 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Artificial microniches for probing mesenchymal stem cell fate in 3D†
Yujie
Ma
*a,
Martin P.
Neubauer
b,
Julian
Thiele
a,
Andreas
Fery
b and
W. T. S.
Huck
*a
aDepartment of Physical Organic Chemistry, Radboud University Nijmegen, Institute for Molecules and Materials, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: y.ma@science.ru.nl; w.huck@science.ru.nl
bDepartment of Physical Chemistry II, University of Bayreuth, Universitätsstraβe 30, D-95447 Bayreuth, Germany
First published on 18th June 2014
Abstract
Droplet microfluidics is combined with bio-orthogonal thiol–ene click chemistry to fabricate micrometer-sized, monodisperse fibrinogen-containing hyaluronic acid hydrogel microbeads in a mild, radical-free procedure in the presence of human mesenchymal stem cells (hMSCs). The gel beads serve as microniches for the 3D culture of single hMSCs, containing hyaluronic acid and additional fibrinogen for cell surface binding, and they are porous and stable in tissue culture medium for up to 4 weeks with mechanical properties right in the range of soft solid tissues (0.9–9.2 kPa). The encapsulation procedure results in 70% viable hMSCs in the microbeads after 24 hours of culture and a very high degree of viability of the cells after long term culture of 2 weeks. hMSCs embedded in the microniches display an overall rounded morphology, consistent with those previously observed in 3D culture. Upon induction, the multipotency and differentiation potential of the hMSCs are characterized by staining of corresponding biomarkers, demonstrating a clear heterogeneity in the cell population. These hydrogel microbeads represent a versatile microstructured material platform with great potential for studying the differences of material cues and soluble factors in stem cell differentiation in a 3D tissue-like environment at the single cell level.
Introduction
Mesenchymal stem cells (MSCs) that can be isolated from a wide variety of tissues1 show great potential in regenerative medicine in delivering new therapies for many diseases.2 It is well-known that MSCs can differentiate into various tissue specific lineages ranging from neurons to osteoblasts,1 which may reflect their interactions with their microenvironment or the so-called stem cell niche.3 The stem cell niche provides multiple effectors to modulate stem cell fate decision including soluble growth factors, the extracellular matrix (ECM) and biochemical signals through cell–cell contact with neighbouring cells.4 In response, cells actively interact with the ECM and remodel the niche they reside in.2 In order to understand this complex regulatory network, advanced cell culture platforms are being developed. Pioneering work by Discher et al. showed the crucial effect of substrate elasticity in directing MSC fate on 2 dimensional (2D) polyacrylamide gels.5 However, three dimensional (3D) cell culture is gaining popularity as it mimics the natural 3D tissue organization more closely.6,7 Hydrogels have become the prime candidate for this purpose due to their ability to mimic many aspects of natural ECM including high water content, tissue-like mechanical properties and adjustable functionalities.8,9 A vast range of hydrogels based on synthetic as well as naturally-derived materials, or combinations thereof, have been utilized to study MSC–matrix interactions in 3D in bulk, with control over several important parameters such as matrix mechanics10,11 and biodegradability.12 However, it is very difficult to separate chemical cues such as material properties (chemical composition, biocompatibility, stiffness, and porosity) from biological cues, for example those originating from cell–cell interactions, using these bulk hydrogel cell culture models. Moreover, bulk studies lack the resolution necessary to reveal cellular heterogeneity.13,14 It is important to understand the signalling mechanisms mediating different stem cell responses at a single cell resolution.2 Therefore, novel in vitro ECM models targeting single cell studies are needed for obtaining an in-depth knowledge of the molecular mechanisms of stem cell/matrix modulation.Micrometer-sized hydrogel microbeads are suitable model systems for the study of 3D cell culture at a single cell level. Recent advances in droplet microfluidics have facilitated the fabrication of uniform, monodisperse cell-laden hydrogel microbeads with precise control over their size, composition and material properties in a high throughput fashion.15–24 In this method, water-in-oil emulsions with cell containing hydrogel precursor solutions as the aqueous phase are first generated microfluidically, and then solidified/cross-linked to form hydrogel microbeads. The obtained microbeads are usually transferred to the aqueous phase for further culturing the cells. Many examples of the successful incorporation of cells into microfluidically generated hydrogel microbeads with preserved cell viability have been shown in recent years. However, all the studies carried out so far were based on simple cell models such as fibroblasts with relatively short periods of culture time (a few days). It is therefore difficult to extend these existing methods to the study of MSCs, which require a prolonged period of time for a clear identification of their lineage commitment. With preserving cell viability as the major consideration, mild gelation conditions are preferred. This means that the cross-linking reactions involved should be bio-orthogonal, non-toxic, relatively fast at or below 37 °C, and preferably free from radicals.18 Hence, the material selection is rather limited for generating cell-laden hydrogel microbeads using droplet microfluidics, and most of the cell-laden hydrogel microbead systems reported so far lack the presence of certain cell-binding moieties (e.g. RGD sequences) that are believed to be crucial for the establishment of proper cell–matrix interactions.10,12,25,26 The fabrication of single MSC-laden hydrogel microbeads based on mild bio-orthogonal cross-linking strategies with full control over cross-linking kinetics, hydrogel mechanical characteristics and desired/tuneable cell-binding functionalities while maintaining a high MSC viability at the same time thus remains a challenge.
Here, we report on the encapsulation of human MSCs (hMSCs) into fibrinogen functionalized hydrogel microbeads based on the naturally derived polysaccharide hyaluronic acid (HA) that are cross-linked through bio-orthogonal thiole–ene chemistry27 using droplet microfluidics. HA is an essential component of the ECM and is involved in many biological processes. It is biodegradable and can be modified to present a variety of functionalities and has been a popular choice for biomedical applications including those involving MSCs.28 In the current study, thiol-modified HA is used (HASH, Fig. 1A).
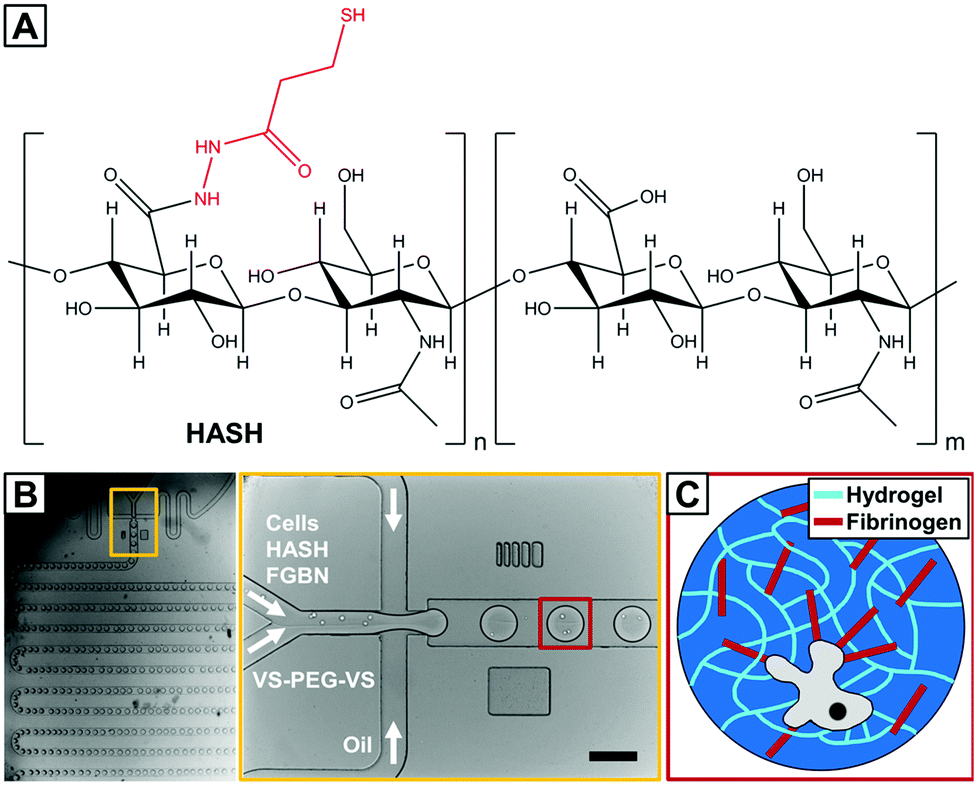 | ||
| Fig. 1 Microfluidic fabrication of hydrogel based microniches for the 3D culture of single human mesenchymal stem cells (hMSCs). (A) Chemical structure of the major components of the hydrogel precursors: thiolated hyaluronic acid (HASH) used in this study. Thiol functional groups were randomly incorporated into the polymer structure with a degree of substitution of 25%. (B) Microfluidic fabrication of water-in-oil droplets templating hydrogel formation using a flow-focusing device with double inlets and meander mixing channels (inset) for the aqueous phase with hydrogel precursors, cells and fibrinogen (FBNG) and one inlet containing the oil phase. (C) Schematic illustration of the obtained cross-linked network of the hydrogel microbeads containing fibrinogen as additional hMSC binding sites. The scale bar denotes 100 μm. | ||
Since the introduction of HASH about a decade ago,29 it has been shown to be an excellent hydrogel base material as it allows both accurate control over hydrogel elasticity and easiness of ligand presentation.30 A particularly attractive chemical feature of HASH for encapsulating sensitive biological substances such as cells is that it can be cross-linked in an extremely mild fashion due to the formation of di-sulfide bridges by itself or through thiol–ene click reaction with biocompatible/bio-inert bi- or multi-functional cross-linkers such as poly(ethylene glycol) diacrylate (PEG-DA)31,32 entirely radical-free. However, di-sulfide hydrogels are not very stable, especially under some reductive cell culture conditions, and HASH-diacrylate hydrogels have been shown to be susceptible to hydrolysis.30 For these reasons, we chose to use PEG di-vinylsulfone (PEG-DVS) as a cross-linker for HASH. The hydrogel is further decorated with fibrinogen (FBNG), the fibrin-forming protein that provides natural binding sites for cell surface integrins,33 through specific HA–FBNG interactions. We first demonstrate the fabrication of FBNG-HASH-PEG hydrogel microbeads by droplet microfluidics with controlled composition, gelling conditions, tunable mechanical properties and sufficient porosity. Next the encapsulation of hMSCs in these microbeads, their long term culture and explorations on their differentiation at a single cell level are described. We show that the FBNG-HASH-PEG hydrogel microbead platform presents great promise in supporting the long-term culture of hMSCs and studying their differentiation in a 3D microenvironment.
Materials and methods
General experimental details
All reagents and chemicals were used as received unless otherwise specified. Dialysis was performed using Spectra/Por® dialysis membranes (MW cut-off: 3500 g mol−1). To determine the exact channel height of the channel network fabricated in SU-8 (Micro Resist Technology GmbH, Germany) via photolithography, differential interference contrast (DIC) microscopy was performed on a Wyco NT1100 optical profiler (Bruker, USA). Bright-field microscopy imaging was performed on an IX71 microscope (Olympus) equipped with a 10× objective (air) and a Phantom MIRO high-speed camera (Vision Research Inc., USA). Confocal microscopy measurements were performed using a Leica SP2 (Leica, Germany) confocal laser scanning microscope (CLSM). Rheology measurements were performed on a TA rheometer (TA Discovery HR-1 Hybrid Rheometer, New Castle, USA) with a parallel plate (diameter 40 mm) geometry. A fixed oscillation frequency of 1 Hz was used in all time sweep experiments, which was checked by a frequency sweep measurement from 0.1 Hz to 10 Hz to be in the rubber plateau region. The excitation/emission wavelengths of the fluorophores used were: fluorescein isothiocyanate (FITC) and Alexa 488 (488 nm/500–575 nm); DAPI (405 nm/420–500 nm); rhodamine dyes (561 nm/580–650 nm); calcein (488 nm/500–550 nm); and EthD-1 (514 nm/580–650 nm).Synthesis of thiolated hyaluronic acid
Thiolated hyaluronic acid was synthesized following a modified procedure as previously reported by Prestwich and coworkers.29 Briefly, 250 mg low molecular weight hyaluronic acid (sodium salt, MW ∼ 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 Lifecore) was dissolved in 25 mL MES buffer (pH 4.75), and 50 mg PDPH (Thermo Scientific) and 300 mg 1-ethyl-3-(3 dimethylaminopropyl)carbodiimide (EDC) were added sequentially as solids. The reaction was carried out with stirring at room temperature for at least 2 hours. The solution was dialyzed against MilliQ to remove excess reactants. Afterwards, 100 mg tris(2-carboxyethyl)phosphine (TCEP) was added and the reaction mixture was stirred at room temperature for another 2 hours. The solution was extensively dialyzed and, in a final step, lyophilized to give thiolated hyaluronic acid as a white solid. The degree of thiolation was measured by 1H NMR (the spectrum is shown in ESI Fig. S1†) and Ellman's test34 to be approximately 25%.
000 g mol−1 Lifecore) was dissolved in 25 mL MES buffer (pH 4.75), and 50 mg PDPH (Thermo Scientific) and 300 mg 1-ethyl-3-(3 dimethylaminopropyl)carbodiimide (EDC) were added sequentially as solids. The reaction was carried out with stirring at room temperature for at least 2 hours. The solution was dialyzed against MilliQ to remove excess reactants. Afterwards, 100 mg tris(2-carboxyethyl)phosphine (TCEP) was added and the reaction mixture was stirred at room temperature for another 2 hours. The solution was extensively dialyzed and, in a final step, lyophilized to give thiolated hyaluronic acid as a white solid. The degree of thiolation was measured by 1H NMR (the spectrum is shown in ESI Fig. S1†) and Ellman's test34 to be approximately 25%.
Synthesis of rhodamine labelled fibrionogen
Fibrinogen (FBNG) was dissolved in sodium carbonate/bicarbonate buffer (100 mM, pH 9) at a concentration of 10 mg mL−1. A 1 mg mL−1 rhodamine B isothiocyanate (RBITC) solution in DMSO was mixed with the above FBNG solution to reach a final RBITC concentration of 60 μg mL−1. The reaction mixture was gently stirred for 2 hours at room temperature in the dark. The excess of RBITC was removed by centrifuge filter units (Amicon Ultra, 10 kDa, Millipore) followed by extensive washing with PBS and centrifugation at 1500g, until the eluent was colorless. The obtained RBITC-FBNG was stored at room temperature in the dark for up to five days.Microfluidic device fabrication and general microfluidic experimental setup
Microfluidic devices were fabricated using combined photo and soft lithography in poly(dimethylsiloxane) (PDMS) (Dow Corning, Germany).35 A negative photoresist (SU-8 25 or SU-8 50, Microchem Co., USA) was spin-coated onto the polished site of a 2-inch silicon wafer (SI-MAT, Germany). A mask aligner (MJB3, Süss MikroTec, Germany) was used to impart the microchannel structure of a transparent photomask (JD Phototools, UK) into the photoresist. We optimized the master device fabrication employing DIC microscopy to obtain microchannels with a very uniform height of 100 μm. The channel width at the droplet forming flow-focusing nozzle was 100 μm. A PDMS replica of the channel design was formed by mixing the PDMS oligomer and cross-linker in a ratio of 10:1 (w/w) and curing the homogeneous, degassed mixture at 65 °C for at least 60 min. Thereafter, access ports were bored into the soft replica with a biopsy needle (outer diameter: 1.0 mm, Pfm, Medical Workshop, USA), and the PDMS replica was bonded to a glass slide after oxygen plasma treatment. The bonding process was completed in an oven at 90 °C for approximately 1 h. Microfluidic devices were connected to high-precision, positive displacement syringe pumps (neMESYS, Cetoni, Germany) via PTFE tubing (inner diameter: 0.56 mm, outer diameter: 1.07 mm, Novodirect, Germany).
Diffusion studies on hydrogel microbeads
The obtained hydrogel microbeads with varying compositions were dispersed in a fluorescein isothiocyanate-dextran (FITC-dextran, Mw 2 × 106 g mol−1) solution (50 μg mL−1) in PBS and incubated at 4 °C for overnight. Subsequently, confocal fluorescence images and differential interference contrast (DIC) images were taken on the equilibrated dispersion on a Leica SP2 confocal laser scanning microscope. Fluorescence signals detected from the inside of the hydrogel microbeads were used to indicate gel permeability.2D cell culture
Human mesenchymal stem cells (MSCs) were thawed from cryopreservation (10% DMSO) and cultured in Dulbecco's modified Eagle's medium (DMEM) low glucose (1 g L−1) medium supplemented with 10% fetal bovine serum (MSC approved FBS, Invitrogen), and 1% penicillin/streptomycin (p/s). The medium was changed every 3 days and cells were passaged at nearly 80% confluency using 0.25 mg mL−1 trypsin:EDTA (Lonza, Switzerland). hMSCs of passage 6 were used for the microfluidic encapsulation experiments.
Fabrication of cell-laden hydrogel microbeads
Thiolated hyaluronic acid (HASH), poly(ethylene glycol) divinylsulfone (PEGDVS, JenKem, Mw 5000 g mol−1), and fibrinogen from bovine plasma (Sigma Aldrich) were dissolved in autoclaved PBS buffer at desired concentrations. All the materials handling was performed in sterile laminar flow hoods and sterile filtered with 0.2 μm filter units when necessary. hMSCs were mixed with HASH and fibrinogen at a final concentration of 1 million cells mL−1 and the mixture was injected into a microfluidic flow-focusing device as one of the inner phases, which was sent together with the PEGDVS solution as the second inner phase. Fluorinated oil (HFE 7500, 3M) containing 2% (w/w) of home-made triblock copolymer surfactant (Krytox–Jeffamine–Krytox) was used as the outer phase.14,21 The flow rates were set to 1200 μL h−1 for the outer phase and 200 μL h−1 for each of the inner phases. The emulsion was collected in an Eppendorf tube covered with Parafilm and subsequently incubated on a thermo shaker at 37 °C for max 20 min to complete the thiol–ene crosslinking reaction. Hydrogel microbeads were obtained by breaking the emulsion with 1H,1H,2H,2H-perfluoro-1-octanol (20% v/v in HFE 7500) and immediately transferring the microbeads into PBS.36 The hydrogel microbead suspension was washed two times with autoclaved PBS, and one time with hMSC proliferation medium, each time followed by centrifugation at 300g for 10 min.Micromechanical characterization
The Young's modulus E of the hydrogel microbeads was obtained by measuring force-displacement curves on an atomic force microscope (AFM, Nanowizard I, JPK Instruments, Germany) with spherical glass probes (with a radius of ∼23 μm) coated with poly(L-lysine)-g-PEG, combined with an inverted optical microscope (Axiovert 200, Zeiss, Germany), as previously reported.20 The spring constant of the utilized cantilever was 0.0281 N m−1 for soft beads (with HASH concentrations of 0.75% and 1.0% w/w) and 0.5216 N m−1 for stiffer beads (with HASH concentrations of 1.5% and 2.0% w/w). All the measurements were carried out at room temperature in PBS. The force-deformation curves were obtained by correcting the original force-displacement data for cantilever bending, which were fitted by the Hertz model to yield corresponding E values. From each batch at least 20 different beads were measured to obtain statistically significant mean values.3D culture of hMSCs in hydrogel microbeads and lineage analysis
hMSC-laden hydrogel microbeads were dispersed in hMSC proliferation medium and incubated at 37 °C (5% CO2) for 24 hours before the medium is replaced with a 1:1 combination of osteogenic and adipogenic chemical supplements. With each change of medium, the hydrogel microbeads were collected by centrifugation at 300g for 10 min. After certain periods (7–14 days) in culture, lineage specification was assessed by in situ staining for alkaline phosphatase (ALP) activity (osteogenic biomarker) and neutral lipid accumulation (functional adipogenesis biomarker) in separate samples. hMSC containing hydrogel microbeads were first fixed with 4% parafomaldehyde (PFA) in PBS, and then washed 2× with PBS before they were embedded in a thin layer of secondary hydrogel made of agarose (2% in PBS) inside Lab-Tek chamber slides (8 wells, Thermo Scientific Nunc). The chamber slides were subsequently kept at 4 °C for at least 1 h for the agarose to solidify. ALP activity was visualized by Fast Blue staining (500 μg mL−1 naphthol-AS-MSC phosphate, NAMP, and 500 μg mL−1 Fast Blue BB, Sigma) in alkaline buffer (100 mM Tris-HCl, 100 mM NaCl, 0.1% Tween-20, 50 mM MgCl2, pH 8.2) and incubation at 37 °C (5% CO2) for 1 h. Accumulated oil droplets in cells were stained with Oil Red O (ORO, 600 μg mL−1 in isopropanol) at room temperature for 1 h. Color images were acquired on a Zeiss inverted microscope fitted with a Coolsnap 5M color camera (Photometrics, USA). Cells with/without specific markers were counted manually. On average, 400–500 beads were collected in each well, out of which approximately 500 cell-containing beads were counted in total to obtain the statistics.
Live/dead, cytoskeleton and nuclear morphology staining
hMSC-laden hydrogel microbeads were cultured in proliferation medium, washed and suspended in PBS and mixed with a Live/Dead Viability/Cytotoxicity kit for mammalian cells (Invitrogen) at room temperature for 1 h at different time points for viability evaluations for short (24 hours) and long terms (14 days). A final concentration of calcein AM of 2 μM and ethidium homodimer-1 (EthD-1) of 4 μM was used. The live/dead stained samples were imaged on a Leica SP2 confocal laser scanning microscope with a 10× objective.Cytoskeletal and nuclear morphology of encapsulated live cells were stained 24 hours after encapsulation. All representative images were taken from the axial plane showing the largest cross-sectional area. F-actin was stained with TRITC-phalloidin (Actin Cytoskeleton/Focal Adhesion Staining Kit, Millipore, 1:1000) and nuclear morphology with 4,6-diamidino-2-phenylindole (DAPI, Actin Cytoskeleton/Focal Adhesion Staining Kit, Millipore, 1:1000) at room temperature for 1 h. Images were taken with a Leica SP2 confocal laser scanning microscope (Leica, Germany) with a 63× oil objective.
Immunostaining
After inducing differentiation (from the start of culture in a 1:1 combination of osteogenic and adipogenic differentiation media) for 7 days, the hMSC-laden hydrogel microbeads were washed with PBS and fixed with 4% paraformaldehyde (PFA)/PBS solution for 10 min at room temperature, washed with PBS and permeabilized using 0.2% Triton X-100/PBS solution for 10 min at room temperature. After extensive washing in PBS, the microbeads were blocked with BSA in PBS (10 mg mL−1) for 1 h at room temperature. Afterwards, the beads were incubated with mouse anti-STRO-1 antibody37 (Invitrogen, 1:100 dilution with 10 mg mL−1 BSA) for 8 hours at 4 °C, washed with PBS (3×), and then incubated with secondary Alexa Fluor-488 goat anti-mouse IgG antibody (Invitrogen, 1:200 dilution with 10 mg mL−1 BSA) for 1 h at room temperature and washed to visualize multipotency biomarkers. DAPI was included in the secondary antibody solution to counter-stain the nuclei. Images were taken with a Leica SP2 confocal laser scanning microscope (Leica, Germany) with a 20× objective.
Results and discussion
Bulk hydrogel mechanical properties and gelling kinetics
As mentioned earlier, matrix mechanics is an important parameter directing hMSC fate. In order to realize a fine-tuning of hydrogel elasticity, the gelling properties of HASH and PEG-DVS with varying concentration combinations in the presence of a fixed concentration of FBNG (1 mg mL−1) were studied in detail by rheology with a parallel plate geometry. In all the experiments, HASH with a fixed degree of thiolation (25%) and PEG-DVS with a fixed Mw of 5000 g mol−1 were used. The reaction was carried out in PBS with a physiological pH value of 7.4 without any additional catalyst or trigger. It should be mentioned that we chose to use PEG-DVS as the crosslinking agent for hMSC encapsulation due to the fact that hydrogel microbeads of similar size obtained through HASH-PEGDA crosslinking degraded within a few days, and thus were not suitable for the long-term culture of hMSCs (unpublished data). This is most likely due to the hydrolysis of the ester bond formed during crosslinking, which appeared to be more pronounced in hydrogels in a microbead format with its higher surface to volume ratio than in bulk. However, this combination may be useful for applications directed at controlled release of living cells.19 Time-dependent rheology measurement provides important information on the bulk gelling kinetics and the final modulus of HASH-PEGDVS-FBNG hydrogels. As shown in Fig. 2, the maximum storage modulus G′ (the plateau region in the curves, measured at a fixed oscillation frequency of 1 Hz) of HASH-PEGDVS-FBNG hydrogels with a fixed concentration of FBNG (1 mg mL−1) increases with increasing the concentration of HASH and PEGDVS in a nearly linear fashion (Fig. 2B). The elastic modulus or Young's modulus E, which is an important parameter in describing hydrogel mechanical properties, can be calculated based on the following equation:| E = 2G′(1 + ν) | (1) |
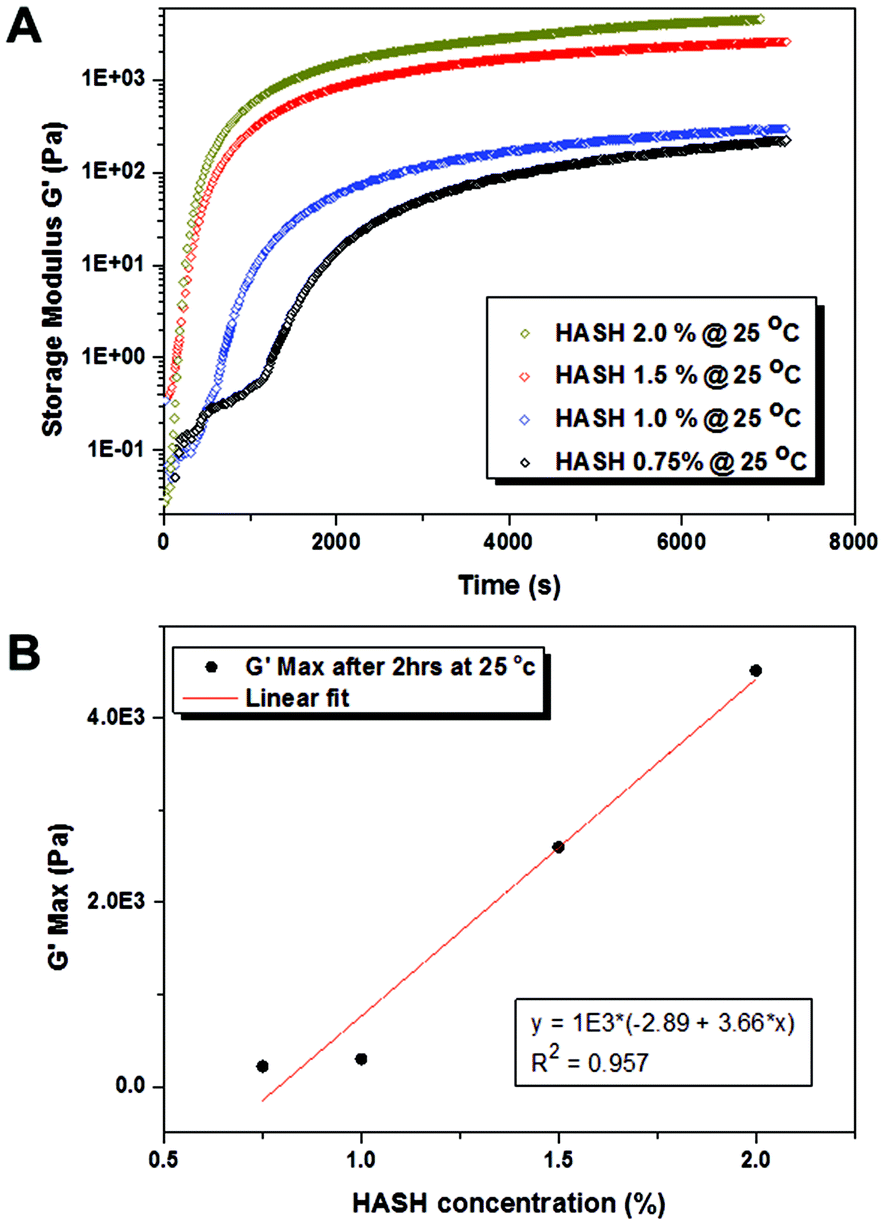 | ||
| Fig. 2 (A) Time-dependent rheological measurements on the storage modulus (G′) of bulk hydrogels made from varying concentrations of thiolated hyaluronic acid (HASH) (corresponding PEGDVS and FBNG concentrations used are listed in Table 1) at 25 °C. (B) The linear increase of plateau storage modulus (G′) with increasing HASH concentrations. | ||
An accurate determination of the gelation time is of great importance for the reliable fabrication of hMSC containing microbeads, as the duration between harvesting cells from 2D culture flasks and re-suspending cell-laden hydrogel microbeads in tissue culture medium should be minimized. The gel point tc was estimated from rheological measurements as the point where the storage modulus (G′) started to increase much more sharply than the loss modulus (G′′), marking the transition from liquid-like to solid-like behaviour (see examples in the ESI Fig. S2†). Table 1 summarizes the gel point of the HASH-PEGDVS-FBNG hydrogels with varying HASH/PEGDVS concentrations. Clearly the gel point decreases with increasing overall polymer concentration, indicating faster crosslinking kinetics with a higher concentration of functional groups. It is noteworthy that even with the lowest polymer concentration used in our study (0.75% w/w HASH), the gelation was fast (30 min) at room temperature. The gelling time can be further reduced by increasing the gelling temperature, for example to 37 °C (ESI Fig. S2†). In the droplet experiment, we used the time at which the gel reached its highest/plateau modulus as a guide for the time required for incubation, after which the emulsion was broken and the beads were transferred into tissue culture medium (vide infra).
| Sample | HASH (mg mL−1) | PEGDVS (mg mL−1) | FBNG (mg mL−1) | t c at 25 °C (min) |
|---|---|---|---|---|
| 1 | 7.5 | 12.5 | 1.0 | 30 |
| 2 | 10.0 | 16.7 | 1.0 | 15 |
| 3 | 15.0 | 25.0 | 1.0 | 7.5 |
| 4 | 20.0 | 33.3 | 1.0 | 7.5 |
Fabrication of hMSC-laden hydrogel microbeads
The hMSC-laden hydrogel microbeads were fabricated in a two-step procedure. First, freshly harvested hMSCs together with hydrogel precursors in PBS buffer were prepared and subsequently injected separately into a microfluidic flow-focusing device with three inlets for microdroplet generation. As shown in Fig. 1C, the continuous oil phase consisted of a fluorinated oil (HFE 7500) containing 1% (w/w) of a biocompatible surfactant (Krytox–Jeffamine–Krytox A–B–A triblock copolymer).14,21 The dispersed, aqueous phase was comprised of two separate streams that met at the flow-focusing junction with one containing HASH of varying concentrations, cells and FBNG as the cell-binding ligand, and the other one containing PEG-DVS of matching concentrations as the crosslinking agent. The final concentrations of HASH were varied between 0.75% and 2% (w/w). With a fixed FBNG concentration of 1 mg mL−1, the PEG-DVS was mixed in with a concentration aiming at the full conversion of the thiol functional groups (see Table 1). After their production, microdroplets flowed through a long, meander channel (see the inset of Fig. 1B) for a thorough mixing of the components before they were collected in sterile Eppendorf tubes. The number of cells in each droplet roughly follows a Poisson distribution,36 with slight deviations due to non-specific clustering of the cells. Approximately 30% of the microbeads obtained contained a single cell and the rest of the beads contained either no cells or multiple cells (probability values and corresponding calculations are presented in ESI Fig. S5† and related text). Subsequently, these hMSC encapsulated hydrogel precursor droplets were gelled at 37 °C for a short period of time (≤20 min) before they were washed and transferred into tissue culture medium for the 3D culture of the cells. The average size of the obtained hydrogel microbeads was 150 μm. A typical transmission microscopy image of an hMSC-laden HASH-PEGDVS-FBNG microbead sample is shown in Fig. 3A. | ||
| Fig. 3 Chemical and physical characteristics of the prepared microniches. (A) Typical wide field optical images of hMSC containing hydrogel microbeads with a uniform size of ∼150 μm. (B) Confocal laser scanning micrograph on hydrogel microbeads made from HASH, PEGDVS and rhodamine B isothiocyanate (RBITC) labelled FBNG. The stable and relatively homogeneous red fluorescence emission from all over the beads suggests the successful incorporation of FBNG and its strong interactions with the hydrogel matrix. (C) Representative confocal laser scanning micrographs showing that the obtained microniches (with a HASH concentration of 1.5% in this case) are permeable to fluorescein labelled dextran with a Mw of 2 × 106 g mol−1 (corresponding hydrodynamic diameter of 54 nm). Left: fluorescence image; right: DIC image. (D) Young's modulus E of hydrogel microbeads obtained by the colloidal probe technique on an atomic force microscope (AFM) plotted against corresponding HASH concentrations. Stiffer gel beads are obtained with a higher concentration of HASH, in good agreement with the above rheological measurements. Scale bars for (A)–(C) are all representing 100 μm. Error bars in (D) correspond to plus/minus one standard deviation. Dotted-line circles were added in C to guide the outer periphery of the gel beads due to the poor visibility originating from their density matching characteristics to water. | ||
Chemical and physical characteristics of HASH-PEGDVS-FBNG hydrogel microbeads
It is known that HA has specific interactions with FBNG.39 It was suggested that the origin of HA–FBNG interaction was due to electrostatic interactions between the anionic polysaccharide and the charged protein, the result of which may be actively involved in the organization of the extracellular space.40 In order to verify the binding of FBNG to the hydrogel microbeads, FBNG was first labelled with rhodamine B isothiocyanate (RBITC) in an amine-free buffer and subsequently hydrogel microbeads were fabricated microfluidically using the RBITC-FBNG conjugate instead of pure FBNG. Fig. 3B shows a confocal fluorescence microscopy image of the obtained microbeads. The red fluorescence signal from all over the microbeads strongly indicates the uniform presence of FBNG in the hydrogel microbeads, which is not removed after washing. The strong binding of FBNG to the hydrogel matrix ensures the availability of cell surface binding sites for the 3D culture of the cells.The porosity of hydrogels is a very important environmental parameter for cell viability. Large pores are generally preferred as they facilitate the efficient transport of nutrients, carbon dioxide, oxygen and even the migration of cells.9 Here, the porosity of the HASH-PEGDVS-FBNG hydrogel microbeads was qualitatively determined by measuring the diffusion of fluorescein isothiocyanate labelled dextran (FITC-dextran) with varying molar masses. Fig. 3C shows that even dextrans with a Mw of 2 × 106 g mol−1 (corresponding hydrodynamic diameter of 54 nm) readily diffused into all the hydrogel microbeads with varying HASH concentrations from 0.75% to 2% (additional images are provided in ESI Fig. S3†), indicating that the average pore size of the hydrogel matrix is sufficiently large for the diffusion of nutrients and waste molecules.
The swelling ratios Q of the hydrogel microbeads were obtained by dividing the swollen mass of the hydrogel microbeads by their corresponding dry mass, based on the initial polymer concentrations, flow rates, droplet generation frequency and final size/volume of the fully swollen beads in MilliQ. Irrespective of the polymer concentration, the HASH-PEGDVS-FBNG displayed Q values of 24–46 (detailed data and calculations can be found in the ESI†).
The elastic/Young's modulus (E) of the hydrogel microbeads was characterized by the colloidal probe technique on an atomic force microscope (AFM), as previously reported.20 The colloidal probe ensures a well-defined tip–sample contact geometry resulting in a uniform sample deformation profile. The measured force-deformation curves with deformations less than 10% of the bead diameters were evaluated by fitting with the Hertz model for the elastic deformation of two spheres in contact. A significant increase in hydrogel stiffness with increasing HASH concentrations can be directly observed from the obtained force-deformation curves (see examples in ESI Fig. S4†). Fig. 3D shows the obtained E values as a function of HASH concentrations. It is observed from the figure that the average E can be varied between ∼0.86 (±0.29) and 9.2 (±2.9) kPa, which is not only well in the range of elasticity of solid tissues but also fitting extremely well the rheological measurements on bulk gels (vide supra).
hMSC viability in hydrogel microbeads
The short- (24 hours) and long-term (2 weeks) viability of encapsulated hMSCs in microbeads with HASH concentrations of 1.5% and 2% were studied in detail by live–dead assays. hMSC-laden microbeads with HASH concentrations below 1.5% degraded within one week in culture, and thus were not studied further for the 3D culture of hMSCs. For the viability studies hMSC-laden hydrogel microbeads were cultured in proliferation medium in order to minimize their differentiation. The short-term viability mainly indicates the influence of the microfluidic microbead fabrication procedure on hMSC viability. These include the toxicity of the chemicals involved, the duration of the procedure as well as the shear force applied on the cells during droplet fabrication. In our study, the short-term viability of hMSCs in the HASH-PEGDVS-FBNG beads is around 70% (see representative images in Fig. 4A), which is comparable to other types of hydrogel microbead systems with encapsulated less sensitive cell types (such as fibroblasts) previously reported.18 The long-term viability and eventually the differentiation of the cells, on the other hand, depend more on the microniche conditions. Fig. 4B shows that after two weeks in culture, only live cells were stained. It is likely that dead cells simply went through disintegration. It was observed that in fact a large number of cells can be kept in culture in the hydrogel microbeads for up to 4 weeks. To our knowledge, this is the first example of a hydrogel microbead system fabricated by droplet microfluidics that can sustain the long-term 3D culture of hMSCs at a single cell level.4,23,41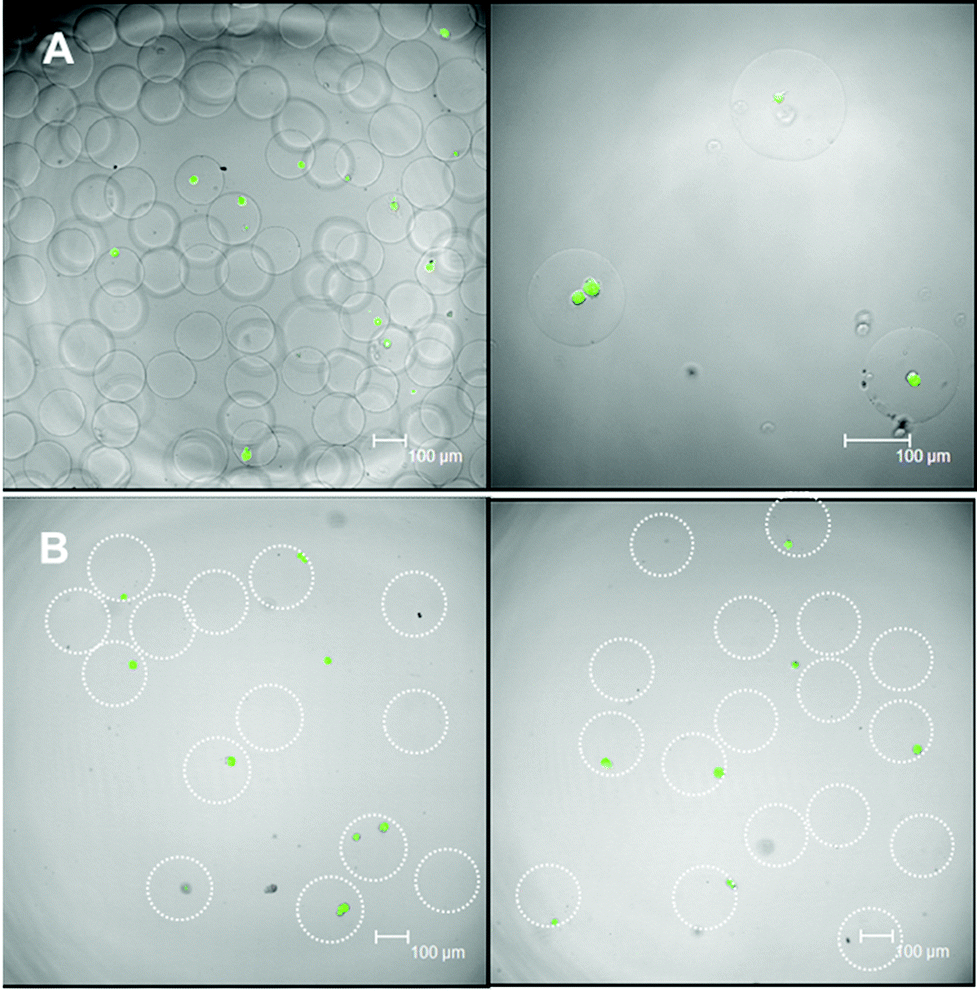 | ||
| Fig. 4 The short (A, 24 hours) and long (B, 2 weeks) term viability of hMSCs in the 3D culture of HASH based microniches characterized by confocal laser scanning microscopy after live–dead staining. The green fluorescence signals of calcein from the inside of many of the encapsulated hMSCs indicate that those are living cells. Scale bars are 100 μm for all images. Dotted-line circles were added in B to guide the outer-periphery of the gel beads due to the poor visibility originating from their density matching characteristics to water. | ||
Single hMSC morphology in 3D
In contrast to 2D culture where matrix rigidity affects hMSC morphology and eventually cell fate,5 it has been shown that this is not the case in 3D. hMSCs appeared to be spherical in 3D culture in a variety of hydrogel matrics,10,12 independent of matrix stiffness. In order to examine the hMSC morphology in our system, TRITC-phalloidin was used to stain the F-actin cytoskeleton of the encapsulated hMSCs, together with DAPI staining of the nucleus. Fig. 5 shows typical examples of stained hMSCs in the hydrogel microniches with varying HASH concentrations of 0.75–2%. Most of the cells embedded in FBNG containing gel beads showed a rounded morphology and micrometer-sized cortical protrusions towards the surrounding matrix. Protrusions were also observed when cells were embedded in control samples with no FBNG present in the microniches (see ESI Fig. S6†), though not as prominent. In fact, HA is known to contain receptors for the cell surface protein CD44.42 However, additional integrin binding sites are usually included in artificial hydrogel matrices made from HA.11,12,28 The hMSCs remained rounded throughout the culture, which suggests a lack of matrix degradation mechanisms in the studied hydrogel system.12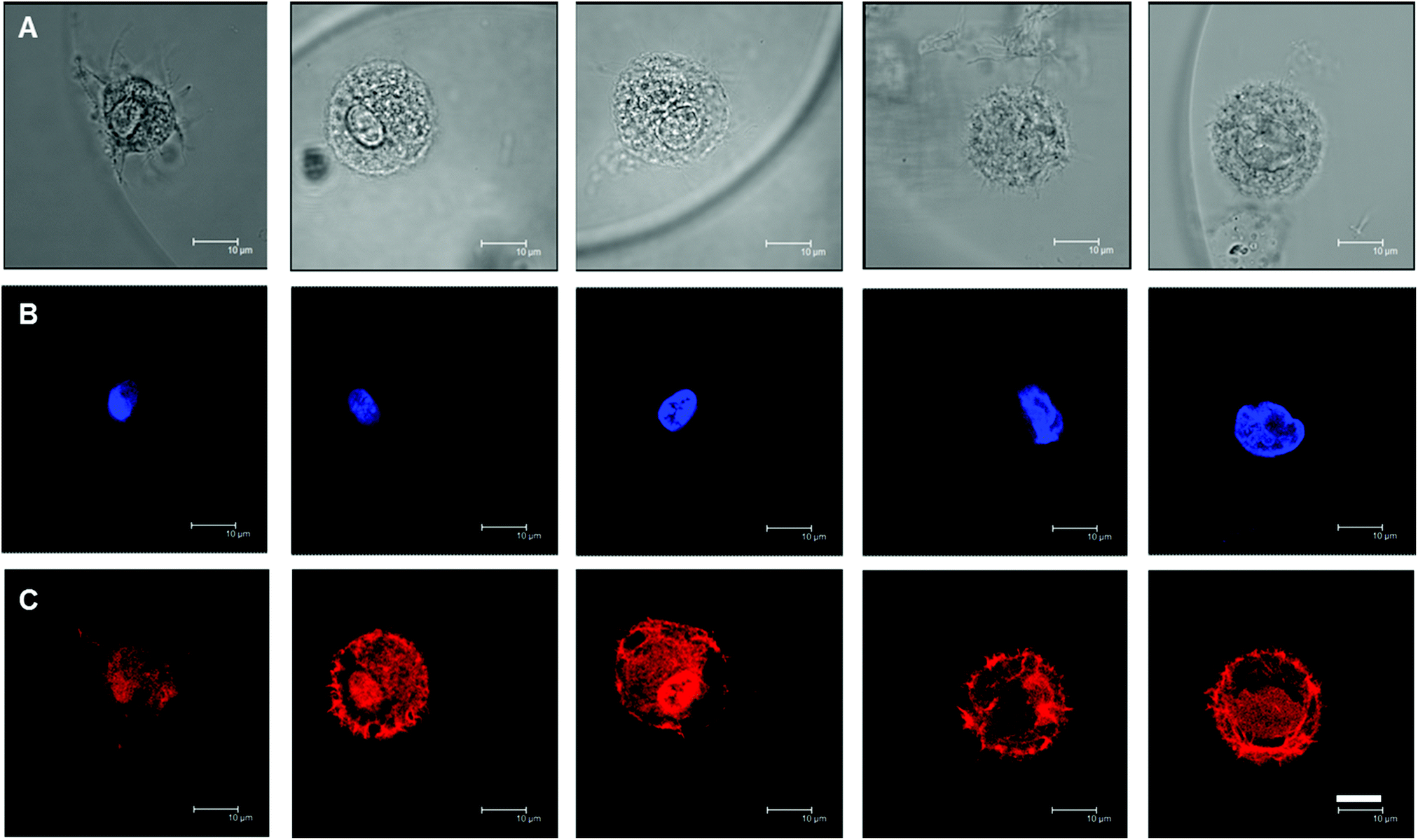 | ||
| Fig. 5 Confocal laser scanning micrographs on cell and nucleus morphologies of encapsulated hMSCs in microniches containing FBNG and varying concentrations of HASH between 0.75% and 2%. DIC images are shown in A. DAPI (B: blue) and TRITC-phalloidin (C: red) were used to stain the nucleus and the F-actin cytoskeleton, respectively. Most of the cells show a rounded morphology and micrometer-sized cortical protrusions on the cells towards the surrounding matrix. No apparent correlation between cell or nucleus morphology with matrix stiffness is observed. Scale bars are 10 μm for all images. | ||
Multipotency of hMSCs in microniches
Sustained hMSC proliferation was observed on all the samples, with microscopy images clearly showing dividing cells in different mitotic phases (see examples in Fig. 6, 7 and ESI Fig. S7†). It is very important to know whether these cells also display a propensity to retain their naïve state. To do so, we first cultured the hMSC containing hydrogel microbeads (with a HASH concentration of 1.5% w/w) in bipotential differentiation medium (a 1:1 combination of osteogenic and adipogenic media) for 7 days, before staining the biomarker STRO-1 signifying hMSC multipotency through recognition of the surface antigen unique to this lineage by immunostaining.37 As shown in Fig. 6, the interior of many cells in beads displayed bright green fluorescence originating from the Alexa 488 labelled secondary antibody attached to the anti-STRO-1 primary antibody. This indicates that a considerable proportion (40–50%) of the hMSCs retained their multipotency at this time point even in the presence of inductions, a somewhat delayed differentiation as compared to those shown in previous studies by Mooney et al.10 This could be due to, for example, a lack of cell–cell communication signal in single cell cultures.3 The fact that cells did maintain their multipotency for a prolonged period of time of 7 days gives additional proof of the suitability of using these hydrogel microbeads as potential microniches for single hMSC differentiation studies.
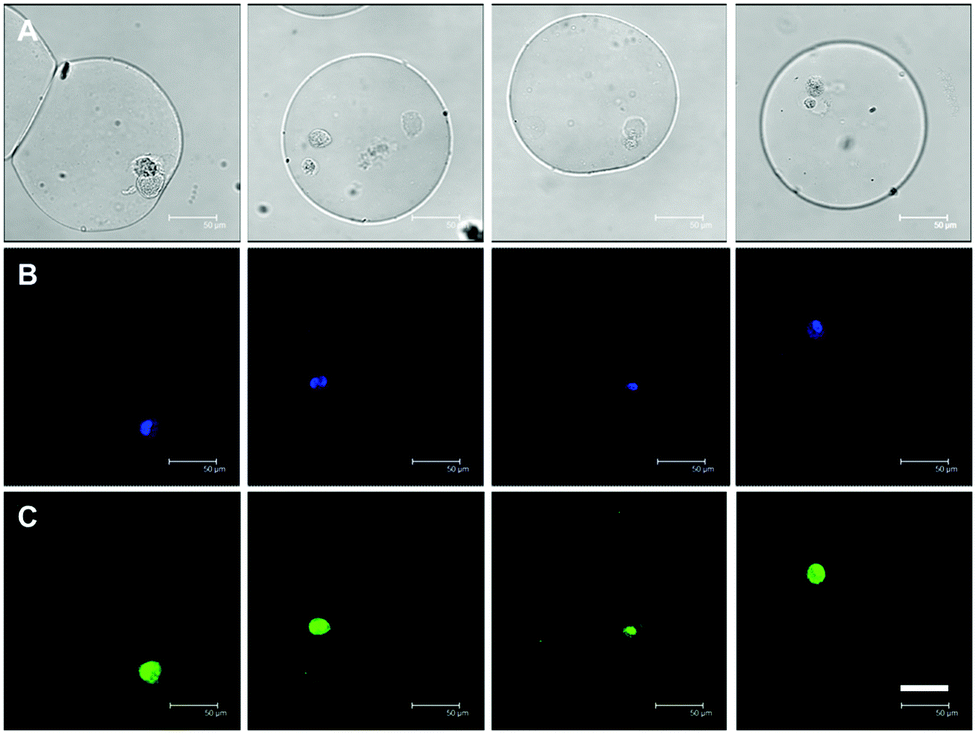 | ||
| Fig. 6 Multipotency of hMSCs embedded in microniches characterized by confocal laser scanning microscopy after immunostaininig. A HASH concentration of 1.5% is used in this experiment. After 7 days of culture in mixed induction medium containing a 1:1 ratio of osteogenic and adipogenic media, cells were stained with anti-STRO-1 (multipotency biomarker) antibody and Alexa 488 (C: green) labelled secondary antibody and the nuclei are stained with DAPI (B: blue). DIC images are shown in A. Scale bars are 50 μm for all images. | ||
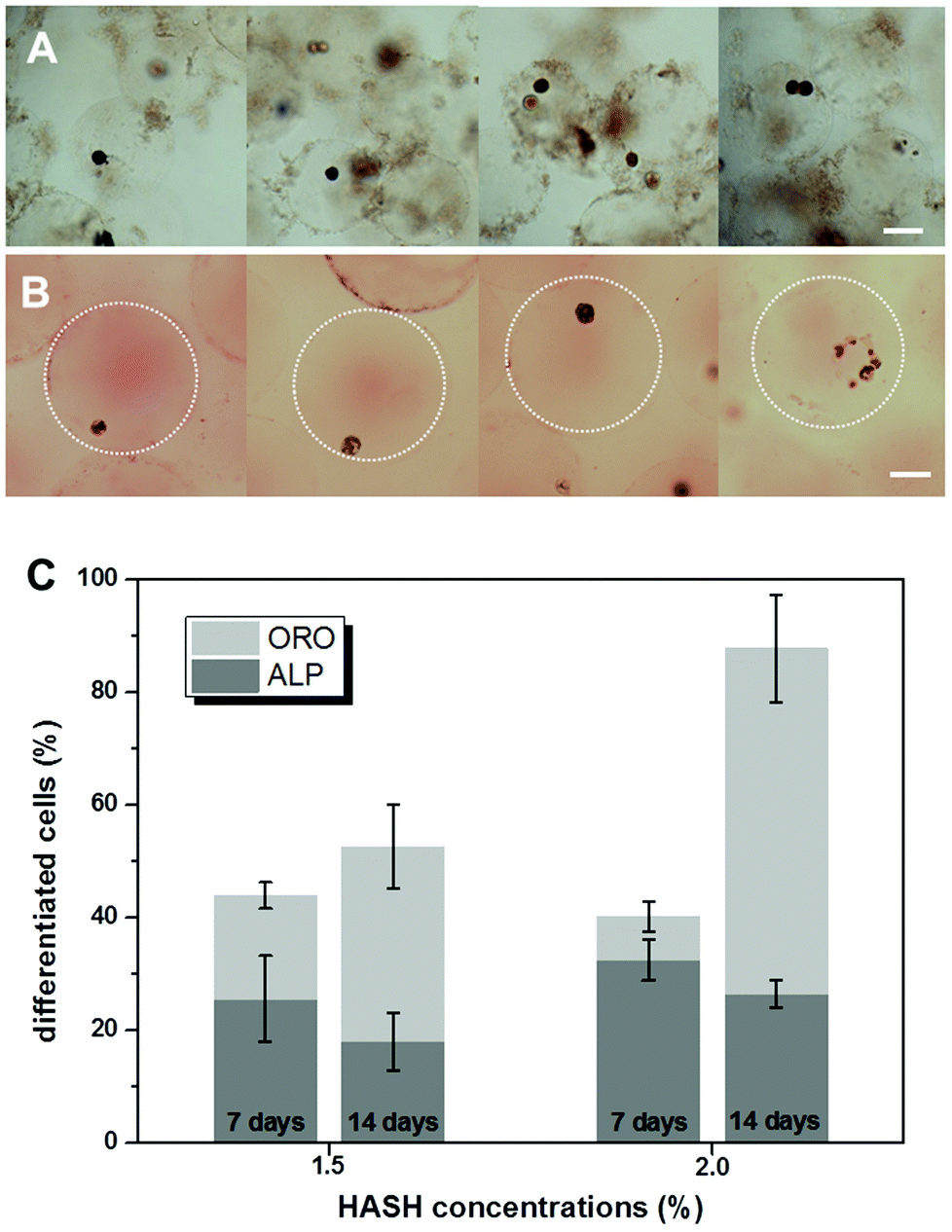 | ||
| Fig. 7
In situ staining of hMSCs embedded in microniches for (A) ALP activity (Fast Blue, osteogenic biomarker, blue) and (B) neutral lipid accumulation (Oil Red O, adipogenic biomarker, red) after 10 days of culture in mixed induction medium containing a 1:1 ratio of osteogenic and adipogenic media. HASH concentrations of 1.5% and 2.0% (w/w) are used in this experiment. Scale bars are 100 μm for all images. Dotted-line circles were added in B to guide the outer periphery of the gel beads due to the poor visibility originating from their density matching characteristics to water. (C) The percentages of differentiated cells embedded in hydrogel microbeads with respect to HASH concentrations and duration of culture (7 days and 14 days) in a 1:1 mixture of osteogenic and adipogenic bipotential differentiation media. The statistics was based on at least 2000 cells in each sample without excluding multi-cell containing hydrogel microbeads. | ||
hMSC differentiation in microniches
Finally the phenotypes of differentiated hMSC in microniches with HASH concentrations of 1.5% and 2.0% (w/w) were analyzed after culturing them in bipotential differentiation medium for 7 and 14 days, respectively. Osteogenesis is reflected by alkaline phosphatase (ALP) activity and adipogenesis by the presence of lipid droplets, which is visualized by Oil Red O (ORO) staining. Typical color micrographs of stained hydrogel microbead samples in suspension are presented in Fig. 7. The dark blue color from the cells in Fig. 7A demonstrates elevated ALP activity, indicating their differentiation into osteoblasts, and the red color from the developed oil droplets in the cells in Fig. 7B shows the differentiation into adipocytes.The microbeads appeared to become larger after ORO staining, probably due to the change of the medium to a mixture of isopropanol and water. A clear heterogeneity of cell differentiation was observed even when separate cells were encapsulated in the same bead. The number of differentiated cells was manually counted in ALP and ORO stained samples and the data are presented in Fig. 7C. The statistics was based on at least 2000 cells for each sample. After 7 days, over 50% of the cells did not show clear differentiation, in accordance with the above immunostaining results on the multipotency of the cells at this stage. For cells cultured in microniches with the same elasticity/HASH concentration, more cells were shown to have differentiated after 14 days. In addition, it is rather obvious that all the hydrogel matrices prepared in our study mainly support the preferential differentiation into adipocytes. For example, in the hydrogel microbeads of a HASH concentration of 2%, over 60% of the cells were stained ORO positive after 14 days. It should be mentioned that the Young's modulus of hydrogel beads prepared from a HASH concentration of 2% of around 10 kPa is just at the border of supporting a preferential osteogenic differentiation under the same induction conditions.9 It is thus not surprising that cells embedded in microbeads from a HASH concentration of 1.5% showed a similar preferential behaviour after 14 days. The apparent slight decrease in the percentage of osteoblasts could result from proliferation of the cells over such a prolonged period of time, whose effect was not studied in detail in the current study. These results agree well with previous bulk studies on hydrogels from other materials with similar mechanical characteristics.
Conclusions and future perspective
Bioorthogonal thiol–ene click chemistry based on thiolated hyaluronic acid (HASH) was successfully combined with droplet microfluidics as a powerful and extremely mild approach to prepare biocompatible hydrogel microbeads containing fibrinogen (FBNG) as additional cell binding sites for the 3D culture of single hMSCs. The mechanical properties of hydrogel microbeads made from HASH, poly(ethylene glycol) divinylsulfone (PEG-DVS) and FBNG of 0.9–9.2 kPa nicely fall in the softer side of the elasticity of solid tissues, which can be tailored by varying the concentration of the polymer precursor solutions. Acting like microniches, these beads are the first examples supporting the long term culture of hMSCs up to 4 weeks. Single hMSCs embedded in the HASH-PEGDVS-FBNG microniches displayed an overall rounded morphology, independent of matrix mechanics. The multipotency and differentiation potential of the hMSCs were characterized separately by various staining procedures, showing the successful preservation of hMSC multipotency in the whole process of fabrication and 3D culture. The soft hydrogel microbeads supported a preferential differentiation of hMSCs into adipocytes after 14 days of culture in osteo/adipo bipotential differentiation medium. A clear heterogeneity in the cell population can be observed in the 3D cell culture samples, which holds great promise for deepening our understanding of stem cell differentiation at the single cell level.As a future perspective, a further and more quantitative study on the proliferation and differentiation of hMSCs in the microniches can be achieved by combining cell encapsulation by droplet microfluidics with fluorescence-activated cell sorting (FACS), either after emulsion breaking or after staining.13 In this way even more sophisticated staining procedures could be applied as well. It would be ideal to study the full differentiation potential of these hMSCs by real time PCR at different stages of differentiation, preferably also at the single cell level. It is believed that the morphology and differentiation behavior of hMSCs are strongly dependent on cytoskeletal contractibility,43 matrix degradability12 and possibly cell–cell interactions.3,44,45 HASH used in our context is particularly adaptable to various (bio)chemical cues, whose effects can be studied separately. The radical-free thiol-Michael addition further provides unprecedented possibilities for identifying the key microenvironmental perturbations in defining stem cell fate as a variety of proteins and peptides can be easily incorporated by the same reaction mechanism. Moreover, HASH hydrogel microbeads used in the current study provide niches with mechanical properties in the soft side of solid tissues, which can be potentially extended to the stiffer region by increasing the degree of thiolation or molar mass of HASH used in the hydrogel precursors. As a natural ECM component, HA has been shown to specifically assist in the chondrolytic differentiation of hMSCs.46 A follow-up study in this direction is currently underway. We believe that when combined with modern cell-sorting platforms, our in vitro model based on hydrogel microbeads can be further improved to study regenerative stem cell bioengineering in a high throughput fashion.
Acknowledgements
The authors would like to thank Ms José M. A. Hendriks for assistance with cell cultures. J. T. is a Feodor-Lynen fellow of the Alexander von Humboldt Foundation. Work in the Huck group was supported by a European Research Council (ERC) Advanced Grant (246812 Intercom), a VICI grant from the Netherlands Organization for Scientific Research (NWO), and by funding from the Ministry of Education, Culture and Science (Gravity program 024.001.035).Notes and references
- F. P. Barry and J. M. Murphy, Int. J. Biochem. Cell Biol., 2004, 36, 568 CrossRef CAS PubMed.
- F. M. Watt and W. T. S. Huck, Nat. Rev. Mol. Cell Biol., 2013, 14, 467 CrossRef CAS PubMed.
- E. Fuchs, T. Tumbar and G. Guasch, Cell, 2004, 116, 769 CrossRef CAS.
- S. Gobaa, S. Hoehnel, M. Roccio, A. Negro, S. Kobel and M. P. Lutolf, Nat. Methods, 2011, 8, 949 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677 CrossRef CAS PubMed.
- T. P. Kraehenbuehl, R. Langer and L. S. Ferreira, Nat. Methods, 2011, 8, 731 CrossRef CAS PubMed.
- M. P. Lutolf and H. M. Blau, Adv. Mater., 2009, 21, 3255 CrossRef CAS PubMed.
- J. A. Burdick and W. L. Murphy, Nat. Commun., 2012, 3, 1269 CrossRef PubMed.
- J. Thiele, Y. Ma, S. M. C. Bruekers, S. Ma and W. T. S. Huck, Adv. Mater., 2014, 26, 125 CrossRef CAS PubMed.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518 CrossRef CAS PubMed.
- M. Guvendiren and J. A. Burdick, Nat. Commun., 2012, 3, 792 CrossRef PubMed.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458 CrossRef CAS PubMed.
- D. W. M. Tan, K. B. Jensen, M. W. B. Trotter, J. T. Connelly, S. Broad and F. M. Watt, Development, 2013, 140, 1433 CrossRef CAS PubMed.
- V. Chokkalingam, J. Tel, F. Wimmers, X. Liu, S. Semenov, J. Thiele, C. G. Figdor and W. T. S. Huck, Lab Chip, 2013, 13, 4740 RSC.
- H. N. Joensson and H. A. Svahn, Angew. Chem., Int. Ed., 2012, 51, 12176 CrossRef CAS PubMed.
- D. Velasco, E. Tumarkin and E. Kumacheva, Small, 2012, 8, 1633 CrossRef CAS PubMed.
- A. Kumachev, E. Tumarkin, G. C. Walker and E. Kumacheva, Soft Matter, 2013, 9, 2959 RSC.
- T. Rossow, J. A. Heyman, A. J. Ehrlicher, A. Langhoff, D. A. Weitz, R. Haag and S. Seiffert, J. Am. Chem. Soc., 2012, 134, 4983 CrossRef CAS PubMed.
- D. Steinhilber, T. Rossow, S. Wedepohl, F. Paulus, S. Seiffert and R. Haag, Angew. Chem., Int. Ed., 2013, 52, 13538 CrossRef CAS PubMed.
- S. H. Ma, M. Natoli, X. Liu, M. P. Neubauer, F. M. Watt, A. Fery and W. T. S. Huck, J. Mater. Chem. B, 2013, 1, 5128 RSC.
- Y. Ma, J. Thiele, L. Abdelmohsen, J. Xu and W. T. S. Huck, Chem. Commun., 2014, 4, 112 RSC.
- P. Agarwal, S. T. Zhao, P. Bielecki, W. Rao, J. K. Choi, Y. Zhao, J. H. Yu, W. J. Zhang and X. M. He, Lab Chip, 2013, 13, 4525 RSC.
- S. Allazetta, T. C. Hausherr and M. P. Lutolf, Biomacromolecules, 2013, 14, 1122 CrossRef CAS PubMed.
- Y. Du, E. Lo, S. Ali and A. Khademhosseini, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9522 CrossRef CAS PubMed.
- B. Trappmann, J. E. Gautrot, J. T. Connelly, D. G. T. Strange, Y. Li, M. L. Oyen, M. A. C. Stuart, H. Boehm, B. J. Li, V. Vogel, J. P. Spatz, F. M. Watt and W. T. S. Huck, Nat. Mater., 2012, 11, 642 CrossRef CAS PubMed.
- R. G. Wylie, S. Ahsan, Y. Aizawa, K. L. Maxwell, C. M. Morshead and M. S. Shoichet, Nat. Mater., 2011, 10, 799 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef CAS PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41 CrossRef CAS PubMed.
- X. Z. Shu, Y. C. Liu, Y. Luo, M. C. Roberts and G. D. Prestwich, Biomacromolecules, 2002, 3, 1304 CrossRef CAS PubMed.
- F. Rehfeldt, A. E. X. Brown, M. Raab, S. S. Cai, A. L. Zajac, A. Zemel and D. E. Discher, Integr. Biol., 2012, 4, 422 RSC.
- J. L. Young and A. J. Engler, Biomaterials, 2011, 32, 1002 CrossRef CAS PubMed.
- J. Thiele, Y. Ma, D. Foschepoth, M. M. K. Hansen, C. Steffen, H. Heus and W. T. S. Huck, Lab Chip, 2014 10.1039/c3lc51427g.
- P. A. Janmey, J. P. Winer and J. W. Weisel, J. R. Soc. Interface, 2009, 6, 1 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70 CrossRef CAS.
- D. B. Wolfe, D. Qin and G. M. Whitesides, Methods Mol. Biol., 2010, 583, 81 CAS.
- L. Mazutis, J. Gilbert, W. L. Ung, D. A. Weitz, A. D. Griffiths and J. A. Heyman, Nat. Protocols, 2013, 8, 870 CAS.
- P. J. Simmons and B. Torok-Storb, Blood, 1991, 78, 55 CAS.
- C. W. Macosko, Rheology: principles, measurements and applications, Wiley-VCH, 1994, ch. 1 Search PubMed.
- R. D. Leboeuf, R. H. Raja, G. M. Fuller and P. H. Weigel, J. Biol. Chem., 1986, 261, 2586 Search PubMed.
- M. Rinaudo, Int. J. Biol. Macromol., 2008, 43, 444 CrossRef CAS PubMed.
- A. Dolatshahi-Pirouz, M. Nikkhah, A. Gaharwar, B. Hashmi, E. Guermani, H. Aliabadi, G. Camci-Unal, T. Ferrante, M. Foss, D. E. Ingber and A. Khademhosseini, Sci. Rep., 2014, 4, 3896 Search PubMed.
- T. C. Laurent, U. B. G. Laurent and J. R. E. Fraser, Immunol. Cell Biol., 1996, 74, A1 CrossRef CAS PubMed.
- K. A. Kilian, B. Bugarija, B. T. Lahn and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4872 CrossRef CAS PubMed.
- C. C. Lin and K. S. Anseth, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6380 CrossRef CAS PubMed.
- M. Andreasson-Ochsner, G. Romano, M. Hakanson, M. L. Smith, D. E. Leckband, M. Textor and E. Reimhult, Lab Chip, 2011, 11, 2876 RSC.
- L. M. Bian, M. Guvendiren, R. L. Mauck and J. A. Burdick, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00104d |
| This journal is © The Royal Society of Chemistry 2014 |