Survival, differentiation, and migration of high-purity mouse embryonic stem cell-derived progenitor motor neurons in fibrin scaffolds after sub-acute spinal cord injury†
D. A.
McCreedy
,
T. S.
Wilems
,
H.
Xu
,
J. C.
Butts
,
C. R.
Brown
,
A. W.
Smith
and
S. E.
Sakiyama-Elbert
*
Department of Biomedical Engineering, Washington University in St. Louis, St. Louis, MO 63112, USA. E-mail: sakiyama@wustl.edu
First published on 30th June 2014
Abstract
Embryonic stem (ES) cells can be differentiated into many neural cell types that hold great potential in cell replacement therapies following spinal cord injury (SCI). Coupling stem cell transplantation with biomaterial scaffolds can produce a unified combination therapy with several potential advantages including enhanced cell survival, greater transplant retention, reduced scarring, and improved integration at the transplant/host interface. Undesired cell types, however, are commonly present in ES-cell derived cultures due to the limited efficiency of most ES cell induction protocols. Heterogeneous cell populations can confound the interaction between the biomaterial and specific neural populations leading to undesired outcomes. In particular, biomaterial scaffolds may enhance tumor formation by promoting survival and proliferation of undifferentiated ES cells that can persist after induction. Methods for purification of specific ES cell-derived neural populations are necessary to recognize the full potential of combination therapies involving biomaterials and ES cell-derived neural populations. We previously developed a method for enriching ES cell-derived progenitor motor neurons (pMNs) induced from mouse ES cells via antibiotic selection and showed that the enriched cell populations are depleted of pluripotent stem cells. In this study, we demonstrate the survival and differentiation of enriched pMNs within three dimensional (3D) fibrin scaffolds in vitro and when transplanted into a sub-acute dorsal hemisection model of SCI into neurons, oligodendrocytes and astrocytes.
Introduction
Spinal cord injury (SCI) is a traumatic event that leads to life-long debilitation. The loss of function following injury is associated with severed ascending and descending tracts, neuronal and oligodendrocyte cell death, and demyelination of spared axons.1–6 Transplantation of neural stem cells (NSCs) or ES cell-derived neural populations can improve remyelination and remodeling of local circuitry following SCI by replacing lost neuronal and oligodendrocyte populations.7–13 Several protocols have been developed for induction of ES cells into specific neural and spinal cord progenitor cell populations for use in the treatment of SCI.14–16 However, cell-based therapies for SCI are often impaired by poor cell survival and the predominant terminal differentiation of transplanted cells into glia.16–19Biomaterial scaffolds have been used as a vehicle for cell transplantation strategies following SCI to enhance cell viability and retention at the injury site.20–23 Many biomaterials, including fibrin, have been shown to reduce scarring at the transplant interface and may enhance integration of cell transplants.24–27 Fibrin scaffolds supplemented with a cocktail of growth factors enhanced the survival of transplanted cells derived from rat embryonic spinal cords, as well as human NSCs, following complete transection SCI in rats.28 Transplanted cells extended long distances into the host spinal cord. When modified with a heparin-based delivery system (HBDS), fibrin scaffolds containing various growth factor combinations have been shown to enhance differentiation of mouse ES cell-derived neural progenitor cells (ESNPCs) into neurons and oligodendrocytes in vitro.29 Fibrin scaffolds modified with the HBDS delivering neurotrophin-3 (NT3) and platelet-derived growth factor (PDGF-AA) improved short-term survival of transplanted ESNPCs and promoted differentiation into neurons and glia in a sub-acute (2 weeks) dorsal hemisection model of SCI.27 These studies suggest that fibrin is a suitable transplantation vehicle that can be engineered to improve survival and differentiation of neural cell populations within the injured cord.
The favorable transplantation environment established by engineered fibrin scaffolds, however, may enhance tumor formation in ES-cell derived transplants. Fibrin scaffolds containing NT3 and PDGF-AA promoted rapid proliferation and tumor formation in ESNPCs at eight weeks post-transplantation.30 Ki-67 is expressed in transplanted cells and often co-labeled with the mouse ES cell marker, stage specific antigen 1 (SSEA-1). Undifferentiated ES cells that persist following induction of ES cells into ESNPCs may be the main tumor-producing cell type. The tumorigenicity of persistent undifferentiated stem cells may explain teratoma formation observed in several other studies.31–33 Methods to purify ES cell-derived neural populations prior to transplantation may be necessary to reach the full potential of combination ES cell-based therapies.
Recently, we demonstrated simple and efficient purification of ES cell-derived progenitor motor neurons (pMNs) and motoneurons by antibiotic selection of transgenic ES cell lines.34,35 Antibiotic resistance driven by the gene regulatory elements of the pMN-associated transcription factor Olig2 was used to enrich cultures for pMNs and their recent progeny following addition of an antibiotic to the culture media. Survival and differentiation within the injured cord of ES-cell derived populations purified by antibiotic selection, however, have yet to be assessed. Furthermore, the effect of enrichment on the interaction between the stem cell transplant population and fibrin scaffolds has not been previously established.
To determine the potential of enriched ES cell-derived pMNs to serve as a potential therapeutic for SCI, we encapsulated cells within 3D fibrin scaffolds containing a combination of NT3 and PDGF-AA, or NT3 and glial-derived neurotrophic factor (GDNF) for two weeks in a sub-acute dorsal hemisection model of SCI. Growth factors were delivered using a HBDS consisting of heparin and a heparin-binding peptide. The bi-domain peptide crosslinks into the fibrin scaffold during polymerization through an N-terminal factor XIIIa substrate. The C-terminus contains an antithrombin III (ATIII)-based sequence to non-covalently bind heparin. Heparin can non-covalently bind to growth factors prolonging their release from fibrin scaffolds.36,37 Enriched pMNs survived within fibrin scaffolds and differentiated into motoneurons, oligodendrocytes, and astrocytes, while no over-proliferation was observed. Minimal scarring was present at the host–graft interface and migration of transplanted cells into the host cord was frequently observed. Furthermore, no undifferentiated ES cells were observed in transplanted cell grafts at two weeks post-transplantation. Antibiotic selection appears to provide a simple and effective method for producing high purity mouse ES-cell derived spinal progenitor cell populations that can be combined with biomaterial scaffolds for use as a potential treatment of SCI.
Results
Puromycin selection removes undifferentiated stem cells
Differentiation of ES cells into pMNs commonly results in a heterogeneous population of cells containing many undesired cell types including persistent undifferentiated ES cells. Using the P-Olig2 ES cell line, which expresses the puromycin resistance enzyme under the control of the Olig2 gene regulatory elements, undesired cell types, such as undifferentiated ES cells, can be depleted to produce high purity populations. P-Olig2 ES cells were genetically modified by random incorporation of a linear plasmid where the chicken beta-actin promoter was used to drive ubiquitous expression of the enhanced green fluorescent protein (GFP). The new cell line, G-POlig2, allowed for visual tracking of transplanted cells and was used for all experiments in this study. To determine the effect of puromycin exposure on EBs, we directed ES cells to form pMNs using a 2−/4+ RA and purmorphamine (Shh agonist) induction protocol in suspension culture as previously described35 (Fig. 1A). In selected cultures, puromycin was present during the last two days of the induction protocol (days 2−/2+–2−/4+). GFP expression was observed in unselected control cultures not receiving puromycin (Fig. 1C) as well as in selected cultures (Fig. 1I). EBs in unselected cultures (no puromycin) contained approximately ∼7500 cells whereas EBs in selected cultures contained ∼4000 cells, suggesting a loss of cells that did not express Olig2 during selection. Large regions devoid of Olig2+ pMNs or Hb9+ immature motoneurons, the recent progeny of pMNs, were observed in unselected cultures (Fig. 1D–G). Flow cytometry was performed on fixed cultures to determine the purity of cells contained within EBs after induction. The percentage of cells expressing Olig2 following the 2−/4+ induction increased significantly from 45.2 ± 3.5% to 67.1 ± 1.2% (Fig. 1N, p < 0.05, n = 3) with puromycin exposure. The percentage of Hb9+ cells at this same time point also increased from 13.9 ± 1.2% to 38.7 ± 1.3% (p < 0.05, n = 3) with puromycin selection. Puromycin exposure did not appear to increase the number of dead cells contained within EBs suggesting that undesired cell types were present at the outer layers of the EBs and were sloughed off during culture, as observed in previous studies (Fig. S1†). Oct4+ cells were occasionally observed within EBs in unselected cultures but were not observed in selected cultures (Fig. 2). Antibiotic selection of the G-POlig2 ES cell line provides a simple technique for purifying ES-cell derived pMNs (and their recent progeny) while depleting undifferentiated ES cells.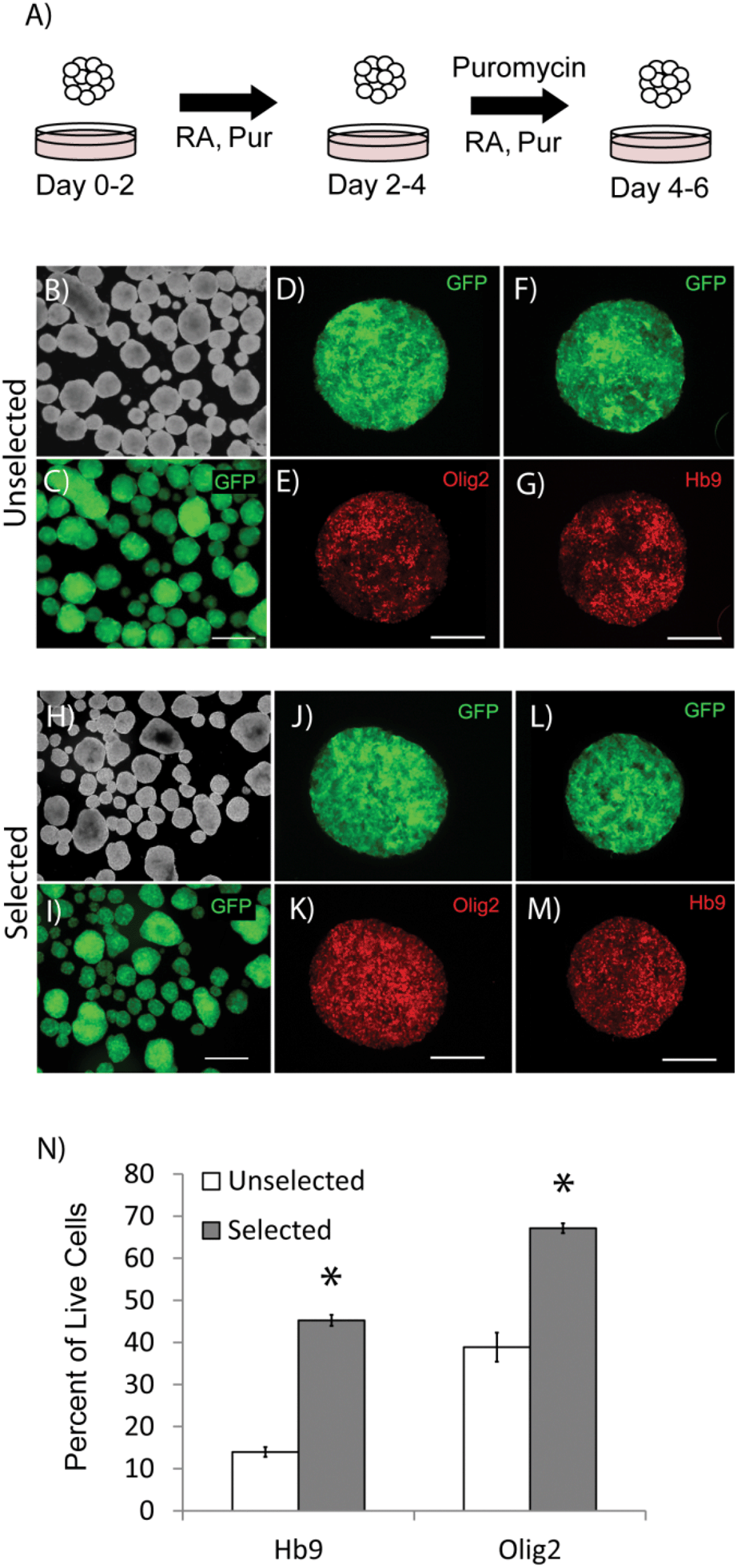 | ||
| Fig. 1 Antibiotic selection during differentiation of embryonic stem cells into progenitor motor neurons. (A) Schematic of the 2−/4+ RA and purmorphamine induction protocol. (B) Morphology of embryoid bodies (EBs) in control (unselected) cultures not receiving puromycin at the end of the 2−/4+ induction. (C) GFP expression in EBs from B. (D and E) GFP and Olig2 expression respectively in an individual unselected EB at the end of the 2−/4+ induction. (F and G) GFP and Hb9 expression respectively in an individual unselected EB. (H and I) Morphology and GFP expression in EBs from selected cultures at the end of the 2−/4+ induction. (J and K) GFP and Olig2 expression respectively in an individual selected EB at the end of the 2−/4+ induction. (L and M) GFP and Hb9 expression respectively in an individual selected EB at the end of the 2−/4+ induction. (N) Flow cytometry data for Olig2 and Hb9 markers at the end of the 2−/4+ induction in unselected and selected cultures. * indicates p < 0.05 compared to unselected controls. Scale bar for B, C, H, I = 500 μM. Scale bar for D–G, J–M = 250 μM. Abbreviations: green fluorescent protein (GFP), retinoic acid (RA), purmorphamine (Pur). | ||
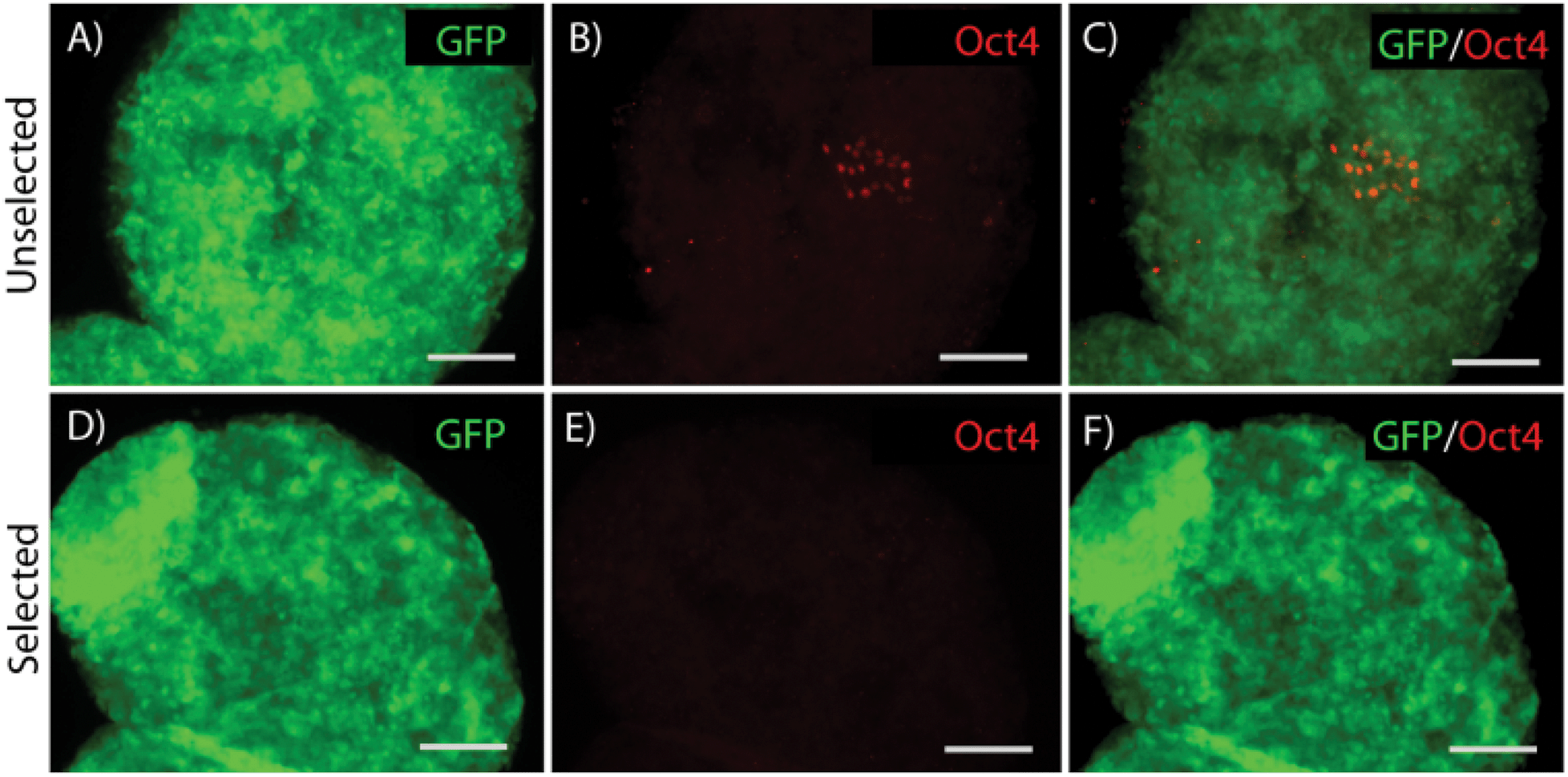 | ||
| Fig. 2 Oct4 expression in EBs at the end of the 2−/4+ induction. (A) GFP expression in a single EB from unselected cultures. (B) Corresponding Oct4 expression from EB in A. (C) Merged view of GFP and Oct4 labeling in the unselected EB. (D) GFP expression in a single EB from selected cultures. (E) Corresponding Oct4 expression from EB in D. (F) Merged view of GFP and Oct4 labeling in the selected EB. Scale bar = 200 μM. Abbreviations: green fluorescent protein (GFP). | ||
High purity pMNs survive and differentiate in fibrin scaffolds in vitro
To determine the effect of antibiotic selection on the subsequent differentiation of selected ES cell-derived pMNs into mature neural cell types (neurons, oligodendrocytes, and astrocytes) within 3D culture, EBs containing pMNs were selected with puromycin in suspension culture to remove undesired cells and further differentiated within fibrin scaffolds for two weeks with a gradual transition to serum-free media (Fig. 3A). The focus of this study was to mimic conditions that might be observed when the selected pMNs are transplanted after spinal cord injury and explore the effects of controlled growth factor delivery on survival and differentiation. Scaffolds contained either unmodified fibrin (Fibrin) or fibrin containing the following delivery system components: heparin and ATIII peptide (DS); heparin, NT3, and PDGF (Hep + NT3 + PDGF); heparin, NT3, and GDNF (Hep + NT3 + GDNF); heparin, ATIII, NT3, and PDGF (DS + NT3 + PDGF); or heparin, ATIII, NT3, and GDNF (DS + NT3 + GDNF) (n = 3 for all conditions). The same fibrin scaffold conditions were repeated for unselected control cells. In all groups and conditions, GFP+ axons and cells were observed extending processes and/or migrating from EBs into the fibrin scaffold (Fig. 3B–E). Glial cells including glial fibrillary acidic protein (GFAP)+ astrocytes and O4+ oligodendrocytes were observed (Fig. 3B and C). NeuN+ neurons and cholinergic axons were also present under all conditions (Fig. 3D and E).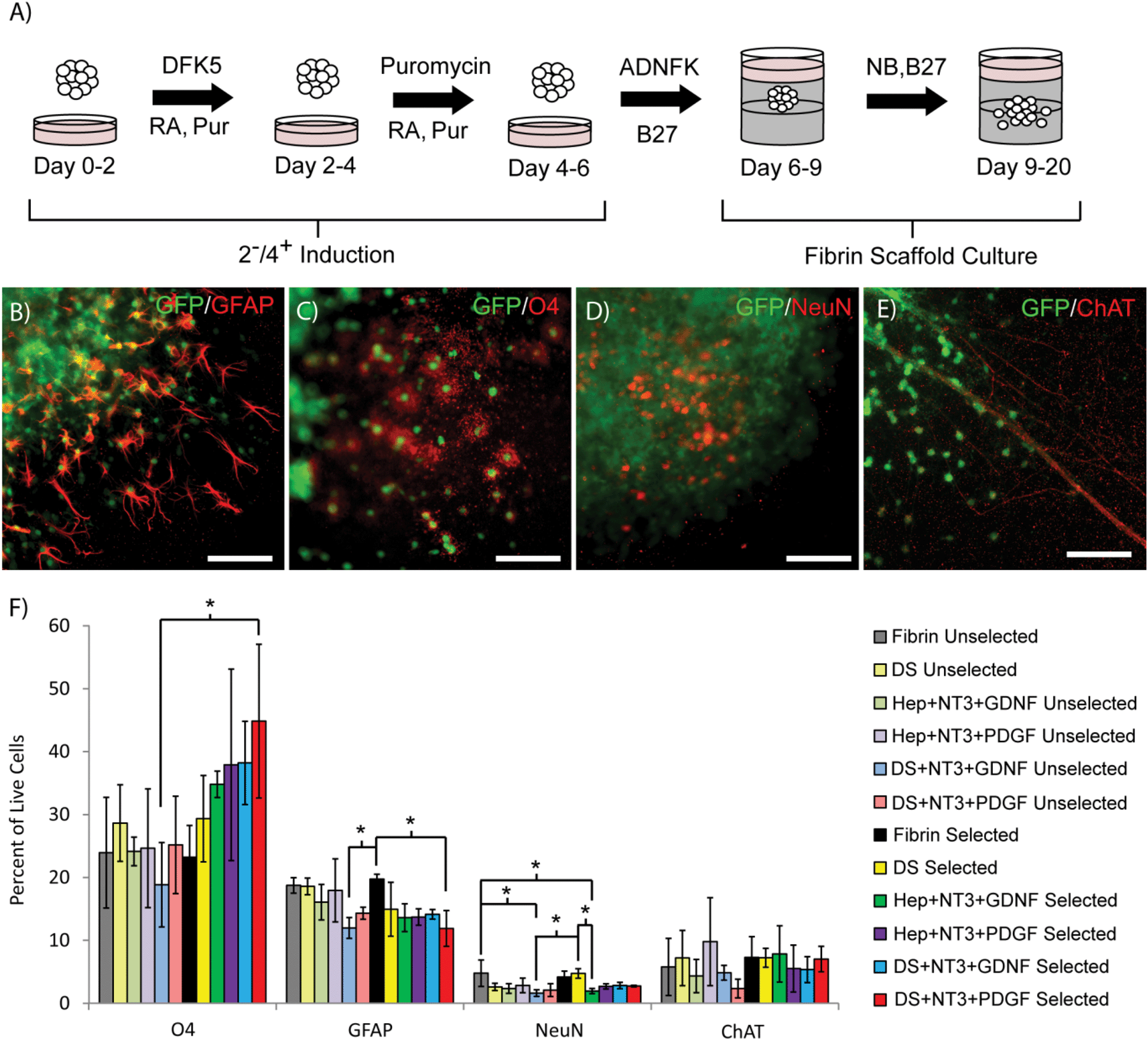 | ||
| Fig. 3 Cell survival, migration, and differentiation following two weeks of in vitro culture within fibrin scaffolds. (A) Schematic of 2−/4+ induction followed by two weeks of differentiation in vitro within 3D fibrin scaffolds. (B–E) Differentiation of pMNs into astrocytes expressing GFAP (B), oligodendrocytes expressing O4 (C), neurons expressing NeuN (D), and motoneurons expressing ChAT (E) in the DS + NT3 + GDNF fibrin group. (F) Flow cytometry results for various fibrin scaffold conditions. * indicates p < 0.05. Scale bar = 100 μM. Abbreviations: purmorphamine (Pur), glial fibrillary acidic protein (GFAP), oligodendrocyte marker 4 (O4), neuronal nuclei (NeuN), choline acetyltransferase (ChAT), green fluorescent protein (GFP), delivery system (DS), heparin (Hep), neurotrophin-3 (NT3), glial-derived neurotrophic factor (GDNF), platelet derived growth factor (PDGF). | ||
To characterize the percentage of cells differentiating into astrocytes, oligodendrocytes, neurons, and motoneurons, flow cytometry was performed following the two week in vitro differentiation period within fibrin scaffolds after induction of the EBs in suspension culture (day 20 in Fig. 3A). Two different combinations of growth factors were tested (NT3 + GDNF and NT3 + PDGF) as these combinations have been shown previously to promote survival and differentiation of neurons and oligodendrocytes. The percentage of cells labeling with ChAT (choline acetyltransferase, motoneurons) was unchanged in selected and unselected groups for all of the fibrin scaffold conditions tested (Fig. 3F). A significant decrease in the percentage of NeuN+ neurons was observed in the unselected Fibrin group and the selected DS group compared to the unselected DS + NT3 + GDNF group and the selected Hep + NT3 + GDNF group. Overall, the percentage of NeuN+ neurons and ChAT+ motoneurons was low (<10%) in all groups. A significant decrease in the percentage of cells staining with GFAP (astrocytes) was observed in the selected Fibrin group compared to the unselected DS + NT3 + GDNF and the selected DS + NT3 + PDGF groups, as well as the unselected DS group. The addition of the two combinations of growth factors tested appeared to decrease differentiation into neurons and astrocytes, and significantly increased O4 (oligodendrocytes) labeling was observed in some cases (the selected DS + NT3 + PDGF group compared to the unselected DS + NT3 + GDNF group). Oligodendrocyte differentiation was unchanged in the unselected groups while the presence of growth factors appeared to improve oligodendrocyte differentiation in selected groups, suggesting that in the absence of other cell populations (e.g. undifferentiated ES cells) growth factors may be beneficial for oligodendrocyte survival.
High purity pMNs survive and migrate in sub-acute dorsal hemisection injury
The ability of ES cell-derived populations purified by antibiotic selection to survive when transplanted in the injured spinal cord has yet to be determined. To investigate the ability of high purity pMNs to survive in vivo when encapsulated in fibrin scaffolds, we examined cell survival post-transplantation in a sub-acute rat dorsal hemisection SCI (Fig. 4A). Cell transplantation was delayed until two weeks after injury to improve cell survival and to mimic the delay in treatment that would occur in a clinical setting. The impact of growth factor delivery using the HBDS on cell survival was also assessed. G-POlig2 cells ubiquitously express GFP under the β-actin promoter to facilitate visualization of transplanted cells. Four fibrin conditions were assessed: unmodified fibrin (Fibrin, n = 6), fibrin scaffolds incorporating the HBDS (DS, n = 7), fibrin scaffolds with DS, NT3, and GDNF (DS + NT3 + GDNF, n = 7), and fibrin scaffolds with the DS, NT3, and PDGF (DS + NT3 + PDGF, n = 8).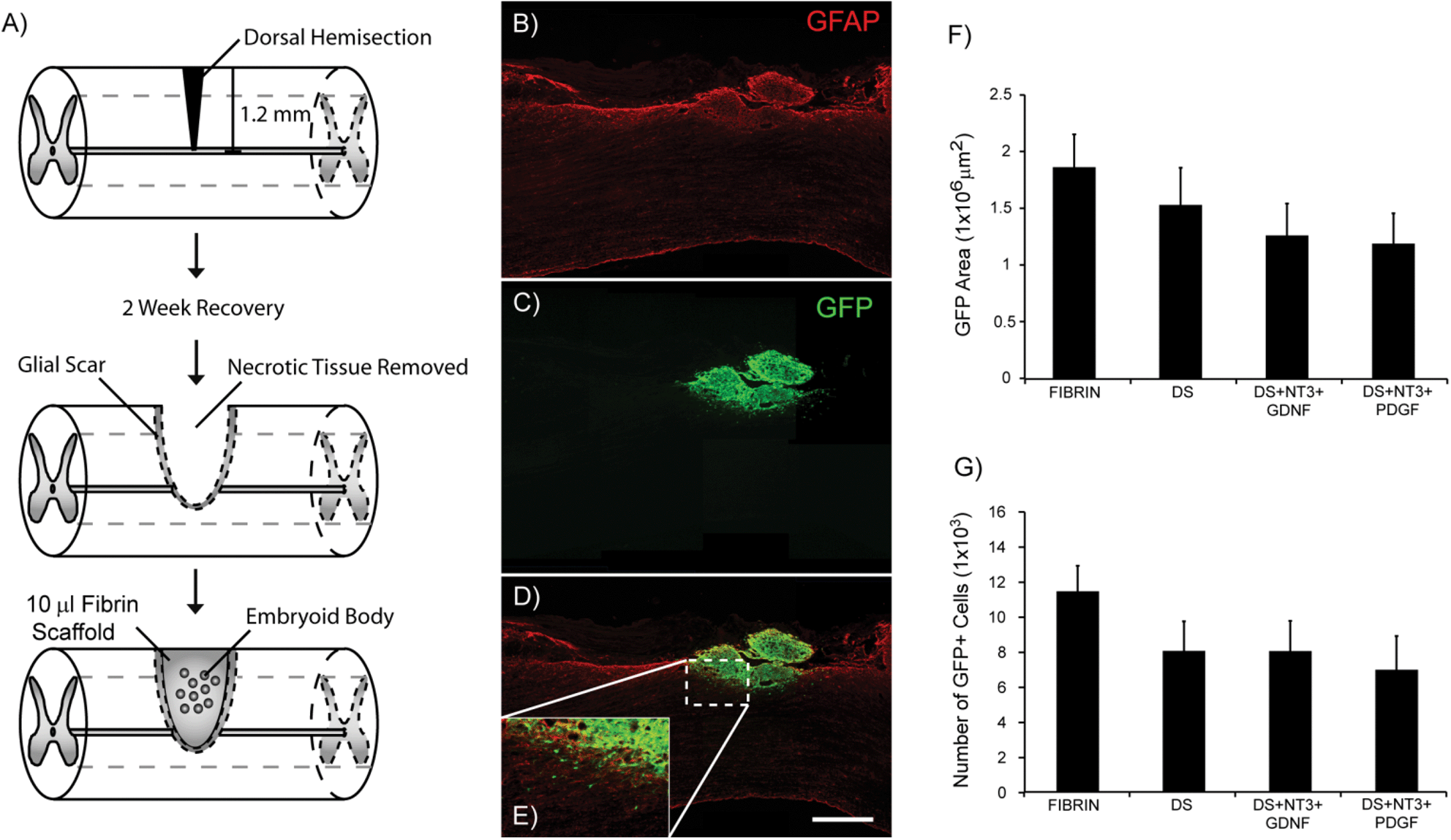 | ||
| Fig. 4 Cell transplantation following a sub-acute dorsal hemisection model of spinal cord injury. (A) Schematic of EB transplantation within a fibrin scaffold into the dorsal hemisection lesion 2 weeks after injury. (B) Compiled overview of the dorsal hemisection injury site in the DS + NT3 + PDGF group at two weeks post-transplantation showing astrocytes in the glial scar labeled with GFAP. (C) Transplanted cells (GFP) located within the injury site. (D) Merged view of GFAP and GFP showing the transplant host interface. (E) Magnified inset of the transplant host border. (F) Average GFP-positive area per subject in the four different fibrin scaffold conditions two weeks post-transplantation. (G) The average number of nuclei contained within GFP-positive areas per subject in each condition two weeks post-transplantation. Scale bar = 500 μm. Abbreviations: glial fibrillary acidic protein (GFAP), green fluorescent protein (GFP), delivery system (DS), heparin (Hep), neurotrophin-3 (NT3), glial-derived neurotrophic factor (GDNF), platelet derived growth factor (PDGF). | ||
Two weeks after transplantation, spinal cords were harvested, and the area inside and around the lesion was examined for scarring, axon density, and survival of transplanted cells. Minimal glial scarring was observed at the transplant–host interface for all groups. The percentage of GFAP+ pixels within the first 500 μM adjacent to the injury site was significantly lower in the DS + NT3 + GDNF group compared to the DS + NT3 + PDGF group (Fig. S2†). No significant differences in the βtubIII staining density were observed between any of the groups within the lesion site, as well as ventral, rostral and caudal to the lesion (Fig. S3†). The area of GFP expression was quantified from ∼30% of the total sections from each subject to assess transplant survival. GFP+ cells were observed in the lesion cavity of all subjects (Fig. 4B and C). In many cases, migration of GFP+ cells into the host spinal cord was observed (Fig. 4D and E, Fig. S4†). No significant differences in the total GFP+ area were found between any of the groups (Fig. 4F). The total cell count, as assessed by nuclei labeling with DAPI within GFP+ areas, was also similar between groups (Fig. 4B).
Residual low level SSEA1 expression typical of recently differentiated ES cell-derived populations was commonly observed in transplant groups (Fig. 5A–D); however, no high density SSEA1+ staining characteristic of undifferentiated ES cells was observed. Host-derived GFP−/SSEA+high cells were observed outside of the transplant region (Fig. 5E–H) in 4/28 subjects. No Oct4+ nuclei were observed within GFP+ areas in any subject confirming the absence of transplanted-derived undifferentiated ES cells. Furthermore, very few GFP+ cells were observed within the cell graft labeled for the proliferative marker Ki67 (Fig. 5M–P). High purity pMNs survive and appear to integrate into the injured host spinal cord. The presence of growth factors did not appear to induce over-proliferation as observed in previous studies.30
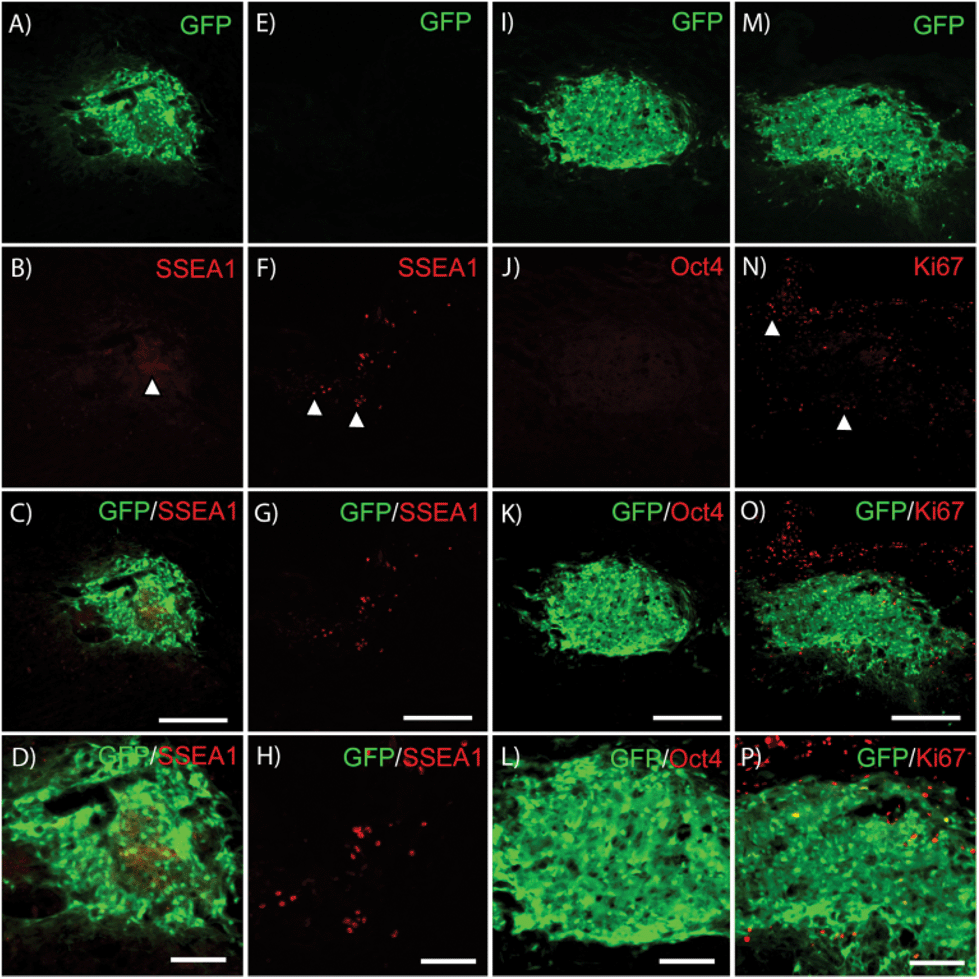 | ||
| Fig. 5 Stem cell and proliferation marker expression at two weeks post-transplantation. (A) GFP and (B) SSEA1 expression in cell transplants (white arrow). (C) Merged view of GFP and SSEA1 (Fibrin). (D) Magnified view of (C). (E) GFP and (F) SSEA1 expression outside of cell transplant area (white arrows; DS + NT3 + PDGF). (G) Merged view of GFP and SSEA1. (H) Magnified view of (G). (I) GFP and (J) Oct4 expression in cell transplants (DS + NT3 + GDNF). (K) Merged view of GFP and Oct4. (L) Magnified view of (K). (M) GFP and (N) Ki67 expression in cell transplants (white arrow; DS + NT3 + PDGF). (O) Merged view of GFP and Ki67. (P) Magnified view of (O). Abbreviations: green fluorescent protein (GFP), stage specific embryonic antigen 1 (SSEA1). Scale bar = 250 μM for A–C, E–G, I–K, M–O. Scale bar = 100 μM for D, H, L, P. | ||
Differentiation of transplanted pMNs following SCI
To determine the ability of high-purity pMNs to differentiate within fibrin scaffolds following transplantation into the injured cord, expression of cell-type specific markers (nestin, neural progenitors; neurons, βtubIII; astrocytes, GFAP; oligodendrocytes, O4; motoneurons, ChAT) was analyzed by immunofluorescence two weeks post-transplantation into the sub-acute dorsal hemisection SCI. A two week time point was chosen to provide sufficient time for differentiation of transplanted cells while still allowing detection of undifferentiated ES cells. The area of each marker of differentiation co-labeled with GFP was quantified for each subject to determine the effect of fibrin scaffold composition on differentiation (Fig. S5†).Nestin expression was low in all groups suggesting that the transplanted progenitor cells had differentiated into mature neural cell types (Fig. 6J–L and 7A). The area of nestin expression was significantly decreased in the DS + NT3 + PDGF group compared to the Fibrin group. βtubIII and NeuN labeling was observed in all groups, demonstrating differentiation into mature neurons (Fig. 6A–C and G–I). No significant differences in βtubIII expression were found between any groups; however, the NeuN+ area was significantly greater in the DS group compared to both the DS + NT3 + PDGF and the DS + NT3 + GDNF groups (Fig. 7B and C).
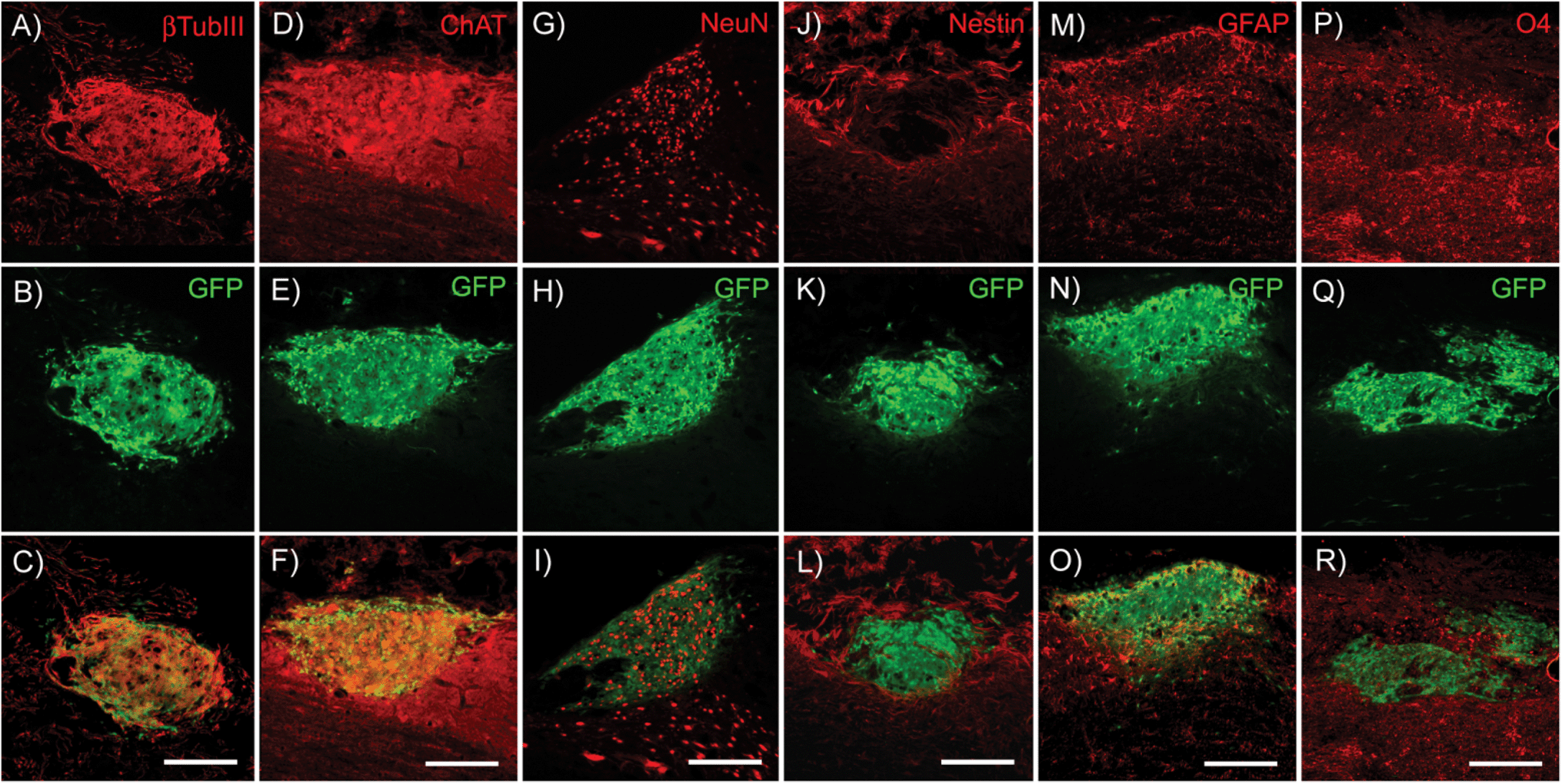 | ||
| Fig. 6 Immunofluorescence images of cell differentiation within the injured spinal cord two weeks post-transplantation. (A) βtubIII labeling and corresponding (B) GFP expression in transplanted cells (Fibrin). (C) Merged view of βtubIII and GFP. (D) ChAT labeling and (E) GFP expression (DS + NT3 + GDNF). (F) Merged view of ChAT and GFP. (G) NeuN labeling and (H) GFP expression (DS). (I) Merged view of NeuN and GFP. (J) Nestin labeling and (K) GFP expression (DS + NT3 + GDNF). (L) Merged view of GFP and nestin. (M) GFAP labeling and (N) GFP expression (DS + NT3 + PDGF). (O) Merged view of GFAP and GFP. (P) O4 labeling and (Q) GFP expression (DS + NT3 + PDGF). (R) Merged view of O4 and GFP. Abbreviations: β-tubulin class III (βtubIII), glial fibrillary acidic protein (GFAP), oligodendrocyte marker 4 (O4), neuronal nuclei (NeuN), choline acetyltransferase (ChAT), green fluorescent protein (GFP). Scale bar = 250 μM. | ||
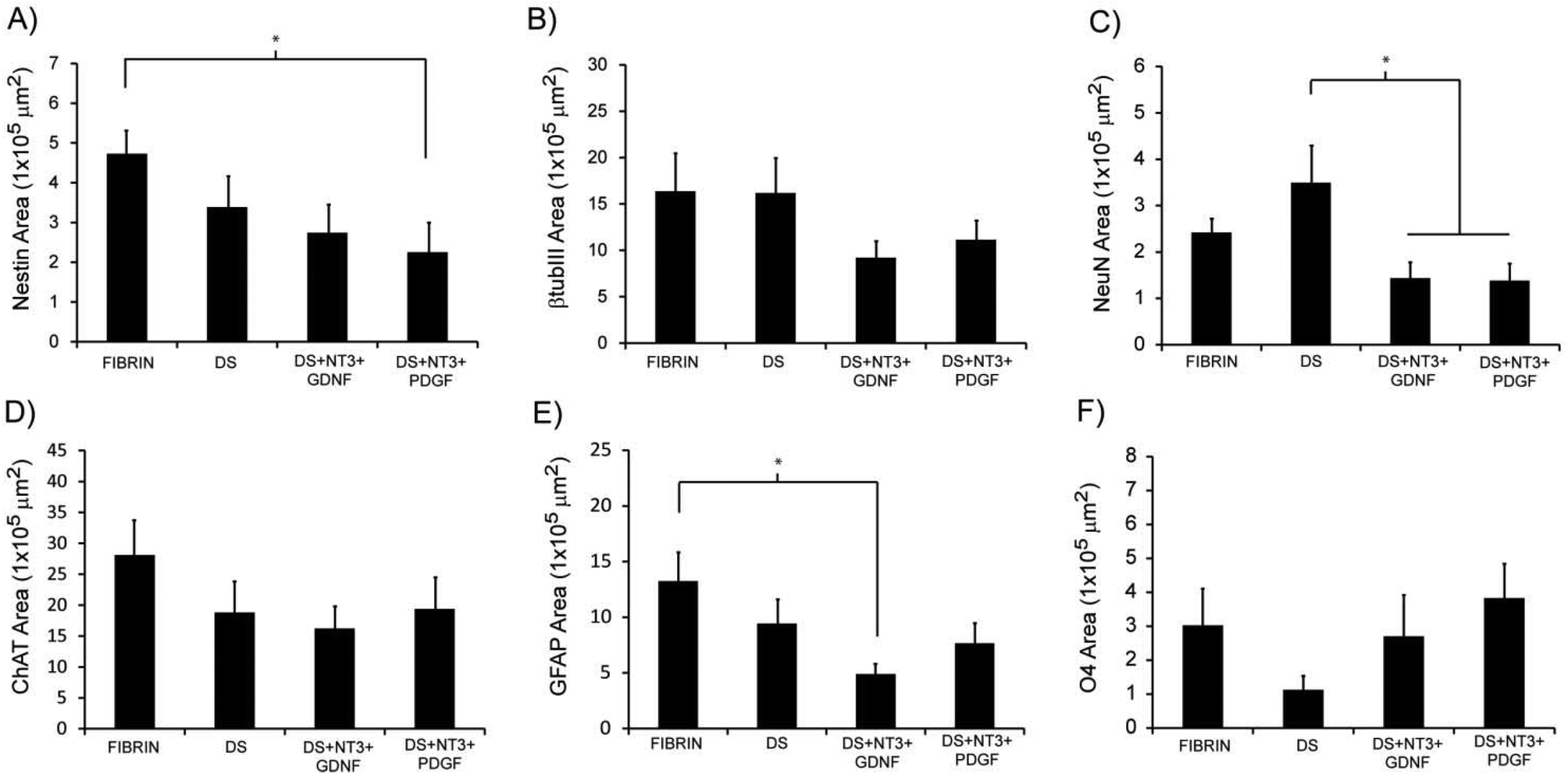 | ||
| Fig. 7 Quantitative analysis of the area of expression for differentiation markers. (A) Average area of nestin (neural progenitors) expression. (B) Average area of βtubIII (immature neurons) expression. (C) Average area of NeuN (mature neurons) expression. (D) Average area of ChAT (motoneurons) expression. (E) Average area of GFAP (astrocytes) expression. (F) Average area of O4 (oligodendrocytes) expression. * indicates p < 0.05. Abbreviations: β-tubulin class III (βtubIII), oligodendrocyte marker 4 (O4), glial fibrillary acidic protein (GFAP), neuronal nuclei (NeuN), choline acetyltransferase (ChAT), delivery system (DS), neurotrophin-3 (NT3), glial-derived neurotrophic factor (GDNF), platelet-derived growth factor (PDGF). | ||
Successful differentiation into motoneurons was observed in every group as evidenced by ChAT staining (Fig. 6D–F). ChAT labeling followed a similar expression pattern to βtubIII with no significant differences between groups (Fig. 7D). Both astrocytes (GFAP) and oligodendrocytes (O4) were observed in all groups (Fig. 6M–R). The area of GFAP expression was significantly lower in the DS + NT3 + GDNF group compared to the Fibrin group (Fig. 7E). No statistical differences were observed in O4+ areas between groups (Fig. 7F). High purity pMN populations not only survive in the injured cord, but differentiate into the expected neural cell types: oligodendrocytes, astrocytes, and motoneurons.
Discussion
The effectiveness of transplanted ES cells is often limited by low differentiation efficiency.38 Heterogeneous transplant populations can contain undesired cell types that confound results and in some cases lead to tumor formation. We have previously demonstrated antibiotic selection as a viable method for enrichment of ES cell-derived pMNs. In this study, we demonstrated that highly enriched pMN populations survive and differentiate into the expected cell types (neurons, oligodendrocytes and astrocytes) within fibrin scaffolds in vitro and within the injured spinal cord in vivo. Furthermore, neuronal differentiation did not appear to be inhibited after injury, as is commonly reported following transplantation after SCI.18,19Appropriate fibrin scaffold conditions have been identified for ES cell-derived neural progenitor cells that yield approximately 70% cell viability.29,39,40 Using similar conditions, axons from EBs containing pMNs extended through the fibrin scaffolds confirming that fibrin scaffolds can provide an appropriate environment for neuronal cell culture in vitro. This is further supported by the frequent migration of oligodendrocytes and astrocytes, as well as axonal extension, out into the scaffold from the EB. GFP expression by the transgenic mouse ES cell line was maintained for two weeks post-transplantation and demonstrated the survival of the transplanted cells. Prior purification of ES-cell derived pMNs by antibiotic selection did not hinder the ability of cells to survive in 3D scaffolds, alleviating concerns that the undifferentiated ES cells were necessary for survival in 3D culture. Thus, fibrin scaffolds appear to be an appropriate vehicle for transplantation of highly enriched pMNs.
Previous studies have demonstrated the ability for growth factor delivery to improve the differentiation of ESNPCs embedded in fibrin scaffolds.29,40 In this study, combinations of NT3 and PDGF-AA or NT3 and GDNF delivered from fibrin scaffolds promoted subtle changes in differentiation of unselected and selected pMN cultures after two week culture in vitro. The percentage of cells differentiating into oligodendrocytes could be increased through a combination of puromycin selection and growth factor delivery. The percentage of neurons was low (<10%) under all conditions; however, this is expected since proliferating glia can quickly dilute post-mitotic neurons in cultures that allow multiple cell doublings. These results suggest that tailoring the 3D culture environment can influence the differentiation of ES cell-derived pMNs. Purification of the ES cell population appeared to remove undesired cell types and enhance the ability of tissue-engineered fibrin scaffolds to direct oligodendrocyte differentiation, which may in turn enhance remyelination after SCI in vivo.
High purity pMN transplants survived in the injured spinal cord and differentiated into expected mature cell types (neurons, oligodendrocytes and astrocytes). Fibrin scaffolds were used to enhance cell retention at the injury site and deliver NT-3 and GDNF or PDGF via a HBDS. The presence of growth factors did not appear to influence survival or proliferation of transplanted cells, which is in contrast to results previously obtained from transplantation of non-enriched heterogeneous populations.27,30 We have previously shown tumor formation and proliferating undifferentiated ES cells in unselected cell populations following transplantation in fibrin scaffolds using the same model of SCI.30 The absence of undifferentiated ES cells may be responsible for the lack of growth factor-dependent proliferation observed in this study and this effect is beneficial in reducing the risk for tumor formation. Similarly, the proliferative potential of the Olig2+ pMNs used in this study appears to be substantially less than other highly proliferative Olig2+ progenitors, such as those commonly associated with malignant glioma.41 In many subjects, cells migrate from the graft into host tissue. This process is likely facilitated by the limited glial scarring observed, which may be a result of the use of fibrin scaffolds that have previously been shown to reduce the glial scar.26 Refining the transplanted cell population to the appropriate spinal neural progenitors may improve incorporation into host spinal tissue.
Neuronal differentiation was observed in all grafts though the greatest NeuN staining was observed in the DS group. Sequestering of endogenous growth factors expressed in the injury site by unoccupied heparin sites present in the scaffold may improve neuronal survival and/or differentiation. Delivering specific growth factor combinations appeared to decrease astrocyte differentiation from transplanted cells, as shown by reduced GFAP labeling in the DS + NT-3 + GDNF group. Modification of the lesion environment through tissue-engineered fibrin scaffolds may promote subtle changes in the differentiation of transplanted cells. Few differences were observed in the surrounding tissue outside of the transplant region. This may have been due to the presence of fibrin and transplanted cells in all groups. Stem cell-derived populations have been shown to secrete multiple neurotrophic factors that support axon extension and the initial differences from the growth factors may have been lost by two weeks post-transplantation.
Conclusions
The development of methods to generate high purity ES cell-derived populations can benefit cell transplantation therapies for SCI. This study demonstrates the feasibility of generating high purity pMN populations for transplantation into the injured spinal cord. Future studies to elucidate the effects of this cell population on functional recovery could demonstrate its potential for therapeutic application. Antibiotic selection appears to reduce growth factor mediated over-proliferation and may be an effective mechanism for purifying ES cell-derived cell transplant populations more generally for many applications.Experimental
Ubiquitous GFP expression
To help visualize transplanted cells, a linear plasmid containing the gene for enhanced GFP driven by the chicken beta-actin promoter was generated and electroporated into the previously established mouse P-Olig2 ES cell line.35,42,43 Clones with the integrated DNA were identified and assessed for ubiquitous GFP fluorescence at the ES cell stage, after induction into pMNs, and following a two week differentiation process to ensure persistent GFP expression in mature cell types. P-Olig2 ES cells expressing GFP are referred to throughout the paper as the G-POlig2 ES cell line.Embryonic stem cell culture
G-POlig2 ES cells were grown in complete media consisting of Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% newborn calf serum (Invitrogen), 10% fetal bovine serum (Invitrogen), 10 μM thymidine (Sigma, St. Louis, MO), and 30 μM of each of the following nucleosides: adenosine, cytosine, guanosine, and uridine (Sigma). Cells were passaged at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio every 2 days and seeded on a new T-25 flask coated with a 0.1% gelatin solution (Sigma). After seeding, 1000 U mL−1 leukemia inhibitory factor (LIF; Millipore, Billerica, MA) and 100 μM β-mercaptoethanol (BME; Invitrogen) were added to the media to maintain the undifferentiated state of the ESCs without the need for a feeder cell layer.35,40
:5 ratio every 2 days and seeded on a new T-25 flask coated with a 0.1% gelatin solution (Sigma). After seeding, 1000 U mL−1 leukemia inhibitory factor (LIF; Millipore, Billerica, MA) and 100 μM β-mercaptoethanol (BME; Invitrogen) were added to the media to maintain the undifferentiated state of the ESCs without the need for a feeder cell layer.35,40
Progenitor motor neuron induction
For pMN induction, ES cells were exposed to retinoic acid (RA; Sigma) and purmorphamine (EMD, Gibbstown, NJ) in a 2−/4+ induction protocol.35 One million ESCs were aggregated into embryoid bodies (EBs) in 100 mm Petri dishes coated with a 0.1% agar solution in DFK5 media consisting of DMEM:F12 base media (Invitrogen) supplemented with 5% knockout serum replacement (Invitrogen), 50 μg mL−1 apo-transferrin (Sigma), 50 μM non-essential amino acids (Invitrogen), 5 μg mL−1 insulin (Sigma), 30 nM sodium selenite (Sigma), 100 μM β-mercaptoethanol, 5 μM thymidine, and 15 μM of the following nucleosides: adenosine, cytosine, guanosine, and uridine. EBs were allowed to form for 2 days in the absence of inducing factors (2−). EBs were then cultured in DFK5 supplemented with 2 μM RA and 1.5 μM purmorphamine for the final four days (4+). Media were changed every 2 days. In selected cultures, 4 ng mL−1 puromycin (Sigma) was added during the final 2 days of induction.353D fibrin scaffold cultures
To prepare fibrin scaffolds, fibrinogen (50 mg mL−1; EMD) was dissolved in tris-buffered saline (TBS) and dialyzed in 4 L TBS overnight. Fibrinogen was sterile filtered and the concentration was measured by UV spectroscopy. The final fibrinogen concentration was adjusted to 20 mg mL−1 with sterile TBS. Fibrin scaffolds (150 μL) were formed by combining 10 mg mL−1 fibrinogen, 2.5 mM CaCl2 (Sigma), and 2 NIH units mL−1 thrombin (Sigma) in individual wells of a 48 well plate as previously described.39,40 For the HBDS, the ATIII bi-domain affinity peptide (GNQEQVSPKβAFAKLAARLYRKA) was synthesized by solid phase Fmoc chemistry.36,37 In delivery system (DS) control groups, 62.5 μM heparin (Sigma) and 0.25 mM ATIII peptide were added to each scaffold.36,37 In growth factor control groups, scaffolds contained 62.5 μM heparin, 62.5 ng NT3 (Peprotech, Rocky Hill, NJ), and 62.5 ng GDNF (Peprotech) (Hep + NT3 + GDNF) or 10 ng PDGF (Peprotech) (Hep + NT3 + PDGF). For growth factor delivery groups, 62.5 μM heparin, 0.25 mM ATIII peptide, 62.5 ng NT3, and 62.5 ng GDNF (DS + NT3 + GDNF) or 10 ng PDGF (DS + NT3 + PDGF) were added to each scaffold. A total of 24 scaffolds were fabricated for each group. All scaffolds were washed 5 times over a 24 hour period with 500 μL TBS per wash to remove unbound delivery system components (heparin and/or growth factors). An individual EB containing pMNs was placed on each fibrin scaffold and covered with a 100 μL fibrin scaffold containing 10 mg mL−1 fibrinogen, 2.5 mM CaCl2 (Sigma), and 2 NIH units mL−1 thrombin (Sigma) such that the EB remained at the interface of the two fibrin layers. Fibrin scaffolds containing the single EB were incubated at 37 °C for 1 hour. 500 μL of modified DFKNB media consisting of a 1:1 ratio of DFK5 media and neural basal (NB) media supplemented with 2% B27 and 5 μg mL−1 aprotinin was added to each well. Cells were cultured for a total of 14 days with a single media change at day 3 to NB media supplemented with 2% B27.
Flow cytometry
Following two weeks of culture, fibrin scaffolds were degraded with 0.25% trypsin–EDTA for 15 min to release cells. Cells from all 24 wells for each group were pooled in 35 mm dishes and triturated prior to quenching with an excess volume of complete media. Cells were centrifuged for 5 min at 230g, the medium was aspirated, and cells were fixed with 1% paraformaldehyde (Sigma). After fixation, the cells were permeabilized with 1% saponin (Sigma) solution for 20 min, and then blocked with 0.5% saponin solution containing 5% normal goat serum (NGS; Sigma). Cell suspensions were then incubated for 30 min in 0.5% saponin solution containing 2% NGS and one of the following primary antibodies: ChAT (Millipore; 1:500), O4 (Millipore; 1:500), GFAP (ImmunoStar, Hudson, WI; 1:50), and NeuN (Millipore; 1:500). Cells were washed with PBS and appropriate Alexa Fluor secondary antibodies (1:200; Invitrogen) diluted in 0.5% saponin with 2% NGS were applied for 30 min. Finally cells were washed with PBS and incubated with Hoechst (1:1000; Invitrogen) for 5 min.
Stained cell suspensions were analyzed using a Canto II flow cytometer (Becton Dickinson, Franklin Lakes, NJ). For each group, 10000 events were recorded. Subsequent analysis was performed using the FloJo software (FloJo, Ashland, OR). Prior to population gating, debris was removed based on forward scatter versus side scatter and Hoechst fluorescence versus forward scatter plots. Flow cytometry control groups, consisting of cells stained with the secondary antibody only, were used to determine quadrant population gating parameters. Flow cytometry results are presented as the percentage of cells staining positive for each marker out of the total live cell population.
Spinal cord injury and scaffold implantation
All experimental procedures on animals complied with the Guide for the Care and Use of Laboratory Animals and were performed under the supervision of the Division of Comparative Medicine at Washington University. Long–Evans female rats (250–275 g) were anesthetized using 5% isoflurane gas and 5 mg kg−1 xylazine. A single incision was created through the skin to expose the back muscle. Parallel incisions were created through the back muscle on each side of the vertebral processes from T5 to T11. A dorsal laminectomy was performed at T8 using fine tip rongeurs to expose the spinal cord. Spinal clamps were placed in the vertebral foramina at T7 and T9 to stabilize the spinal cord. The dura mater was removed from the exposed cord at T8. Vitrectomy scissors mounted to a micromanipulator were lowered by 1.2 mm into the spinal cord. A lateral incision was created across the spinal cord to form a dorsal hemisection. The injury site was covered with a piece of artificial dura and the back muscles were closed using degradable sutures. Finally the skin was stapled close and the animals were treated with buprenorphine.26Two weeks following the initial injury, the lesion site was re-exposed and the scar tissue removed from the spinal cord to create a cavity for scaffold implantation. Fibrin scaffolds were prepared by mixing 10 mg mL−1 fibrinogen, 2.5 mM CaCl2 (Sigma), 2 NIH units mL−1 thrombin (Sigma), 62.5 μM heparin, 0.25 mM ATIII peptide, 125 ng NT3, and 125 ng GDNF or 20 ng PDGF-AA per scaffold (NT3 + GDNF and NT3 + PDGF groups). For fibrin control groups (Fibrin), the following components were omitted: heparin, ATIII peptide, NT3, GDNF, and PDGF. For delivery system (DS) control groups all growth factors were omitted. A 10 μL fibrin scaffold with 10 EBs containing pMNs was slowly ejected from a 10 μL pipette tip to form a spherical scaffold with the EBs at the center. The scaffold was allowed to polymerize at the end of the pipette tip for 5 min prior to implantation into the injury site. The pre-polymerized scaffold was then pressed into the spinal cord lesion. A second 10 μL fibrin scaffold consisting of 10 mg mL−1 fibrinogen, 2.5 mM CaCl2 (Sigma), and 2 NIH units mL−1 thrombin was allowed to polymerize in situ to hold the first scaffold containing EBs within the lesion site. The implantation site was then covered with artificial dura, the overlying back muscle was closed using degradable sutures, and the skin was stapled close.
Immediately following each surgery, animals were given cefazolin (25 mg kg−1) and buprenorphine (0.04 mg kg−1). Cefazolin was continued twice daily for 5 days with carprofen tablets. Bladders were manually expressed twice a day for the entire study. Following the second surgery, immune suppression was accomplished by daily injections of cyclosporine-A (10 mg kg−1) to reduce immune rejection of mouse cells. Two weeks following cell transplantation, animals were euthanized by an overdose of euthasol. Spinal cords were harvested following transcardial perfusion with 4% paraformaldehyde and post-fixed in 4% paraformaldehyde overnight. The following day, spinal cords were cryoprotected by immersion in 30% sucrose in PBS. Prior to embedding, 2 cm sections of the spinal cords with the injury site in the center were cut and frozen on dry ice. Cords were embedded in the Tissue-Tek OCT compound and cut into 20 μm sagittal sections with a cryostat.
Immunofluorescence
To assess the presence of pMNs and recent progeny, EBs were collected and fixed for 1 hour in 4% paraformaldehyde in PBS. Cells within EBs were then permeabilized with 0.1% Triton X-100 for 30 min and blocked overnight in 5% NGS in PBS. Primary antibodies against Olig2 (Millipore, 1:500), Hb9 (Iowa Hybridoma Bank, Iowa City, IA; 1:500), and Oct3/4 (Oct4, Santa Cruz Biotech, Santa Cruz, CA; 1:500) were applied overnight at 4 °C in 2% NGS in PBS. EBs were washed 3× with PBS over 24 hours. Alexafluor secondary antibodies (1:200 in 2% NGS in PBS) were applied overnight and then washed 3× with PBS over 24 hours. Stained EBs were placed on microscope slides and flattened by mounting a coverslip onto the slide.
To visualize differentiation within fibrin scaffolds in vitro, scaffolds were fixed in 4% paraformaldehyde in PBS for 30 min. Scaffolds were then removed and placed in 0.1% Triton X-100 for 30 min and blocked for 4 hours in 5% NGS in PBS. Small sections of the fibrin scaffolds containing the EB were dissected and placed in the following primary antibody solutions in 2% NGS in PBS overnight at 4 °C: ChAT (1:500), O4 (1:500), GFAP (1:50), NeuN (1:500). Sections were then washed 3× with PBS over 24 hours. Sections were then placed in Alexafluor secondary antibody solutions (1:200 in 2% NGS in PBS) overnight followed by 3 washes with PBS over 24 hours. Sections were then placed on microscope slides and mounted using the FluorMount (Invitrogen).
To determine expression of differentiated cell markers in transplanted pMNs, immunofluorescence was performed on 6 equidistance spinal cord sections per animal. OCT from spinal cord sections was washed with PBS. Sections were permeabilized with 0.1% Triton X-100 for 15 min and blocked with 10% bovine serum albumin and 2% NGS. The following primary antibodies were applied overnight at 4 °C in 2% NGS in PBS: β-tubulin III (βtubIII, Millipore, 1:400), ChAT (1:500), O4 (1:500), GFAP (1:50), NeuN (1:500), nestin (1:25), Oct3/4 (Oct4, 1:1000), SSEA1 (DSHB, 1:100), Olig2 (Millipore, 1:500), and Ki67 (AbCam, Cambridge, MA; 1:125). Primary antibody staining was followed by 3 washes with PBS. Appropriate Alexafluor secondary antibodies (Invitrogen) in 2% NGS in PBS were applied for 1 hour at room temperature followed by additional 3 washes in PBS. Cell nuclei were stained with the nuclei binding dye DAPI (1:1000). Sections were mounted using the FluorMount.
Image analysis of differentiation markers
To quantify the staining of differentiation markers on transplanted EBs containing pMNs, every twentieth section from each spinal cord was analyzed for six markers of differentiation (nestin, βtubIII, GFAP, O4, NeuN, and ChAT). A series of 100× images spanning the lesion site were captured using a MICROfire camera attached to an Olympus IX70 inverted microscope. GFP+ pixels were identified in each image and the total area of GFP+ pixels was counted per image using the Matlab code adapted from ref. 44 (Mathworks, Natick, MA). Fluorescence staining for one of the six markers of differentiation was assessed in each of the GFP+ pixels to determine a total area of staining. In addition, nuclei counts were performed within GFP+ areas to determine the total number of GFP+ cells in each image.Image analysis of glial scarring
To quantify scarring following spinal cord injury, every twentieth section was stained for GFAP and a series of 40× images spanning the section were compiled to create one continuous image. Using Photoshop (Adobe, San Jose, CA), the area of the lesion was selected and expanded 500 μM into the tissue. GFAP+ pixels within this region were identified using Matlab and the ratio of GFAP+ pixels to the total number of pixels was recorded for each section.Analysis of βtubIII staining
The neurite density in and around the injury site was measured by the intensity threshold of βtubIII immunofluorescence. Every twentieth section was stained for βtubIII and a series of 40× images spanning the section were compiled to create one continuous image. Using Photoshop, the area of the lesion was selected along with the area directly ventral to the injury site as well as the first 300 μM rostral and caudal to each respective edge of the lesion boundary. For each area, the βtubIII+ pixels were identified by the intensity threshold using Matlab and the ratio of βtubIII+ pixels to the total number of pixels was recorded.Statistical analysis
Statistical significance was determined by one way analysis of variance (ANOVA) using the Statistica Software (StatSoft, Tulsa, OK). Statistical significance was set at p < 0.05. For in vitro experiments, a Tukey's HSD test was used to determine significance (p < 0.05). Statistical significance for in vivo studies was determined by the planned comparisons and post hoc tests (p < 0.05).Acknowledgements
The authors were funded by the NIH RO1 grant 5R01NS051454. Support was provided for Dylan McCreedy by the NSF GRFP DGE-1143954.References
- M. J. Crowe, J. C. Bresnahan, S. L. Shuman, J. N. Masters and M. S. Beattie, Nat. Med., 1997, 3, 73–76 CrossRef CAS PubMed.
- J. D. Guest, E. D. Hiester and R. P. Bunge, Exp. Neurol., 2005, 192, 384–393 CrossRef CAS PubMed.
- N. L. Banik, E. L. Hogan, J. M. Powers and L. J. Whetstine, Neurochem. Res., 1982, 7, 1465–1475 CrossRef CAS.
- P. A. Schumacher, R. G. Siman and M. G. Fehlings, J. Neurochem., 2000, 74, 1646–1655 CrossRef CAS.
- X. Z. Liu, X. M. Xu, R. Hu, C. Du, S. X. Zhang, J. W. McDonald, H. X. Dong, Y. J. Wu, G. S. Fan, M. F. Jacquin, C. Y. Hsu and D. W. Choi, J. Neurosci., 1997, 17, 5395–5406 CAS.
- S. Casha, W. R. Yu and M. G. Fehlings, Neuroscience, 2001, 103, 203–218 CrossRef CAS.
- B. J. Cummings, N. Uchida, S. J. Tamaki, D. L. Salazar, M. Hooshmand, R. Summers, F. H. Gage and A. J. Anderson, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14069–14074 CrossRef CAS PubMed.
- S. Nori, Y. Okada, A. Yasuda, O. Tsuji, Y. Takahashi, Y. Kobayashi, K. Fujiyoshi, M. Koike, Y. Uchiyama, E. Ikeda, Y. Toyama, S. Yamanaka, M. Nakamura and H. Okano, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16825–16830 CrossRef CAS PubMed.
- H. S. Keirstead, G. Nistor, G. Bernal, M. Totoiu, F. Cloutier, K. Sharp and O. Steward, J. Neurosci., 2005, 25, 4694–4705 CrossRef CAS PubMed.
- J. Sharp, J. Frame, M. Siegenthaler, G. Nistor and H. S. Keirstead, Stem Cells, 2010, 28, 152–163 CAS.
- A. J. Mothe, I. Kulbatski, A. Parr, M. Mohareb and C. H. Tator, Cell Transplant., 2008, 17, 735–751 Search PubMed.
- S. Karimi-Abdolrezaee, E. Eftekharpour, J. Wang, C. M. Morshead and M. G. Fehlings, J. Neurosci., 2006, 26, 3377–3389 CrossRef CAS PubMed.
- A. M. Parr, I. Kulbatski, T. Zahir, X. Wang, C. Yue, A. Keating and C. H. Tator, Neuroscience, 2008, 155, 760–770 CrossRef CAS PubMed.
- G. Bain, D. Kitchens, M. Yao, J. E. Huettner and D. I. Gottlieb, Dev. Biol., 1995, 168, 342–357 CrossRef CAS PubMed.
- H. Wichterle, I. Lieberam, J. A. Porter and T. M. Jessell, Cell, 2002, 110, 385–397 CrossRef CAS.
- S. Erceg, M. Ronaghi, M. Oria, M. G. Rosello, M. A. Arago, M. G. Lopez, I. Radojevic, V. Moreno-Manzano, F. J. Rodriguez-Jimenez, S. S. Bhattacharya, J. Cordoba and M. Stojkovic, Stem Cells, 2010, 28, 1541–1549 CrossRef PubMed.
- S. Liu, Y. Qu, T. J. Stewart, M. J. Howard, S. Chakrabortty, T. F. Holekamp and J. W. McDonald, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6126–6131 CrossRef CAS.
- Q. L. Cao, Y. P. Zhang, R. M. Howard, W. M. Walters, P. Tsoulfas and S. R. Whittemore, Exp. Neurol., 2001, 167, 48–58 CrossRef CAS PubMed.
- Q. L. Cao, R. M. Howard, J. B. Dennison and S. R. Whittemore, Exp. Neurol., 2002, 177, 349–359 CrossRef CAS.
- V. Patel, G. Joseph, A. Patel, S. Patel, D. Bustin, D. Mawson, L. M. Tuesta, R. Puentes, M. Ghosh and D. D. Pearse, J. Neurotrauma, 2010, 27, 789–801 CrossRef PubMed.
- G. Bozkurt, A. J. Mothe, T. Zahir, H. Kim, M. S. Shoichet and C. H. Tator, Neurosurgery, 2010, 67, 1733–1744 CrossRef PubMed.
- H. Itosaka, S. Kuroda, H. Shichinohe, H. Yasuda, S. Yano, S. Kamei, R. Kawamura, K. Hida and Y. Iwasaki, Neuropathology, 2009, 29, 248–257 CrossRef PubMed.
- A. Iwata, K. D. Browne, B. J. Pfister, J. A. Gruner and D. H. Smith, Tissue Eng., 2006, 12, 101–110 CrossRef CAS PubMed.
- S. Woerly, V. D. Doan, N. Sosa, J. de Vellis and A. Espinosa-Jeffrey, J. Neurosci. Res., 2004, 75, 262–272 CrossRef CAS PubMed.
- S. J. Taylor, E. S. Rosenzweig, J. W. McDonald 3rd and S. E. Sakiyama-Elbert, J. Control. Release, 2006, 113, 226–235 CrossRef CAS PubMed.
- P. J. Johnson, S. R. Parker and S. E. Sakiyama-Elbert, J. Biomed. Mater. Res., Part A, 2010, 92, 152–163 CrossRef PubMed.
- P. J. Johnson, A. Tatara, A. Shiu and S. E. Sakiyama-Elbert, Cell Transplant., 2010, 19, 89–101 Search PubMed.
- P. Lu, Y. Wang, L. Graham, K. McHale, M. Gao, D. Wu, J. Brock, A. Blesch, E. S. Rosenzweig, L. A. Havton, B. Zheng, J. M. Conner, M. Marsala and M. H. Tuszynski, Cell, 2012, 150, 1264–1273 CrossRef CAS PubMed.
- S. M. Willerth, A. Rader and S. E. Sakiyama-Elbert, Stem Cell Res., 2008, 1, 205–218 CrossRef CAS PubMed.
- P. J. Johnson, A. Tatara, D. A. McCreedy, A. Shiu and S. E. Sakiyama-Elbert, Soft Matter, 2010, 6, 5127–5137 RSC.
- N. Amariglio, A. Hirshberg, B. W. Scheithauer, Y. Cohen, R. Loewenthal, L. Trakhtenbrot, N. Paz, M. Koren-Michowitz, D. Waldman, L. Leider-Trejo, A. Toren, S. Constantini and G. Rechavi, PLoS Med., 2009, 6, e1000029 Search PubMed.
- L. M. Bjorklund, R. Sanchez-Pernaute, S. Chung, T. Andersson, I. Y. Chen, K. S. McNaught, A. L. Brownell, B. G. Jenkins, C. Wahlestedt, K. S. Kim and O. Isacson, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2344–2349 CrossRef CAS PubMed.
- F. Erdo, C. Buhrle, J. Blunk, M. Hoehn, Y. Xia, B. Fleischmann, M. Focking, E. Kustermann, E. Kolossov, J. Hescheler, K. A. Hossmann and T. Trapp, J. Cereb. Blood Flow Metab., 2003, 23, 780–785 Search PubMed.
- D. A. McCreedy, C. R. Brown, J. C. Butts, H. Xu, J. E. Huettner and S. E. Sakiyama-Elbert, Biotechnol. Bioeng., 2014 DOI:10.1002/bit.25260.
- D. A. McCreedy, C. R. Rieger, D. I. Gottlieb and S. E. Sakiyama-Elbert, Stem Cell Res., 2012, 8, 368–378 CrossRef CAS PubMed.
- S. E. Sakiyama-Elbert and J. A. Hubbell, J. Control. Release, 2000, 65, 389–402 CrossRef CAS.
- S. E. Sakiyama-Elbert and J. A. Hubbell, J. Control. Release, 2000, 69, 149–158 CrossRef CAS.
- D. Anderson, T. Self, I. R. Mellor, G. Goh, S. J. Hill and C. Denning, Mol. Ther., 2007, 15, 2027–2036 CrossRef CAS PubMed.
- S. M. Willerth, K. J. Arendas, D. I. Gottlieb and S. E. Sakiyama-Elbert, Biomaterials, 2006, 27, 5990–6003 CrossRef CAS PubMed.
- S. M. Willerth, T. E. Faxel, D. I. Gottlieb and S. E. Sakiyama-Elbert, Stem Cells, 2007, 25, 2235–2244 CrossRef CAS PubMed.
- D. H. Meijer, M. F. Kane, S. Mehta, H. Liu, E. Harrington, C. M. Taylor, C. D. Stiles and D. H. Rowitch, Nat. Rev. Neurosci., 2012, 13, 819–831 CrossRef CAS PubMed.
- W. W. Quitschke, Z. Y. Lin, L. DePonti-Zilli and B. M. Paterson, J. Biol. Chem., 1989, 264, 9539–9546 CAS.
- A. Seiler-Tuyns, J. D. Eldridge and B. M. Paterson, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 2980–2984 CrossRef CAS.
- A. W. Smith, J. D. Hoyne, P. K. Nguyen, D. A. McCreedy, H. Aly, I. R. Efimov, S. Rentschler and D. L. Elbert, Biomaterials, 2013, 34, 6559–6571 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5. See DOI: 10.1039/c4bm00106k |
| This journal is © The Royal Society of Chemistry 2014 |