight aggregation of the probe was noticed (Fig. S1†), which may be due to low serum content during the culture and loading.
Possible toxic effects of the probe are of major concern for physiological experiments. This was assessed by analysing total ATP content (Fig. 2E). Significant effects (<80% viability after exposure to the conjugate) were only seen at concentrations above 10 μM. As a more sensitive and selective toxicity test, we also analysed changes in glycolysis and Krebs-cycle activities by measuring lactate-related and total ECA.15Fig. 2F and Fig. S2† show that both conjugates at 5 μM concentration had no detectable impact on tested cells. These results demonstrate minimal effect of PtPFPP conjugates on cell viability and bioenergetics at ≤10 μM concentration. Therefore, both Pt-Glc and Pt-Gal are suitable for use as O2 imaging probes.
3.3. Evaluation of PtPFPP conjugates in PLIM microscopy
The efficient intracellular staining of many cell types and compatibility with PLIM microscopy make Pt-Glc and Pt-Gal conjugates attractive for use with 2D and 3D cell and tissue models. First, we evaluated their brightness and photostability with 2D cultures of MEF cells (Fig. 3). Although significantly less bright than the nanoparticle-based NanO2 probes, Pt-Glc and Pt-Gal were still much brighter than the small molecule PtCP-based conjugate, PEPP338 (Fig. 3A). Thus, under optimal cell loading conditions the signal to noise ratio (S:N) was ≤1 for the PEPP3 conjugate and for the PtPFPP conjugates it was 2–5. Photobleaching experiments demonstrated similar rates for Pt-Glc and NanO2 probes (Fig. 3B). Therefore, with 2D cultures such as MEF cells, the new conjugates are significantly better than the previously reported small-molecule O2 probes such as PtCP-based PEPP3, but still not as bright as the nanoparticle PtPFPP based probe NanO2.
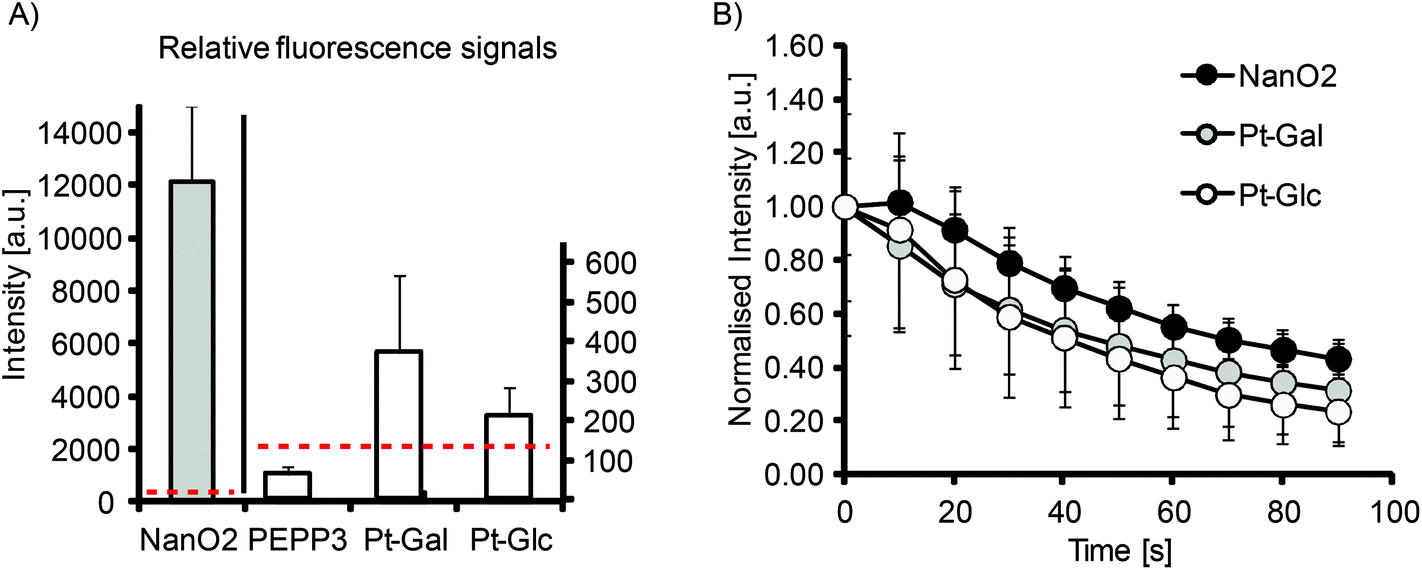 | ||
| Fig. 3 Relative brightness and photostability of Pt-Glc and Pt-Gal. (A) Intensity signals (background corrected) produced by the different probes in MEF cells: Pt-Glc, Pt-Gal, PEPP3 – 2.5 μM, NanO2 – 5 μg ml−1, loading time – 16 h. Dashed lines show background signals. (B) Bleaching of Pt-Glc, Pt-Gal and NanO2 in MEF cells under constant illumination (normalised intensity signals). | ||
Next, Pt-Glc conjugate was tested in PLIM imaging multi-cellular aggregates formed from PC12 cells at high serum content (10% HS and 5% FBS). Although not considered as spheroids, such structures having sizes 50–200 μm provide an optimal micro-environment for proliferating cancer cells.5 Active respiration of PC12 cells39,53 leads to the formation of micro-gradients of O2 in these aggregates.33 We therefore stained PC12 aggregates with Pt-Glc and Calcein Green (cell tracer) probes and analysed them by PLIM. Fig. 4A shows that even in large aggregates Pt-Glc provides efficient and uniform staining of cells and localises in the cytoplasm, comparable with Calcein Green dye. Fig. 4B shows PLIM images at rest and after treatment with the drugs that alter cell respiration and O2: FCCP, which activates mitochondrial respiration; potassium chloride, which depolarises membrane potential; and sulfite, which chemically deoxygenates the sample. Such treatments produce robust responses in 2D cultures of PC12 cells that can be measured on a fluorescence plate reader.39 Analysis of PLIM images and selected regions of interest (ROI1-3, Fig. 4B–D) showed good agreement with these early observations. Importantly, PLIM analysis can inform on heterogeneity of sample oxygenation and spatial micro-gradients of O2, unlike the average effects of cell populations measured on a plate reader. Thus, for the aggregates having irregular shape the heterogeneous O2 distribution was observed under resting and stimulated conditions. We also performed treatment of PC12 aggregates with inhibitors of respiration (antimycin A and rotenone) and V-ATPase inhibitor Bafilomycin A1 which possesses K+-ionophore activity.57 The results presented in Fig. S3† show that the Pt-Glc probe allows for monitoring of O2 in cultures of respiring mammalian cells and heterogeneous 3D samples such as multi-cellular aggregates by the live cell PLIM technique.
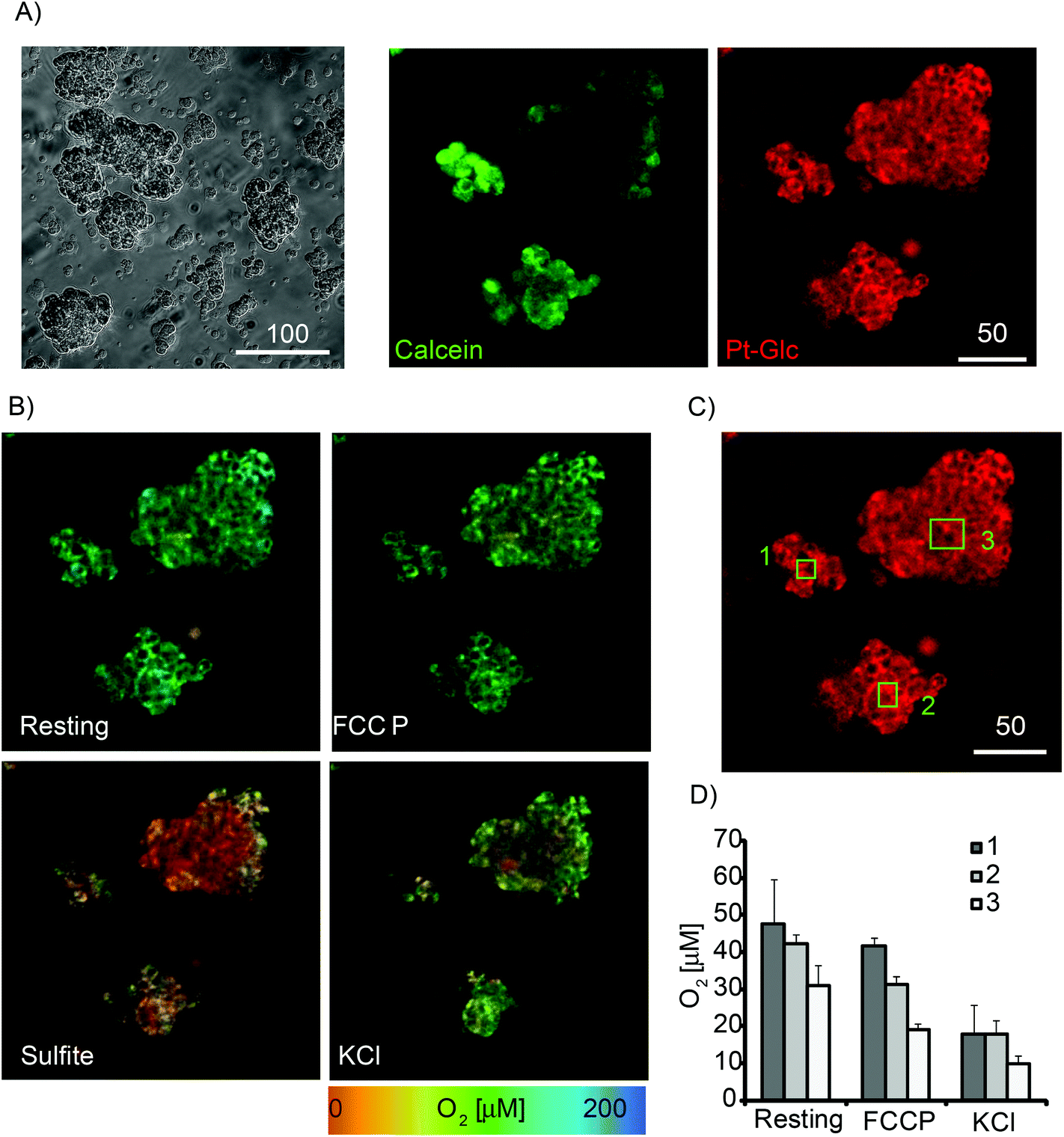 | ||
| Fig. 4 Images of multi-cellular aggregates of PC12 under different metabolic conditions. Staining with Pt-Glc conjugate (2.5 μM, 16 h) and imaged on the confocal TCSPC (PLIM) microscope. Single optical sections are shown. (A) Transmission light and fluorescence intensity images of Calcein Green (1 μM, 0.5 h) and Pt-Glc. (B) Images of O2 concentration (determined from PLIM) under resting conditions and upon treatment with 4 μM FCCP, then 0.1 M KCl and sulphite. (C) Phosphorescence images of several spheroids with indicated ROI (1–3). (D) Average O2 levels for selected ROIs under different conditions. Scale bar is in μm. N = 3. | ||
3.4. Monitoring of oxygenation in spheroid models
Multi-cellular spheroids are a useful 3D model of tissue for studying the development and differentiation of cancer and stem cells.5 We evaluated Pt-Glc with neurospheres produced from embryonic rat brain (Fig. 5). Compared to tumour cell spheroids, neurospheres are more difficult to stain and prolonged incubation (up to 72 h) is required for many nanoparticle probes.34,43 In contrast, 5 μM Pt-Glc provided efficient staining of neurosphere core regions after 16 h incubation, with spheroids up to 500 μm diameter (Fig. 5A and B). Importantly, Pt-Glc remained inside the cells after fixation and immunostaining of neurospheres, i.e. >48 h after probe removal from the medium. Analysis of shorter staining times confirmed (Fig. S4†) that Pt-Glc accumulates in the neurospheres more rapidly (16–24 h) and provides deeper staining than TMRM (a rhodamine based probe for mitochondrial membrane potential). The localisation of Pt-Glc in the neurosphere was compared with the distribution of GFAP-positive cells (immunostained developing astrocytes in the peripheral layers of spheroid) and the nuclear stain, DAPI. Fig. 5C shows that in neurospheres Pt-Glc is confined to cell bodies, rather than neuritis or sprouts. A similar localisation was seen in PC12 cells (see section 3.3).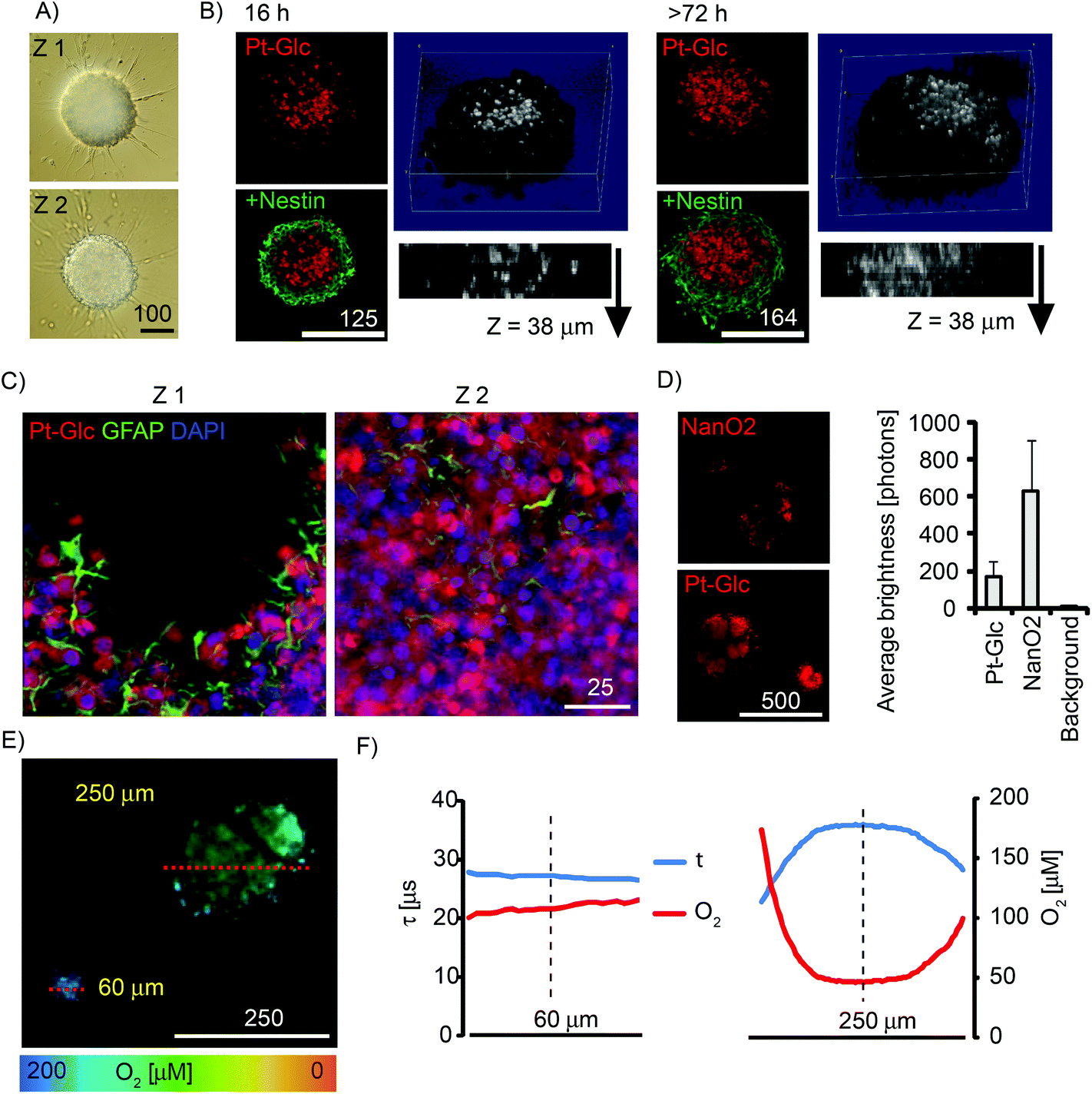 | ||
| Fig. 5 Phosphorescent staining and oxygenation of cultured neurospheres. Neurospheres from E18 rat cortices were grown for 5–7 days in vitro under proliferating conditions. (A) DIC images of a neurosphere at Z (focal) positions with 100 μm distance. (B) Immunofluorescence images of fixed neurospheres stained for different periods of time with Pt-Glc (5 μM, red) and counter-stained with anti-nestin antibodies (neural progenitor cells, green). Single optical sections (2 μm) for each staining time are shown on the left, 3D reconstructions of Pt-Glc distribution with side views – on the right. (C) High-resolution images showing localisation of Pt-Glc in the cytoplasm of neurosphere cells. Glial cells were visualised with an anti-GFAP antibody using immunofluorescence. Nuclei stained with DAPI are shown in blue distance between optical sections 10 μm. (D) Comparison of neurosphere staining with NanO2 and Pt-Glc probes. Left: probe distribution (intensity images). Right: average brightness of stained regions. N = 6. (E, F) PLIM images (E) and O2 profiles (F) for the cross-sections (shown with red lines) of the ∼60 and 250 μm large neurospheres. Scale bar is in μm. | ||
Using neurospheres as a 3D model of animal tissue, we compared staining efficiency and average brightness of Pt-Glc with the NanO2 probe. Fig. 5D shows the patchy distribution of NanO2,31 and the more uniform and continuous distribution of Pt-Glc in the neurosphere. Using the Pt-Glc probe, we also managed to identify the micro-regions with increased brightness and lifetime values (τ > 35 μs), and attributed them to hypoxic niches within the neurosphere.20 Under optimised conditions, staining efficiency and brightness of Pt-Glc in the neurospheres was only 4–5 times lower than for the NanO2 probe (compare with Fig. 3A), thus reflecting its better accumulation in multi-cellular aggregates. PLIM experiments with live neurospheres of different sizes revealed (Fig. 5E and F) that small spheroids (∼60 μm) did not possess a hypoxic core, while larger structures did. Efficient staining with Pt-Glc facilitates the mapping of hypoxic or anoxic micro-regions in neurospheres by PLIM. In line with our previous findings,34 spheroids produced in static cultures demonstrated the significant heterogeneity and size dependence of their oxygenation (not shown).
The so-called “hypoxia stains” are commonly used to visualise the low O2 environment in in vivo and in vitro models.15,19,58 However imaging information provided by these stains is difficult to interpret and also prone to artefacts. We performed comparative multiplexed analysis of hypoxia in the neurospheres using the Pt-Glc probe (live cell imaging) and immunofluorescent pimonidazole and HIF-2a stains (Fig. 6). In this case, the same neurospheres were grown for 5 days in vitro (DIV) under proliferating conditions, then attached to gridded glass bottom dishes under differentiating conditions (1 DIV) and analysed by confocal (3D) fluorescence microscopy. It was found that in situ imaging with the Pt-Glc probe produced the anticipated O2 patterns, with a clearly identifiable hypoxic core inside and increased oxygenation towards the surface of the neurosphere.
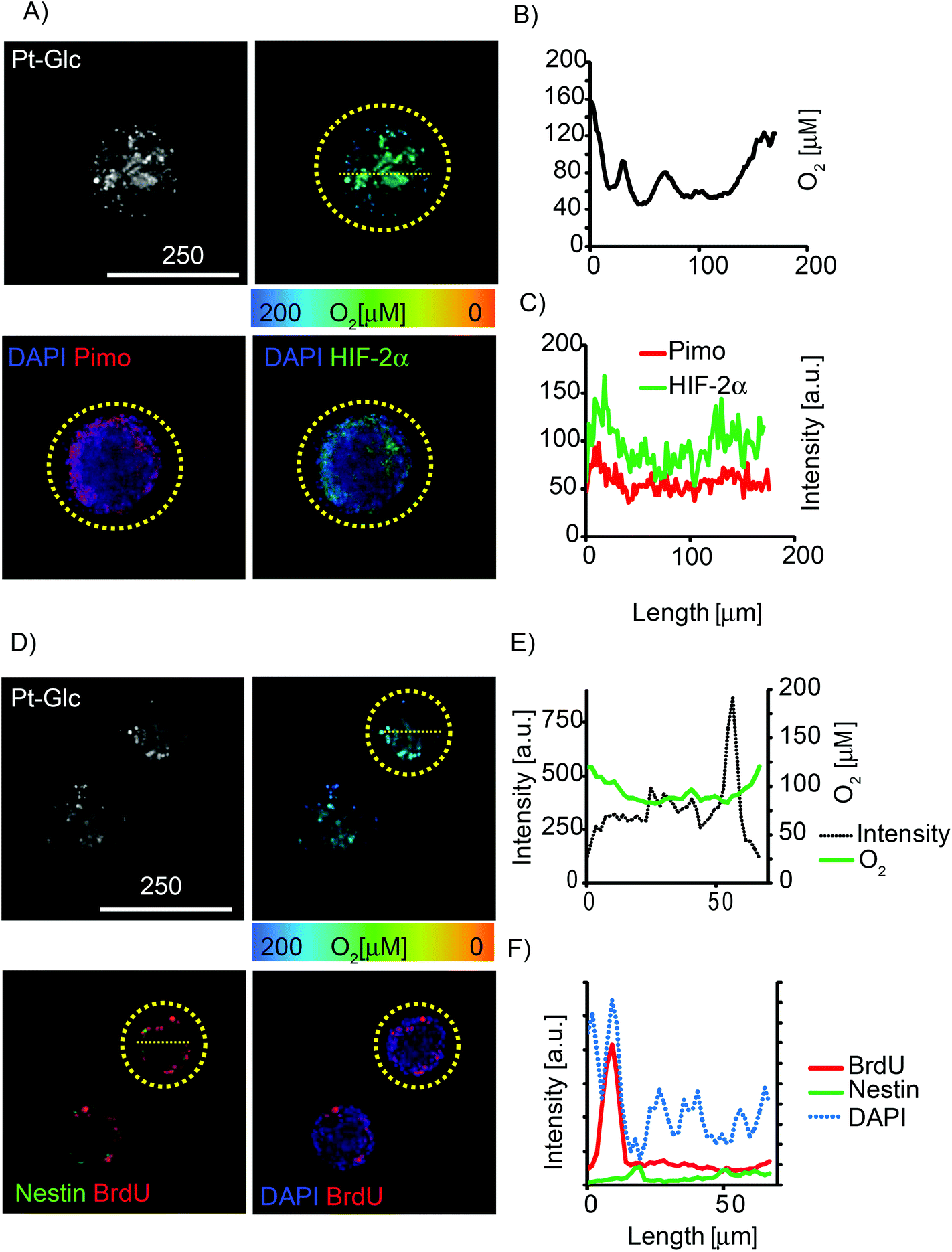 | ||
| Fig. 6 Multiplexed analysis of neurosphere oxygenation with different hypoxia stains. Neurospheres were grown under proliferating (5DIV) and differentiating conditions (1DIV), stained with Pt-Glc (1 μM, >72 h), pimonidazole (200 μM, 24 h) or BrdU (0.2 μM, >18 h), analysed by live cell PLIM and immunofluorescence. (A) Live cell intensity (gray scale) and PLIM (false-color) images of Pt-Glc (top) and corresponding fluorescent images of nuclear (DAPI, blue), pimonidazole (Pimo, red) and HIF-2α (green) (immuno)stains (bottom). (B) O2 profile (indicated by yellow dashed line) across one optical section of the neurosphere produced with live cell PLIM. (C) Matching intensity profiles for pimonidazole and HIF-2α stains. (D) Live cell intensity (gray scale) and PLIM (false-color) images of Pt-Glc (top) and the corresponding immunofluorescent images of nuclei (DAPI, blue), BrdU (red) and nestin (green) (bottom). (E) Intensity and O2 profiles across the spheroid produced by the Pt-Glc probe. (F) Intensity profiles for DAPI, BrdU and nestin staining. Analysed neurospheres are highlighted by yellow circles. Scale bar is in μm. | ||
In contrast, the two antibody-based “hypoxia stains” produced strikingly different patterns, showing higher probe staining of the surface (thus suggesting its deeper oxygenation) and a low degree of staining of the core. However, careful analysis revealed that staining with pimonidazole and HIF-2a was simply restricted to ∼50 μm depth (Fig. 6A and D) and the resulting pattern of tissue oxygenation was greatly “skewed” by diffusion limitations for antibodies during immunostaining (and possibly by pimonidazole diffusion and reduction rates). Potentially, the performance of immunostaining can be improved by fixing neurospheres, embedding and (cryo)sectioning (10–50 μm thick), staining with antibodies and analysis; however, such experimental design makes it difficult to trace and compare individual spheroids. In addition, the immunofluorescent stains assess hypoxia qualitatively and their threshold response to O2 is hard to quantify.
Multiplexing potential of the Pt-Glc probe was further demonstrated by fluorescence microscopy imaging of proliferating and neural stem cells within the neurosphere. It revealed that these cell types co-localise close to the surface layers of the neurosphere (BrdU and nestin staining shown in Fig. 6D–F) and are possibly confined to regions with specific O2.
In summary, we found that the Pt-Glc probe works much better for imaging O2 in live spheroids than antibody-based assays, providing information across their entire volume which is generally independent of intensity (Fig. 6E). At the same time, indirect and antibody-based end-point hypoxia stains such as pimonidazole and HIF-1/2α are limited by diffusion, provide unreliable information on hypoxia within neurospheres and make multiplexing difficult.
3.5. O2 imaging in live brain tissue slices
Tissue slices are an important 3D ex vivo model that conserves the cytoarchitecture of the tissue and closely resemble native tissue.1 Compared to the neurospheres, brain tissue slices are thicker and can have a lifespan in culture of 1–2 weeks or longer.59 They have been used in studies of brain cell function under different (patho)physiological conditions such as cancer, excitotoxicity, drug treatments,60–63 but rarely with reliable in situ control of O2. To cope with the limited diffusion of O2, tissue sections are often maintained in carbogen atmosphere (95% O2, 5% CO2),64 which may also cause oxidative damage of peripheral cell layers.We assessed cortical tissue slices from embryonic (E16, E18) and postnatal (P7, P21) rat brain (300–400 μm thick, 9–45 mm2) for staining efficiency with Pt-Glc under ambient culturing conditions (20% O2, 5% CO2) (Fig. 7). To reduce the harmful effects of high O2 and prevent formation of anoxic and necrotic cell layers deep inside tissue, tissue slices were incubated for 3–24 h on porous membranes in contact with the atmosphere and stained with the Pt-Glc probe. When analysed visually and with cholera toxin stain (CTX, live neuronal cell tracer, Fig. 7A), TMRM (marker of active mitochondria, not shown) and propidium iodide (marker of toxicity),65 such tissue slices showed staining patterns very similar to those of unstained control slices (not shown), and no detrimental effects of the Pt-Glc probe on tissue viability were seen. Similar to PC12 cells and neurospheres, staining of tissue slices with Pt-Glc was very efficient but without significant co-localisation with cholera toxin (Fig. 7A). Staining of brain tissue having different age and structure (e.g. E16 and P21) was also very comparable. In contrast, the MM2 probe only stained the surface of brain sections (Fig. 7A).
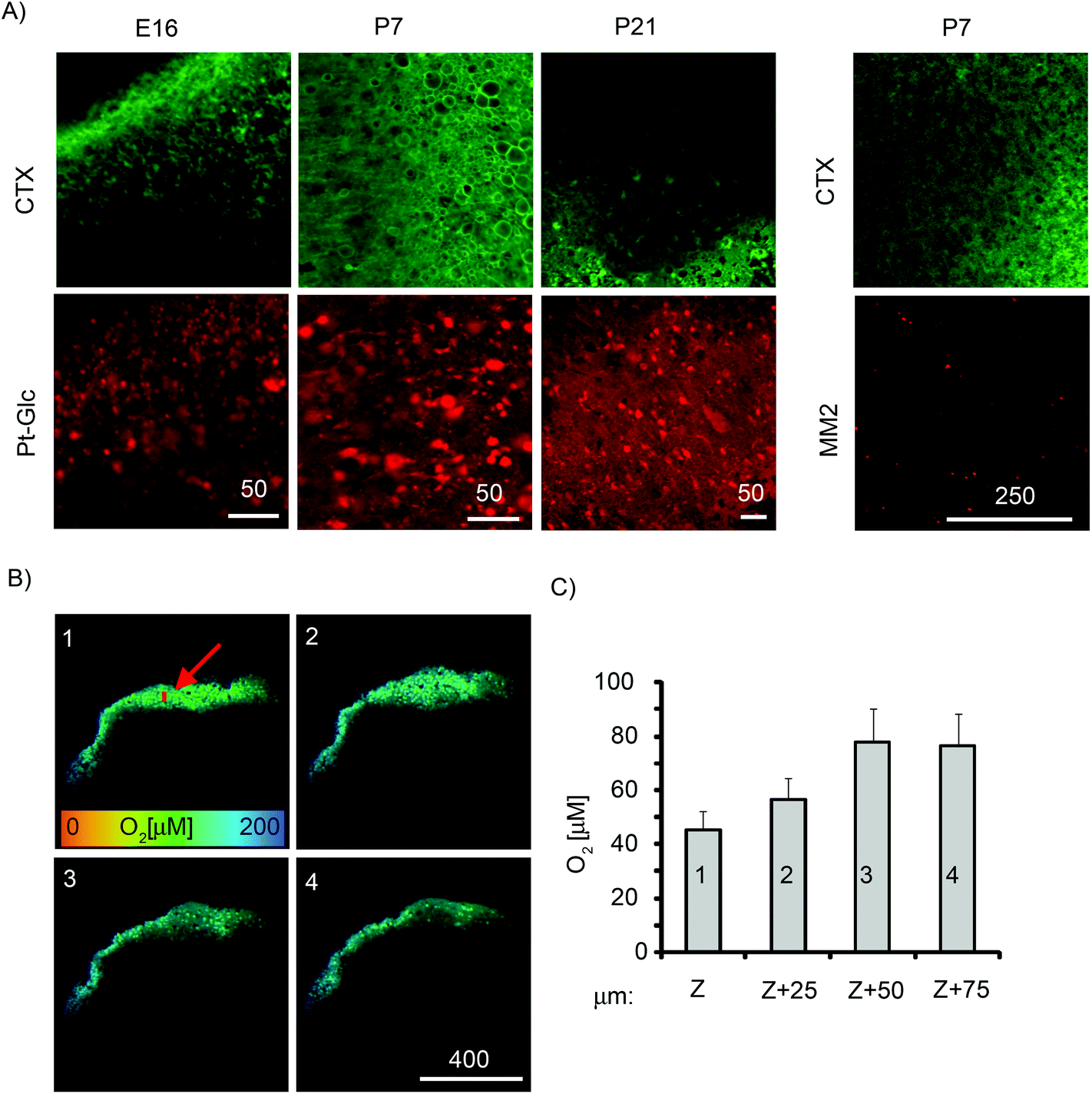 | ||
| Fig. 7 Staining of live brain slices. Slices (400 μm thick) of E16, P7 and P21 rat brain stained with Pt-Glc (10 μM) or MM2 (25 μg ml−1) probes and cholera toxin (2 ng ml−1, CTX) for 3 h and analysed by PLIM. (A) Localisation of Pt-Glc (left) and MM2 (right) in tissue sections of different ages. (B) Oxygenation maps produced from PLIM images (left) for the four optical sections of a P7 brain slice stained with Pt-Glc (25 μm step along the Z-axis; 1 = 75 μm deep and 4 = tissue surface), and (C) average O2 levels at different depths for the indicated region (red arrow) (right). Scale bar unit is μm. | ||
We then imaged the O2 distribution in P7 tissue slices at different depths using the PLIM technique and confocal sectioning (Fig. 7B). The sections at the surface of the tissue (optical section 4) and at 25 μm depth (optical section 3) showed O2 levels of ∼90 μM, while the sections located 50 μm (section 2) and 75 μm (section 1) below the surface showed decreased O2 levels. In the early postnatal brain, neurons possess active respiration and mitochondrial function,66 and our results confirm that brain slices contain a large number of live cells which can deoxygenate deep regions of tissue slices. Even without carbogen atmosphere (95% O2), our tissue was exposed to O2 levels much higher than physiological levels found in live rat cortex (5–10 μM67). Our imaging data suggest that control of O2 in experimental tissue slices and their environment (atmosphere, medium) should be used to maintain adequate oxygenation conditions close to in vivo levels. Such O2 monitoring systems may also require circulation of medium or microfluidics to mimic O2 supply by blood and vasculature.15
4. Discussion
In this study, we synthesized several new derivatives of the highly photostable phosphorescent dye PtPFPP with short peptides and monosaccharides (hexoses). Compared to the previously reported small molecule O2 probes, these conjugates were seen to combine improved photostability,25 efficient cell penetrating properties, high brightness and ease of use, particularly with 3D tissue models. The simple click modification with thiols has allowed mono- and tetra-substitution of PtPFPP and preparation of conjugates with peptides and hexoses with high yields and purity. One-step synthesis was achieved using thio-glucose or thio-galactose (Fig. 1), whereas amino-coupling of Arg-containing peptides and purification of the resulting conjugates were more laborious, less efficient and produced probe structures with sub-optimal spectral and O2 sensing characteristics. We therefore focused on Pt-Glc and Pt-Gal conjugates and evaluated them with several different cell and tissue models.Pt-Glc and Pt-Gal showed efficient staining of mammalian cells in 2D cultures. Eight different cell lines were tested (see Fig. 2, S1†) and all showed efficient intracellular staining with relatively fast kinetics (4–6 h for MEF cells). Compared to the nanoparticle based probes31 and PtCP conjugates38 that remain within cells for more than 72 h, a weaker cell retention was observed for the Pt-Glc and Pt-Gal probes with a half-leakage time of <24 h. On the other hand, the new probes provided much better in-depth penetration and staining of multi-cellular structures and 3D tissue models, including aggregates of PC12 cells (tumour spheroids), neurospheres from primary cells and slices of live brain tissue (Fig. 4–7, S4†). This may be due to the gradual leak of the probes and transfer between the neighbouring cells in the tissue. At the same time, Pt-Glc remained within samples for prolonged periods of time (>48 h), proven to be sufficient for imaging experiments.
The improved penetration of Pt-Glc and Pt-Gal conjugates across cell membranes may also be due to their different internalisation mechanisms, likely mediated by glucose uptake transporters, GluTs.46 We also found (Fig. S5†) that both conjugates demonstrated >10 times higher intracellular accumulation when the cells were deprived of glucose and O2, i.e. upon activation of glucose uptake.68 On the other hand, we observed no marked differences in the uptake of Pt-Glc and Pt-Gal by different cell types (Fig. S1†) and upon inhibition of glucose transport by 2-deoxyglucose (not shown). This suggests that minor structural differences in hexose conjugates are not critical for their uptake. On the other hand, GluT-independent uptake and endocytic mechanisms of cell entry cannot be ruled out. The different intracellular distribution of the conjugates in different cells types (from lysosomal-ER-like in MEF cells to whole cell staining in Caco-2 cells, Fig. S1†) also supports this.
We observed no significant cyto- or phototoxicity of Pt-Glc and Pt-Gal at loading concentrations ≤10 μM. Cellular ATP levels, glycolytic and total acidification rates remained unchanged (Fig. 2, S3†), suggesting that cellular function was largely unaffected by probe staining. These features contrast with the high phototoxicity of the other porphyrin–carbohydrate conjugates developed for PDT (e.g. TPFPP48,54,69), which can be explained by the differences in the doses and cell line (MDA-MB-231 breast cancer cells) used, intracellular distribution and efficiency of singlet oxygen photo-generation for these drug compounds.
Relative brightness and photostability of Pt-Glc and Pt-Gal were somewhat weaker than for the nanoparticle PtPFPP-based probe, NanO2,31 but better than the small molecule IrOEP or PtCP conjugates38,40 (Fig. 3). In 2D cultures (MEF cells), the new probes were 10–20 times less bright than NanO2, but in 3D tissue models their signals became comparable due to their better in-depth penetration. Additionally, the staining of 3D structures showed a more uniform cytosolic distribution across the whole spheroid, as opposed to the patchy patterns produced by NanO2 (Fig. 5) or hypoxia stains providing staining mainly of surface (peripheral) layers (Fig. 6). We confirmed that the penetration depth of antibodies is highly limited by the diffusion (approx. 50–100 μm). This method is difficult to use with live tissue and in 3D, unlike the PLIM method which operates in real time and with live spheroids.
Due to the lack of phosphor shielding in small molecule probes (unlike the nanoparticle structures), probe lifetime can be influenced by its microenvironment (e.g. by serum proteins or inside the cell). Nonetheless, Pt-Glc and Pt-Gal conjugates displayed satisfactory performance in PLIM measurements (Fig. 2–5, S3†) and stable characteristics in different cell types (e.g. calibrations in PC12 cells, spheroids).
Using the Pt-Glc probe, we analysed the dynamic changes in O2 distribution in PC12 cell aggregates, with sub-cellular spatial resolution (Fig. 4). Stimulated cells demonstrated the responses consistent with the measurements using the PEPP0 probe on a microplate reader39 or the NanO2 probe on a wide-field PLIM microscope.31 For the neurosphere model the performance of the Pt-Glc conjugate was comparable to if not better than NanO2. Again, Pt-Glc showed more uniform distribution across the spheroid and allowed quantitative and accurate mapping of hypoxic regions within its structure (Fig. 5). The probe was also successfully used to stain live brain slices and monitor their oxygenation during culturing (Fig. 6).
Overall, phosphorescent Pt-Glc and Pt-Gal conjugates demonstrate good potential for high-resolution mapping of O2 concentration in 3D tissue models and related biomaterials using the PLIM technique or emerging approaches such as light-sheet microscopy. They allow easy synthesis by click modification and coupling with other targeting vectors to optionally tune their delivery. Their advantages are small size, moderate brightness, good photostability, characteristic transport mechanisms in tissue and convenient spectral characteristics (excitation with 390, 405 or 534 nm lasers/LEDs and emission at 650 nm). They are well-suited for PLIM applications and look favourably over the nanoparticle-based O2 probes, particularly for 3D tissue models where efficient staining throughout the tissue is required. Structural analogues of these probes can also be produced, particularly with increased sensitivity to O2 (e.g. derivatives of PdPFPP) or red-shifted spectra.70
Abbreviations
| 3D | Three-dimensional |
| Arg | Arginine |
| BrdU | 5-Bromo-2′-deoxyuridine |
| CTX | Cholera toxin, subunit B |
| DIV | Days in vitro |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| HBSS | Hank's balanced salt solution |
| ER | Endoplasmic reticulum |
| HIF | Hypoxia inducible factor |
| HXT | Hoechst 33342 |
| iO2 | Intracellular O2 |
| PBS | Phosphate buffered saline |
| PDL | Poly-D-lysine |
| PDT | Photodynamic therapy |
| PLIM | Phosphorescence lifetime imaging microscopy |
| PtPFPP | Pt(II)-tetrakis(pentafluorophenyl)porphine |
| ROI | Region of interest |
| RP-HPLC | Reverse phase high-performance liquid chromatography |
| ROS | Reactive oxygen species |
| RT | Room temperature |
| TBST | Tris-buffered saline, Tween 20 |
| TCSPC | Time-correlated single photon counting |
| TFA | Trifluoroacetic acid. |
Acknowledgements
This work was supported by Science Foundation Ireland, grant 12/TIDA/B2413, and by the European Commission FP7 Program, grant FP7-HEALTH-2012-INNOVATION-304842-2. We thank T. Foley (Department of Anatomy and Neuroscience, UCC) and Dr U. Anilkumar (Royal College of Surgeons in Ireland) for help with primary neuronal cultures, and D. Aigner and P. W. Zach (Graz University of Technology, Austria) for help with probe synthesis and characterization.References
- F. Pampaloni, E. G. Reynaud and E. H. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS PubMed.
- A. Astashkina, B. Mann and D. W. Grainger, Pharmacol. Ther., 2012, 134, 82–106 CrossRef CAS PubMed.
- N. Kramer, A. Walzl, C. Unger, M. Rosner, G. Krupitza, M. Hengstschläger and H. Dolznig, Mutat. Res., Rev. Mutat. Res., 2013, 752, 10–24 CrossRef CAS PubMed.
- D. Huh, Y.-s. Torisawa, G. A. Hamilton, H. J. Kim and D. E. Ingber, Lab Chip, 2012, 12, 2156–2164 RSC.
- G. Mehta, A. Y. Hsiao, M. Ingram, G. D. Luker and S. Takayama, J. Controlled Release, 2012, 164, 192–204 CrossRef CAS PubMed.
- H. Page, P. Flood and E. G. Reynaud, Cell Tissue Res., 2013, 1–9 Search PubMed.
- A. Williamson, S. Singh, U. Fernekorn and A. Schober, Lab Chip, 2013, 13, 3471–3480 RSC.
- B. A. Wilt, L. D. Burns, E. T. W. Ho, K. K. Ghosh, E. A. Mukamel and M. J. Schnitzer, Annu. Rev. Neurosci., 2009, 32, 435 CrossRef CAS PubMed.
- F. Pampaloni, N. Ansari and E. H. Stelzer, Cell Tissue Res., 2013, 1–17 Search PubMed.
- W. Supatto, T. V. Truong, D. Débarre and E. Beaurepaire, Curr. Opin. Genet. Dev., 2011, 21, 538–548 CrossRef CAS PubMed.
- K. Si, R. Fiolka and M. Cui, Sci. Rep., 2012, 2 Search PubMed.
- K. K. Ghosh, L. D. Burns, E. D. Cocker, A. Nimmerjahn, Y. Ziv, A. E. Gamal and M. J. Schnitzer, Nat. Methods, 2011, 8, 871–878 CrossRef CAS PubMed.
- T. Ha and P. Tinnefeld, Annu. Rev. Phys. Chem., 2012, 63, 595–617 CrossRef CAS PubMed.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2009, 110, 2620–2640 CrossRef PubMed.
- D. B. Papkovsky and R. I. Dmitriev, Chem. Soc. Rev., 2013, 42, 8700–8732 RSC.
- A. Melvin and S. Rocha, Cell. Signal., 2012, 24, 35–43 CrossRef CAS PubMed.
- J. D. Webb, M. L. Coleman and C. W. Pugh, Cell. Mol. Life Sci., 2009, 66, 3539–3554 CrossRef CAS PubMed.
- O. Toussaint, G. Weemaels, F. Debacq-Chainiaux, K. Scharffetter-Kochanek and M. Wlaschek, J. Cell. Physiol., 2011, 226, 315–321 CrossRef CAS PubMed.
- G. L. Semenza, Physiology, 2009, 24, 97–106 CrossRef CAS PubMed.
- A. Mohyeldin, T. Garzón-Muvdi and A. Quiñones-Hinojosa, Cell Stem Cell, 2010, 7, 150–161 CrossRef CAS PubMed.
- L. C. Clark, R. Wolf, D. Granger and Z. Taylor, J. Appl. Physiol., 1953, 6, 189–193 CAS.
- S. Kazmi, A. J. Salvaggio, A. D. Estrada, M. A. Hemati, N. K. Shaydyuk, E. Roussakis, T. A. Jones, S. A. Vinogradov and A. K. Dunn, Biomed. Opt. Express, 2013, 4, 1061–1073 CrossRef PubMed.
- S. Walenta, J. Doetsch, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Histochem. Cytochem., 2000, 48, 509–522 CrossRef CAS PubMed.
- J. M. Vanderkooi, G. Maniara, T. J. Green and D. F. Wilson, J. Biol. Chem., 1987, 262, 5476–5482 CAS.
- R. I. Dmitriev and D. B. Papkovsky, Cell. Mol. Life Sci., 2012, 69, 2025–2039 CrossRef CAS PubMed.
- X.-d. Wang, J. A. Stolwijk, T. Lang, M. Sperber, R. J. Meier, J. Wegener and O. S. Wolfbeis, J. Am. Chem. Soc., 2012, 134, 17011–17014 CrossRef CAS PubMed.
- J. Lecoq, A. Parpaleix, E. Roussakis, M. Ducros, Y. G. Houssen, S. A. Vinogradov and S. Charpak, Nat. Med., 2011, 17, 893–898 CrossRef CAS PubMed.
- D. F. Wilson, Am. J. Physiol. Heart Circ. Physiol., 2008, 294, H11–H13 CrossRef CAS PubMed.
- A. Devor, S. Sakadzic, V. J. Srinivasan, M. A. Yaseen, K. Nizar, P. A. Saisan, P. Tian, A. M. Dale, S. A. Vinogradov, M. A. Franceschini and D. A. Boas, J. Cereb. Blood Flow Metab., 2012, 32, 1259–1276 CrossRef CAS PubMed.
- R. I. Dmitriev, A. V. Zhdanov, G. V. Ponomarev, D. V. Yashunski and D. B. Papkovsky, Anal. Biochem., 2010, 398, 24–33 CrossRef CAS PubMed.
- A. Fercher, S. M. Borisov, A. V. Zhdanov, I. Klimant and D. B. Papkovsky, ACS Nano, 2011, 5, 5499–5508 CrossRef CAS PubMed.
- R. I. Dmitriev, A. V. Zhdanov, G. Jasionek and D. B. Papkovsky, Anal. Chem., 2012, 84, 2930–2938 CrossRef CAS PubMed.
- A. V. Zhdanov, V. I. Ogurtsov, C. T. Taylor and D. B. Papkovsky, Integr. Biol., 2010, 2, 443–451 RSC.
- R. I. Dmitriev, A. V. Zhdanov, Y. M. Nolan and D. B. Papkovsky, Biomaterials, 2013, 34, 9307–9317 CrossRef CAS PubMed.
- D. Lambrechts, M. Roeffaers, G. Kerckhofs, S. J. Roberts, J. Hofkens, T. Van de Putte, H. Van Oosterwyck and J. Schrooten, Biomaterials, 2013, 34, 922–929 CrossRef CAS PubMed.
- L. Karamchand, G. Kim, S. Wang, H. J. Hah, A. Ray, R. Jiddou, Y.-E. Koo Lee, M. A. Philbert and R. Kopelman, Nanoscale, 2013, 5, 10327–10344 RSC.
- H. Lütcke, D. J. Margolis and F. Helmchen, Trends Neurosci., 2013, 36, 375–384 CrossRef PubMed.
- R. I. Dmitriev, H. M. Ropiak, G. V. Ponomarev, D. V. Yashunsky and D. B. Papkovsky, Bioconjugate Chem., 2011, 22, 2507–2518 CrossRef CAS PubMed.
- R. I. Dmitriev, H. M. Ropiak, D. V. Yashunsky, G. V. Ponomarev, A. V. Zhdanov and D. B. Papkovsky, FEBS J., 2010, 277, 4651–4661 CrossRef CAS PubMed.
- K. Koren, R. I. Dmitriev, S. M. Borisov, D. B. Papkovsky and I. Klimant, ChemBioChem, 2012, 13, 1184–1190 CrossRef CAS PubMed.
- J. Napp, T. Behnke, L. Fischer, C. Würth, M. Wottawa, D. M. Katschinski, F. Alves, U. Resch-Genger and M. Schäferling, Anal. Chem., 2011, 83, 9039–9046 CrossRef CAS PubMed.
- Y.-E. Koo Lee, E. E. Ulbrich, G. Kim, H. Hah, C. Strollo, W. Fan, R. Gurjar, S. Koo and R. Kopelman, Anal. Chem., 2010, 82, 8446–8455 CrossRef CAS PubMed.
- A. V. Kondrashina, R. I. Dmitriev, S. M. Borisov, I. Klimant, I. O'Brien, Y. M. Nolan, A. V. Zhdanov and D. B. Papkovsky, Adv. Funct. Mater., 2012, 22, 4931–4939 CrossRef CAS.
- I. Dunphy, S. A. Vinogradov and D. F. Wilson, Anal. Biochem., 2002, 310, 191–198 CrossRef CAS.
- N. W. Choi, S. S. Verbridge, R. M. Williams, J. Chen, J.-Y. Kim, R. Schmehl, C. E. Farnum, W. R. Zipfel, C. Fischbach and A. D. Stroock, Biomaterials, 2012, 33, 2710–2722 CrossRef CAS PubMed.
- X. Chen, L. Hui, D. A. Foster and C. M. Drain, Biochemistry, 2004, 43, 10918–10929 CrossRef CAS PubMed.
- D. Samaroo, C. E. Soll, L. J. Todaro and C. M. Drain, Org. Lett., 2006, 8, 4985–4988 CrossRef CAS PubMed.
- D. Samaroo, M. Vinodu, X. Chen and C. M. Drain, J. Comb. Chem., 2007, 9, 998–1011 CrossRef CAS PubMed.
- K. KumaráPasunooti, K. MeiáChing and H. SupáYoon, MedChemComm, 2011, 2, 371–377 RSC.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- K. Koren, S. M. Borisov and I. Klimant, Sens. Actuators, B, 2012, 169, 173–181 CrossRef CAS PubMed.
- J. Hynes, T. C. O'Riordan, A. V. Zhdanov, G. Uray, Y. Will and D. B. Papkovsky, Anal. Biochem., 2009, 390, 21–28 CrossRef CAS PubMed.
- A. V. Zhdanov, R. I. Dmitriev, A. V. Golubeva, S. A. Gavrilova and D. B. Papkovsky, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 3553–3569 CrossRef CAS PubMed.
- S. Singh, A. Aggarwal, S. Thompson, J. o. P. Tomé, X. Zhu, D. Samaroo, M. Vinodu, R. Gao and C. M. Drain, Bioconjugate Chem., 2010, 21, 2136–2146 CrossRef CAS PubMed.
- M. Quaranta, S. M. Borisov and I. Klimant, Bioanal. Rev., 2012, 4, 115–157 CrossRef PubMed.
- W. R. Algar, D. E. Prasuhn, M. H. Stewart, T. L. Jennings, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2011, 22, 825–858 CrossRef CAS PubMed.
- A. Zhdanov, R. Dmitriev and D. Papkovsky, Cell. Mol. Life Sci., 2011, 68, 903–917 CrossRef CAS PubMed.
- M. A. Varia, D. P. Calkins-Adams, L. H. Rinker, A. S. Kennedy, D. B. Novotny, W. C. Fowler and J. A. Raleigh, Gynecol. Oncol., 1998, 71, 270–277 CrossRef CAS PubMed.
- B. H. Gähwiler, M. Capogna, D. Debanne, R. A. McKinney and S. M. Thompson, Trends Neurosci., 1997, 20, 471–477 CrossRef.
- J. Zimmer, B. W. Kristensen, B. Jakobsen and J. Noraberg, Amino Acids, 2000, 19, 7–21 CrossRef CAS.
- T. Ohnishi, H. Matsumura, S. Izumoto, S. Hiraga and T. Hayakawa, Cancer Res., 1998, 58, 2935–2940 CAS.
- H. Kim, E. Kim, M. Park, E. Lee and K. Namkoong, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2012, 41, 36–43 CrossRef PubMed.
- K. Albus, U. Heinemann and R. Kovács, J. Neurosci. Methods, 2013, 217, 1–8 CrossRef PubMed.
- J. Gredell, P. Turnquist, M. Maciver and R. Pearce, Br. J. Anaesth., 2004, 93, 810–817 CrossRef CAS PubMed.
- J. Noraberg, B. W. Kristensen and J. Zimmer, Brain Res. Protoc., 1999, 3, 278–290 CrossRef CAS.
- M. Tsatmali, E. C. Walcott and K. L. Crossin, Brain Res., 2005, 1040, 137–150 CrossRef CAS PubMed.
- M. Erecińska and I. A. Silver, Respir. Physiol., 2001, 128, 263–276 CrossRef.
- M. Bershteyn and A. R. Kriegstein, Cell, 2013, 155, 19–20 CrossRef CAS PubMed.
- S. Thompson, X. Chen, L. Hui, A. Toschi, D. A. Foster and C. M. Drain, Photochem. Photobiol. Sci., 2008, 7, 1415–1421 CAS.
- G. Khalil, M. Gouterman, S. Ching, C. Costin, L. Coyle, S. Gouin, E. Green, M. Sadilek, R. Wan and J. Yearyean, J. Porphyrins Phthalocyanines, 2002, 6, 135–145 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60272a |
| This journal is © The Royal Society of Chemistry 2014 |