DOI:
10.1039/C4BM00115J
(Paper)
Biomater. Sci., 2014,
2, 1275-1286
Oligoamine-tethered low generation polyamidoamine dendrimers as potential nucleic acid carriers†
Received
9th April 2014
, Accepted 27th May 2014
First published on 23rd June 2014
Abstract
In recent years, dendrimers have emerged as the most widely explored materials for theranostics emphasizing their potential in therapeutic delivery and diagnostics as well as in pharmaceutical technology. Amongst them, PAMAM dendrimers have been extensively studied for their prospects in various biomedical applications due to their defined structures and distinctive features such as monodispersity, uniformity and amenability to functionalization. Here, low generation PAMAM dendrimers (G2–G4) have been modified via Michael addition reaction followed by amidation with an oligoamine linker, tetraethylenepentamine (TEPA). Subsequently, these modified dendrimers were characterized by physicochemical techniques and evaluated for their capability to transfer nucleic acids in vitro. The results displayed significant improvements in the transfection efficiency in both HeLa and A549 cells maintaining higher cell viability. Sequential delivery of GFP-specific siRNA resulted in ∼73% suppression of the target gene. Flow cytometry results revealed that one of the formulations, mG3–pDNA complex, exhibited the highest gene transfection (∼49–68%) outperforming pDNA complexes of native dendrimers and the standard transfection reagent, Superfect (∼32–36%). All these results ensure the potential of the modified dendrimers as effective vectors for future gene delivery applications.
1. Introduction
Dendrimers are highly branched and regular synthetic macromolecules with well-defined structures, which are finding widespread use as functional nano-biomaterials.1,2 The unique architecture and properties have made them useful for various biomedical applications including drug and gene delivery, imaging and diagnostics.3,4 Recently, gene therapy has shown potential to treat various diseases ranging from inherited disorders to acquired diseases.5–8 Nonetheless, challenges associated with gene therapy such as gene delivery efficiency, gene expression/regulation, and toxicity still exist mainly due to the lack of safe and efficient gene vectors. Viruses have been shown to overcome these challenges;9,10 however, severe complications such as immunogenic response, pathogenicity and difficulties in large scale production have led to lethal outcomes, which limit their applications. Non-viral gene delivery vectors, on the other hand, suggest structural and chemical versatility for manipulating their properties to modulate the gene carrying capacity.11–13 The rich collection of polymeric vectors and their designing strategies are receiving attention of the researchers nowadays. Various synthetic and natural vectors have been shown to possess safety and scale-up capability, though they suffer from low transfection efficiency and high cytotoxicity.14–18 Of these, branched polyethylenimine (bPEI, 25 kDa) has been the most widely used vector and considered as a ‘gold standard’ for the delivery of therapeutic genes to various cells.13,16 PAMAM dendrimers have also been demonstrated as potential candidates in the design and synthesis of efficient and biocompatible gene delivery vectors.19–23 The high degree of branching, high charge density, globular architecture, well defined molecular weight and tunable surface functional groups of dendrimers allow efficient interactions with the therapeutic molecules. Moreover, primary amines on the outer surface of the dendrimers allow electrostatic interactions with the negatively charged nucleic acids and condense them into nano-sized complexes (dendriplexes) as well as protect the cargo against nucleases. In contrast, tertiary amines in the core get protonated at acidic pH and facilitate the endosomal escape through the proton-sponge mechanism. PAMAM dendrimers of higher generations (G5–G10) are more toxic than lower generation ones (G2–G4) but exhibit higher transfection efficiency. It is well reported that the cytotoxicity of higher generation cationic PAMAM dendrimers is the result of interactions between positively charged dendrimers and negatively charged molecules located on the cell surface. Therefore, several modifications have been suggested to reduce the toxicity and enhance their gene transfer efficacy.24–31 Additionally, to reduce the cost and time of the production of higher generation dendrimers, non-toxic lower generation PAMAM dendrimers have been modified by tethering polyethylenimine (PEI), diethylenetriamine, histidine, guanidine and spermine or by crosslinking using the disulfide linker. These modifications have resulted in improved transfection efficiency compared to their native counterparts but almost comparable to Lipofectamine or branched PEI (25 kDa).
In the present investigation, low generation PAMAM dendrimers (G2–G4) have been modified via Michael addition of methyl acrylate onto the surface amine groups followed by amidation reaction with a flexible oligoamine linker, TEPA, to improve the transfection efficiency of the modified dendrimers without compromising on cytotoxicity. Modified PAMAM dendrimers (mG2–mG4) were characterized physicochemically and then evaluated for their gene carrying capacity and cell viability. The best formulation among all was assessed for its ability to deliver siRNA by quantifying the suppression of the target gene expression. The results were compared with native dendrimers and commercially available efficient transfection reagents, Lipofectamine 2000 and Superfect.
2. Materials and methods
2.1. Synthesis of modified PAMAM dendrimers (mG2–mG4)
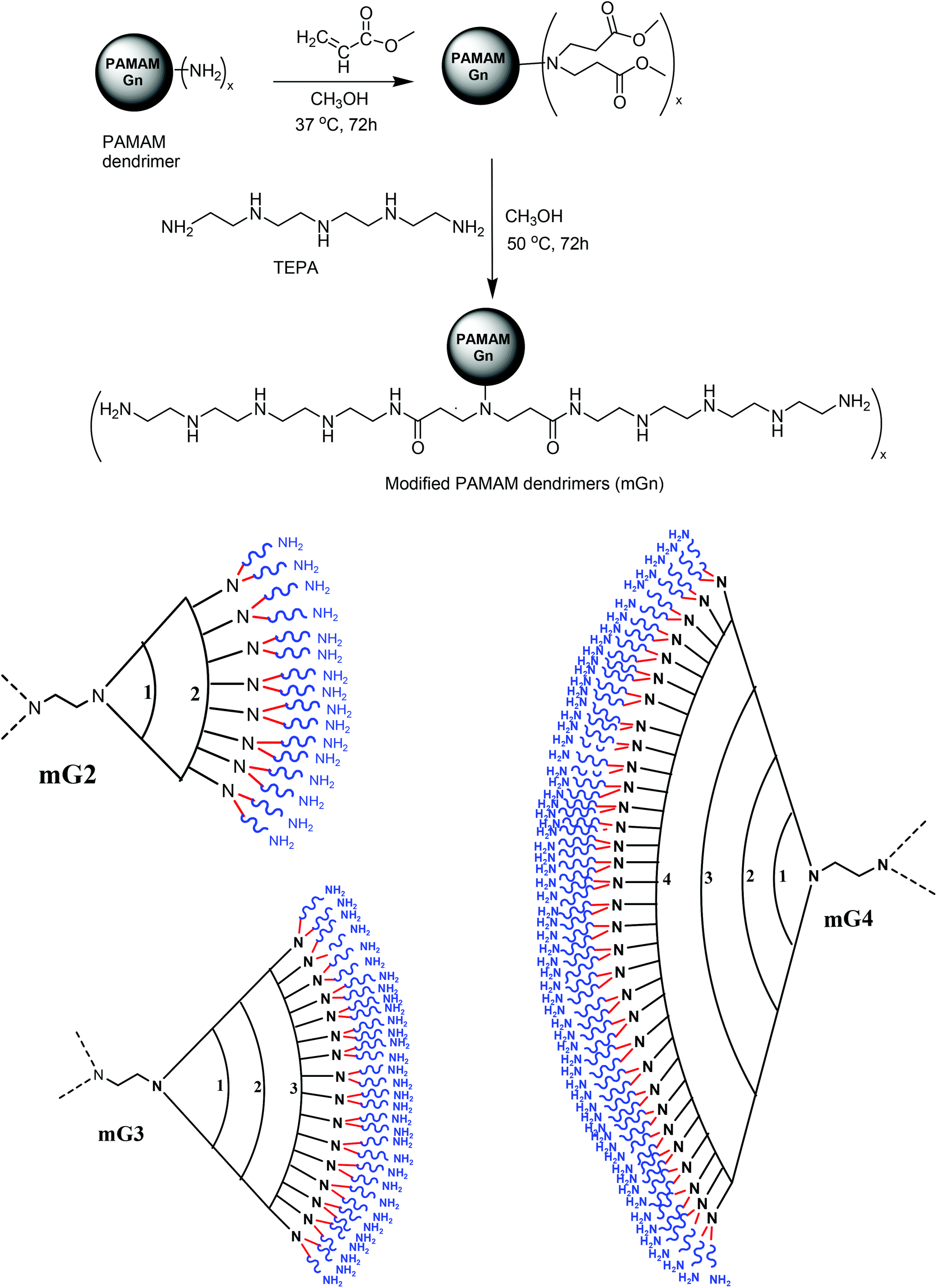
Poly(amidoamine) (PAMAM) dendrimers (G2–G4) were conjugated with tetraethylenepentamine (TEPA) in a two-step process following a reported method.32 Briefly, to an ice-cold solution of PAMAM dendrimer (G2, 50 mg, 0.015 mmol) in methanol (2.5 ml), a large excess of methyl acrylate (5.76 mmol), taken up in methanol (2.5 ml), was added dropwise and the reaction mixture was stirred for 72 h at 37 °C. The excess methanol and methyl acrylate were removed under reduced pressure on a rotary evaporator to obtain a viscous syrupy residue (G2.5), which was again dissolved in methanol (5 ml) and cooled in an ice-salt bath. A pre-ice-cold solution of TEPA (1.2 g, dissolved in 10 ml of methanol) was added dropwise over a period of 15 min and the reaction mixture was stirred for 72 h at 50 °C. Then the solvent was evaporated on a rotary evaporator, the syrupy residue, so obtained, was dissolved in deionized water (dH2O) and subjected to dialysis against water for 48 h with intermittent changes of water. The dialyzed sample was collected and lyophilized in a speed vac to obtain the modified G2 (mG2) dendrimer in ∼80% yield. Similarly, PAMAM G3 and G4 dendrimers were modified to obtain mG3 and mG4 in ∼77% yield. These modified dendrimers were subjected to characterization by 1H-NMR. 1H-NMR (D2O) δ (ppm): 2.25 (–CH2CONH–), 2.5 (>N–CH2–), 2.65 (–NCH2–), 2.85 (–NCH2, TEPA), 3.15 (–NCH2CH2CO). Percent substitution of TEPA onto G2–G4 dendrimers was determined by the standard TNBS method.33
2.2. Preparation of modified dendrimers (mG2, mG3, mG4)-pDNA complexes
Aqueous solutions of mG2/mG3/mG4 (1 mg ml−1 of each) were separately mixed with 0.3 μg pDNA at various w/w ratios (0.16, 0.33, 0.5 and 0.66) in the presence of 20% dextrose (5 μl) and the final volume was made up to 20 μl with deionized water. The resulting samples were then vortexed and incubated for 30 min at room temperature (25 ± 1 °C) prior to their use in biophysical studies.
2.3. DNA binding assay by gel electrophoresis
The DNA binding ability of PAMAM dendrimers (G2, G3, G4) and modified PAMAM dendrimers (mG2, mG3, mG4) was determined by gel electrophoresis. All the native and modified dendrimer–pDNA complexes were prepared at w/w ratios of 0.16, 0.33, 0.50 and 0.66 keeping the amount of pDNA fixed (300 ng). These complexes were vortexed, incubated for 30 min at 37 °C, mixed with 2 μl Orange G dye and loaded into the wells of 0.8% agarose gel pre-treated with ethidium bromide. The samples were electrophoresed (100 V, 1 h) and the bands were visualized in the Gel Doc System (G:box UV illuminator, Syngene, UK).
2.4. Acid–base titrations
Buffering capacity is usually measured by acid–base titration. Here, in this study, we have compared the buffering capacity of native (G2–G4) and modified dendrimers (mG2–mG4). Each dendrimer (3 mg) was dissolved in 0.1 M NaCl solution (30 ml, 0.1 mg ml−1) and pH was adjusted to 10.0 by the addition of 0.1 N NaOH. Then, the pH of each of the solution was brought to 3.0 by adding 25 μl aliquots of 0.1 N HCl (titrant) and after each addition, pH of the solution was recorded. Finally, a graph was plotted between pH and the amount of aq. HCl used (μl).
2.5. Stability of native and modified dendrimer–pDNA complexes
Native PAMAM dendrimers (G2–G4) and their modified analogs (mG2–G4) (1 mg ml−1) were complexed with pDNA at their best working w/w ratios (at which these complexes exhibited the highest transfection efficiency) and incubated for 30 min. Subsequently, heparin, a strong polyanion, was added in increasing amounts from 0 to 3.0 U, which competed with pDNA and released it from native and modified PAMAM dendrimers. The samples were further incubated for 30 min, mixed with 2 μl Orange G dye, electrophoresed (100 V, 1 h) in 0.8% agarose gel containing EtBr and bands were visualized in the Gel Doc System (G:box UV illuminator). Percent release of pDNA from the complexes was determined by densitometry.
2.6.
In vitro transfection assay
HeLa and A549 cells were seeded into 96-well culture plates in DMEM with 10% FBS and incubated at 37 °C in a humidified 5% CO2 environment. Transfection was carried out at ∼70–75% confluency in the absence and presence of serum. Native PAMAM–pDNA and modified PAMAM–pDNA complexes were formed at various w/w ratios (1.66, 3.33, 5.0, 6.66, 10.0 and 16.66) and diluted separately with complete and incomplete media. The bPEI/pDNA complex was prepared at a w/w ratio of 1.6. Similarly, pDNA complexes were also prepared with commercially available transfection reagents, Lipofectamine 2000 and Superfect, according to manufacturers' protocols. These complexes were then gently added onto the cells and the plates were kept at 37 °C in a CO2 incubator. After 3 h, the media was aspirated out and fresh complete medium was added onto the cells in each well. Post-36 h, the cells were observed for GFP expression under an inverted fluorescence microscope at 10× magnification.
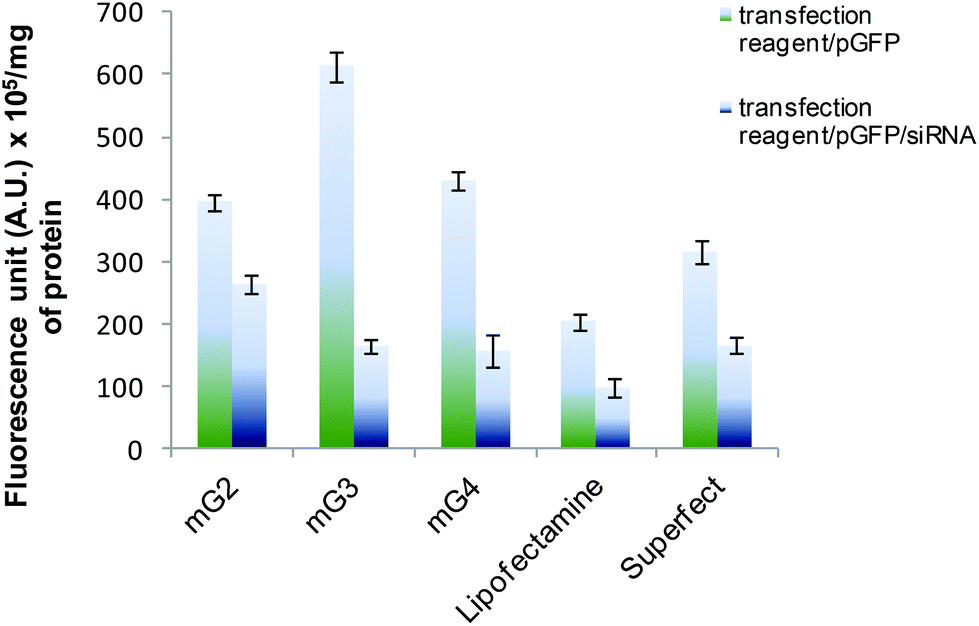
In another experiment, siRNA was sequentially delivered by the modified PAMAM dendrimers (mG2–mG4) and the results were compared with Lipofectamine- and Superfect-mediated siRNA delivery. HeLa cells were first treated with modified PAMAM (mGn)–pDNA complexes at a w/w ratio of 6.66 and after 3 h of incubation, the complex was aspirated out followed by addition of the modified PAMAM (mGn)-siRNA complex (0.3 μM μg−1 of mGn). Again, after 3 h, it was replaced by complete medium (DMEM with 10% FBS). The plate was kept in an incubator for 36 h and then suppression levels were monitored by GFP quantification. All the experiments were repeated three times.
2.7. Quantification of GFP expression and the total protein content
Quantitative analysis of green fluorescent protein (GFP) in the transfected cells was performed using a Nanodrop ND-3000 spectrofluorometer. Post-transfection, the cells were washed with 1× PBS and incubated with cell lysis buffer for 45 min at 37 °C. After centrifugation, 2 μl of the cell lysate from each well was dropped on Nanodrop to estimate the protein spectrofluorometrically. 1× PBS was used as a blank to calibrate the spectrofluorometer to zero reading and corrected for background. The total protein content was estimated at 590 nm using the Bradford reagent (BioRad) with BSA (Bangalore Genei, India) as a standard. Coomassie Brilliant Blue G dye present in the Bradford reagent binds to proteins and results in the shifting of the absorption maxima. The fluorescence intensity was estimated in triplicates and expressed as arbitrary fluorescence units per mg protein.
2.8. Cytotoxicity assay
The MTT colorimetric assay was performed to evaluate the cytotoxicity of the native and modified dendrimer–pDNA complexes on HeLa and A549 cells and the results were compared with pDNA complexes of bPEI (25 kDa), Lipofectamine and Superfect. Transfection was carried out as described above and after 36 h of incubation, MTT (100 μl, 1 mg ml−1) was added on to cells in each well and the plate was kept at 37 °C in an incubator under a humidified 5% CO2 environment for 2 h. Then, the supernatant was aspirated out and the formazan crystals, so formed, were dissolved in 100 μl isopropanol containing 0.5% SDS and 0.06 M HCl. The color intensity was then measured spectrophotometrically at 540 nm on an ELISA plate reader. Untreated cells with 100% viability were taken as control. The IC50 value or the concentration of native and modified dendrimer–pDNA complexes, at which the HeLa cells' viability reached 50%, was determined at their w/w ratios of 10.0 and 6.66, respectively. All the experiments were carried out in triplicates.
2.9. Hemolytic activity
To examine the activity of modified PAMAM dendrimers on human red blood cells (hRBCs), the hemolysis assay was performed. Human blood was collected and RBCs were isolated. These were washed with PBS (pH 7.2) thrice and then suspended as 4% (v/v) solution in PBS. 100 μl of hRBC suspension was added into each well of a sterilized 96-well plate and treated with varying concentrations (5, 10, 20, 40, 50 and 100 μg ml−1) of modified dendrimers and their native counterparts. The plates were incubated for 1 h at 37 °C and centrifuged at 800g for 10 min. Furthermore, aliquots of 100 μl of supernatant were transferred to fresh 96-well plates and hemoglobin release was monitored using an ELISA plate reader by measuring the absorbance at 540 nm. Triton X-100 (2%, w/v) was used as a control causing 100% hemolysis.
2.10. Enzymatic assay
To examine the ability of modified PAMAM dendrimers to provide protection to the condensed pDNA against nucleases, DNase I protection assay was carried out. mGn–pDNA complexes were separately incubated with DNase I in a buffer (100 mM Tris, 25 mM MgCl2 and 5 mM CaCl2) at 37 °C for 0.25, 0.5, 1.0, and 2.0 h. Complexes in PBS alone served as controls. After the stipulated time of incubation, 5 μl EDTA (100 mM) was added to the reaction mixture to quench the activity of DNase I followed by heating to 75 °C for 10 min. The mixture was further incubated for 1 h at ambient temperature with 10 U of heparin to release the bound pDNA from mGn-pDNA complexes. Subsequently, the samples were mixed with 2 μl Orange G dye, electrophoresed (100 V, 1 h) in 0.8% agarose gel containing EtBr (2 μl per 100 ml gel) and bands were visualized on a Gel Doc System (G:box UV illuminator). The amount of pDNA released from complexes was estimated by densitometry.
2.11. Fluorescence activated cell sorting (FACS) analysis
For examining GFP expression at the individual cell level, FACS analysis was performed post-36 h of incubation. Briefly, HeLa and A549 cells after seeding in 24-well plates for 24 h were washed with 1× PBS (pH 7.4) followed by removal of the media. pDNA (1.5 microgram) complexes of native PAMAM (G2, G3 and G4) dendrimers were prepared at w/w ratios of 6.66, 10.0 and 16.66, while the modified ones (mGn) were complexed at w/w ratios of 3.33, 6.66, 10.0 and 16.66. Thereafter, these complexes were incubated for 30 min at room temperature. Similarly, pDNA complexes were prepared with bPEI (w/w ratio 1.6) and Superfect (following the manufacturer's protocol). These complexes, diluted with DMEM with and without 10% FBS, were added gently onto the cells for 3 h at 37 °C. Then, the media was replaced by complete media (containing 10% FBS) and the plates were kept in an incubator. After 36 h, the plates were observed under a fluorescence microscope and cells were subjected to trypsinization. An equal volume of complete medium (DMEM with 10% FBS) was added to each well and centrifuged at 5000 rpm for 5 min at 4 °C. The supernatant was removed and the pellet was washed with 1× PBS. Subsequently, the pellet was resuspended in 1× PBS (1 ml), transferred to a cuvette and subjected to flow cytometric analysis. The cells without sample treatment (i.e. non-transfected cells) were used as a negative control with their fluorescence set to 1%. For each sample, 5000 events were counted using a flow cytometer (Guava easyCyte™Plus Flow Cytometry System, USA) equipped with the Cytosoft software.
3. Results and discussion
Synthetic polymers with a defined structure, shape and size are receiving increasing attention in the development of drug and gene delivery systems. The unique structure and shape of PAMAM dendrimers have received the most attention to develop as potential gene and drug delivery systems. These molecules possess a highly branched structure, modifiable functional groups on the surface, mono-dispersity, a fixed molecular weight and the ability to bind negatively charged nucleic acids and protect them from degradation; however, their gene carrying capacity and cytotoxicity are generation dependent, i.e. higher generation PAMAM dendrimers (G5–G10) transfer these biomolecules more efficiently but they are cytotoxic as compared to lower generation ones (G2–G4), which are relatively non-toxic but exhibit poor transfection. Therefore, modifications have been incorporated into their structures to improve the transfection efficiency and cell viability. In the present investigation, we assumed that the transfection efficiency of the lower generation (G2–G4) dendrimers could be enhanced by tethering a flexible oligoamine linker, tetraethylenepentamine (TEPA). The projected commercially available linker was selected mainly for the following reasons, viz., (i) on conjugation, it would provide primary amines on the surface away from the branched structure for interaction with negatively charged nucleic acids, (ii) the secondary amines (3 per linker molecule) in the backbone would not only improve the buffering capacity but also, to an extent, participate in the cooperative binding of nucleic acids via electrostatic interactions, and (iii) the incorporated secondary amines would assist in controlling the cytotoxicity of the modified dendrimers. The cooperative binding would strengthen the DNA binding ability and suppress the degradation in the cellular milieu. Keeping these points in mind, we synthesized TEPA conjugated G2, G3 and G4 dendrimers (mG2, mG3 and mG4) via a two-step reaction, as depicted in Scheme 1.
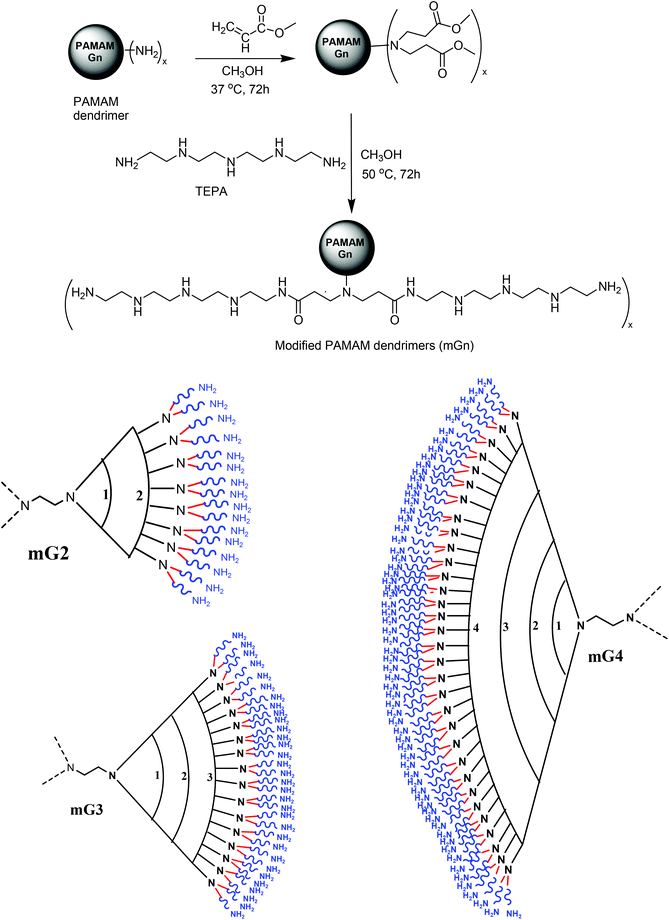 |
| | Scheme 1 Schematic representation of synthesis of modified PAMAM dendrimers (mGn) n = 2, x = 16; n = 3, x = 32; n = 4, x = 64. | |
Preparation of modified dendrimers begins with Michael addition reaction involving methyl acrylate and surface amine groups of PAMAM dendrimers (G2, G3 and G4) following the standard method.32 Each terminal nitrogen atom serves as a branching point. The reaction resulted in the generation of G2.5, G3.5 and G4.5 dendrimers, which were subsequently subjected to reaction with TEPA in large excess to yield a new set of surface primary amine groups and secondary amines in the backbone of the flexible oligoamine linker (Table 1).
Table 1 Density of primary, secondary and tertiary amines in native and modified PAMAM dendrimers
| Types of amine groups |
Dendrimer gen. |
| G2 |
G3 |
G4 |
mG2 |
mG3 |
mG4 |
| Primary |
16 |
32 |
64 |
32 |
64 |
128 |
| Secondary |
— |
— |
— |
96 |
192 |
384 |
| Tertiary |
8 |
16 |
32 |
24 |
48 |
96 |
The attempted conjugation was determined by the TNBS method using UV-VIS spectrophotometry.33,34 A soluble yellow-colored product was formed between 1° amines of mGn dendrimers and TNBS, which was measured at 335 nm. Analysis results revealed quantitative generation of TEPA-conjugated G2, G3 and G4 dendrimers. Modified dendrimers were also characterized by 1H-NMR; a peak at δ 2.85 due to –NCH2 in TEPA confirmed the incorporation of the linker into the modified dendrimers (Fig. S1†).
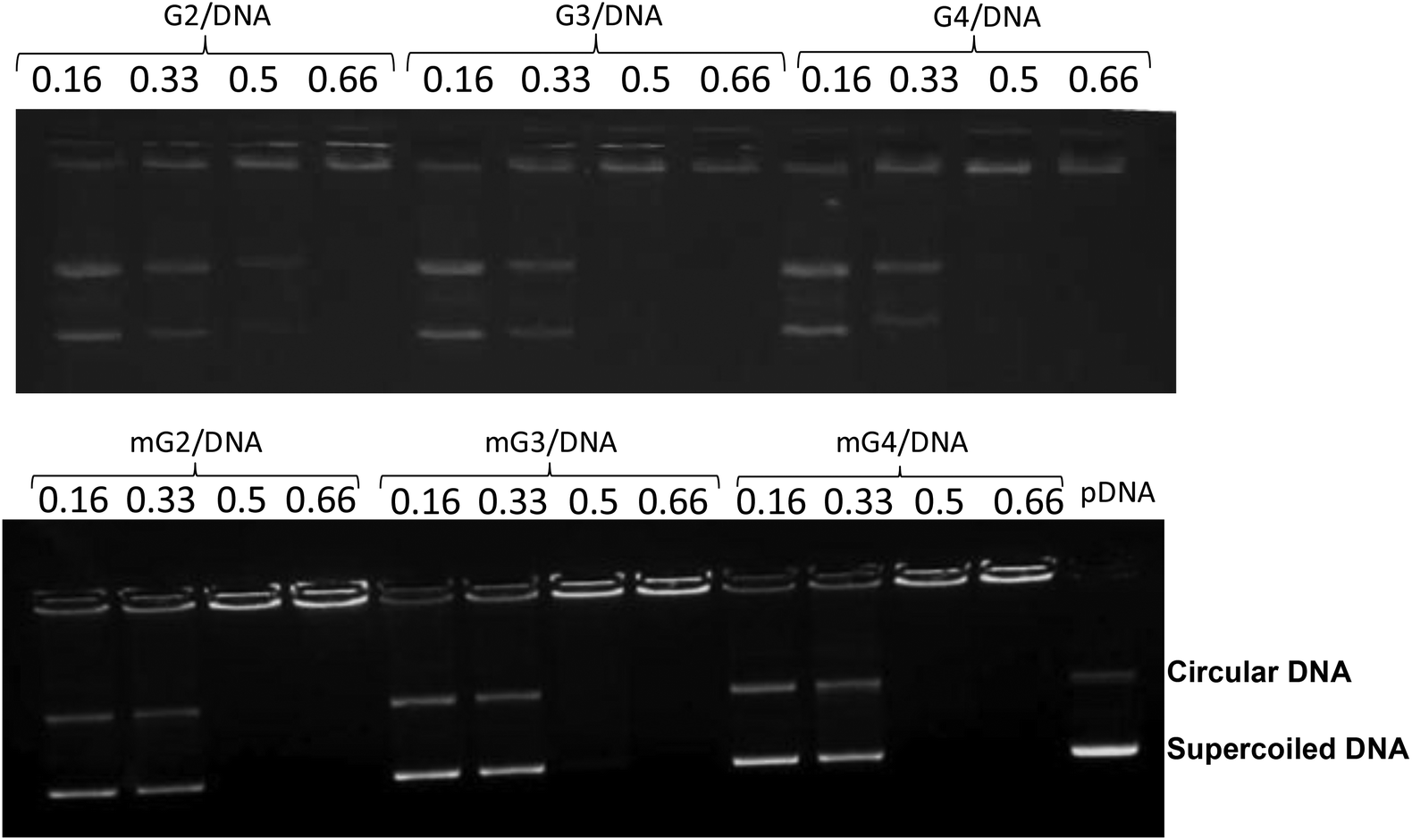
3.1. Mobility shift assay
The positive charge on native PAMAM (G2, G3 and G4) and modified PAMAM (mG2, mG3 and mG4) dendrimers and the negatively charged backbone of DNA interact with each other through electrostatic interactions and form nanosized complexes (dendriplexes). The DNA retardation assay was executed to find out the amount of cationic polymer required to completely neutralize the charge on the fixed amount of DNA. The mixing was done at different w/w ratios of dendrimers and pDNA and the samples were run on the gel. Fig. 1 shows DNA binding to PAMAM dendrimers at different w/w ratios. The complete retardation of the mobility of 0.3 μg of pDNA by the PAMAM (G2)–pDNA complex occurred at a w/w ratio of 0.66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while PAMAM (G3 and G4) and PAMAM (mG2, mG3 and mG4)–pDNA complexes retarded at a w/w ratio of 0.5:1. This might be due to the fact that after amidation, pDNA interacted mainly with the surface amine groups and the modified dendrimers behaved in a similar fashion to the unmodified ones. Furthermore, to determine the binding affinity between dendrimers and pDNA, the apparent equilibrium dissociation constant (Kd) was calculated using the gel retardation assay following a reported protocol.35 As expected, the results revealed the highest affinity of mG4 and it decreased from mG4–mG3–mG2–G4–G3–G2 on the basis of their Kd values (Table S1†).
:1, while PAMAM (G3 and G4) and PAMAM (mG2, mG3 and mG4)–pDNA complexes retarded at a w/w ratio of 0.5:1. This might be due to the fact that after amidation, pDNA interacted mainly with the surface amine groups and the modified dendrimers behaved in a similar fashion to the unmodified ones. Furthermore, to determine the binding affinity between dendrimers and pDNA, the apparent equilibrium dissociation constant (Kd) was calculated using the gel retardation assay following a reported protocol.35 As expected, the results revealed the highest affinity of mG4 and it decreased from mG4–mG3–mG2–G4–G3–G2 on the basis of their Kd values (Table S1†).
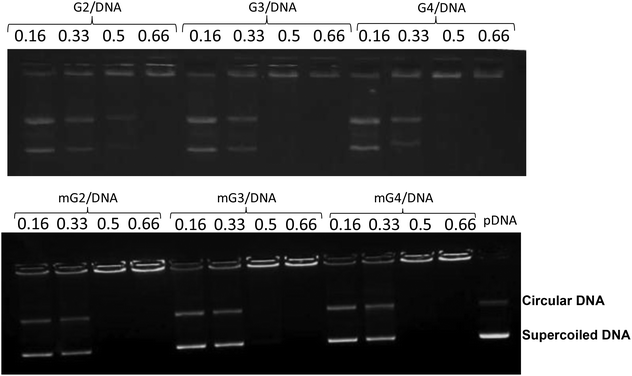 |
| | Fig. 1 DNA mobility shift assay of native and modified PAMAM dendrimer–pDNA complexes. Dendrimers were mixed with pDNA at various weight ratios and electrophoresed in 0.8% agarose gel. | |
3.2.
In vitro transfection in mammalian cells
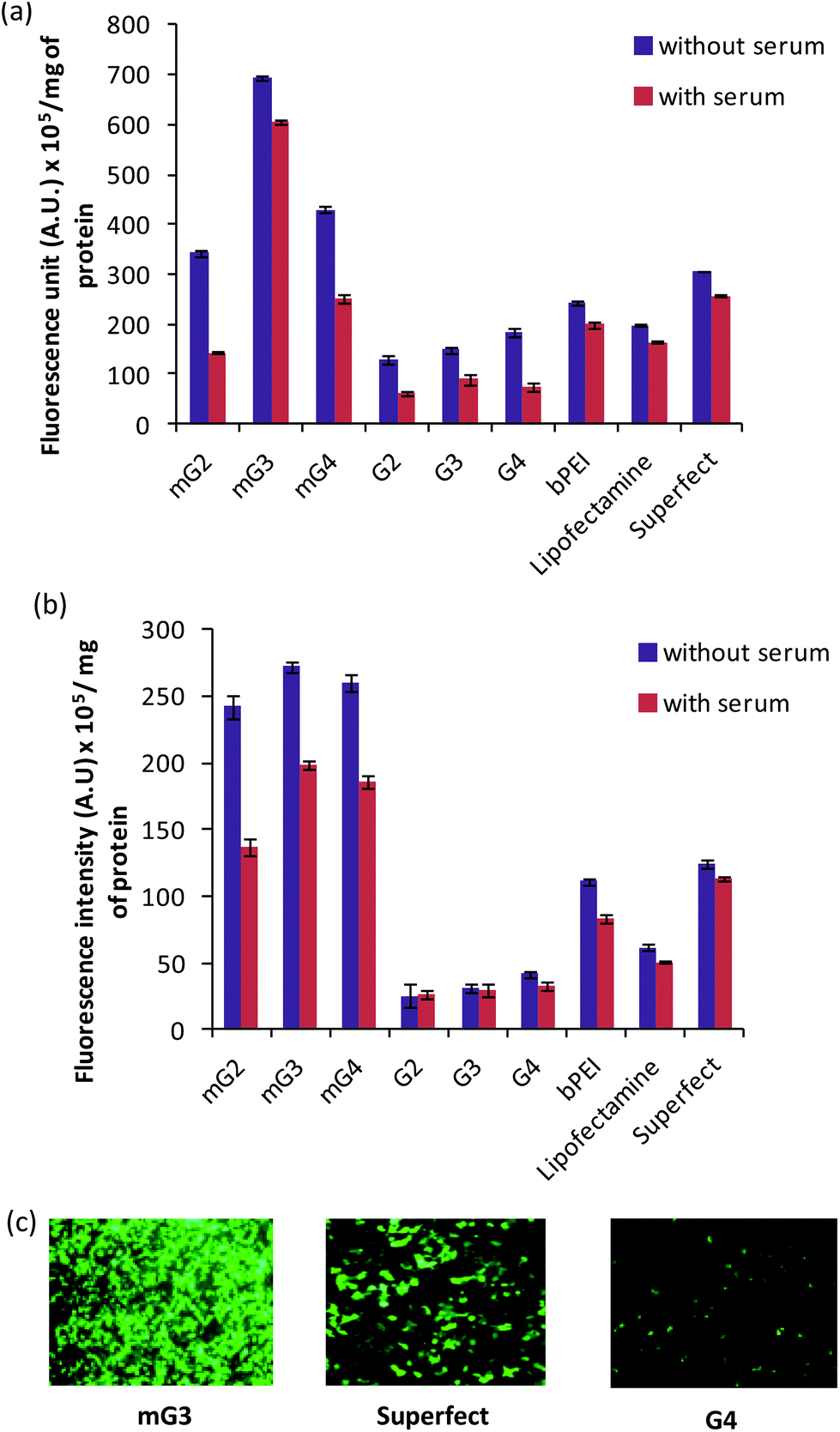
The gene delivery efficacy of the modified PAMAM dendrimers (mG2, mG3 and mG4) was evaluated by in vitro transfection experiments on HeLa and A549 cell lines. The results were compared with the native PAMAM dendrimers (G2–G4) and commercial transfection reagents, viz., Lipofectamine and Superfect. The assay was performed at different w/w ratios of dendrimer (modified dendrimers)–pDNA complexes slightly higher than those used in the gel retardation assay so as to keep the overall charge positive on the complexes. The transfection results on A549 and HeLa cell lines, in the absence and presence of serum (10%), are presented in Fig. 2(a–c). It was observed that pDNA complexes of modified dendrimers showed significantly enhanced transfection efficiency (∼2.2–8.6 folds) than pDNA complexes of native PAMAM dendrimers, bPEI and commercially available Lipofectamine and Superfect. The transfection efficiency was found to be cell line dependent and showed higher transfection in HeLa cells (Fig. 2a). Also, the transfection efficiency varied with the w/w ratio of dendrimer:pDNA in the complexes. Native and modified PAMAM dendrimer–pDNA complexes exhibited the highest transfection efficiency at a w/w ratio of 6.66:1 in A549 cells while, in HeLa cells, native and modified dendrimer-pDNA complexes displayed the same at w/w ratios of 10.0 and 6.66, respectively. Initially, the transfection efficiency, in A549 cells, increased with an increase in the w/w ratio from 1.66 to 6.66 and after attaining the maximum at this ratio, it started decreasing. Among the modified mGn–pDNA complexes, the mG3–pDNA complex showed the highest transfection efficiency in both the cell lines. The mG3–pDNA complex showed ∼3.5 and 2.3 fold higher transfection efficiency than Lipofectamine–pDNA and Superfect–pDNA complexes, respectively, in HeLa cells, while in A549 cells, it was ∼4.4 and 2.2 fold higher than Lipofectamine–pDNA and Superfect–pDNA complexes, respectively. Compared to the native G3–pDNA complex, the mG3–pDNA complex showed ∼8.6 and 4.7 fold higher transfection efficiency in A549 and HeLa cells, respectively. In the presence of serum, a similar trend was observed; however, as expected, the transfection efficiency decreased marginally, which might be attributed to non-specific interactions between the positively charged complexes and negatively charged serum proteins. Still, the mG3–pDNA complex performed the best and exhibited ∼1.8–3.9 fold higher transfection efficiency as compared to Lipofectamine–pDNA and Superfect–pDNA complexes in both HeLa and A549 cells. With respect to native dendrimer–pDNA complexes, the mG3–pDNA complex showed ∼6.8 fold higher gene delivery activity in both the cell lines. Surprisingly, in HeLa cells, the decrease in transfection efficiency of the native G3–pDNA complex was higher than that observed in the mG3–pDNA complex. In both the absence and presence of serum, the modified dendrimers showed considerably higher transfection efficiency compared to their unmodified (native) counterparts. In A549 cells, a surprising result was obtained in the absence and presence of serum, both mG3–pDNA and mG4–pDNA complexes exhibited comparable transfection efficiency (Fig. 2b).
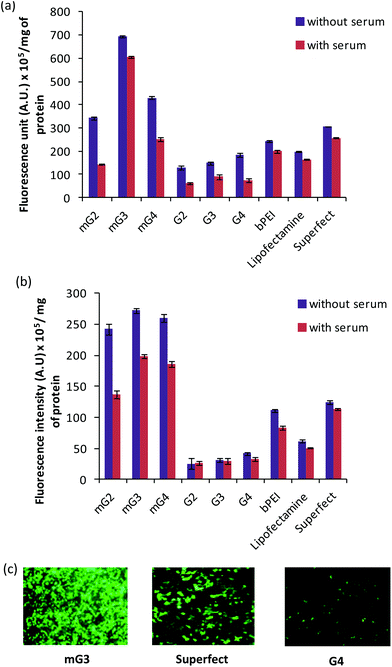 |
| | Fig. 2
In vitro transfection efficiency of native and modified dendrimer–pDNA complexes in the absence and presence of serum in (a) HeLa cells (w/w ratios of native dendrimer–pDNA complexes 10 and modified dendrimer–pDNA complexes 6.66), and (b) A549 cells (w/w ratios of native dendrimers and modified dendrimer–pDNA complexes 6.66). The fluorescence intensity of GFP in the cell lysate was measured using a NanoDrop spectrofluorometer and the results are expressed in terms of arbitrary units (A.U.) per mg of total protein. The results represent the mean of three independent experiments. (c) Fluorescence microscopic images, captured at 10×, of transfected HeLa cells with mG3, Superfect and G4–pDNA complexes. Cells were incubated with pDNA complexes of mG3, Superfect and G4 for 3 h and expression of GFP was monitored post 36 h of incubation. | |
The higher transfection efficiency shown by the modified dendrimer–pDNA complexes might be attributed to several factors such as higher buffering capacity, improved binding affinity, stability under the cellular environment (i.e. enzymatic stability) and higher charge density. Conversion of terminal primary amines of the native dendrimers to tertiary amines as well as incorporation of secondary amines of the oligoamine linker improved the buffering capacity marginally but good enough to enhance the endosomal escape capacity.36 Previously, it has also been demonstrated that upon modification of polypropylenimine with oligoethylenimine, the transfection efficiency increased, which might be due to the change in the molecular weight and the number of secondary and tertiary amines incorporated post-modification implying enhanced proton sponge effects.37,38 Besides, higher charge density (i.e. zeta potential) is also important during interactions with the cell membrane and endosomal release of the complexes. It has been shown that increased zeta potential enhances cell association to promote the cellular uptake and thereby the transfection efficiency.39–41 Internalization and accumulation of positively charged complexes in the endosomes induce protonation of the surface amine groups via the buffering mechanism, which eventually triggers the entry of Cl− ions into the endosomes resulting in an increase in the endosomal osmotic pressure followed by its rupturing to release the complexes into the cytoplasm of the target cells.42 All these factors are cumulatively responsible for the enhanced transfection efficiency of the modified dendrimer complexes outperforming the efficacy of the native dendrimers and commercial transfection reagents.
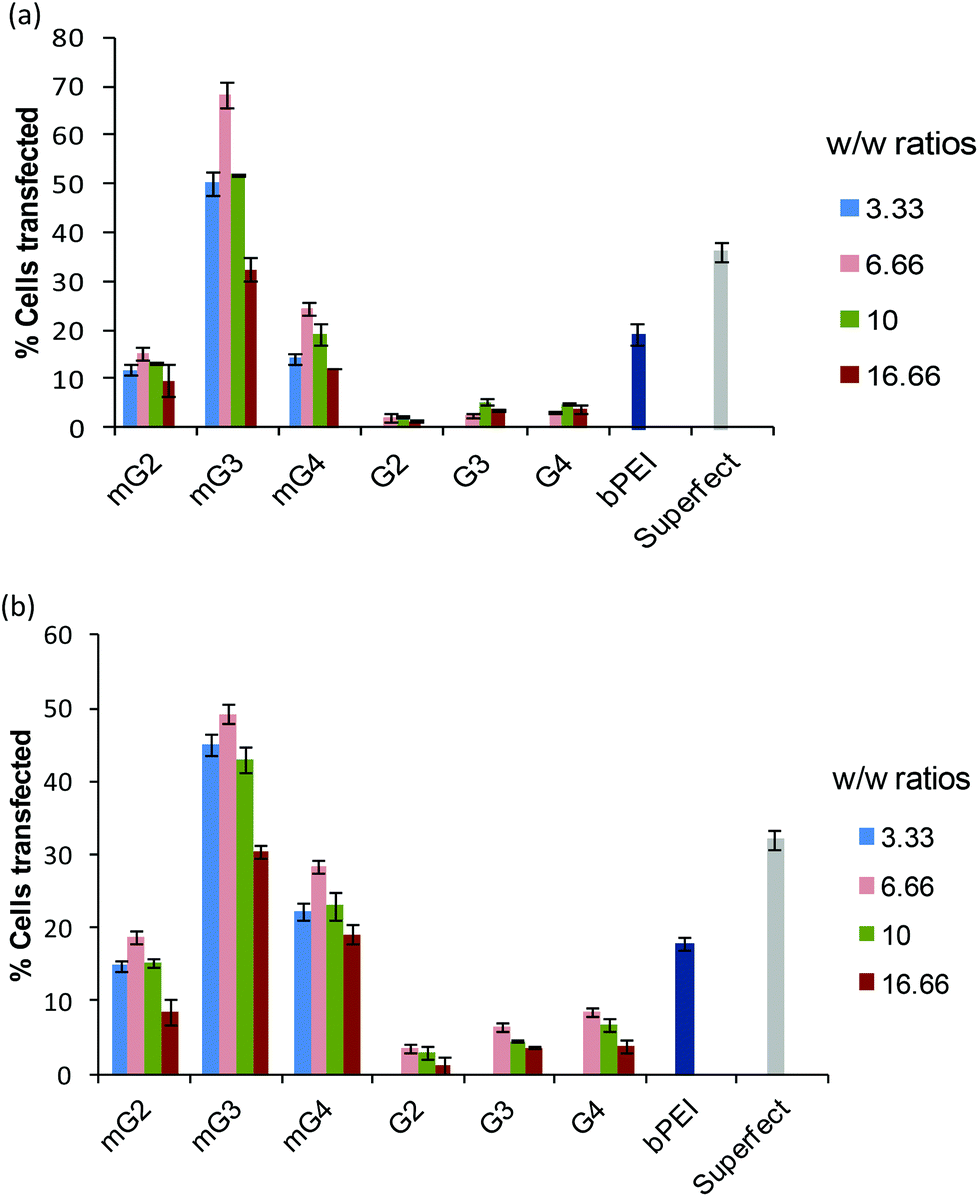
Furthermore, in order to determine the percentage of cells transfected with pDNA complexes of native dendrimers, modified dendrimers, bPEI (25 kDa) and Superfect, FACS analysis was carried out. It was found that modified dendrimer–pDNA complexes transfected higher percentage of cells as compared to native dendrimer–pDNA complexes. Among modified dendrimers, the mG3–pDNA complex transfected the highest percentage of cells (∼49–68% cells) at a w/w ratio of 6.66, while PAMAM G4–pDNA, bPEI–pDNA and Superfect–pDNA complexes could transfect only ∼2–8%, ∼18–20% and ∼32–36% cells, respectively (Fig. 3), in both the cells. Even mG2–pDNA and mG4–pDNA complexes transfected a higher number of cells (∼13–28%) compared to unmodified dendrimer–pDNA complexes in both HeLa and A549 cells. Interestingly, in contrast to FACS results for A549 cells, wherein mG2–pDNA, mG3–pDNA and mG4–pDNA complexes showed a marked difference in the number of transfected cells (∼18%, 49% and 28%, respectively), quantification results (using spectrofluorometry) of GFP showed more or less comparable transfection (Fig. 2b). This anomaly could be explained that once transfected, mG2 and mG4 facilitated more efficient gene expression than mG3 specifically in A549 cells, i.e. higher GFP expression in less number of cells. The results suggest the potential of these modified dendrimers as promising gene carriers.
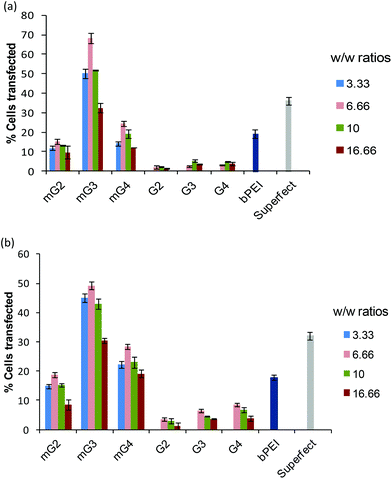 |
| | Fig. 3 Percent transfection efficiency of pDNA complexes of native and modified dendrimer–pDNA complexes at various weight ratios in (a) HeLa and (b) A549 cells. Results were compared with bPEI–pDNA (w/w 1.6) and Superfect–pDNA complexes. The experiment was repeated three times and error bars represent the standard deviation. | |
3.3. siRNA delivery
The versatility of the mG3 vector was further established by sequential delivery of siRNA onto HeLa cells. The results showed that siRNA delivered by mG3 dendrimers suppressed expression of GFP by ∼73% (Fig. 4), while Superfect and Lipofectamine 2000 mediated sequential delivery of siRNA resulted in ∼47% suppression in the expression of the GFP gene implying that the modified G3 dendrimer can be used for efficient delivery of nucleic acids (DNA and RNA) in vivo.
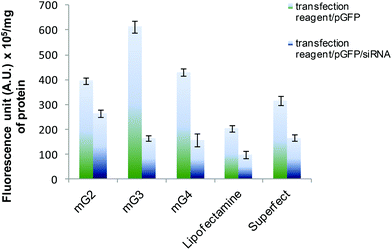 |
| | Fig. 4 Sequential delivery of GFP-specific siRNA using mGn dendrimers in HeLa cells. Percent suppression of EGF expression by mGn mediated delivery was compared with the efficacy of Lipofectamine and Superfect-mediated sequential delivery. Experiments were repeated three times and error bars represent the standard deviation. | |
3.4. Particle size, morphology and zeta potential measurements
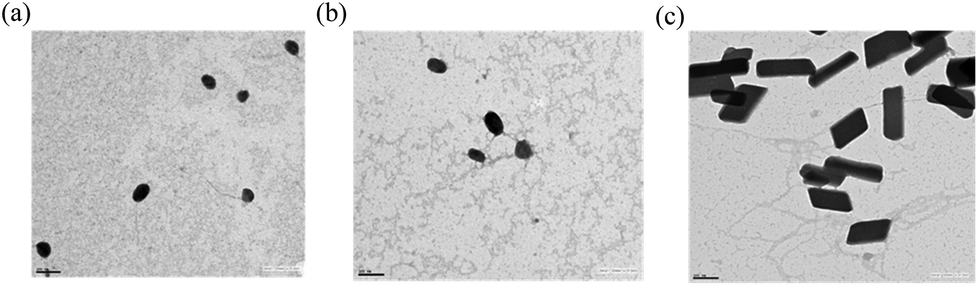
The particle size and zeta potential of pDNA complexes of G2, G3, G4, mG2, mG3 and mG4 at their best w/w ratios are shown in Table 2. An interaction with pDNA yielded particles in the nano-size range. It was observed that the size of modified mGn–pDNA complexes was higher in comparison with their respective native dendrimers, which might be due to tethering of an oligoamine linker, TEPA, with a higher degree of branching in modified dendrimers. On complexation with pDNA, the particle size was found to be in the range 157–211 nm and in the presence of 10% serum, the size of the complexes further decreased due to more compaction and ranged between 37 and 52 nm. The morphology and size of pDNA complexes of mG2–mG4 were also examined by TEM, which revealed that mG2 and mG3–pDNA complexes were almost spherical/oval in shape with sizes ∼40 and 50 nm, respectively (Fig. 5). However, surprising results were obtained for the mG4–pDNA complex; it exhibited rectangular shaped particles of dimensions 162 nm × 72 nm. The particles in DLS were found to have bigger size as compared to that observed in TEM images. This could be due to the measurement of the hydrodynamic diameter in DLS, whereas TEM provides the size of the complex in the dry state.11,12,16
 |
| | Fig. 5 Transmission electron microscopy (TEM) images of pDNA complexes of (a) mG2, (b) mG3 and (c) mG4. Scale bar: 100 nm. | |
Table 2 Size and zeta potential measurements of native and modified dendrimer–pDNA complexes
| Samples |
Average size ± S.D. (nm) |
Average zeta potential ± S.D. (mV) |
| pDNA complex in H2O |
pDNA complex in 10% FBS |
pDNA complex in H2O |
pDNA complex in 10% FBS |
| G2 |
210.6 ± 8.0 |
36.7 ± 1.8 |
27.9 ± 2.9 |
−22.3 ± 2.6 |
| G3 |
167.8 ± 6.7 |
38.5 ± 1.6 |
30.1 ± 3.5 |
−22.2 ± 3.1 |
| G4 |
194.2 ± 6.2 |
41.2 ± 1.9 |
30.2 ± 1.6 |
−21.6 ± 2.2 |
| mG2 |
156.7 ± 5.7 |
49.8 ± 1.7 |
32.6 ± 2.1 |
−17.0 ± 3.3 |
| mG3 |
170.1 ± 7.0 |
51.8 ± 1.8 |
33.6 ± 6.2 |
−15.1 ± 1.2 |
| mG4 |
194.1 ± 7.5 |
51.4 ± 1.9 |
34.5 ± 4.4 |
−9.2 ± 1.7 |
Zeta potential studies showed a slightly higher +ve charge on the pDNA complexes of modified dendrimers as compared to pDNA complexes of native dendrimers. However, in the presence of 10% FBS, the polarity of the complexes reversed, i.e. all the complexes became negatively charged due to adsorption of the serum proteins around +vely charged complexes.
3.5. Buffering capacity
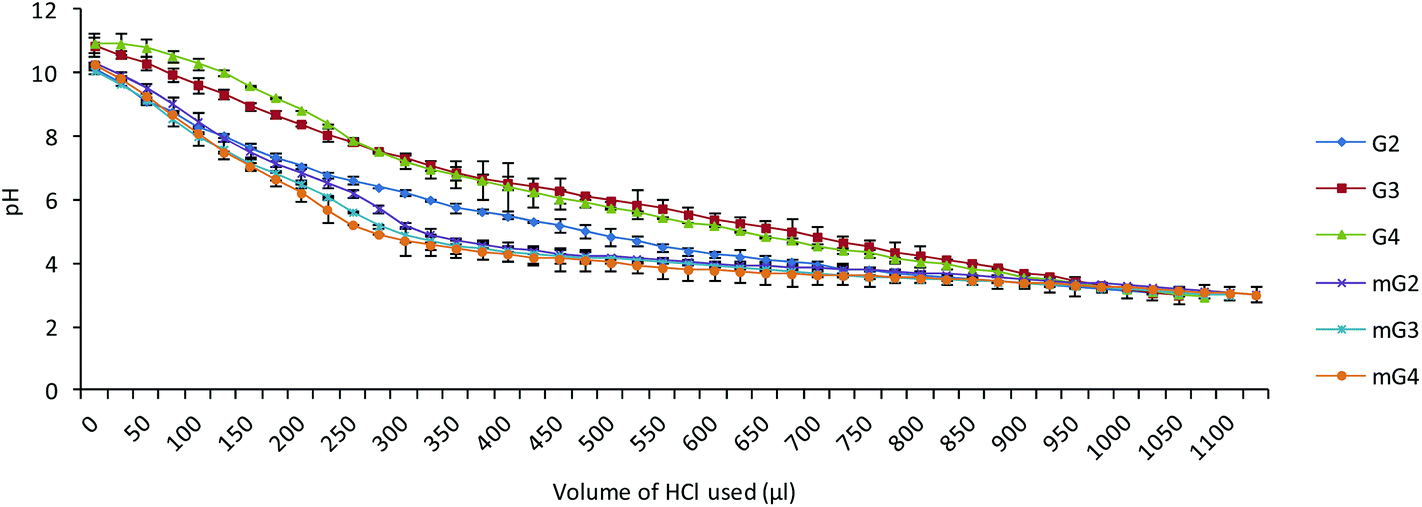
The buffering capacity of the cargo is an essential factor for endosomal escape of the complexes into the cytoplasm for nuclear translocation of the plasmid. Endosomal escape is generally accomplished by endosomal disruption by protonation of polymers (proton sponge effect) in endosomal compartments (low pH). The buffering capacity of the native PAMAM (G2–G4) and modified PAMAM (mG2–mG4) dendrimers was determined by an acid–base titration method in the pH range 3–10. From the plot (Fig. 6), it was found that the proton capturing tendency of the modified PAMAM (mG2, mG3 and mG4) dendrimers slightly increased compared to native PAMAM (G2, G3 and G4) dendrimers. This could be due to the facts, viz. (i) during Michael addition, the primary amines were converted into tertiary amines, and (ii) after reaction with the tetraethylenepentamine, secondary amines were introduced. Incorporation of secondary and tertiary amines into the modified dendrimers might have contributed a bit towards an increase in the overall buffering capacity, which destabilized the endosomal membrane at acidic pH facilitating the effective endosomal escape.
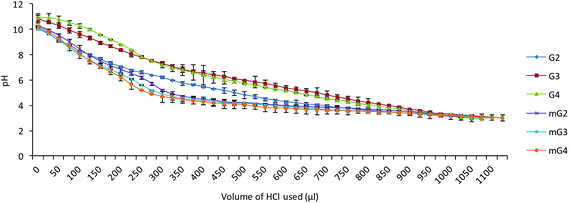 |
| | Fig. 6 Acid−base titration curves of native and modified PAMAM dendrimers. | |
3.6. Stability of pDNA complexes of modified dendrimers
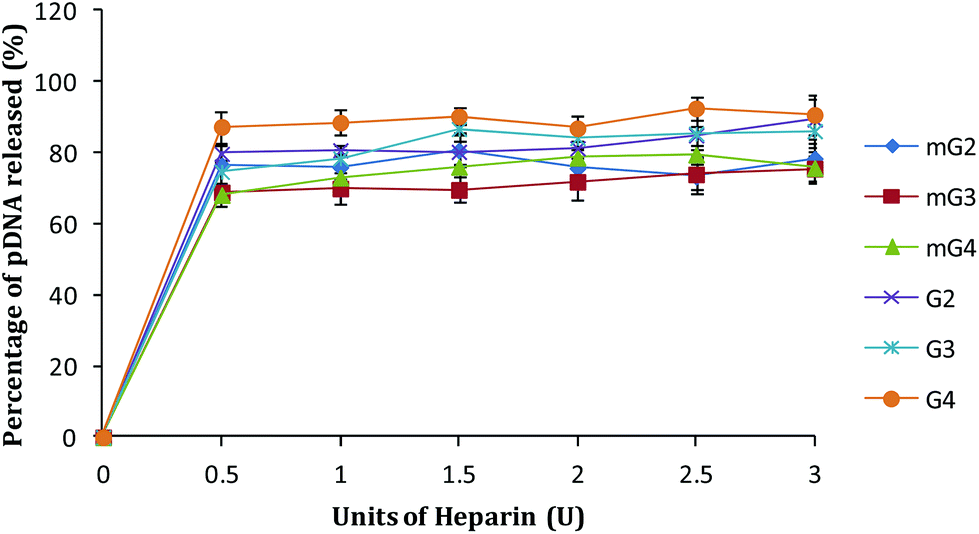
A cationic polymer capable of binding pDNA tightly significantly affects its release inside the cells. An ideal vector should bind pDNA strongly enough so that it can carry it to the target inside the cell without exposing it for degradation in the cellular environment. To mimic the release of pDNA inside the cells in vitro, pDNA complexes of native and modified dendrimers were exposed to a highly negatively charged polymer, heparin, and by increasing its concentration (in terms of units), the release pattern of pDNA was obtained. As shown in Fig. 7, ∼86–90% pDNA was released from the native dendrimer (G2–G4) complexes. Under similar conditions, modified PAMAM (mG2–mG4) dendrimer–pDNA complexes released ∼75–78% of the complexed DNA. Interestingly, the binding ability of mGn–pDNA complexes was observed to be higher, which could be due to higher charge and branching in the modified dendrimers enabling better interactions with the complexed pDNA, whereas loose or inefficient binding of pDNA with G2–G4 dendrimers might be one of the reasons for showing lower transfection.
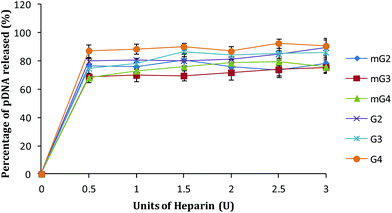 |
| | Fig. 7 DNA release assay of native and modified PAMAM dendrimer–pDNA complexes. Complexes were treated with increasing amounts of heparin (0–3.0 U) and the amount of pDNA released was estimated by densitometry. | |
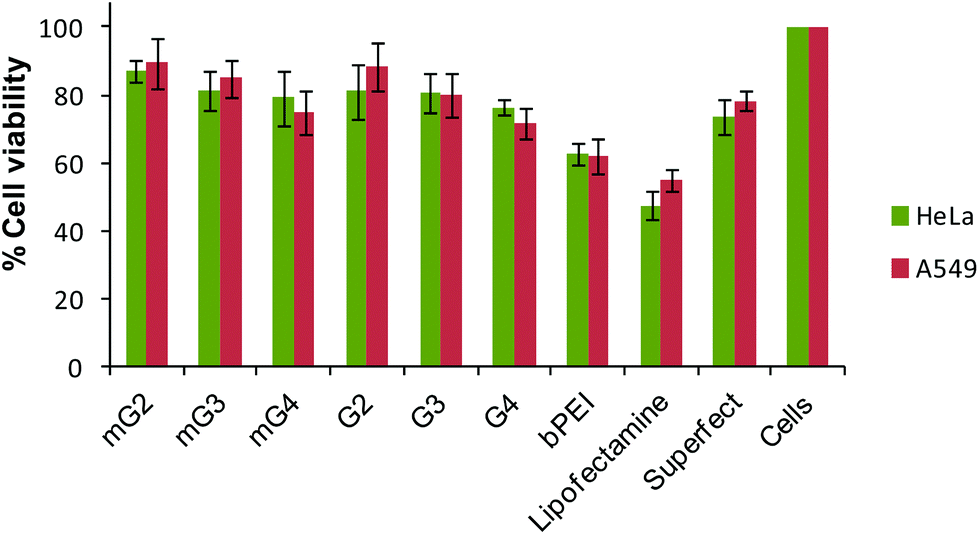
3.7. Cell viability assay
To demonstrate the cytotoxicity profiling of modified PAMAM dendrimers, the MTT assay was carried out and results were compared with the native dendrimers, bPEI and the commercial transfection reagents, Lipofectamine and Superfect. It was observed that the cell viability did not change significantly after incorporation of the oligoamine linker, TEPA. Modified PAMAM dendrimer–pDNA complexes exhibited a cell viability ∼75–89% comparable to the PAMAM counterparts (∼71–88%), but significantly higher than Lipofectamine–pDNA (∼48–55%) and bPEI–pDNA (∼62%) complexes. The Superfect–pDNA complex exhibited 73–77% cell viability, which is an activated G5-PAMAM dendrimer (Fig. 8). As reported in the literature, higher generation PAMAM dendrimers exhibit higher toxicity due to strong interactions with the cell membranes. Here, in the present study, our major goal was to improve the transfection efficiency of the non-toxic lower generation PAMAM dendrimers without compromising on the cytotoxicity. As expected, the conjugation of TEPA did not result in the enhanced cytotoxicity of the modified dendrimers even after the density of primary amines increased. This could be due to a simultaneous increase in the density of secondary and tertiary amines in the modified dendrimers, which, to an extent, took care of the effect of an increase in the cytotoxicity due to an increment in the density of primary amines. The literature also has the record of cationic polymers, such as linear polyethylenimine,43 end-modified poly(β-aminoesters),44 linear poly(amido amine)s,45 which have secondary or tertiary amines or both, exhibiting minimal cytotoxicity. Even branched polyethylenimine (25 kDa), which is highly toxic, shows very low toxicity when the primary amine density is converted into secondary or tertiary amines.16,46,47 The IC50 values further revealed that the native and modified dendrimers were non-toxic (Table S2†).
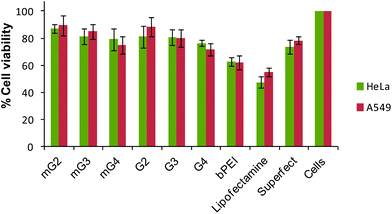 |
| | Fig. 8 Cell viability profiles of native and modified PAMAM dendrimers, bPEI (w/w 1.6), Lipofectamine and Superfect–pDNA complexes at their respective best working w/w or v/w ratios in HeLa and A549 cells (HeLa cells – w/w ratios of native dendrimer–pDNA complexes 10 and modified dendrimer–pDNA complexes 6.66; A549 cells – w/w ratios of native dendrimers and modified dendrimer–pDNA complexes 6.66). Cells were treated with pDNA complexes and their viability was determined by the MTT assay. Percent viability of cells is expressed relative to control cells. Each point represents the mean of three independent experiments performed in triplicate. | |
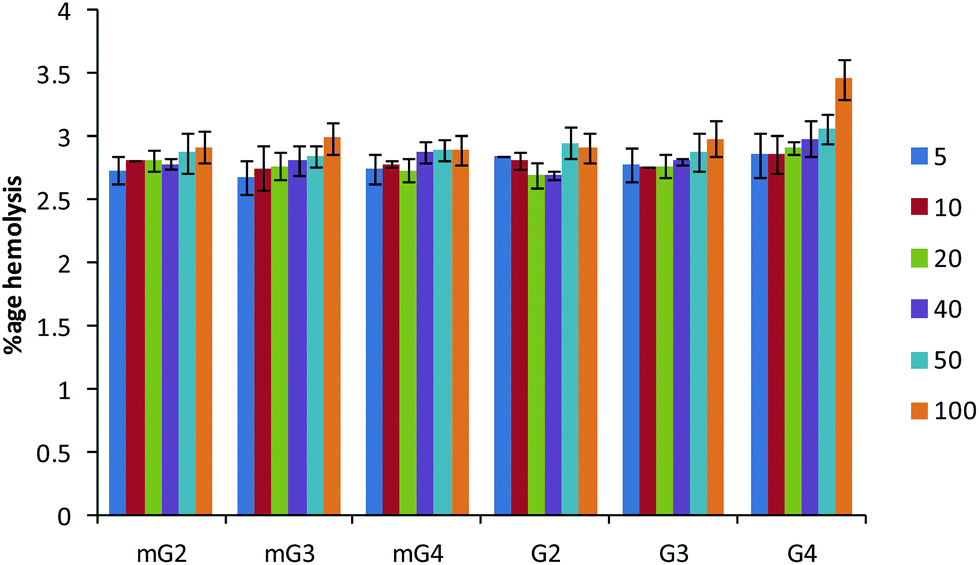
3.8. Hemolytic activity
The hemolysis of human red blood cells was measured at varying concentrations of dendrimers and the results are presented in Fig. 9. All the modified dendrimers (mG2–mG4) showed low hemolytic activity up to 100 μg ml−1 with less than 3% of human erythrocytes being lysed. The hemolytic activity was found to be almost similar to the activity of native PAMAM dendrimers (G2–G4). It was also noted that modification did not result in the increase in the membrane disrupting effect, supporting the endosomal membrane disruption by the proton-sponge mechanism. The low hemolytic activity at high dendrimer concentrations along with their enhanced gene delivery efficiency suggests the suitability of these vectors for future gene delivery applications.
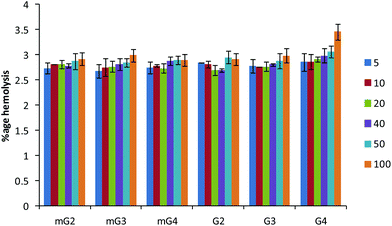 |
| | Fig. 9 Hemolysis assay. Percent hemolysis caused by native and modified PAMAM dendrimers at various concentrations (μg ml−1). Triton X-100 was used as a control causing 100% lysis. The experiment was carried out in triplicate. | |
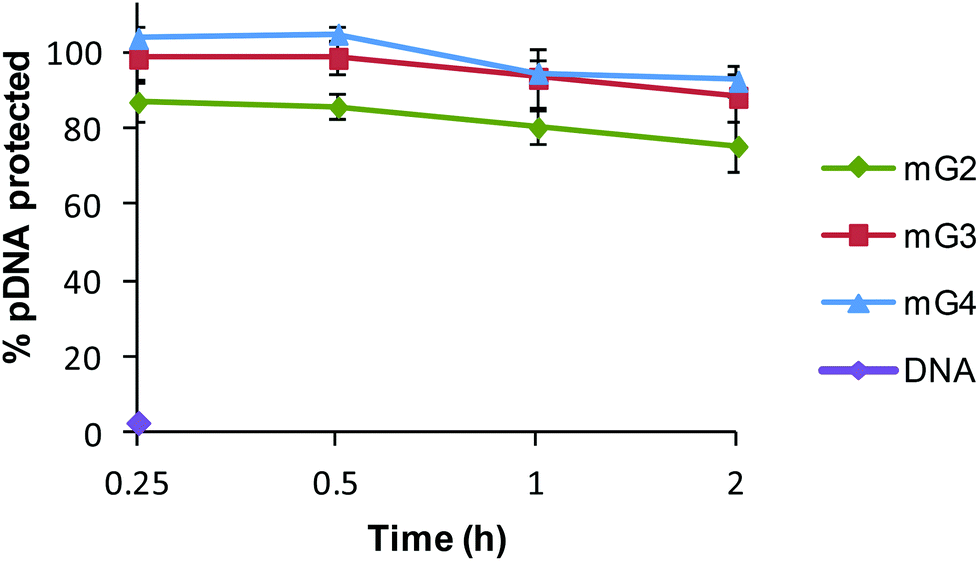
3.9. Protection of pDNA against nucleases
The protection of DNA is a prime requisite for successful entry of DNA inside the cells, as cellular milieu contains nucleases, which rapidly degrade native DNA. Therefore, the DNase I assay was performed to reveal the ability of modified dendrimers (mG2, mG3 and mG4) to protect the bound pDNA against nucleases. Naked DNA was found to be digested by DNase I within 15 min, whereas mG2, mG3 and mG4 dendrimers protected ∼75%, 88% and 92%, respectively, of bound pDNA even after 2 h of treatment (Fig. 10). These results clearly imply that modified dendrimers have the ability to provide protection to nucleic acids in the cellular environment and can serve as efficient vectors for nucleic acids to carry them into the cells.
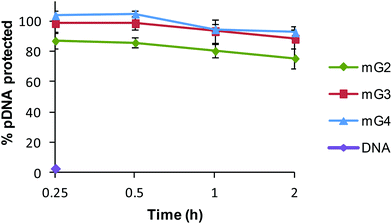 |
| | Fig. 10 DNase I protection assay of mGn–pDNA complexes at a w/w ratio of 6.66. | |
4. Conclusions
In the present study, low generation PAMAM dendrimers (G2–G4) were conjugated with a flexible oligoamine linker via Michael addition of methyl acrylate followed by amidation to improve the transfection efficiency without compromising on the cell viability. These modified dendrimers not only showed improved buffering capacity, which facilitated their endosomal escape but also exhibited significantly higher transfection efficiency with the cell viability almost comparable to native dendrimers. Increased positive charge density on the modified dendrimers was the prime factor for enhanced transfection efficiency which aided in binding of pDNA and protecting it against nucleases. The surface modification of dendrimers with the projected linker can be a simple and effective method to develop a proficient carrier system for the successful delivery of nucleic acids in vitro and in vivo.
Abbreviations
| PAMAM | Poly(amidoamine) |
| G2, G3, G4 | Generation 2, 3 and 4 |
| TEPA | Tetraethylenepentamine |
| bPEI | Branched polyethylenimine |
| GFP | Green fluorescent protein |
| pDNA | Plasmid DNA |
| EtBr | Ethidium bromide |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| TNBS | 2,4,6-Trinitrobenzene sulfonic acid |
| FBS | Fetal bovine serum |
| DMEM | Dulbecco's modified Eagle's medium |
| SDS | Sodium dodecyl sulfate |
| RBCs | Red blood cells |
| EDTA | Ethylenediaminetetraacetate |
Acknowledgements
Financial support from the CSIR network project (BSC 0112) is gratefully acknowledged. RB also thanks the Council of Scientific and Industrial Research (CSIR), New Delhi, India, for the award of a senior research fellowship to carry out this work. The authors thank Mr M. P. Singh for his help in TEM analysis.
References
- A. C. Grimsdale and K. Mullen, Angew. Chem., Int. Ed., 2005, 44, 5592–5629 CrossRef CAS PubMed.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS PubMed.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2005, 19, 311–330 CrossRef CAS PubMed.
-
(a) L. Jin, X. Zeng, M. Liu, Y. Deng and N. He, Theranostics, 2014, 4, 240–255 CrossRef CAS PubMed;
(b) T. Xiao, W. Hou, X. Cao, S. Wen, M. Shen and X. Shi, Biomater. Sci., 2013, 1, 1172–1180 RSC;
(c) Y. Yue and C. Wu, Biomater. Sci., 2013, 1, 152–170 RSC;
(d) M. Byrne, D. Victory, A. Hibbitts, M. Lanigan, A. Heise and S. A. Cryan, Biomater. Sci., 2013, 1, 1223–1234 RSC;
(e) P. M. Carlson, J. G. Schellinger, J. A. Pahang, R. N. Johnson and S. H. Pun, Biomater. Sci., 2013, 1, 736–744 RSC.
- S. Somanathan, E. Breous, P. Bell and J. M. Wilson, Mol. Ther., 2010, 18, 977–982 CrossRef CAS PubMed.
- H. S. Jung, W. H. Kong, D. K. Sung, M. Y. Lee, S. E. Beack, D. H. Keum, K. S. Kim, S. H. Yun and S. K. Hahn, ACS Nano, 2014, 8, 260–268 CrossRef CAS PubMed.
- W. L. Hwu, S. Muramatsu, S. H. Tseng, K. Y. Tzen, N. C. Lee, Y. H. Chien, R. O. Synder, B. J. Byrne, C. H. Tai and R. M. Wu, Sci. Transl. Med., 2012, 4, 134ra61 Search PubMed.
- D. L. Coutu, J. Cuerquis, R. E. I. Ayoubi, K. A. Forner, R. Roy, M. François, M. Griffith, D. Lillicrap, A. M. Yousefi, M. D. Blostein and J. Galipeau, Biomaterials, 2011, 32, 295–305 CrossRef CAS PubMed.
- A. Paul, A. Hasan, L. Rodes, M. Sangaralingam and S. Prakash, Adv. Drug Delivery Rev., 2014, 71, 115–130 CrossRef CAS PubMed.
- J. W. Bainbridge, A. J. Smith, S. S. Barker, S. Robbie, R. Henderson, K. Balaggan, A. Viswanathan, G. E. Holder, A. Stockman, N. Tyler, S. Petersen-Jones, S. S. Bhattacharya, A. J. Thrasher, F. W. Fitzke, B. J. Carter, G. S. Rubin, A. T. Moore and R. R. Ali, N. Engl. J. Med., 2008, 358, 2231–2239 CrossRef CAS PubMed.
- R. Goyal, S. K. Tripathi, S. Tyagi, K. Ravi Ram, K. M. Ansari, Y. Shukla, D. K. Chaowdhuri, P. Kumar and K. C. Gupta, Eur. J. Pharm. Biopharm., 2011, 79, 3–14 CrossRef CAS PubMed.
-
(a) R. Bansal, S. K. Tripathi, K. C. Gupta and P. Kumar, J. Mater. Chem., 2012, 22, 25427–25436 RSC;
(b) A. El-Fiqi, T. H. Kim, M. Kim, M. Eltohamy, J. E. Won, E. J. Leeab and H. W. Kim, Nanoscale, 2012, 4, 7475–7488 RSC;
(c) M. S. Shim and Y. J. Kwon, Polym. Chem., 2012, 3, 2570–2577 RSC;
(d) C. Li, Y. W. Yang, Z. Liang, G. Wu and H. Gao, Polym. Chem., 2013, 4, 4366–4374 RSC;
(e) J. Sun, F. Zeng, H. Jian and S. Wu, Polym. Chem., 2013, 4, 5810–5818 RSC.
- R. Goyal, R. Bansal, S. Tyagi, Y. Shukla, P. Kumar and K. C. Gupta, Mol. BioSyst., 2011, 7, 2055–2065 RSC.
- H. Lee, Y. Kim, P. G. Schweikert and S. F. Konieczny, Biomaterials, 2014, 35, 1040–1049 CrossRef CAS PubMed.
-
(a) D. Lin, Q. Cheng, Q. Jiang, Y. Huang, Z. Yang, S. Han, Y. Zhao, S. Guo, Z. Liang and A. Dong, Nanoscale, 2013, 5, 4291–4301 RSC;
(b) Z. Liang, X. Wu, Y. W. Yang, C. Li, G. Wu and H. Gao, Polym. Chem., 2013, 4, 3514–3523 RSC.
- M. Arif, S. K. Tripathi, K. C. Gupta and P. Kumar, J. Mater. Chem. B, 2013, 1, 4020–4031 RSC.
- Y. Tao, Z. Li, E. Ju, J. Ren and X. Qu, Nanoscale, 2013, 5, 6154–6160 RSC.
- J. Khandare, M. Calderon, N. M. Dagiaa and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- C. V. Kelly, M. G. Liroff, L. D. Triplett, P. R. Leroueil, D. G. Mullen, J. M. Wallace, S. Meshinchi, J. R. Baker Jr., B. G. Orr and M. M. Banaszak Holl, ACS Nano, 2009, 3, 1886–1896 CrossRef CAS PubMed.
- D. G. Mullen, M. Fang, A. Desai, J. R. Baker, B. G. Orr and M. M. Banaszak Holl, ACS Nano, 2010, 4, 657–670 CrossRef CAS PubMed.
- A. Kwok, G. A. Eggimann, J. L. Reymond, T. Darbre and F. Hollfelder, ACS Nano, 2013, 7, 4668–4682 CrossRef CAS PubMed.
- S. Biswas and V. P. Torchilin, Pharmaceuticals, 2013, 6, 161–183 CrossRef CAS PubMed.
- J. Lee, J. Jung, Y. J. Kim, E. Lee and J. S. Choi, Int. J. Pharm., 2014, 459, 10–18 CrossRef CAS PubMed.
- Y. J. Tsai, C. C. Hu, C. C. Chu and T. Imae, Biomacromolecules, 2011, 12, 4283–4290 CrossRef CAS PubMed.
- A. Lakshminarayanan, V. K. Ravi, R. Tatineni, Y. B. R. D. Rajesh, V. Maingi, K. S. Vasu, N. Madhusudhan, P. K. Maiti, A. K. Sood, S. Das and N. Jayaraman, Bioconjugate Chem., 2013, 24, 1612–1623 CrossRef CAS PubMed.
- M. T. P. Favaroa, M. A. S. de Toledo, R. F. Alves, C. A. Santos, L. L. Beloti, R. Janissen, L. G. de la Torre, A. P. Souza and A. R. Azzoni, J. Biotechnol., 2014, 173, 10–18 CrossRef PubMed.
- G. M. Pavan, P. Posocco, A. Tagliabue, M. Maly, A. Malek, A. Danani, E. Ragg, C. V. Catapano and S. Pricl, Chemistry, 2010, 16, 7781–7795 CrossRef CAS PubMed.
- J. L. Santos, H. Oliveira, D. Pandita, J. Rodrigues, A. P. Pego, P. L. Granja and H. Tomas, J. Controlled Release, 2010, 144, 55–64 CrossRef CAS PubMed.
-
(a) S. Pan, D. Cao, R. Fang, W. Yi, H. Huang, S. Tian and M. Feng, J. Mater. Chem. B, 2013, 1, 5114–5127 RSC;
(b) J. Li, D. Cheng, T. Yin, W. Chen, Y. Lin, J. Chen, R. Li and X. Shuai, Nanoscale, 2014, 6, 1732–1740 RSC;
(c) S. Yu, J. Chen, R. Dong, Y. Su, B. Ji, Y. Zhou, X. Zhu and D. Yan, Polym. Chem., 2012, 3, 3324–3329 RSC.
- L. Albertazzi, B. Storti, L. Marchetti and F. Beltram, J. Am. Chem. Soc., 2010, 132, 18158–18167 CrossRef CAS PubMed.
- G. S. Yu, Y. M. Bae, H. Choi, B. Kong, I. S. Choi and J. S. Choi, Bioconjugate Chem., 2011, 22, 1046–1055 CrossRef CAS PubMed.
- E. M. M. de Brabander-van den Berg and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1993, 32, 1308–1311 CrossRef.
- A. Grotzky, Y. Manaka, S. Fornera, M. Willeke and P. Walde, Anal. Methods, 2010, 2, 1448–1455 RSC.
- S. K. Tripathi, N. Gupta, M. Mahato, K. C. Gupta and P. Kumar, Colloids Surf., B, 2014, 115, 79–85 CrossRef CAS PubMed.
- Y. Li, Z. Jiang, H. Chen and W. J. Ma, J. Biochem. Biophys. Methods, 2004, 60, 85–96 CrossRef CAS PubMed.
- G. Navarro, G. Maiwald, R. Haase, A. L. Rogach, E. Wagner, C. T. de Ilarduya and M. Ogris, J. Controlled Release, 2010, 146, 99–105 CrossRef CAS PubMed.
- V. Russ, H. Elfberg, C. Thoma, J. Kloeckner, M. Ogris and E. Wagner, Gene Ther., 2008, 15, 18–29 CrossRef CAS PubMed.
- V. Russ, M. Günther, A. Halama, M. Ogris and E. Wagner, J. Controlled Release, 2008, 132, 131–140 CrossRef CAS PubMed.
- J. Shi, J. G. Schellinger, R. N. Johnson, J. L. Choi, B. Chou, E. L. Anghel and S. H. Pun, Biomacromolecules, 2013, 14, 1961–1970 CrossRef CAS PubMed.
- Y. N. Xue, M. Liu, L. Peng, S. W. Huang and R. X. Zhuo, Macromol. Biosci., 2010, 10, 404–414 CrossRef CAS PubMed.
- N. D. Sonawane, F. C. Szoka Jr. and A. S. Verkman, J. Biol. Chem., 2003, 278, 44826–44831 CrossRef CAS PubMed.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acd. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- P. Zhang, N. Xu, L. Zhou, X. Xu, Y. Wang, K. Li, Z. Zeng, X. Wang, X. Zhang and C. Bai, Transl. Respir. Med., 2013, 1, 2 CrossRef.
- G. T. Zugates, N. C. Tedford, A. Zumbuehl, S. Jhunjhunwala, C. S. Kang, L. G. Griffith, D. A. Lauffenburger, R. Langer and D. G. Anderson, Bioconjugate Chem., 2007, 18, 1887–1896 CrossRef CAS PubMed.
- C. Lin, Z. Zhong, M. C. Lok, X. Jiang, W. E. Hennink, J. Feijen and J. F. J. Engbersen, J. Controlled Release, 2006, 116, 130–137 CrossRef CAS PubMed.
- A. Swami, R. Goyal, S. K. Tripathi, N. Singh, N. Katiyar, A. K. Mishra and K. C. Gupta, Int. J. Pharm., 2009, 374, 125–138 CrossRef CAS PubMed.
- S. K. Tripathi, S. Yadav, K. C. Gupta and P. Kumar, Mol. BioSyst., 2012, 8, 1426–1434 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4bm00115j |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.