Comparative studies of thermogels in preventing post-operative adhesions and corresponding mechanisms
Lin
Yu†
a,
Hongtao
Hu†
b,
Lin
Chen
a,
Xiaogang
Bao
b,
Yuzhuo
Li
a,
Liang
Chen
a,
Guohua
Xu
*b,
Xiaojian
Ye
b and
Jiandong
Ding
*a
aState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200433, China. E-mail: jdding1@fudan.edu.cn; Fax: +86-21-65640293; Tel: +86-21-65643506
bDepartment of Orthopedic Surgery, Changzheng Hospital, Second Military Medical University of the Chinese People's Liberation Army, Shanghai 200003, China. E-mail: xuguohuamail@163.com
First published on 28th March 2014
Abstract
Post-surgical peritoneal adhesions constitute a classic problem in surgery, and thus anti-adhesion materials are much required. In this study, a series of polyester–PEG–polyester triblock copolymers with different biodegradable polyester compositions were synthesized, their properties were examined, and the in vivo efficacies as anti-adhesion biomaterials were evaluated in a comparative way for the first time. These samples not only exhibited various morphologies in the bulk state, but also possessed different stabilities in the sol state. All the polymer aqueous solutions with appropriate compositions and concentrations underwent sol–gel transitions with increase of temperature and formed semi-solid hydrogels at body temperature. The efficacy of PEG/polyester thermogels (25 wt%) for preventing post-operative abdominal adhesions was investigated and compared in a rabbit model of sidewall defect-bowel abrasion. Different efficacies of anti-adhesion were observed, possible mechanisms were discussed, and the importance of viscoelasticity was suggested for the first time. These results illustrated that appropriate properties of PEG/polyester thermogels including viscoelastic matrix, hydrophilic surface and moderate in vivo persistence played crucial roles in enabling an effective device to prevent post-surgical peritoneal adhesions.
Introduction
One of the common and serious complications in surgery is the formation of post-operative peritoneal adhesions, which can cause a series of secondary effects such as severe pain, infertility, intestinal obstruction and even death.1–3 Incidence of adhesions is as high as 80% after general surgical procedures.3 In particular, almost all patients suffer from intestinal adhesions after abdominal and pelvic surgery.4 Post-operative adhesions affect millions of individuals worldwide, and induce huge expense in re-operative interventions.Over recent decades, numerous pharmacological and barrier-based approaches have been tried to prevent post-operative adhesions.5–8 Barrier systems in the form of polymer solutions or solid membranes are currently one of the most effective approaches for reducing adhesion formation.9–12 They separate the injured regions during the critical period of adhesion development 3–5 days after surgery. This separation is achieved via the hydroflotation effect of polymer solutions or direct physical isolation with the use of solid membranes. For polymer solutions such as chitosan, hyaluronic acid (HA) and carboxymethylcellulose (CMC),9,10 the residual time at the injured tissues is short; for solid membranes including HA–CMC (Seprafilm, Genzyme, Cambridge, MA) and polylactide,13–15 it is difficult to completely cover the affected tissues.
Recently, in situ-forming hydrogels have been developed as injectable biomaterials for drug delivery,16–22 tissue repair23–25 and submucosal injection substances in endoscopic submucosal dissection,26,27 and other biomedical applications.28,29 Such a class of biomaterials are low-viscous sols before administration and turn into semi-solid gels under physiological conditions once injected into the target site. Meanwhile, these materials offer the ability to form any desired implant shape, and the use of organic solvents in the operation is completely avoided. Some in situ chemically cross-linked hydrogels based on HA,30,31 dextran,32 gelatin11 or macromonomers of poly(ethylene glycol) (PEG)33 have been applied for preventing the formation of post-operative peritoneal adhesion. Nevertheless, in situ gelation triggered by chemical modification or cumbersome ultraviolet (UV) illumination may bring with it some biocompatibility problems, and the relative long gelation time also restricts the practical clinical applications. In situ physically crosslinked hydrogels have become an alternative choice.4,8,34–36 For example, Guardix-SG (Biorane, Seoul, Korea) is a temperature-sensitive physical gel consisting of Pluronics/alginate/CaCl2 and has successfully prevented pericardial adhesion in a rabbit model.36
As unique injectable physical hydrogels, thermogels have gained increasing attention recently. Some amphiphilic block copolymers are not only dissolved in water at low or room temperature, but also undergo, under appropriate composition and concentration, a reversible sol–gel transition upon heating, as illustrated in Fig. 1a, and thus are called thermogels.37–42 Thermogelling copolymers composed of PEG and aliphatic polyesters are particularly interesting and important because both blocks have been approved by the US Food and Drug Administration (FDA). To date, a variety of biodegradable polyester chains such as poly(D,L-lactic acid-co-glycolic acid) (PLGA),43–46 poly(ε-caprolactone) (PCL),47,48 poly(ε-caprolactone-co-D,L-lactic acid) (PCLA),49,50 and poly(ε-caprolactone-co-D,L-lactic acid) (PCGA),51 have been introduced as the hydrophobic blocks into PEG/polyester copolymers to form thermogels. The basic mechanism of the thermogelation of PEG/polyester copolymers is the formation of a percolated micelle network.40,52,53
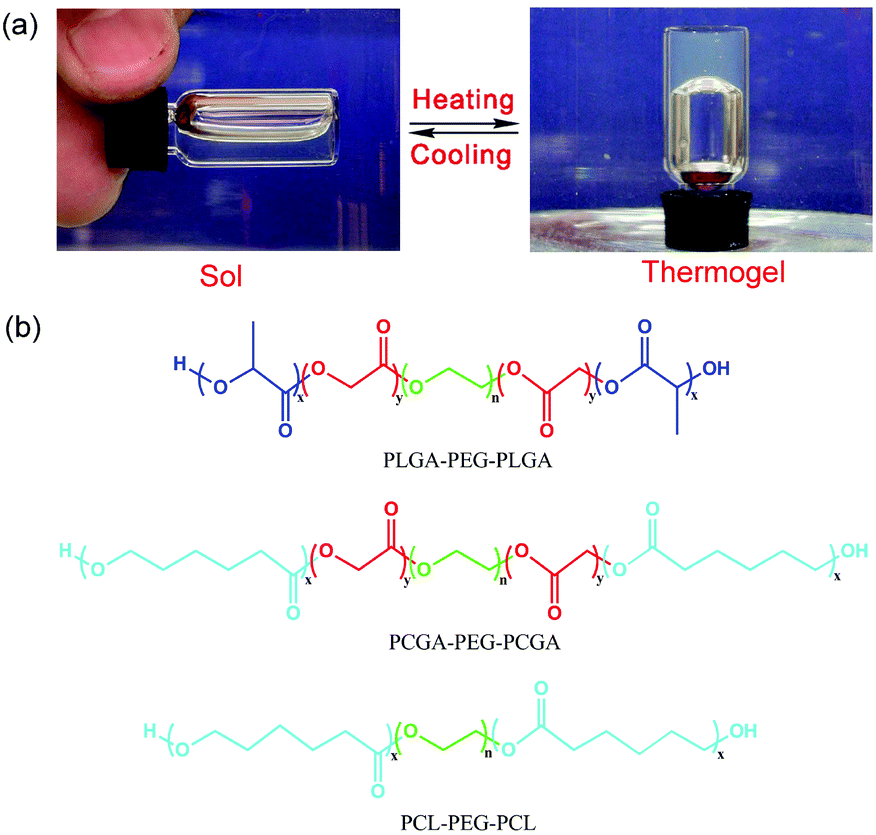 | ||
| Fig. 1 (a) Presentation of the temperature-induced reversible sol-thermogel transition. (b) The molecular structures of three thermogelling block copolymers composed of polyester and polyether: PLGA–PEG–PLGA, PCGA–PEG–PCGA, and PCL–PEG–PCL. | ||
In 2011, our group suggested and confirmed that the biodegradable and thermoreversible PCLA–PEG–PCLA hydrogel could serve as a physical barrier for prevention of post-operative adhesions,54 and thus the important and facile medical applications of such a kind of interesting soft matter were explored. As a result, a series of fundamental questions are triggered. For instance, is the thermogel with the most popular composition, namely, PLGA–PEG–PLGA, feasible as an anti-adhesion biomaterial? Do viscoelastic properties and biodegradable periods of thermogels with different polyester compositions influence the in vivo efficacy of prevention of post-operative adhesions? Answering of these questions should be based upon comprehensive work, and the corresponding efforts are meaningful for guiding the potential medical applications of the thermogels.
Herein, three thermogelling ABA-type PEG/polyester triblock copolymers with commonly used polyester compositions were synthesized. Fig. 1b shows the molecular structures of the three PEG/polyester triblock copolymers synthesized in this study. We selected the three compositions based upon the following considerations. PLGA has been clinically applied and widely investigated as biomaterials. PLGA–PEG–PLGA thermogels have also been extensively studied by several groups,38,44,55 yet it has never been reported as a barrier device for post-operative anti-adhesion. Hence, it seems necessary to examine the feasibility of PLGA–PEG–PLGA thermogels. PCL–PEG–PCL is a typical thermogel with a crystal structure.47 Yet again, its feasibility as an anti-adhesion biomaterial has not been examined. Recently, based on molecular parameter design, our group achieved block copolymers of PCGA–PEG–PCGA in a powder form in a dry state and observed a temperature-induced sol–gel transition in water without unexpected gelling prior to heating.51 Therefore, we would like to examine this system together with PLGA–PEG–PLGA and PCL–PEG–PCL. These samples have been proved to be biocompatible and biodegradable.55–58 Their different viscoelasticities and degradation rates have also been reported.47,51,57,59 The novelty of the present study is to evaluate the efficacy of prevention of post-surgical tissue adhesions with the use of these thermogels for the first time and shed some insight into designing a thermogel-based barrier system for post-surgical anti-adhesions. Meanwhile, the thermogelling properties of these copolymers have also been investigated together in a comparative way for the first time.
Materials and methods
Materials
DL-Lactide (LA, Purac), glycolide (GA, Purac), ε-caprolactone (CL, Aldrich), stannous octoate (Aldrich), and PEG with molecular weight (MW) 1500 (Aldrich) were used as received without further purification. Medical chitosan (CHS) was provided by Shanghai Qisheng Biological Fabrication Co., Ltd, China.Animals
Male New Zealand rabbits with body weight 2.0 ± 0.2 kg were supplied by the Experimental Animal Center of the Second Military Medical University of the Chinese People's Liberation Army (SMMU, Shanghai, China). The animals were raised at a temperature of 25 ± 2 °C and a relative humidity of 70 ± 5% with cycles of 12 h light and 12 h dark at least for 1 week before experiments. All the animal experiments were conducted with approval from the ethics committee of Changzheng Hospital for animal investigation.Synthesis of polyester–PEG–polyester triblock copolymers
The ABA-type PEG/polyester triblock copolymers were synthesized via bulk ring-opening polymerization. The detailed procedure has been described elsewhere.46,60 To synthesize the PLGA–PEG–PLGA, PEG (20 g) was added into a three-necked flask and heated under vacuum at 150 °C for 3 h. Next, GA and LA monomers were added after the flask was cooled to room temperature. The replacement of argon in the flask was carried out three times to eliminate the residual moisture in the monomers. Then, the reaction system was heated under an argon atmosphere at 150 °C for 12 h after the addition of stannous octoate (0.2 wt% of monomers). Crude polymers were washed using 80 °C water at least three times. The residual water was removed via lyophilization and the final products were stored at −20 °C until use. The other two specimens PCGA–PEG–PCGA and PCL–PEG–PCL were prepared by a similar procedure but with different monomers.1H NMR characterization
1H NMR spectra were recorded at ambient temperature with a Bruker spectrometer (DMX500) operating at 500 MHz to study the chemical structure and composition of the polymers. CDCl3 was used as the solvent, and chemical shifts (δ) were given in ppm using tetramethylsilane as an internal standard.Gel permeation chromatography
Gel permeation chromatography (CPC) measurements were performed on an Agilent apparatus (Agilent1100) equipped with a refractive index detector. Tetrahydrofuran was used as the mobile phase at a flow rate of 1.0 mL min−1 at 35 °C. A 1.0% (w/v) polymer solution (20 μL) was injected for each measurement. MWs were calibrated by monodispersed polystyrene standards.Differential scanning calorimetry
A differential scanning calorimeter (DSC Q2000, TA) was used to measure the melting and crystallization temperatures of the polymers in the temperature range from −20 to 80 °C. 5.0 mg of polymer was loaded in a cell for each analysis and DSC thermograms were recorded with a heating and cooling rate of 5 °C min−1.X-ray diffraction analysis
X-ray diffraction (XRD) was performed using a PANalytical X′Pert PRO diffractometer equipped with a Cu Kα radiation source. The diffraction patterns of a scan range between 5° to 50° were recorded with a scanning rate of 5° min−1 at room temperature. The voltage was set at 40 kV and the current was fixed at 40 mA.Rheological measurements
A stress-controlled rheometer (Malvern, Kinexus) equipped with a 60 mm steel cone (1 degree) using a gap size of 0.03 mm was used to study the sol–gel transition of polymer aqueous solutions as the temperature increased. Solutions of 25 wt% copolymer in normal saline solution (NS) were transferred onto the rheometer. In stress-controlled dynamic rheological measurements, a sinusoidal shear stress with a given frequency ω was exerted on the sample, and the corresponding time-dependent shear strain was detected. We controlled small oscillatory amplitudes to guarantee the range of the linear viscoelasticity, and thus the shear strain was also changed sinusoidally but probably with a phase different from that of the shear stress if the material is not fully elastic. The plate was heated at a rate of 0.5 °C min−1 and the oscillatory frequency ω was fixed at 10 rad s−1. The viscoelastic properties of the polymer aqueous solutions, namely, the dynamic shear storage modulus (G′) and loss or dissipative modulus (G′′) were recorded as a function of temperature. The complex modulus (G*) was determined based on the equation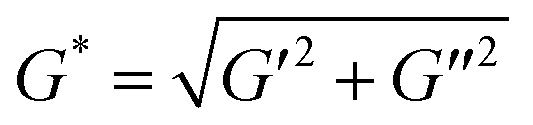 , and the phase angle δ was obtained via tan δ = G′′/G′.
, and the phase angle δ was obtained via tan δ = G′′/G′.
In vivo anti-adhesion tests
A rabbit model of sidewall defect-bowel abrasion30,31 was used to evaluate the anti-adhesion efficacy of the PEG/polyester thermogels with different polyester blocks. Briefly, after anesthetizing the animal with 3% sodium pentobarbital solution, the enterocoelia was opened. A 3 × 4 cm2 defect on the right lateral abdominal wall was created and the corresponding cecal haustra was abraded using a surgical brush. And thus a model of peritoneal intestinal adhesion was established.The rabbits were randomly divided into five groups (six per group): control (without treatment), PLGA–PEG–PLGA (4 mL of the PLGA–PEG–PLGA solution (25 wt% in NS) was pipetted onto the defects, and spontaneous gelation happened rapidly due to contact with the warmer surroundings), PCGA–PEG–PCGA (administration of 4 mL of the PCGA–PEG–PCGA solution), PCL–PEG–PCL (administration of 4 mL of the PCL–PEG–PCL solution) and CHS (administration of 4 mL of the biomedical chitosan solution). Finally, the peritoneum and abdominal wall were sutured using 3-0 silk sutures, and the skin was sutured using 4-0 silk sutures.
The rabbits were euthanized 30 days after surgery. Autopsies were performed to assess the post-operative adhesion and the tissue regeneration at the defect. If intestinal adhesion occurred, the adhesion area of the sidewall injury was measured. The tenacity of the intestinal adhesion found was also defined as follows: score 0: no adhesion; score 1: mild intestinal adhesion and separation was facile; score 2: moderate intestinal adhesion and blunt dissection was needed; score 3: severe intestinal adhesion and sharp dissection was needed. Specimens were harvested, fixed in 10% formalin, embedded in paraffin, and cut into slices of thickness 4 μm. The sections were routinely stained with haematoxylin-eosin (HE) and observed with a light microscope.
Results
Synthesis and characterization of polyester–PEG–polyester triblock copolymers
Three polyester–PEG–polyester triblock copolymers were prepared via the ring-opening polymerization of different monomers in the presence of α,ω-dihydroxyl terminated PEG using stannous octoate as catalyst. Typical 1H NMR spectra of the triblock copolymers with their chemical structures are presented in Fig. 2. All the characteristic signals are assigned on the spectra. For PLGA–PEG–PLGA, the peaks at 5.20 ppm (–COCH(CH3)O–), 4.80 ppm (–COCH2O–) and 3.65 ppm (–CH2CH2O–) were used to calculate the number-average MW (Mn).40 In the case of PCGA–PEG–PCGA and PCL–PEG–PCL, the peaks around 4.60 ppm (–COCH2O–), 3.65 ppm (–CH2CH2O–) and 1.39 ppm (–COCH2CH2CH2CH2CH2O–) were used to determine Mn.47,51 The obtained results are summed in Table 1.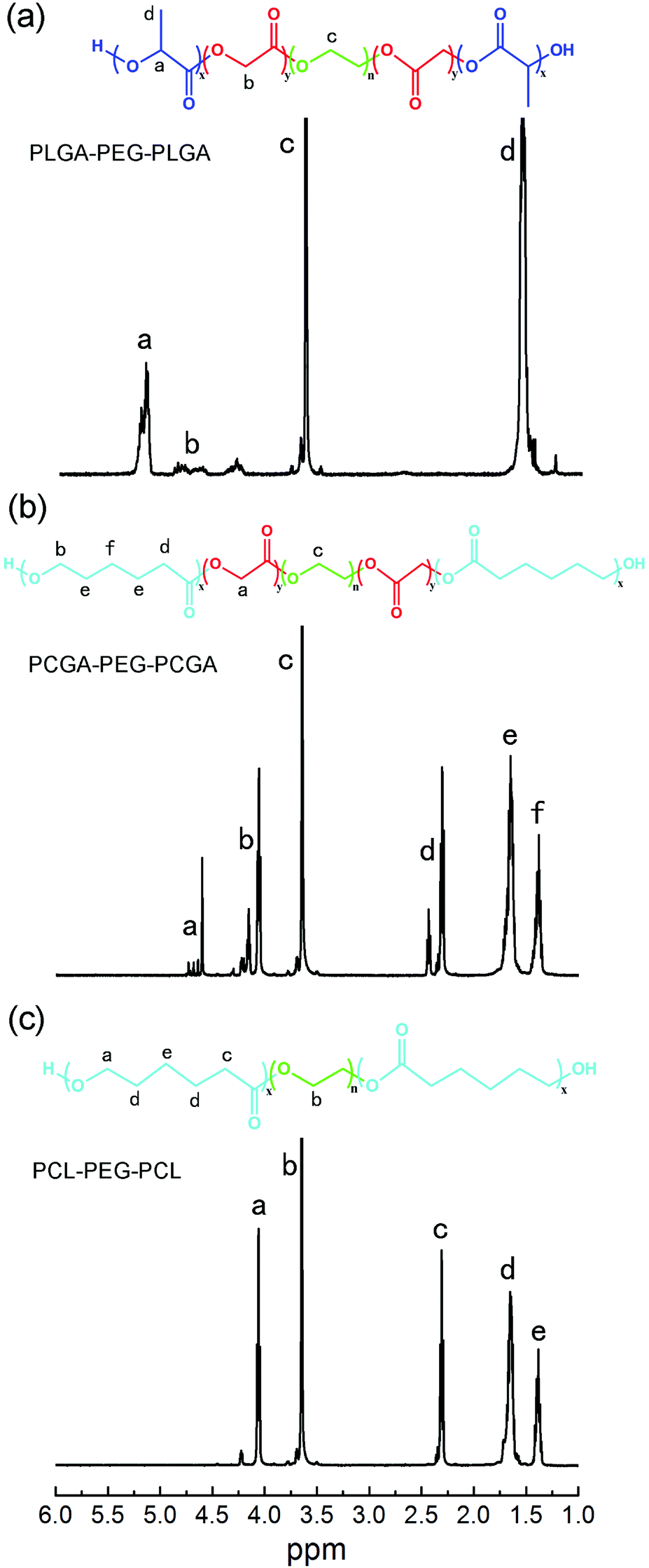 | ||
| Fig. 2 1H NMR spectra of triblock copolymers in CDCl3 (a) PLGA–PEG–PLGA, (b) PCGA–PEG–PCGA, (c) PCL–PEG–PCL. | ||
| Sample ID |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Monomer ratioa (mol mol−1) |
M
nb |
(Mw/Mn)b | Morphology |
|---|---|---|---|---|---|
| a The number-averaged MW, Mn of the central block PEG was provided by Aldrich, and Mn of each polyester block was calculated by 1H-NMR. b Measured via GPC. | |||||
| PLGA–PEG–PLGA | 1675–1500–1675 | LA/GA = 10/1 | 6520 | 1.23 | Sticky paste |
| PCGA–PEG–PCGA | 1725–1500–1725 | CL/GA = 10/1 | 7570 | 1.27 | Powder |
| PCL–PEG–PCL | 1510–1500–1510 | / | 6980 | 1.26 | Powder |
The triblock copolymers were further measured via GPC to determine MWs and their distributions. Fig. 3 displays the results of GPC analysis. A symmetric peak with a relative narrow MW distribution was observed. The polydispersity index, defined as weight-average MW over number-average MW (Mw/Mn), was lower than 1.3 for all of the specimens. Such a result indicated that purity was sufficiently high to investigate their physical–chemical properties. All the quantitative data on Mn and polydispersity index of the copolymers obtained from GPC are also listed in Table 1.
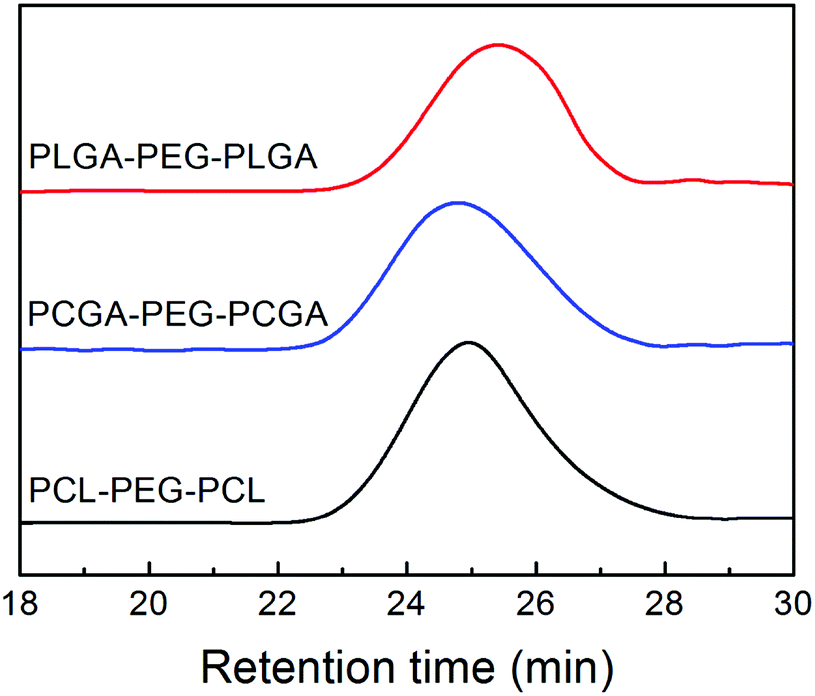 | ||
| Fig. 3 GPC traces of the indicated triblock copolymers. | ||
Thermal properties of polyester–PEG–polyester triblock copolymers in the bulk state and the stability of their aqueous solutions
The thermal properties of synthesized copolymers were examined via DSC characterizations. Fig. 4a shows DSC thermograms of polyester–PEG–polyester triblock copolymers with various polyester compositions. PCL–PEG–PCL presented two melting transitions at 46 °C and 51 °C in the heating curve and also two crystallization peaks at 7 °C and 28 °C in the cooling curve, which were attributed to the blocks of PCL and PEG, respectively.47 Due to the incorporation of GA into the PCL block, the crystallization of both PCL and PEG blocks in PCGA–PEG–PCGA was interrupted. Consequently, the melting and crystallization peaks of PCGA–PEG–PCGA were shifted to lower temperatures. For PLGA–PEG–PLGA, neither melting peak nor crystallization peak was detected during the heating and cooling cycles, suggesting that the polymer exhibits an amorphous state.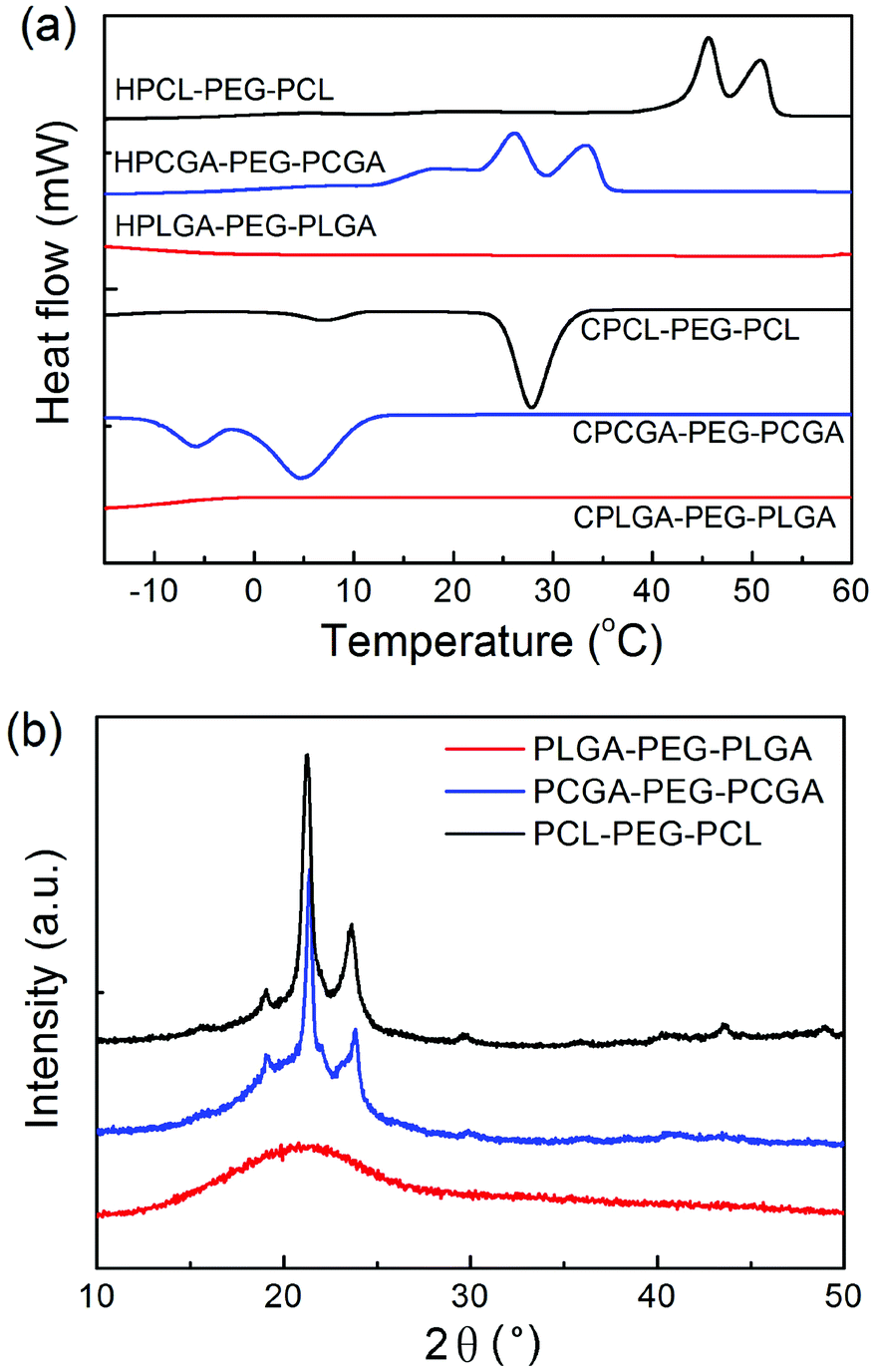 | ||
| Fig. 4 (a) DSC thermograms of the indicated triblock copolymers in the second heating run and the first cooling run. HPLGA–PEG–PLGA, HPCGA–PEG–PCGA and HPCL–PEG–PCL indicate heating curves, while CPLGA–PEG–PLGA, CPCGA–PEG–PCGA and CPCL–PEG–PCL refer to cooling curves. The heating and cooling rate was 5 °C min−1. (b) XRD patterns of the indicated triblock copolymers. | ||
The crystalline or amorphous behavior of three triblock copolymers was further analyzed via XRD measurements. Fig. 4b shows their XRD patterns. In the case of PCL–PEG–PCL, two strong diffraction peaks at 21.2° and 23.8° were observed, which came from the crystallization of the PCL component.51 PCGA–PEG–PCGA presented a similar pattern to PCL–PEG–PCL. The XRD pattern of PLGA–PEG–PLGA just exhibited a broad amorphous peak. These features coincided with the DSC results.
The three samples had different morphologies. PLGA–PEG–PLGA presented a sticky paste in the bulk state, while the other two samples had a powder form, as shown in Fig. 5a. Different from sticky PLGA–PEG–PLGA, powder PCGA–PEG–PCGA and PCL–PEG–PCL allowed comfortable handling in weighing and transferring, and different approaches were used to prepare their aqueous solutions. PLGA–PEG–PLGA was dissolved in water using stirring at low or room temperature and several hours were needed. The aqueous solution of PCGA–PEG–PCGA was easily prepared by heating the polymer/water system at 55 °C for several minutes followed by quenching in an ice bath for 30 min. The PCL–PEG–PCL aqueous solution was obtained via a similar method but using a higher temperature (70 °C) to melt the polymers. Moreover, varying stabilities of their aqueous solutions at ambient temperature were observed. When the aqueous solution of PCL–PEG–PCL was left at room temperature (20 °C) for one hour, an opaque gel was spontaneously formed, which was attributed to the crystallization of PCL.51 In contrast, the aqueous solutions of the other two samples maintained their sol states under the same conditions, with the results displayed in Fig. 5b.
 | ||
| Fig. 5 (a) Bulk morphologies of the indicated triblock copolymers. (b) The stability of their aqueous solutions. While PLGA–PEG–PLGA and PCGA–PEG–PCGA systems maintained stable sols, PCL–PEG–PCL spontaneously formed a crystal gel when their aqueous solutions were stored at room temperature for one hour. | ||
Sol–gel transition of polyester–PEG–polyester triblock copolymers in water
All the polyester–PEG–polyester triblock copolymers in the sol state were free-flowing liquids and exhibited a sol–gel transition in response to an increase of temperature. The sol–gel transitions of their aqueous solutions (25 wt% in NS) as a function of temperature were quantitatively evaluated using a stress-controlled rheometer with the results shown in Fig. 6a–d. The storage modulus G′, loss modulus G′′, complex modulus G* and phase angle δ of the polymeric aqueous systems abruptly changed as the temperature increased, indicating the formation of in situ thermogels.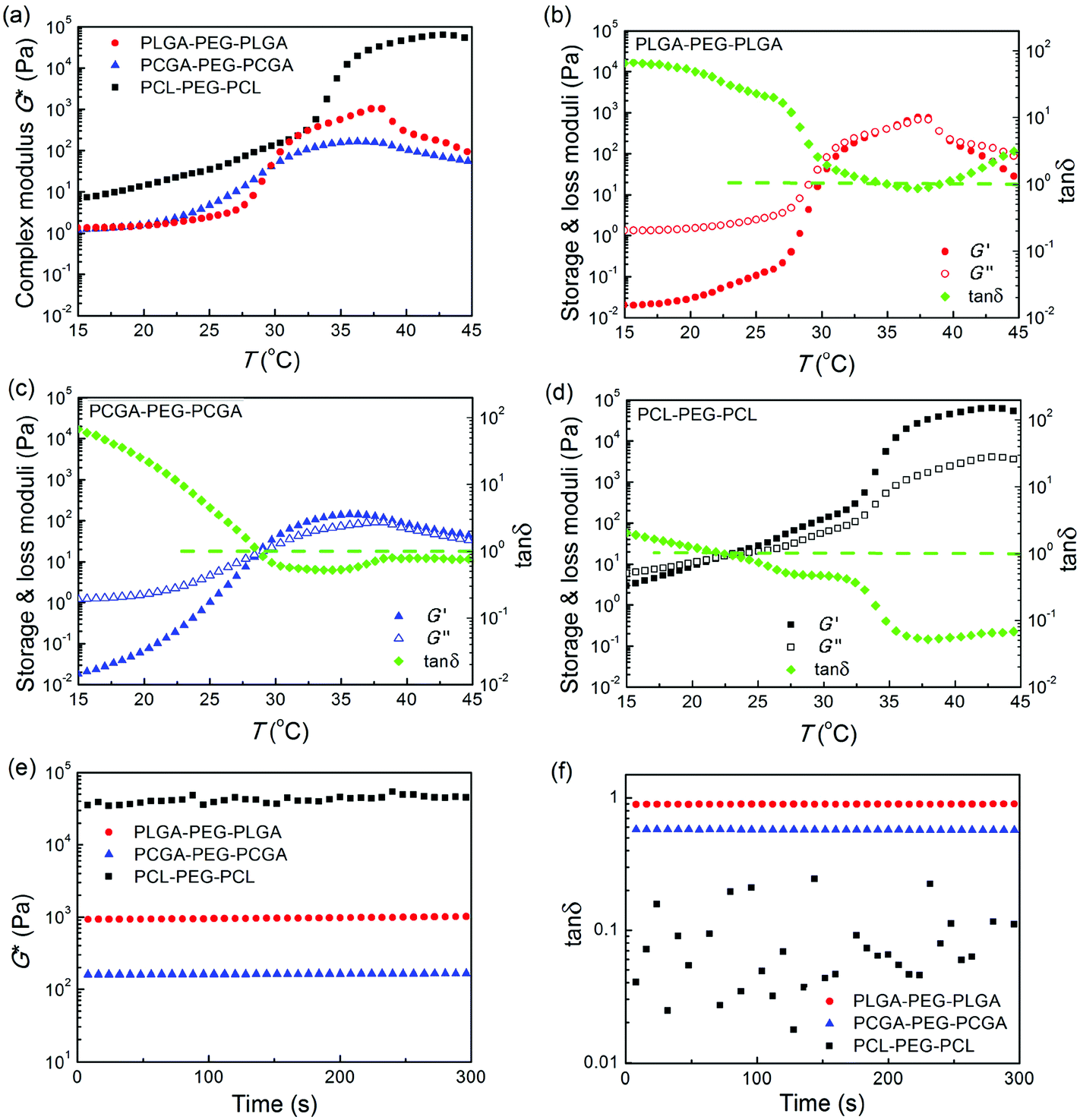 | ||
| Fig. 6 (a) Complex modulus (G*) of the indicated three triblock copolymers in NS as a function of temperature. Heating rate: 0.5 °C min−1; oscillatory frequency: 10 rad s−1. (b–d) Corresponding storage modulus G′, loss modulus G′′ and the tangent of the phase angle tan δ of the indicated triblock copolymers in NS as a function of temperature. The dashed lines represent tan δ = 1, where the effect of viscoelasticity was the most significant. (e–f) Time dependence of G* and tan δ of the indicated thermogels with a frequency of 10 rad s−1 at a given temperature (37 °C). Polymer concentrations in all cases were 25 wt%. | ||
Their G* values of the three samples exhibited different orders of magnitude at physiological temperature (Fig. 6e). The G* of PCL–PEG–PCL was the highest, and that of PCGA–PEG–PCGA was the lowest. In contrast, the phase angle δ obeyed the order of PLGA–PEG–PLGA > PCGA–PEG–PCGA > PCL–PEG–PCL, and all of the values at 37 °C were less than 45° or tan δ < 1, as seen in Fig. 6f.
In vivo evaluation of prevention of abdominal adhesions
A model of sidewall defect and bowel abrasion in rabbits was used to create peritoneal intestinal adhesions. Fig. 7 shows the process of establishment of animal model (a–c) and administration of the thermogel (d). The animals in the control group were not treated with any barrier substance on the defect after surgery; the thermogels or CHS solution was painted on the injured sites in the other groups. Compared with the CHS solution, the more viscous thermogels adhered to the defects easily.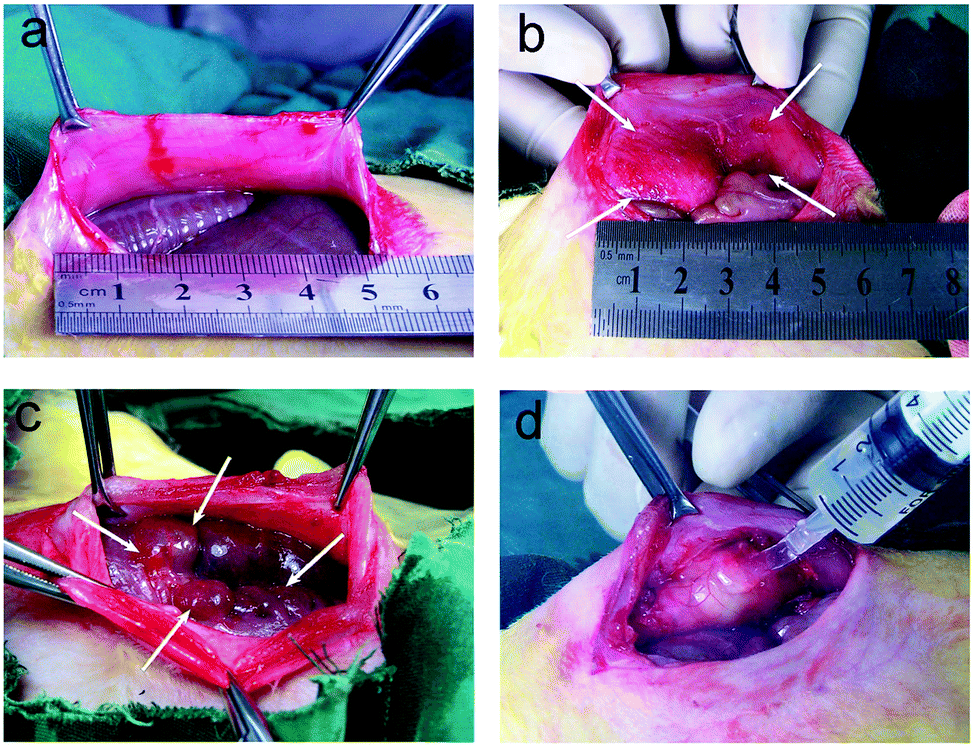 | ||
| Fig. 7 Photographs of experimental procedure of creating post-surgical peritoneal adhesions in a rabbit model. (a) The enterocoelia of a rabbit was opened. (b) A defect (4 × 3 cm2) comprising the peritoneum and a layer of muscle (∼1 mm thickness) was created locating 1 cm from the midline of peritoneal wall. (c) The corresponding site on bowel was abraded via a surgical brush resulting in bleeding. (d) The defect was covered by thermogel. Arrows are used to display the defect boundaries. | ||
One month after surgery, all of the animals were euthanized and autopsies were carried out to determine the abdominal adhesions. The gross inspection did not show any noticeable PLGA–PEG–PLGA and PCGA–PEG–PCGA polymer gels. In contrast, some PCL–PEG–PCL gel residues were observed around the adhesion tissues in a part of animals, as typically illustrated in Fig. 8.
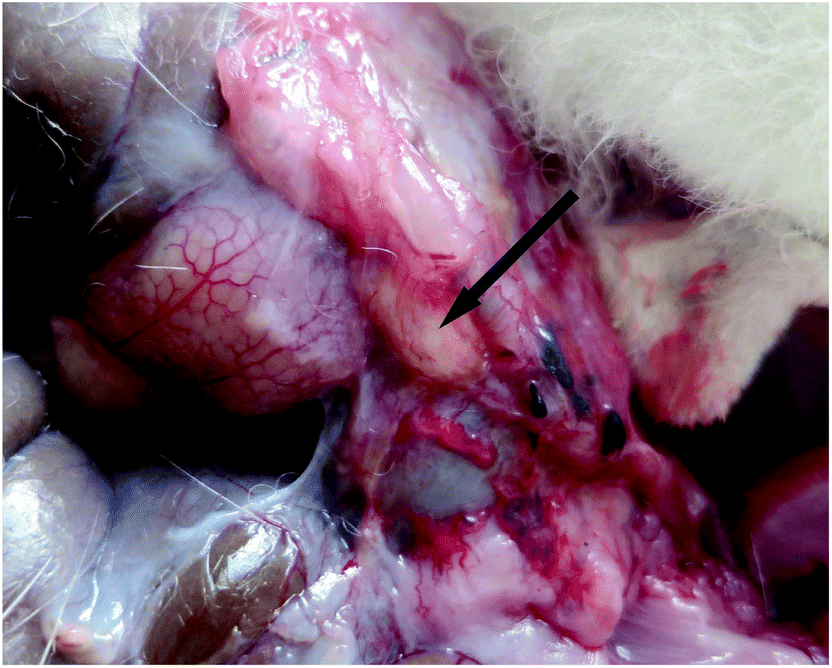 | ||
| Fig. 8 PCL–PEG–PCL residual was detected at the application site one month after surgery. The arrow indicates the residual PCL–PEG–PCL thermogel. | ||
The efficacies of thermogels against adhesion were evaluated based on the acknowledged protocol.30,31Fig. 9a shows the visual results of abdominal adhesion with various adhesion tenacities, and the percentage of animals with each adhesion score in all the groups is presented in Fig. 9b. The control group without any treatment suffered from severe abdominal adhesions, and score 3 adhesions developed in 83% of animals. The administration of PLGA–PEG–PLGA and PCGA–PEG–PCGA thermogels markedly decreased abdominal adhesions. In particular, with the use of PLGA–PEG–PLGA thermogel, adhesions were completely prevented in 50% of tested rabbits, and the injured sites were almost healed within one month; the remaining 50% of animals just suffered from mild intestinal adhesion.
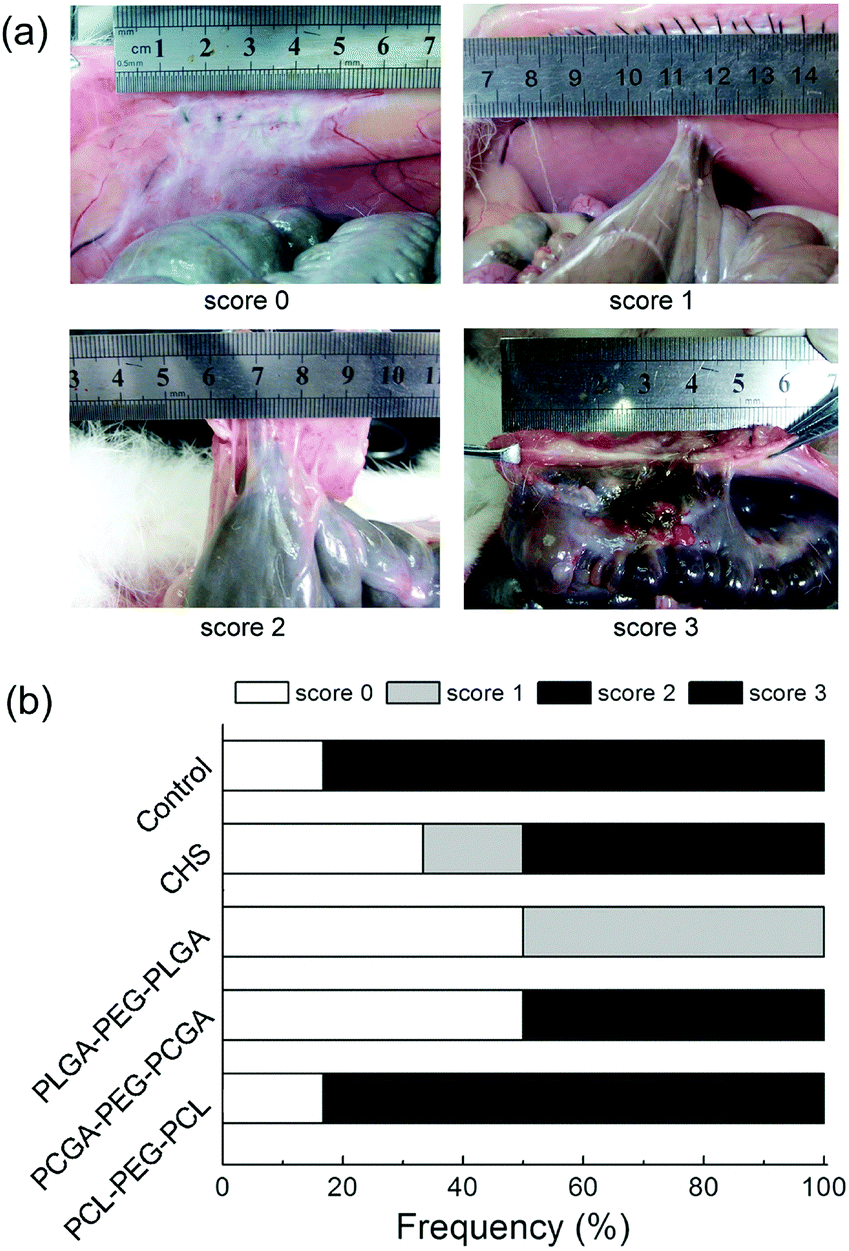 | ||
| Fig. 9 (a) Photographs of gross observations of the adhesion of defected tissues after one month. The scores demote adhesion tenacity. Score 0: no adhesion; score 1: mild intestinal adhesion and separation was facile; score 2: moderate intestinal adhesion and blunt dissection was needed; score 3: severe intestinal adhesion and sharp dissection was needed. (b) Distribution of adhesion tenacity scores of rabbits treated with the indicated thermogels. | ||
The anti-adhesion efficacy was also quantified via measurement of adhesion areas. The scores of adhesion extent and statistical results of the adhered area are displayed in Fig. 10. PLGA–PEG–PLGA and PCGA–PEG–PCGA thermogels significantly reduced both adhesion scores and area, and their adhesion area was statistically different from that of the control group. The adhesion area for PCL–PEG–PCL gel and CHS solution were slightly lower than that of the control group but no statistical difference was observed.
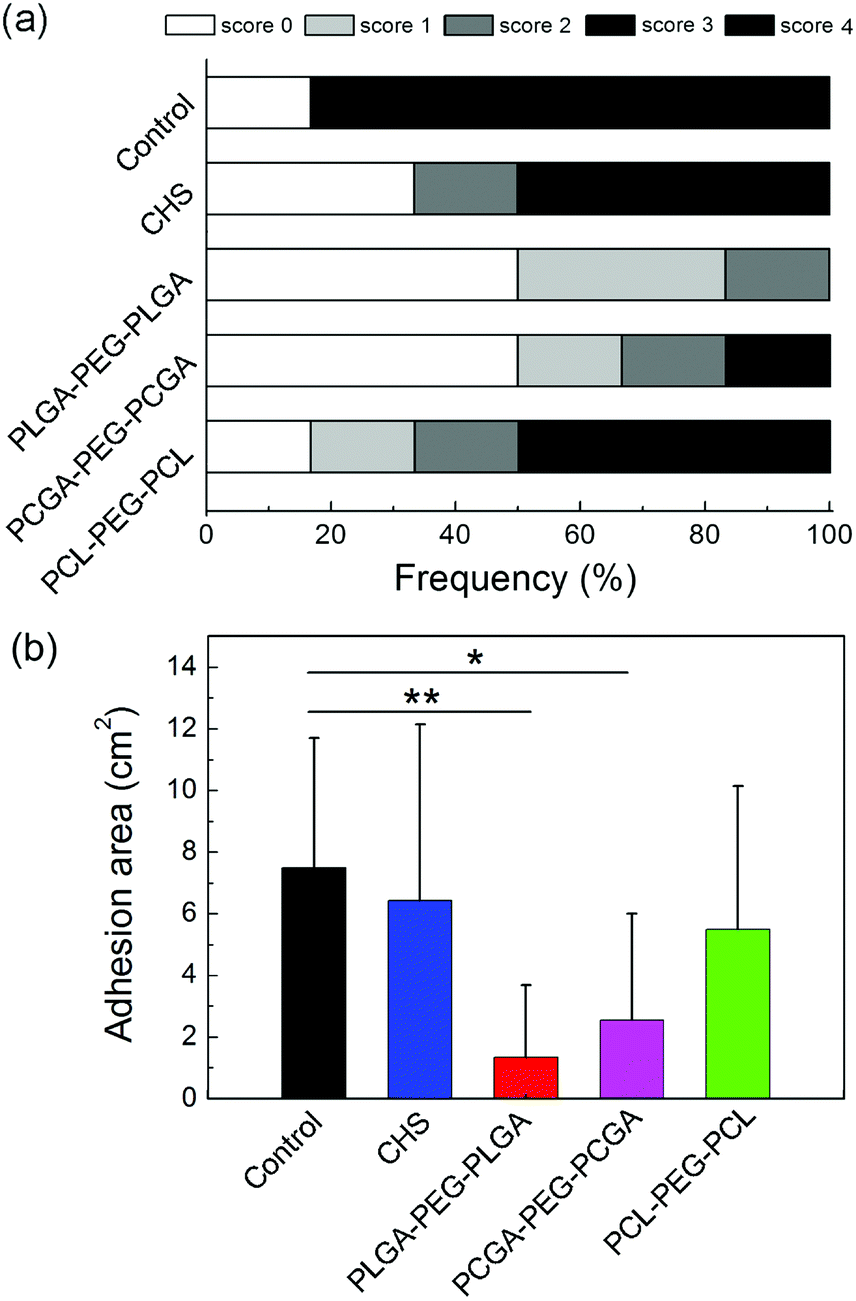 | ||
| Fig. 10 (a) Distribution of adhesion-scope scores of rabbits treated with the indicated thermogels. Score 0: no adhesion; score 1: adhesion area is between 0–25% of the area of initial abrasion damage; score 2: 25–50% of the area of initial abrasion damage; score 3: 50–75% of the area of initial abrasion damage; score 4: 75–100% of the area of initial abrasion damage. The area of initial abrasion damage was 12 cm2. (b) Statistic results of adhesion area of defect in the abdominal wall. *: p < 0.05; **: p < 0.01. | ||
Histological observations of the sites of injury were further examined. The surface of the peritoneal defects was completely covered with new mesotheliums in animals without post-operative adhesions (Fig. 11a) and the healed tissue was similar to the normal peritoneal tissue.3 In contrast, specimens that suffered from tissue adhesions displayed close apposition of the smooth muscle layers of the bowel to the abdominal wall musculature with different thickness of connected tissue (Fig. 10c–d), which reflected the adhesion tenacity as shown in Fig. 9a.
 | ||
| Fig. 11 Histological examination of (a) a healed peritoneal surface, (b) a mild abdominal adhesion, (c) a moderate abdominal adhesion, (d) a severe abdominal adhesion. The scores denote adhesion tenacity. Me: mesothelial layer; CE: cecal mucosa; SM: visceral smooth muscle; AW: abdominal wall. | ||
Discussion
Peritoneal adhesion is one of the principal causes of post-surgical morbidity and mortality.1–3 Barrier devices such as viscous polymer solutions, solid membranes and in situ-forming hydrogels have been designed to physically separate injured tissue surfaces and thus reduce the contact between affected organs to prevent adhesion.9–12 Polymer solutions are faced with a short duration at the application site; it is difficult to achieve complete coverage of damaged tissues using solid membranes; in situ chemical hydrogels triggered by in vivo chemical reactions might bring with biocompatibility problems. These disadvantages reduce their efficacies and thus limit their applications. Therefore, an ideal barrier should not only provide complete coverage of the traumatized surfaces and maintain effectiveness during peritoneal healing, but also be biocompatible, biodegradable and conveniently administrated via a laparoscope and open surgical procedures.Thermogelling polymers have great potential as novel barrier devices for preventing abdominal adhesions. In this study, three polyester–PEG–polyester triblock copolymers with different polyester compositions were synthesized and their efficacies of prevention of abdominal adhesion were assessed for the first time. These polymers were dissolved in water and formed micelles at low or ambient temperature.47,51,52 The hydrophilic PEG block is mainly located in the corona of micelle, and the hydrophobic polyester block occupies the core. As the temperature increases, spontaneous physical gelation occurs due to the formation of a percolated micelle network via micellar aggregation.40,51,52
The processes of fibrinous adhesion, fibroblast invasion and collagen deposition play critical roles in adhesion formation.54 It is well-known that PEG is a famous anti-adhesion agent against proteins and cells including fibroblasts.61–63 The thermogels used here were formed via the micellar aggregation and their surfaces were evidently rich in the PEG blocks, resulting in an enhancement of the hydroflotation effect and thus excellent anti-adhesion efficacies.
As far a comparison between the thermogelling samples is concerned, these thermogels exhibited different gel moduli at body temperature with the sequence of PCL–PEG–PCL > PLGA–PEG–PLGA > PCGA–PEG–PCGA (Fig. 6e). The efficacy of prevention of abdominal adhesions did not follow this sequence. From the animal experiments, PLGA–PEG–PLGA was the best, and PCL–PEG–PCL was the least successful. This finding suggested that gel strength was not the crucial element for a physical barrier system. On the other hand, the phase angle in Fig. 6f followed the same sequence as that of the anti-adhesion efficacy. It seems worthy of noting that a larger phase angle does not necessarily lead to more significant anti-adhesion. The phase angle measures the phase lag between stress and strain in a dynamical mechanical experiment. For a full elastic material, δ = 0; for a fully viscous material, δ = 90°; for any viscoelastic material, δ ∈ (0, 90°). It could be roughly regarded that δ = 45° or tan δ = 1 corresponds to the most significant viscoelastic material. Hence, the ideal phase angle for an excellent anti-adhesion material might be 45°, and the PLGA–PEG–PLGA thermogel was very close to this state under physiological conditions.
Besides the hydrophilic surface and the viscoelastic matrix, another important issue comes from the in vivo retention time of a material. These thermogels exhibited different degradation periods due to various hydrolysis rates of polyester segments. The in vivo persistence of PLGA–PEG–PLGA and PCGA–PEG–PCGA thermogels at the target sites was only several weeks after the subcutaneous injection into SD rats,51,56 whereas the in vivo integrity of PCL–PEG–PCL system at the administration site was maintained over many months due to the crystallization of the PCL block.57 What's more, the enterocoelia is a relative open system and the thermogels are rapidly absorbed at such an administration site.34 The previous studies have also demonstrated that the material residues of barrier devices with prolonged periods at the application sites (over 7 days) could not only negatively affect the remesothelialization of peritoneal defects, but also induce foreign body reactions, resulting in the formation of adhesion.11,64 In this study, no residual PLGA–PEG–PLGA and PCGA–PEG–PCGA thermogels were observed at the abdominal space one month after surgery. In fact, PLGA–PEG–PLGA and PCGA–PEG–PCGA thermogels in the abdominal cavity were completely absorbed within one week (data not shown). On the contrary, residual PCL–PEG–PCL gel was encapsulated as foreign body at the application site (Fig. 8). This result further compromised the efficacy of the PCL–PEG–PCL system for prevention of post-operative adhesion. The long retention of a barrier device over several weeks might not be helpful for prevention of post-operative abdominal adhesions.
Conclusions
Three thermogelling polyester–PEG–polyester triblock copolymers were tried as physical barrier devices for preventing post-operative peritoneal adhesions in a rabbit model. These polymers were prepared via ring-opening polymerization of commonly used monomers. Polyester compositions affected their bulk morphology in the dry state, and also sol stability and gel modulus in water. All of these thermogels were applied for prevention of post-surgical adhesion. The three thermogels with the same polymer concentration (25 wt%) exhibited different efficacies. The PLGA–PEG–PLGA thermogel was the most effective in reducing the formation of intraperitoneal adhesion, whereas PCL–PEG–PCL system presented a lower efficacy. Hence, gel modulus at body temperature was not vital; the viscoelasticity of a gel and its appropriate in vivo persistence in the peritoneal cavity played critical roles in prevention of post-surgical adhesions. The present study is meaningful for guiding the design of a physical barrier device to prevent post-surgical adhesions.Acknowledgements
The group was supported by NSF of China (grants no. 51273217, no. 91127028, no. 81271954, and no. 21034002), Chinese Ministry of Science and Technology (973 Program no. 2011CB606203) and Science and Technology Developing Foundation of Shanghai (grants no. 12JC1402600, no. 13XD1401000 and no. 12qh1402700).References
- M. A. Weibel and G. Majno, Am. J. Surg., 1973, 126, 345 CrossRef CAS PubMed.
- G. S. Dizerega, Fertil. Steril., 1994, 61, 219 CAS.
- Y. Yeo and D. S. Kohane, Eur. J. Pharm. Biopharm., 2008, 68, 57 CrossRef CAS PubMed.
- C. Z. Wei, C. L. Hou, Q. S. Gu, L. X. Jiang, B. Zhu and A. L. Sheng, Biomaterials, 2009, 30, 5534 CrossRef CAS PubMed.
- H. Orita, W. Girgis and G. S. Dizerega, Drug Dev. Res., 1987, 10, 97 CrossRef CAS.
- T. Segura, H. Schmokel and J. A. Hubbell, J. Surg. Res., 2007, 141, 162 CrossRef CAS PubMed.
- P. N. Zawaneh, S. P. Singh, R. F. Padera, P. W. Henderson, J. A. Spector and D. Putnam, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11014 CrossRef CAS PubMed.
- N. Ishiyama, T. Moro, K. Ishihara, T. Ohe, T. Miura, T. Konno, T. Ohyama, M. Kimura, M. Kyomoto, K. Nakamura and H. Kawaguchi, Biomaterials, 2010, 31, 4009 CrossRef CAS PubMed.
- J. M. Burns, K. Skinner, J. Colt, A. Sheidlin, R. Bronson, Y. Yaacobi and E. P. Goldberg, J. Surg. Res., 1995, 59, 644 CrossRef CAS PubMed.
- J. M. Seeger, L. D. Kaelin, E. M. Staples, Y. Yaacobi, J. C. Bailey, S. Normann, J. W. Burns and E. P. Goldberg, J. Surg. Res., 1997, 68, 63 CrossRef CAS PubMed.
- S. Matsuda, N. Se, H. Iwata and Y. Ikada, Biomaterials, 2002, 23, 2901 CrossRef CAS PubMed.
- N. M. Paulo, M. Silva, A. M. Moraes, A. P. Rodrigues, L. B. de Menezes, M. P. Miguel, F. G. de Lima, A. D. Faria and L. M. L. Lima, J. Biomed. Mater. Res., Part B, 2009, 91, 221 CrossRef PubMed.
- J. M. Becker, M. T. Dayton, V. W. Fazio, D. E. Beck, S. J. Stryker, S. D. Wexner, B. G. Wolff, P. L. Roberts, L. E. Smith, S. A. Sweeney and M. Moore, J. Am. Coll. Surg., 1996, 183, 297 CAS.
- J. Iliopoulos, G. B. Cornwall, R. O. N. Evans, C. Manganas, K. A. Thomas, D. C. Newman and W. R. Walsh, J. Surg. Res., 2004, 118, 144 CrossRef CAS PubMed.
- W. R. Walsh, R. O. N. Evans, J. Iliopoulos, G. B. Cornwall and K. A. Thomas, J. Biomed. Mater. Res., Part B, 2006, 79, 166 CrossRef PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853 CrossRef CAS PubMed.
- G. T. Chang, T. Y. Ci, L. Yu and J. D. Ding, J. Controlled Release, 2011, 156, 21 CrossRef CAS PubMed.
- L. Yu, T. Y. Ci, S. C. Zhou, W. J. Zeng and J. D. Ding, Biomater. Sci., 2013, 1, 411 RSC.
- K. Li, L. Yu, X. J. Liu, C. Chen, Q. H. Chen and J. D. Ding, Biomaterials, 2013, 34, 2834 CrossRef CAS PubMed.
- T. Y. Ci, L. Chen, T. Li, G. T. Chang, L. Yu and J. D. Ding, Biomater. Sci., 2013, 1, 1235 RSC.
- L. Yu, K. Li, X. J. Liu, C. Chen, Y. C. Bao, T. Y. Ci, Q. H. Chen and J. D. Ding, J. Pharm. Sci., 2013, 102, 4140 CrossRef CAS PubMed.
- D. Y. Ko, U. P. Shinde, B. Yeon and B. Jeong, Prog. Polym. Sci., 2013, 38, 672 CrossRef CAS.
- R. Jin, L. S. M. Teixeira, P. J. Dijkstra, Z. Y. Zhong, C. A. van Blitterswijk, M. Karperien and J. Feijen, Tissue Eng. A, 2010, 16, 2429 CrossRef CAS PubMed.
- H. K. Kim, W. S. Shim, S. E. Kim, K. H. Lee, E. Kang, J. H. Kim, K. Kim, I. C. Kwon and D. S. Lee, Tissue Eng. A, 2009, 15, 923 CrossRef CAS PubMed.
- V. X. Truong, M. P. Ablett, H. T. J. Gilbert, J. Bowen, S. M. Richardson, J. A. Hoyland and A. P. Dove, Biomater. Sci., 2014, 2, 167 RSC.
- I. Kumano, M. Ishihara, S. Nakamura, S. Kishimoto, M. Fujita, H. Hattori, T. Horio, Y. Tanaka, K. Hase and T. Maehara, Gastrointest. Endosc., 2012, 75, 841 CrossRef PubMed.
- L. Yu, W. Xu, W. J. Shen, L. P. Cao, Y. Liu, Z. S. Li and J. D. Ding, Acta Biomater., 2014, 10, 1251 CrossRef CAS PubMed.
- D. Gyawali, P. Nair, H. K. W. Kim and J. Yang, Biomater. Sci., 2013, 1, 52 RSC.
- T. D. Johnson, J. A. DeQuach, R. Gaetani, J. Ungerleider, D. Elhag, V. Nigam, A. Behfar and K. L. Christman, Biomater. Sci., 2014 10.1039/c3bm60283d.
- Y. Yeo, C. B. Highley, E. Bellas, T. Ito, R. Marini, R. Langer and D. S. Kohane, Biomaterials, 2006, 27, 4698 CrossRef CAS PubMed.
- Y. Yeo, E. Bellas, C. B. Highley, R. Langer and D. S. Kohane, Biomaterials, 2007, 28, 3704 CrossRef CAS PubMed.
- T. Ito, Y. Yeo, C. B. Highley, E. Bellas and D. S. Kohane, Biomaterials, 2007, 28, 3418 CrossRef CAS PubMed.
- J. L. West and J. A. Hubbell, Biomaterials, 1995, 16, 1153 CrossRef CAS PubMed.
- B. Yang, C. Y. Gong, Z. Y. Qian, X. Zhao, Z. Y. Li, X. R. Qi, S. T. Zhou, Q. A. Zhong, F. Luo and Y. Q. Wei, BMC Biotechnol., 2010, 10, 65 CrossRef PubMed.
- Z. Zhang, J. Ni, L. Chen, L. Yu, J. W. Xu and J. D. Ding, J. Biomed. Mater. Res., Part B, 2012, 100, 1599 CrossRef PubMed.
- J. H. Hong, J. W. Choe, G. Y. Kwon, D. Y. Cho, D. S. Sohn, S. W. Kim, Y. C. Woo, C. J. Lee and H. Kang, J. Surg. Res., 2011, 166, 206 CrossRef CAS PubMed.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860 CrossRef CAS PubMed.
- L. Yu and J. D. Ding, Chem. Soc. Rev., 2008, 37, 1473 RSC.
- X. J. Loh, S. H. Goh and J. Li, Biomacromolecules, 2007, 8, 585 CrossRef CAS PubMed.
- L. Yu, Z. Zhang, H. Zhang and J. D. Ding, Biomacromolecules, 2009, 10, 1547 CrossRef CAS PubMed.
- C. D. Liu, Z. Zhang, K. L. Liu, X. Ni and J. Li, Soft Matter, 2013, 9, 787 RSC.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860 RSC.
- B. Jeong, Y. H. Bae and S. W. Kim, Macromolecules, 1999, 32, 7064 CrossRef CAS.
- D. S. Lee, M. S. Shim, S. W. Kim, H. Lee, I. Park and T. Y. Chang, Macromol. Rapid Commun., 2001, 22, 587 CrossRef CAS.
- C. Chen, L. Chen, L. P. Cao, W. J. Shen, L. Yu and J. D. Ding, RSC Adv., 2014, 4, 8789 RSC.
- L. Yu, Z. Zhang and J. D. Ding, Biomacromolecules, 2011, 12, 1290 CrossRef CAS PubMed.
- S. J. Bae, J. M. Suh, Y. S. Sohn, Y. H. Bae, S. W. Kim and B. Jeong, Macromolecules, 2005, 38, 5260 CrossRef CAS.
- J. S. Kwon, S. M. Yoon, D. Y. Kwon, D. Y. Kim, G. Z. Tai, L. M. Jin, B. Song, B. Lee, J. H. Kim, D. K. Han, B. H. Min and M. S. Kim, J. Mater. Chem. B, 2013, 1, 3314 RSC.
- Z. Zhang, Y. X. Lai, L. Yu and J. D. Ding, Biomaterials, 2010, 31, 7873 CrossRef CAS PubMed.
- A. Petit, B. Muller, P. Bruin, R. Meyboom, M. Piest, L. M. J. Kroon-Batenburg, L. G. J. de Leede, W. E. Hennink and T. Vermonden, Acta Biomater., 2012, 8, 4260 CrossRef CAS PubMed.
- L. Yu, W. J. Shen, D. C. Yang and J. D. Ding, Macromol. Res., 2013, 21, 207 CrossRef CAS.
- L. Yu, G. T. Chang, H. Zhang and J. D. Ding, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1122 CrossRef CAS.
- H. Zhang, L. Yu and J. D. Ding, Macromolecules, 2008, 41, 6493 CrossRef CAS.
- Z. Zhang, J. Ni, L. Chen, L. Yu, J. W. Xu and J. D. Ding, Biomaterials, 2011, 32, 4725 CrossRef CAS PubMed.
- G. M. Zentner, R. Rathi, C. Shih, J. C. McRea, M. H. Seo, H. Oh, B. G. Rhee, J. Mestecky, Z. Moldoveanu, M. Morgan and S. Weitman, J. Controlled Release, 2001, 72, 203 CrossRef CAS PubMed.
- L. Yu, Z. Zhang, H. Zhang and J. D. Ding, Biomacromolecules, 2010, 11, 2169 CrossRef CAS PubMed.
- Y. M. Kang, S. H. Lee, J. Y. Lee, J. S. Son, B. S. Kim, B. Lee, H. J. Chun, B. H. Min, J. H. Kim and M. S. Kim, Biomaterials, 2010, 31, 2453 CrossRef CAS PubMed.
- L. Yu, Z. Zhang and J. D. Ding, Macromol. Res., 2012, 20, 234 CrossRef CAS.
- L. Yu, G. T. Chang, H. Zhang and J. D. Ding, Int. J. Pharm., 2008, 348, 95 CrossRef CAS PubMed.
- L. Yu, H. Zhang and J. D. Ding, Angew. Chem., Int. Ed., 2006, 45, 2232 CrossRef CAS PubMed.
- X. Yao, R. Peng and J. D. Ding, Adv. Mater., 2013, 25, 5257 CrossRef CAS PubMed.
- X. Wang, C. Yan, K. Ye, Y. He, Z. H. Li and J. D. Ding, Biomaterials, 2013, 34, 2865 CrossRef CAS PubMed.
- C. Yan, J. G. Sun and J. D. Ding, Biomaterials, 2011, 32, 3931 CrossRef CAS PubMed.
- M. Wallwiener, S. Brucker, H. Hierlemann, C. Brochhausen, E. Solomayer and C. Wallwiener, Fertil. Steril., 2006, 86, 1266 CrossRef PubMed.
Footnote |
| † These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |