Synthesis of cholic acid-core poly(ε-caprolactone-ran-lactide)-b-poly(ethylene glycol) 1000 random copolymer as a chemotherapeutic nanocarrier for liver cancer treatment
Wei
Tao†
ab,
Xiaowei
Zeng†
ab,
Jinxie
Zhang
ab,
Huijun
Zhu
ab,
Danfeng
Chang
bc,
Xudong
Zhang
ab,
Yongfeng
Gao
ab,
Jiao
Tang
d,
Laiqiang
Huang
*ab and
Lin
Mei
*ab
aSchool of Life Sciences, Tsinghua University, Beijing 100084, P.R. China
bThe Ministry-Province Jointly Constructed Base for State Key Lab- Shenzhen Key Laboratory of Chemical Biology, the Shenzhen Key Lab of Gene and Antibody Therapy, and Division of Life and Health Sciences, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, P.R. China. E-mail: huanglq@sz.tsinghua.edu.cn; mei.lin@sz.tsinghua.edu.cn; Tel: +86 75526036052, +86 75526036736
cDepartment of Chemistry, Tsinghua University, Beijing 100084, P.R. China
dSchool of Medicine, Tsinghua University, Beijing 100084, P.R. China
First published on 18th June 2014
Abstract
A star-shaped random copolymer, cholic acid functionalized poly(ε-caprolactone-ran-lactide)-b-poly(ethylene glycol) 1000 (CA-(PCL-ran-PLA)-b-PEG1k), was synthesized by a core-first approach involving three stages of chemical reactions, and was characterized by hydrogen-1 nuclear magnetic resonance (1H NMR), gel permeation chromatography and thermogravimetric analysis. The docetaxel-loaded nanoparticles (NPs) were prepared by a modified nano-precipitation method. The formation and characterization of these NPs were confirmed through dynamic light scattering, zeta potential measurements, field emission scanning electron microscopy, and transmission electron microscopy. The in vitro release profiles indicated that CA-(PCL-ran-PLA)-b-PEG1k NPs had excellent sustained and controlled drug release properties. Both confocal laser scanning microscope and flow cytometric results showed that the coumarin-6 loaded CA-(PCL-ran-PLA)-b-PEG1k NPs had the highest cellular uptake efficiency compared with PEG1k-b-(PCL-ran-PLA) NPs and CA-(PCL-ran-PLA) NPs in human hepatic carcinoma cells. The docetaxel-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs were also proved to have the highest drug loading content, encapsulation efficiency, and the best anti-tumor efficacy both in vitro and in vivo. In conclusion, the star-shaped CA-(PCL-ran-PLA)-b-PEG1k copolymer was successfully synthesized and could be used as a promising drug-loaded biomaterial for liver cancer chemotherapy.
1. Introduction
It has been reported that liver cancer (hepatocellular carcinoma) resulted in 754 000 deaths worldwide as of 2010, making it the 3rd leading cause of cancer death after lung cancer and stomach cancer.1 The current clinical treatments for liver cancer mainly involve surgery followed by radiotherapy, chemotherapy, and some emerging modalities (e.g. gene therapy, immunotherapy, phototherapy, and thermal therapy). However, each of these single modalities does not provide complete treatment because of their dose limitations and the resistance of cancer cells to the modality.2 For example, chemotherapy is non-specific and is accompanied by side effects, due to the lack of drug targeting, resistance of the cancer cells and problems with the permeability, solubility and stability of anticancer drugs.3Nanomedicine, the application of nanotechnology to medicine, has shown significant prospects for solving these problems in cancer chemotherapy. In particular, formulations containing biodegradable polymeric nanoparticles (NPs), could enable the controlled and sustained delivery of anticancer drugs, and are expected to fundamentally change the landscape of pharmaceutical and biotechnology industries.4–6 Due to the pathophysiological conditions and anatomical changes caused by cancer, polymeric NPs designed for drug delivery systems can be exploited for the passive targeting of drugs.7 Thus, an enhanced permeability and retention (EPR) effect of polymeric NPs can be found in tumors as a result of the increased vascular permeability coupled with the impaired lymphatic drainage. Moreover, the high drug loading capacity, high stability, excellent tolerability, protection of incorporated labile drugs from degradation, controlled release and feasibility of variable routes of administration (e.g. parenteral, oral, dermal, ocular, pulmonary, and rectal) are important technological advantages of drug-loaded NPs.8 Polymeric NPs could also reduce the multidrug resistance characterized by many anticancer drugs (e.g. docetaxel) through endocytosis of the drugs,9 and prevent drug efflux from cells mediated by the P-glycoprotein.10 Compared with conventional chemotherapy, drug delivery systems based on polymeric NPs have the advantage of overcoming many of the problems, such as the toxicity of drugs to normal tissues, the limited aqueous solubility, the short circulation half-life in plasma and non-selectivity, associated with conventional drug delivery systems.11
Biodegradable polymers approved by the FDA, such as poly(ε-caprolactone) (PCL) and poly(lactide) (PLA), are widely used nowadays in the development of polymeric nanoformulations.12–15 PCL has attracted considerable interest owing to its permeability to drugs, its non-cytotoxicity, its miscible properties with other polymers and its biocompatibility.16,17 However, PCL crystallizes easily, which limits its compatibility with soft tissues and makes it degrade much slower than PLA, thus limiting its application in nanomedicine.18 Nevertheless, this problem can be settled through the copolymerization of ε-caprolactone with other monomers.19,20 PLA is a thermoplastic aliphatic polyester and can be used for the preparation of medical implants because it is biodegradable.21,22 Copolymerization between the lactide enantiomers used in the synthesis of PLA results in variable stereoregularity, and is a useful approach for adjusting the degradability of the polymer as well as for adjusting its mechanical and physical properties. It has been reported that PEGylation, chemical modification with poly(ethylene glycol) (PEG), is an effective way of improving the biological potency of various pharmaceutical formulations in vivo,23 and the conjugation of drugs to PEG or formulation in PEG copolymer nanocarriers can enhance the solubility, permeability, stability and thus oral bioavailability of drugs.24,25 Novel polymers with desired parameters derived from the parent polymers could be obtained by random copolymerization, which is valuable for nanoparticle-based drug delivery systems.26–28 Cholic acid (CA), which is composed of a steroid unit with three hydroxyl groups and one carboxyl group, is the main bile acid in the body. Due to its biological origin, CA has been selected as a polyhydroxy initiator, and the resulting polymers incorporated with a CA moiety were shown to have better biocompatibility than those without a CA moiety.2,29 Moreover, it has been reported that CA functionalized copolymers exhibit a faster hydrolytic degradation rate compared with PCL, and the existence of a CA moiety in the drug delivery system could also significantly enhance both cell proliferation and adherence.30 Branched polymers with useful rheological and mechanical properties, such as star-shaped polymers, hyperbranched polymers and dendrimers, have attracted a great deal of attention by research groups all over the world.31–33 With several branches extending from a single point,34 star-shaped polymers applied in drug carriers have advantages such as a lower solution viscosity, a smaller hydrodynamic radius, a higher encapsulation efficiency and loading content compared with linear polymers in a drug delivery system.32,35–37 Therefore, star-shaped block copolymers based on PCL, PLA, PEG1k and CA could potentially be used to prepare excellent biodegradable drug-carriers.38,39 In this study, we successfully synthesized a star-shaped random copolymer CA-(PCL-ran-PLA)-b-PEG1k with three branch arms for docetaxel (DTX) delivery. The DTX-loaded NPs exhibited satisfactory drug loading content, encapsulation efficiency, and achieved significant in vivo therapeutic effects for the treatment of liver cancer.
2. Materials and methods
2.1. Materials
2.2. Synthesis of star-shaped copolymer CA-(PCL-ran-PLA)-b-PEG1k
The star-shaped copolymer CA-(PCL-ran-PLA)-b-PEG1k was synthesized as described in the literature,2,5 and included three chemical reactions: ring opening polymerization of ε-caprolactone and lactide monomer initiated by cholic acid, carboxylation of PEG, and esterification of CA-(PCL-ran-PLA) and CPEG.2.3. Characterization of star-shaped CA-(PCL-ran-PLA)-b-PEG1k copolymer
The structures of the synthesized CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k were confirmed by NMR (Bruker AMX 500) with CDCl3 as the solvent. Gel permeation chromatography (Waters GPC analysis system with RI-G1362A refractive index detector, Milford, USA) was used to measure the molecular weight and molecular weight distribution. THF was used as the eluent at a flow rate of 1 ml min−1 to calibrate the data against polystyrene standards. The thermal properties of the star-shaped CA-(PCL-ran-PLA)-b-PEG1k coploymer were studied by thermogravimetric analysis (TGA, TGA Q500 thermogravimetric analyzer, USA). During TGA measurement, approximately 10 mg of copolymer sample was heated from 40 °C to 600 °C at a rate of 20 °C min−1. The weight loss pattern in the copolymer thermogram could be related to the composition of the polymer.2.4. Formulation of DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs
A modified nano-precipitation method with an acetone–water system was used to prepare the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs.40 20 mg of DTX powder and 200 mg of CA-(PCL-ran-PLA)-b-PEG1k copolymer were dissolved in 16 ml of acetone, and then the organic solution was injected into 200 ml of TPGS aqueous solution (0.03%, w/v) with gentle stirring. Afterwards, the mixture was stirred at a speed of 800 rpm overnight to completely remove the acetone (at room temperature). The resulting particles suspension was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min (4 °C), and the precipitates were washed three times to get rid of the emulsifier and unencapsulated DTX. The resulting particles were resuspended in 10 ml of deionized water, and stored at −80 °C in a refrigerator overnight. Finally, the solid was freeze-dried for two days. Drug-loaded CA-(PCL-ran-PLA) and PEG1k-b-(PCL-ran-PLA) NPs were fabricated using the same method. The fluorescent coumarin-6 loaded CA-(PCL-ran-PLA)-b-PEG1k NPs were prepared in the same way except coumarin-6 was encapsulated instead of DTX.
000 rpm for 20 min (4 °C), and the precipitates were washed three times to get rid of the emulsifier and unencapsulated DTX. The resulting particles were resuspended in 10 ml of deionized water, and stored at −80 °C in a refrigerator overnight. Finally, the solid was freeze-dried for two days. Drug-loaded CA-(PCL-ran-PLA) and PEG1k-b-(PCL-ran-PLA) NPs were fabricated using the same method. The fluorescent coumarin-6 loaded CA-(PCL-ran-PLA)-b-PEG1k NPs were prepared in the same way except coumarin-6 was encapsulated instead of DTX.
2.5. Nanoparticles characterization
For observation of the surface morphology of the NPs, field emission scanning electron microscopy (FESEM, S-4800, Hitachi, Tokyo, Japan), carried out at a 15.0 kV accelerating voltage, was used in this experiment. To prepare the samples for FESEM the particles were fixed on a copper plate using double-sided sticky conductive adhesives and then coated with a platinum layer using a JCF-1300 automatic fine platinum coater. Transmission electron microscopy (TEM, Tecnai G2 20, FEI Company, Hillsboro, Oregon, USA) was used to further study the resultant NPs. Sample was dropped onto a carbon-coated-on lacey support film and the lacey support film was allowed to dry before characterization.
:50, v/v). A nitrogen stream was introduced to evaporate the DCM for about 20 min, and then a clear solution was obtained for HPLC analysis. The flow rate of the mobile phase was set at 1.0 ml min−1. A reverse-phase C18 column (150 × 4.6 mm, 5 μm, C18, Agilent Technologies, CA, USA) was used in this study, and detection was carried out at 227 nm using a UV/VIS detector. The drug loading content and encapsulation efficiency of the DTX-loaded NPs were calculated by the following equations respectively.20 Measurements were carried out three times for each batch.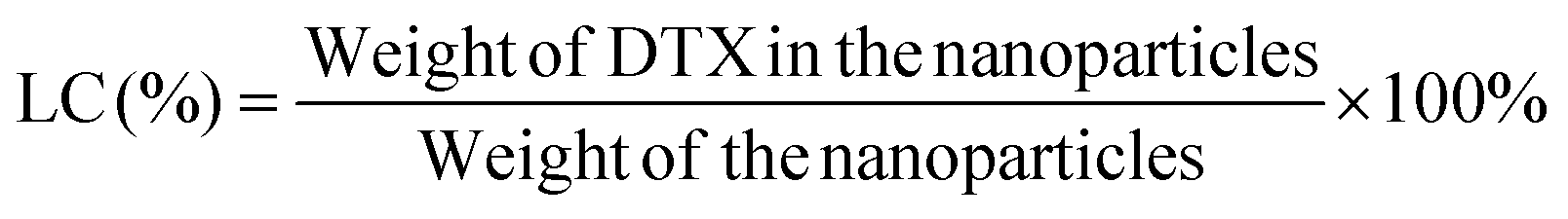
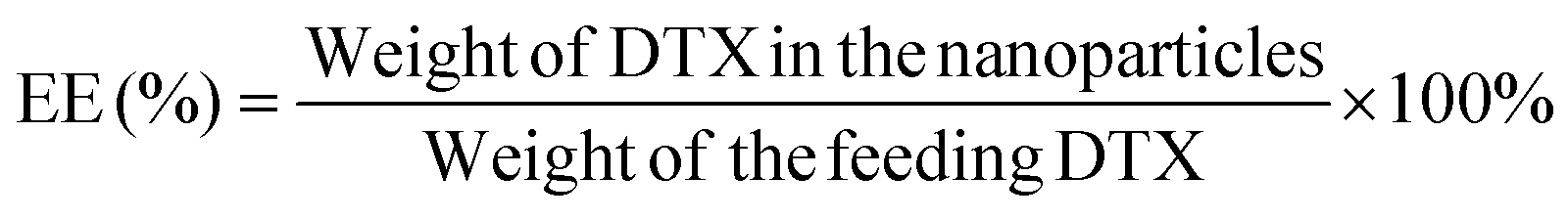
2.6. Cellular uptake of fluorescent NPs
The HepG2 cells were cultivated in a chambered cover glass system in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 100 U ml−1 penicillin, and 100 mg ml−1 streptomycin in 5% CO2 at 37 °C. A fluorescent probe (coumarin-6) was used to replace the DTX in the nanoformulation for the observation and analysis of cellular uptake of NPs.42,43 After the cells were incubated with 250 μg ml−1 of coumarin-6 loaded CA-(PCL-ran-PLA)-b-PEG1k NPs at 37 °C for 4 h, the cells were rinsed three times with cold PBS and then fixed with cold methanol for 20 min. The nuclei were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI, Fluka, Buche, Switzerland) for 10 min, then the stained cells were washed three times again with PBS to get rid of free DAPI. In order to visualize the HepG2 cells, the chambers were mounted onto the confocal laser scanning microscope (CLSM, Olympus Fluoview FV-1000, Tokyo, Japan) with imaging software. Images of the cells were determined with a differential interference contrast channel. The nuclei of the cells were stained with DAPI and coumarin-6 loaded NPs were recorded with the following channels: a blue channel with excitation at 340 nm and a green channel with excitation at 485 nm.For flow cytometric (FCM) assay, the HepG2 cells were incubated with 250 μg ml−1 of coumarin-6 loaded CA-(PCL-ran-PLA), PEG1k-b-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k NPs at 37 °C for 1 h, respectively. The cells were collected and washed with PBS, and the intracellular fluorescence of coumarin-6 was detected by FCM after excitation at 488 nm. Fluorescence emission at 530 nm from 10000 cells were collected, amplified and scaled to generate a single parameter histogram.
For quantitative analysis, HepG2 cells (initial density of 1 × 104 cells per well) were plated in 96-well plates and cultivated overnight.44,45 Hank's buffered salt solution (HBSS) was used to equilibrate the cells at 37 °C for 1 h, then coumarin-6 loaded NPs were added at concentrations of 100, 250 and 500 μg ml−1, respectively. After a 2 h-incubation, the medium was removed and the wells were washed three times with 50 μl of cold PBS solution. Finally, 50 μl of 0.5% Triton X-100 in 0.2 N sodium hydroxide was added to each sample well to lyse the cells.
2.7. In vitro cell viability assay
HepG2 cells were seeded in 96-well plates at a density of 5000 viable cells per well, and incubated for 24 h to allow cell attachment. After that, the cells were incubated with DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs, PEG1k-b-(PCL-ran-PLA) NPs suspension, commercial Taxotere® at 0.25, 2.5, 12.5 and 25 μg ml−1 equivalent DTX concentrations and drug-free CA-(PCL-ran-PLA)-b-PEG1k NPs suspension with the same amount of NPs for 24, 48 and 72 h, respectively. The formulations were replaced with DMEM containing MTT (5 mg ml−1) at a determined time, and incubated cells for an additional 4 h. Then MTT was aspirated off and DMSO was added to dissolve the formazan crystals (incubated for 2 h at 37 °C in the dark). A microplate reader (Bio-Rad Model 680, UK) was used to measure the absorbance at 570 nm, and untreated cells were taken as the control with 100% viability, while cells without the addition of MTT were used as the blank to calibrate the spectrophotometer to zero absorbance. IC50, the drug concentration which inhibited 50% of cell growth, was calculated by curve fitting of the cell viability data in comparison with that of the control samples.2.8. Anti-tumor efficacy study with a xenograft tumor model
HepG2 tumor cells (2 × 106 cells per mouse) in the culture medium were implanted into the subcutaneous space of BALB/c nude mice (15–20 g) at the right axilla and the tumor growth in each mouse was observed frequently. The tumor size was measured using a vernier caliper and its volume (V) was calculated as V = d2 × D/2, where d and D represent the shortest and the longest diameter of the tumor in mm, respectively. Animals were closely observed for clinical signs and behavior. At the moment the tumor volume reached around 50 mm3 (designated as the 0 day), treatments were performed. The mice were randomly divided into three groups (each group was made up of 5 animals, n = 5). With saline as the control, the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs and Taxotere® were intraperitoneally injected at a single dose of 10 mg DTX kg−1 in saline on days 0, 4, 8 and 12. Mice were sacrificed by cervical decapitation after 14 days’ treatment. The terminal tumor weight was measured and utilized to evaluate the anti-tumor activity.2.9. Statistical methodology
All the experiments were repeated at least three times unless otherwise stated. The results are expressed as mean ± SD, and the statistical significance of all the results was determined by the Student's t-test. P < 0.05 was considered to be significant.3. Results and discussion
3.1. Synthesis and characterization of CA-(PCL-ran-PLA)-b-PEG1k copolymer
With Sn(Oct)2 as the bulk catalyst, the star-shaped random copolymer CA-(PCL-ran-PLA) was synthesized via the ring-opening copolymerization of ε-caprolactone (CL) and D,L-lactide (LA) initiated by multifunctional CA at 160 °C. Studies have demonstrated that ring-opening polymerization is an efficient method for preparing aliphatic polyesters with high molecular weights.46 With the help of initiators possessing hydroxyl groups, controlled architectures, such as linear, comb-like and star-shaped, can be obtained for copolymers.47 As shown in Fig. 1, the synthesized CA-(PCL-ran-PLA) was connected to carboxylated PEG through an esterification reaction with DCC and DMAP as catalysts, ultimately generating a three-armed CA-(PCL-ran-PLA)-b-PEG1k copolymer. | ||
| Fig. 1 Schematic diagram of synthesis of star-shaped CA-(PCL-ran-PLA)-b-PEG1k random copolymer. | ||
In order to confirm the formation of the star-shaped random copolymer, a 1H NMR spectrum was recorded and the detailed assignment of the signals is shown in Fig. 2. Typical signals from the CL and LA monomer repeating units and the CA moiety can be observed in the spectrum of the CA functionalized star-shaped polymer CA-(PCL-ran-PLA). 1H NMR (CDCl3): a (δ = 5.21 ppm, LA repeating unit: –CHCH3), b (δ = 4.05 ppm, CL repeating unit: –CH2OCO–), c (δ = 2.31 ppm, CL repeating unit: –COCH2–), d + g (δ = 1.55–1.70 ppm, CL repeating unit: –(CH2)5- and LA repeating unit: –CH3), e (δ = 1.38 ppm, CL repeating unit: –(CH2)5–). The successful coupling of star-shaped CA-(PCL-ran-PLA) and CPEG was confirmed by the appearance of peak f (δ = 3.65 ppm, methoxyl protons of PEG: –CH2CH2O–) and peak h (δ = 3.38 ppm, methoxyl protons of PEG: –OCH3) in the spectra. The 1H NMR spectrum provides evidence that the star-shaped copolymers CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k with a well-defined three-branched structure were synthesized successfully. The actual molecular weights (data shown in Table 1) of the star-shaped copolymers CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k were calculated from the peak areas integral ratio of a (δ = 5.21 ppm), b (δ = 4.05 ppm) and f (δ = 3.65 ppm).
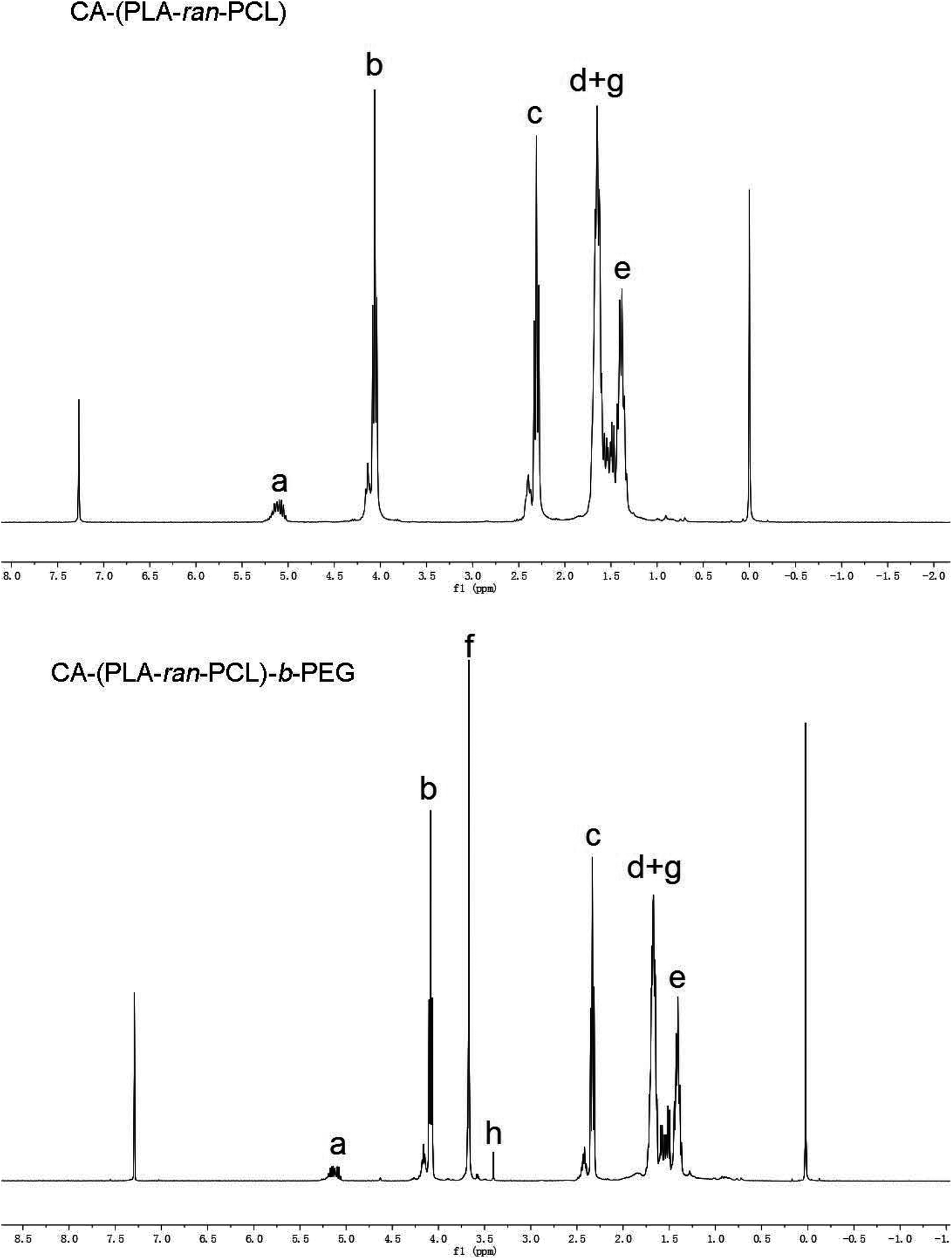 | ||
| Fig. 2 Typical 1H NMR spectra of copolymer CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k. | ||
| Sample | M n (NMRa) | M n (GPCb) | M w (GPCb) | PDIc |
|---|---|---|---|---|
| a Determined by 1H NMR. b Determined by GPC. c Polydispersity index (Mw/Mn) determined by gel permeation chromatography. M n: Number-average molecular weight. Mw: Weight-average molecular weight. | ||||
| CA-(PCL-ran-PLA) | 14361.86 |
12045.32 |
15177.47 |
1.26 |
| CA-(PCL-ran-PLA)-b-PEG1k | 17535.43 |
14972.69 |
17816.68 |
1.19 |
| PEG1k-b-(PCL-ran-PLA) | 18142.09 |
18376.71 |
22602.07 |
1.23 |
For the purpose of investigating the thermal properties, TGA was conducted on the synthesized random copolymer CA-(PCL-ran-PLA)-b-PEG1k. Typical thermal decomposition profiles for carboxyl-terminated PEG, CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k are displayed in Fig. 3. The thermal decomposition process of the star-shaped copolymer CA-(PCL-ran-PLA)-b-PEG1k involved two stages of weight loss, whereas CA-(PCL-ran-PLA) and CPEG only had a single step of weight loss. The combustion of a new component in the copolymer was marked by each turning point: the first stage (approximately 250–380 °C) could be attributed to the decomposition of CA-(PCL-ran-PLA) segments, while the second stage (approximately 380–450 °C) could be assigned to the decomposition of PEG segments, further proving the presence of two blocks in the star-shaped copolymer CA-(PCL-ran-PLA)-b-PEG1k.
 | ||
| Fig. 3 Thermogravimetric analysis of CPEG, CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k copolymers. | ||
The molecular weights of the obtained copolymers were measured by 1H NMR spectrometry and GPC, respectively. The detailed results are presented in Table 1. For the star-shaped copolymers CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k, the Mn determined by 1H NMR spectrometry were higher than those determined by GPC (14361.86 vs. 12045.32, 17535.43 vs. 14972.69, respectively). This could be attributed to the fact that the molecular weight estimated by GPC analysis used a linear polymer for calibration. The star-shaped copolymer has a smaller hydrodynamic volume than the linear polymer with a similar molecular weight, and hardly expands in solution. Furthermore, the molecular weight polydispersity of the copolymer is rather narrow.
3.2. Formulation and characterization of NPs
As displayed in Fig. 4(A), the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs were formulated by a modified nano-precipitation method with acetone as the acceptable solvent, which provided a facile, mild, and energy-saving pathway for drug encapsulation of polymeric NPs. Random copolymers and DTX without any chemical modification could be completely dissolved in acetone to generate an homogenous and clear solution. The formulation process was as follows: acetone solution was injected into a continuously stirred aqueous solution, and the water-insoluble DTX was immediately precipitated. In the meantime the hydrophobic –(PCL-ran-PLA)– segment of the star-shaped CA-(PCL-ran-PLA)-b-PEG1k also precipitated rapidly, resulting in the spontaneous formation of DTX into NPs.48 In order to obtain a stable DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs aqueous dispersion, the mixed solution was stirred overnight to volatilize the organic solvent acetone. Eventually, NPs with a hydrophilic stabilization shell (PEG segment), a core–shell structure (hydrophobic PCL-ran-PLA segment) and a hydrophobic core (entrapped DTX) were formed. | ||
| Fig. 4 (A) Schematic representation of the preparation technique for DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs; (B) DLS size distribution of DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs; (C) FESEM image of DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs (scale bar is 100 nm); (D) TEM image of DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs (scale bar is 100 nm). | ||
| Polymer | Size (nm) | PDI | ZP (mV) | LC (%) | EE (%) |
|---|---|---|---|---|---|
| PDI = polydispersity index, ZP = zeta potential, LC = loading content, EE = encapsulation efficiency, n = 3. | |||||
| PEG1k-b-(PCL-ran-PLA) | 140.6 ± 7.2 | 0.237 | −12.5 ± 2.1 | 8.67 ± 0.3 | 81.57 ± 4.5 |
| CA-(PCL-ran-PLA) | 127.2 ± 6.1 | 0.203 | −20.3 ± 2.5 | 9.27 ± 0.2 | 89.36 ± 2.9 |
| CA-(PCL-ran-PLA)-b-PEG1k | 95.1 ± 4.3 | 0.167 | −8.3 ± 1.6 | 10.13 ± 0.4 | 97.98 ± 1.9 |
The zeta potential of the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs was observed to be −8.3 mV, a slightly increase compared with that of DTX-loaded PEG1k-b-(PCL-ran-PLA) NPs (−12.5 mV) and CA-(PCL-ran-PLA) NPs (−20.3 mV), as shown in Fig. 2. The slightly negative charge of the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs can reduce the undesirable clearance by the reticuloendothelial system (e.g. liver) and improve the blood compatibility, thus facilitating the accumulation of NPs at tumor sites.51
The surface morphology of the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs was investigated by FESEM and TEM. Fig. 4 illustrates the FESEM (C) and TEM (D) image of the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs. The NPs seemed to have a near-spherical shape with a rough surface and were around 60 nm in diameter, reaching a significantly smaller size than that obtained from DLS experiments. This difference may be caused by a tendency to shrink and collapse while the NPs are in the dry state.51
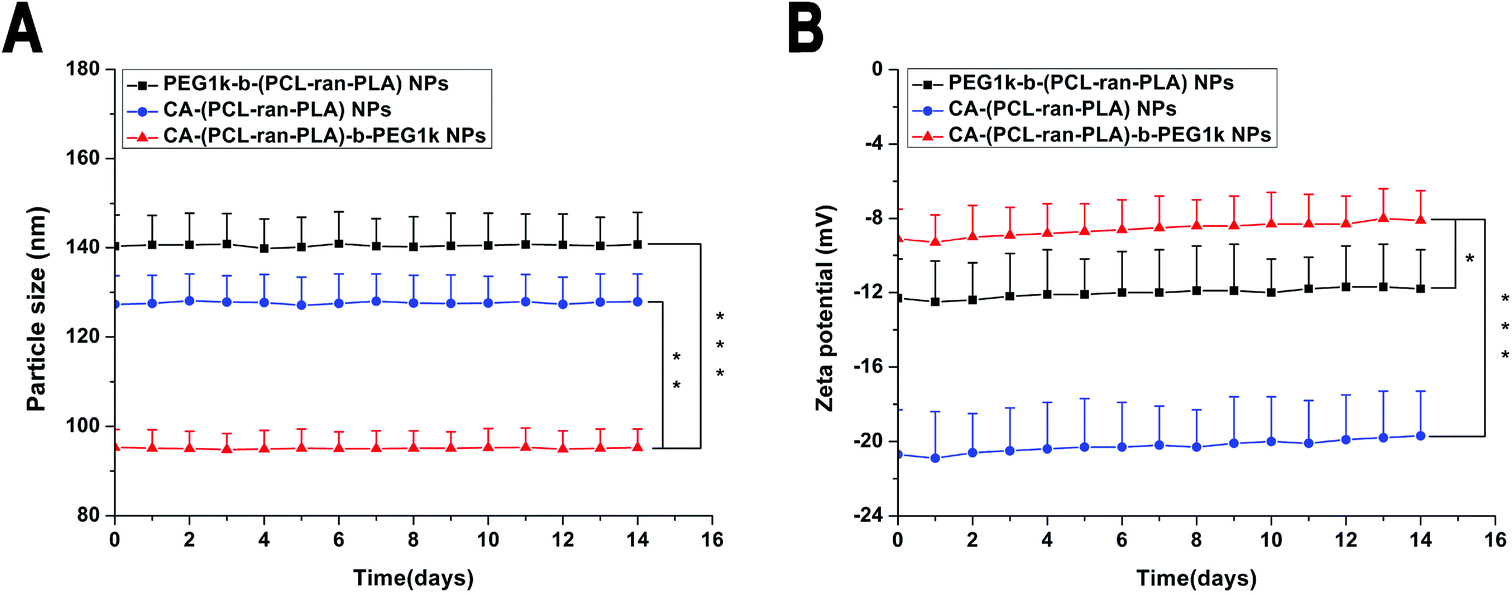 | ||
| Fig. 5 In vitro stability of DTX-loaded NPs: (A) size distribution and (B) zeta potential of the DTX-loaded PEG1k-b-(PCL-ran-PLA), CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k NPs for two-week storage at 4 °C (*p < 0.05, **p < 0.01, ***p < 0.001, n = 3). | ||
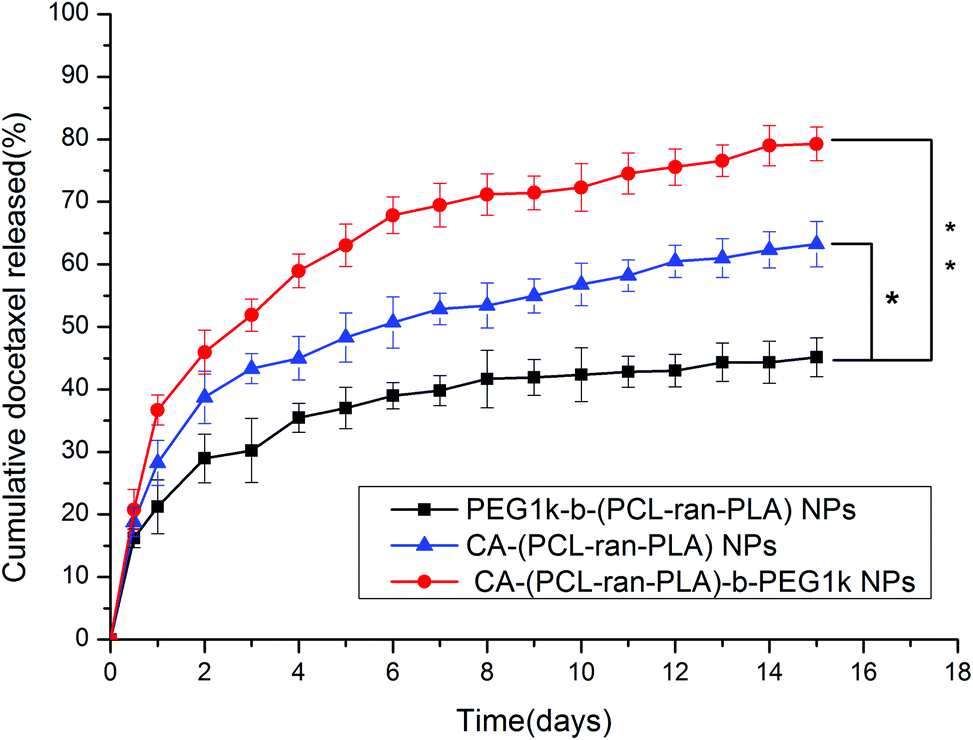 | ||
| Fig. 6 The in vitro release profiles of DTX-loaded PEG1k-b-(PCL-ran-PLA) NPs, CA-(PCL-ran-PLA) NPs and CA-(PCL-ran-PLA)-b-PEG1k NPs at 37 °C (*p < 0.05, **p < 0.01, n = 5). | ||
3.3. Cellular uptake of fluorescent NPs by HepG2 cells
In order to visualize and analyze cellular uptake of the NPs, a fluorescent probe called coumarin-6 was used to represent the DTX in the NPs and confocal laser scanning microscopy (CLSM) was used to observe this process and the distribution of NPs in cells. Fig. 7 displays CLSM images of HepG2 cells after 4 h incubation with the coumarin-6 loaded CA-(PCL-ran-PLA)-b-PEG1k NPs suspension in the Dulbecco's modified Eagle medium (DMEM) at a concentration of 250 μg ml−1 of the NPs. The images were obtained from (A) the DAPI channel (blue); (B) the EGFP channel (green); (C) the overlay of the two channels. It can be seen from the figure that the coumarin-6 loaded CA-(PCL-ran-PLA)-b-PEG1k NPs (green) were closely located around the nuclei (blue, stained by DAPI), demonstrating that the fluorescent NPs had been internalized into the HepG2 cells.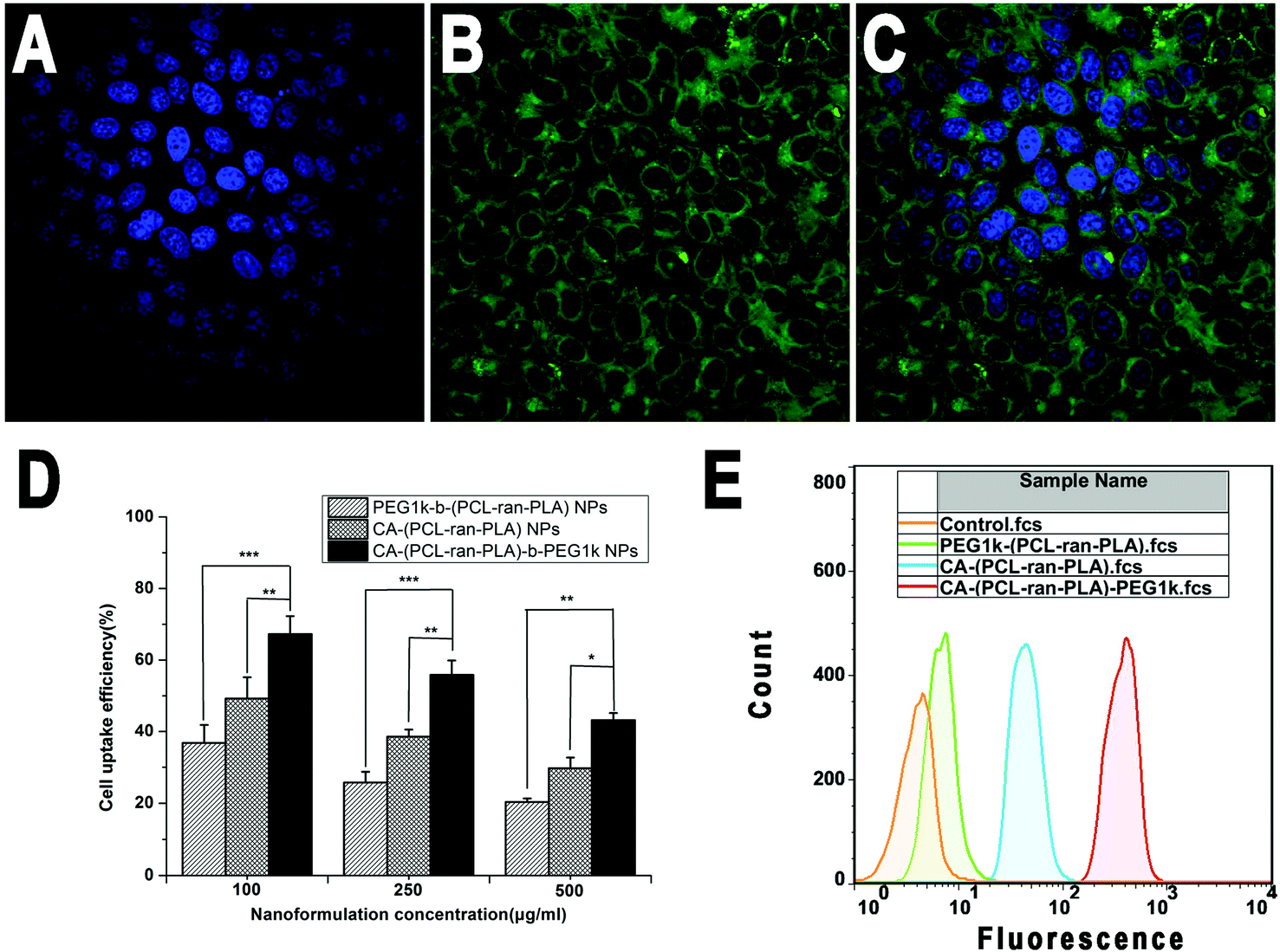 | ||
| Fig. 7 CLSM images of HepG2 cells after 4 h incubation with coumarin-6 loaded CA-(PCL-ran-PLA)-b-PEG1k NPs at 37 °C. The cells were stained with DAPI (blue) and the coumarin-6 loaded CA-(PCL-ran-PLA)-b-PEG1k NPs are green. The cellular uptake was visualized by overlaying images obtained by DAPI filter and EGFP filter: (A) image from DAPI channel; (B) image from EGFP channel; (C) image from combined DAPI channel and EGFP channel. (D) Cellular uptake efficiency of coumarin-6 loaded NPs (*p < 0.05, **p < 0.01, ***p < 0.001, n = 5). (E) FCM histograms for coumarin-6 loaded NPs on HepG2 cells after 1 h incubation. | ||
The cellular internalization and sustained retention properties of the NPs were demonstrated to play an important role on the therapeutic effects of the drug-loaded nanoformulation.52 In order to evaluate the internalization and sustained retention properties of PEG1k-b-(PCL-ran-PLA), CA-(PCL-ran-PLA) and CA-(PCL-ran-PLA)-b-PEG1k NPs, the cellular uptake efficiency of the coumarin-6 loaded NPs was tested through 2 h incubation. As presented in Fig. 7(D), the cellular uptake efficiency of all coumarin-6 loaded NPs decreased with an increase of the incubated NPs concentration from 100 to 500 μg ml−1. It has been reported that NPs can be internalized into cells, but the endocytic process of NPs can be influenced by particle size.53,54 In agreement with these reports, our data showed that the cellular uptake efficiency of CA-(PCL-ran-PLA)-b-PEG1k NPs was 1.83-, 2.17-, and 2.12-fold higher than that of PEG1k-b-(PCL-ran-PLA) NPs at the incubated NPs concentration of 100, 250, and 500 μg ml−1, respectively. Furthermore, the cellular uptake efficiency of both CA-(PCL-ran-PLA)-b-PEG1k and CA-(PCL-ran-PLA) NPs was higher than that of PEG1k-b-(PCL-ran-PLA) NPs at any concentration.
The cellular uptake of coumarin-6 loaded NPs was also confirmed by FCM assay. As shown in Fig. 7(E), the fluorescence intensity of cells treated with CA-(PCL-ran-PLA)-b-PEG1k NPs was the highest, followed by those treated with CA-(PCL-ran-PLA) NPs. The fluoresence intensity of cells treated with PEG1k-b-(PCL-ran-PLA) NPs was the lowest after 1 h incubation, further indicating that the size distribution of CA-(PCL-ran-PLA)-b-PEG1k NPs was appropriate for cellular internalization and sustained retention, which could also lead to excellent therapeutic effects.
3.4. In vitro cell viability assay
For the purpose of investigating the in vitro cytotoxicity of DTX-loaded PEG1k-b-(PCL-ran-PLA) NPs, DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs and drug-free CA-(PCL-ran-PLA)-b-PEG1k NPs, an MTT assay was conducted of the HepG2 cell line and the results were compared with those obtained for a clinically available DTX formulation, i.e. Taxotere®. The DTX-loaded NPs were sterilized using gamma rays to exclude any contamination before being suspended and then cultured with HepG2 cells.Fig. 8 shows the in vitro cell viability of HepG2 cells after 24, 48, and 72 h-incubation with DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs, DTX-loaded PEG1k-b-(PCL-ran-PLA) NPs and Taxotere® at equivalent DTX concentrations of 0.25, 2.5, 12.5 and 25 μg ml−1, respectively. Drug-free CA-(PCL-ran-PLA)-b-PEG1k NPs were used in the control groups. As can be concluded from the figure, no obvious cytotoxic activity was observed for the drug-free CA-(PCL-ran-PLA)-b-PEG1k NPs at various concentrations from 0.25 μg ml−1 to 25 μg ml−1, indicating that the synthesized star-shaped CA-(PCL-ran-PLA)-b-PEG1k copolymer could be biocompatible and non-toxic to tissues and cells. The cellular viability decreased with the prolonged incubation time for both Taxotere® and DTX-loaded NPs, exhibiting a dose-dependent and time-dependent effect, especially for drug-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs. DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs exhibited the best cytotoxicity efficacy against HepG2 cells, compared with Taxotere® and PEG1k-b-(PCL-ran-PLA) NPs. For example, the HepG2 cellular viability after 72 h incubation at the 12.5 μg ml−1 drug concentration was 43.96% for Taxotere®, 38.27% for PEG1k-b-(PCL-ran-PLA) NPs, and 18.36% for CA-(PCL-ran-PLA)-b-PEG1k NPs. Furthermore, in contrast to Taxotere®, the cytotoxicity of the HepG2 cell was higher than 30.36% (**p < 0.01, n = 5) and 25.60% (**p < 0.01, n = 5) for DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs after 72 h of incubation at a drug concentration of 2.5 μg ml−1 and 12.5 μg ml−1, respectively. The cytotoxicity of the drug-loaded star-shaped copolymer CA-(PCL-ran-PLA)-b-PEG1k NPs also exhibited better cytotoxicity efficacy than the HepG2 cells in comparison with DTX-loaded linear copolymer PEG1k-b-(PCL-ran-PLA) NPs, demonstrating that the star-shaped copolymer may have a more superior performance than the linear copolymer in a drug delivery system.
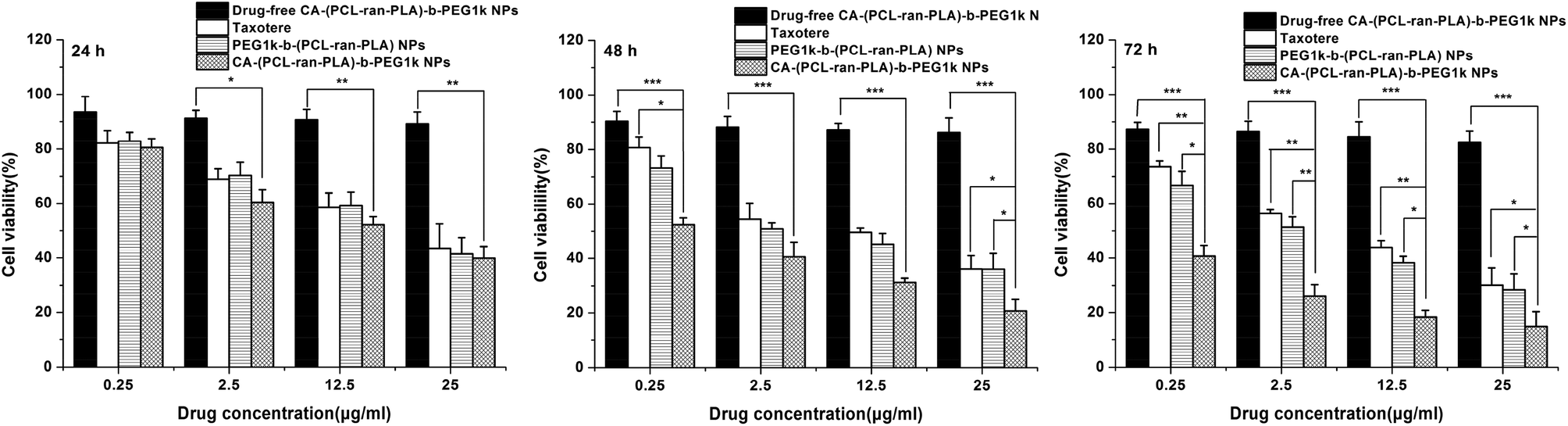 | ||
| Fig. 8 Viability of HepG2 cells incubated with DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs in comparison with those incubated with Taxotere® at the same DTX concentration and that of PEG1k-b-(PCL-ran-PLA) NPs, drug-free CA-(PCL-ran-PLA)-b-PEG1k NPs with the same amount of NPs. Compared with Taxotere® and drug-loaded PEG1k-b-(PCL-ran-PLA) NPs, a significant reduction in the cell number for drug-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs can be observed (*p < 0.05, **p < 0.01, ***p < 0.001, n = 5). | ||
Drug carriers based on star-shaped polymers have a lot of advantages, such as a lower solution viscosity, a smaller hydrodynamic radius, a higher drug loading content (LC) and a higher drug encapsulation efficiency (EE) than that of linear polymers.20,46 In addition, star-shaped DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs have a faster drug release rate and a higher cell uptake efficiency than linear PEG1k-b-(PCL-ran-PLA) NPs. Therefore it is reasonable that the in vitro therapeutic effect of star-shaped DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs is better than that of linear PEG1k-b-(PCL-ran-PLA) NPs.
Table 3 lists the IC50 values of HepG2 cells after 24, 48 and 72 h incubation with the DTX formulation Taxotere®, drug-loaded PEG1k-b-(PCL-ran-PLA) NPs and CA-(PCL-ran-PLA)-b-PEG1k NPs. The IC50 value, which could quantitatively evaluate the in vitro therapeutic effects of a pharmaceutical formulation, is defined as the drug inhibitory concentration needed to cause 50% tumor cell mortality in a designated period. As shown in the table, the IC50 value of DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs for HepG2 cells was 10.31 μg ml−1, a little higher than Taxotere® after 24 h incubation. However, the IC50 value for HepG2 cells was decreased from 7.04 to 4.25 μg ml−1 for Taxotere®, from 4.64 to 2.30 μg ml−1 for DTX-loaded PEG1k-b-(PCL-ran-PLA) NPs, and from 0.456 to 0.069 μg ml−1 for DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs after 48 and 72 h incubation, respectively. The advantages in tumor cell inhibition followed the order: CA-(PCL-ran-PLA)-b-PEG1k NPs formulation > PEG1k-b-(PCL-ran-PLA) NPs formulation > Taxotere® formulation, thus demonstrating that the DTX-loaded NPs had better in vitro therapeutic effects for HepG2 cells, and the star-shaped CA-(PCL-ran-PLA)-b-PEG1k was superior to the linear PEG1k-b-(PCL-ran-PLA) as a drug carrier for cancer chemotherapy.
| Incubation time (h) | IC50 (μg ml−1) | ||
|---|---|---|---|
| Taxotere® | PEG1k-b-(PCL-ran-PLA) NPs | CA-(PCL-ran-PLA)-b-PEG1k NPs | |
| 24 | 19.76 ± 1.83 | 18.63 ± 1.61 | 10.31 ± 1.54 |
| 48 | 7.04 ± 0.48 | 4.64 ± 0.52 | 0.456 ± 0.36 |
| 72 | 4.25 ± 0.43 | 2.30 ± 0.17 | 0.069 ± 0.05 |
3.5. In vivo anti-tumor efficacy study
Considering the satisfactory in vitro cytotoxicity against HepG2 cells, it is possible that CA-(PCL-ran-PLA)-b-PEG1k NPs could be developed as a promising deliverable vehicle for liver cancer chemotherapy. To further investigate the in vivo anti-tumor efficacy of CA-(PCL-ran-PLA)-b-PEG1k NPs formulation of DTX vs. Taxotere®, BALB/c nude mice were subcutaneously (s.c.) inoculated into the right flank with 2 × 106 HepG2 cells in 100 μl culture medium, and the results showed that 95% of the mice injected developed a tumor with an average volume of ∼50 mm3 after one week. Then the mice were randomized into 3 groups (n = 5 for each group) and treated with DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k nanoformulation or Taxotere®, respectively at 10 mg kg−1 DTX dose at day 0, 4, 8, 12 through intraperitoneal injection. Physiological saline was used as the control, and the tumors size of the mice was recorded every 2 days until the 14th day. After two-week's therapy, all the mice were sacrificed and the tumors were separated from the bodies at the experimental terminal.The morphology of the tumors of each group is presented in Fig. 9(A). The images clearly show that the tumor size for the saline group was the largest, in-between for the Taxotere® group, and smallest for the CA-(PCL-ran-PLA)-b-PEG1k NPs formulation group. Fig. 9(B) shows the tumor growth surveyed in the mice after intraperitoneal injection with DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs, Taxotere® and physiological saline. The tumor volume for the saline group increased obviously during the experimental period, while the tumor size for the CA-(PCL-ran-PLA)-b-PEG1k nanoformulation group was significantly inhibited. The weights of the tumors in each group showed in Fig. 9(C) also indicated that the advantages of the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs vs. Taxotere® in suppressing tumors were significant. In summary, it can be confirmed that DTX nanoformulation in CA-(PCL-ran-PLA)-b-PEG1k NPs could maintain its pharmacological activity and significantly inhibit the tumor growth compared to Taxotere® at the same dose.
 | ||
| Fig. 9 Anti-tumor efficacy of DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs, Taxotere® and saline on the BALB/c nude mice bearing HepG2 cells: (A) images of tumors in each group taken out from the sacrificed mice at the end point of research; (B) tumor-growth curve of SCID mice after intraperitoneal injection with DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs, Taxotere® and saline (*p < 0.05, **p < 0.01, n = 5); (C) tumor weight of each group taken out from the sacrificed mice at the end of the study. Compared with the saline and Taxotere® groups, a significant reduction in the tumor weight for CA-(PCL-ran-PLA)-b-PEG1k NPs could be observed (*p < 0.05, ***p < 0.001, n = 5). | ||
4. Conclusions
In this research, a novel CA functionalized star-shaped CA-(PCL-ran-PLA)-b-PEG1k NPs system was developed for the sustained and controlled delivery of DTX for application in liver cancer therapy. The DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs were prepared by a modified nano-precipitation technique. The hydrodynamic size of the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs was about 90 nm, much smaller than that of CA-(PCL-ran-PLA) and linear PEG1k-b-(PCL-ran-PLA) NPs. Compared with PEG1k-b-(PCL-ran-PLA) and CA-(PCL-ran-PLA) NPs, the drug loading content and encapsulation efficiency of the star-shaped CA-(PCL-ran-PLA)-b-PEG1k NPs were significantly improved. Moreover, the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs could achieve a much faster drug release speed, as well as a higher cellular uptake efficiency and cytotoxicity by human liver carcinoma cells (HepG2). Both in vitro cell viability experiments and in vivo anti-tumor efficacy studies demonstrated that the DTX-loaded CA-(PCL-ran-PLA)-b-PEG1k NPs exhibited significantly superior anti-tumor efficiency than the linear PEG1k-b-(PCL-ran-PLA) NPs and commercially available Taxotere® formulation. In conclusion, the star-shaped CA-(PCL-ran-PLA)-b-PEG1k is a novel and promising biodegradable copolymer for nanoformulations applied in liver cancer treatment.Appendix
Abbreviation
| 1H NMR | Proton nuclear magnetic resonance |
| GPC | Gel permeation chromatography |
| TGA | Thermogravimetric analysis |
| DLS | Dynamic light scattering |
| FESEM | Field emission scanning electron microscopy |
| TEM | Transmission electron microscopy |
| CLSM | Confocal laser scanning microscope |
| FCM | Flow cytometric |
| HPLC | High performance liquid chromatography |
| LC | Loading content |
| EE | Encapsulation efficiency |
| PDI | Polydispersity index |
| CA | Cholic acid |
| PCL | Poly (ε-caprolactone) |
| PLA | Poly(lactide) |
| PEG | Poly(ethylene glycol) |
| DTX | Docetaxel |
| EPR | Enhanced permeability and retention |
| SCID | Severe combined immunodeficient |
| DCC | 1,3-Diisopropylcarbodiimide |
| DCU | Dicyclohexylcarbodiurea |
| DMAP | 4-(Dimethylamino)pyridine |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| DAPI | 4′,6-Diamidino-2-phenylindole dihydrochloride |
| HBSS | Hank's buffered salt solution |
| DMEM | Dulbecco's Modified Eagle's Medium |
Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (no. 31270019, 51203085), the Natural Science Foundation of Guangdong Province (no. S2012010010046, S2012040006820), the Science, Technology & Innovation Commission of Shenzhen Municipality (no. JCYJ20120616213729920, JCYJ20120614191936420, KQC201105310021A, JC201005270308A), and the Program for New Century Excellent Talents in University (NCET-11-0275).References
- R. Lozano and M. Naghavi, et al. , Lancet, 2012, 380, 2095–2128 CrossRef.
- Y. D. Ma, L. Q. Huang, C. X. Song, X. W. Zeng, G. Liu and L. Mei, Polymer, 2010, 51, 5952–5959 CrossRef CAS PubMed.
- Y. Mi, X. Liu, J. Zhao, J. Ding and S. S. Feng, Biomaterials, 2012, 33, 7519–7529 CrossRef CAS PubMed.
- J. Shi, Z. Xiao, N. Kamaly and O. C. Farokhzad, Acc. Chem. Res., 2011, 44, 1123–1134 CrossRef CAS PubMed.
- M. Colilla, B. Gonzalez and M. Vallet-Regi, Biomater. Sci., 2013, 1, 114–134 RSC.
- M. Sinn Aw, M. Kurian and D. Losic, Biomater. Sci., 2014, 2, 10–34 RSC.
- E. Wagner, Biomater. Sci., 2013, 1, 804–809 RSC.
- W. E. Bawarski, E. Chidlowsky, D. J. Bharali and S. A. Mousa, Emerging nanopharmaceuticals, Nanomed.: Nanotechnol., Biol. Med., 2008, 4, 273–282 CrossRef CAS PubMed.
- D. B. Buxton, Nanomedicine, 2009, 4, 331–339 CrossRef CAS PubMed.
- H. Onyuksel, E. Jeon and I. Rubinstein, Cancer Lett., 2009, 274, 327–330 CrossRef PubMed.
- Y. Q. Zhang, L. Tang, L. L. Sun, J. B. Bao, C. X. Song, L. Q. Huang, K. X. Liu, Y. Tian, G. Tian, Z. Li, H. F. Sun and L. Mei, Acta Biomater., 2010, 6, 2045–2052 CrossRef CAS PubMed.
- L. Mei, Z. P. Zhang, L. Y. Zhao, L. Q. Huang, X. L. Yang, J. T. Tang and S. S. Feng, Adv. Drug Delivery Rev., 2013, 65, 880–890 CrossRef CAS PubMed.
- J. Wang, K. Yao, C. Wang, C. Tang and X. Jiang, J. Mater. Chem. B, 2013, 1, 2324–2332 RSC.
- Y. Yue and C. Wu, Biomater. Sci., 2013, 1, 152–170 RSC.
- B. H. Tan, H. Hussain, Y. W. Leong, T. T. Lin, W. W. Tjiu and C. He, Polym. Chem., 2013, 4, 1250–1259 RSC.
- L. Q. Huang, H. B. Chen, Y. Zheng, X. S. Song, R. Y. Liu, K. X. Liu, X. W. Zeng and L. Mei, Integr. Biol., 2011, 3, 993–1002 RSC.
- Y. Wan, Z. Gan and Z. Li, Polym. Chem., 2014, 5, 1720–1727 RSC.
- Y. Yang, J. Bugno and S. Hong, Polym. Chem., 2013, 4, 2651–2657 RSC.
- V. Delplace, P. Couvreur and J. Nicolas, Polym. Chem., 2014, 5, 1529–1544 RSC.
- W. Tao, X. Zeng, T. Liu, Z. Wang, Q. Xiong, C. Ouyang, L. Huang and L. Mei, Acta Biomater., 2013, 9, 8910–8920 CrossRef CAS PubMed.
- L. Mei, Z. Zhang and S. S. Feng, J. Controlled Release, 2013, 172, e55 CrossRef PubMed.
- Z. Yue, Z. You, Q. Yang, P. Lv, H. Yue, B. Wang, D. Ni, Z. Su, W. Wei and G. Ma, J. Mater. Chem. B, 2013, 1, 3239–3247 RSC.
- K. Fujii, J. Nagai, T. Sawada, R. Yumoto and M. Takano, Bioconjugate Chem., 2009, 20, 1553–1558 CrossRef CAS PubMed.
- S. M. Kelleher, Z. Zhang, A. Lobus, C. Strehmel and M. C. Lensen, Biomater. Sci., 2014, 2, 410–418 RSC.
- J. Wang, K. Yao, C. Wang, C. Tang and X. Jiang, J. Mater. Chem. B, 2013, 1, 2324–2332 RSC.
- B. Newland, E. Dowd and A. Pandit, Biomater. Sci., 2013, 1, 556–576 RSC.
- Z. Mao, X. Zhou and C. Gao, Biomater. Sci., 2013, 1, 896–911 RSC.
- C. M. Yang, L. Q. Jiang, S. J. Bu, L. H. Zhang, X. J. Xie, Q. G. Zeng, D. W. Zhu and Y. Zheng, J. Biomed. Nanotechnol., 2013, 9, 1617–1623 CrossRef CAS PubMed.
- T. Zou, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, J. Biomed. Mater. Res., Part B, 2007, 82, 400–407 CrossRef PubMed.
- H. L. Fu, L. Yu, H. Zhang, X. Z. Zhang, S. X. Cheng and R. X. Zhuo, J. Biomed. Mater. Res., Part A, 2007, 81, 186–194 CrossRef PubMed.
- C. V. Synatschke, F. A. Plamper and A. H. E. Muller, Nachr. Chem., 2013, 61, 1008–1012 CAS.
- M. Q. Lv, Y. Shi, W. T. Yang and Z. F. Fu, J. Appl. Polym. Sci., 2013, 128, 332–339 CrossRef CAS.
- A. Swinarew, B. Piekarnik, Z. Grobelny, A. Stolarzewicz, J. Simokaitiene, E. Andrikaityte and J. V. Grazulevicius, Macromol. Symp., 2011, 308, 8–11 CrossRef CAS.
- A. J. Inglis, P. Pierrat, T. Muller, S. Brase and C. Barner-Kowollik, Soft Matter, 2010, 6, 82–84 RSC.
- D. G. Kim, H. S. Sohn, S. K. Kim, A. Lee and J. C. Lee, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3618–3627 CrossRef CAS.
- L. M. de Espinosa, M. Winkler and M. A. R. Meier, Macromol. Rapid Commun., 2013, 34, 1381–1386 CrossRef CAS PubMed.
- D. Ma, Z. H. Liu, Q. Q. Zheng, X. Y. Zhou, Y. Zhang, Y. F. Shi, J. T. Lin and W. Xue, Macromol. Rapid Commun., 2013, 34, 548–552 CrossRef CAS PubMed.
- M. R. Nabid, S. J. T. Rezaei, R. Sedghi, H. Niknejad, A. A. Entezami, H. A. Oskooie and M. M. Heravi, Polymer, 2011, 52, 2799–2809 CrossRef CAS PubMed.
- T. H. Li, R. Y. Han, M. Z. Wang, C. B. Liu, X. B. Jing and Y. B. Huang, Macromol. Biosci., 2011, 11, 1570–1578 CAS.
- P. M. Valencia, M. H. Hanewich-Hollatz, W. W. Gao, F. Karim, R. Langer, R. Karnik and O. C. Farokhzad, Biomaterials, 2011, 32, 6226–6233 CAS.
- L. Mei, Y. Q. Zhang, Y. Zheng, G. Tian, C. X. Song, D. Y. Yang, H. L. Chen, H. F. Sun, Y. Tian, K. X. Liu, Z. Li and L. Q. Huang, Nanoscale Res. Lett., 2009, 4, 1530–1539 CrossRef CAS PubMed.
- K. L. Hu, J. W. Li, Y. H. Shen, W. Lu, X. L. Gao, Q. Z. Zhang and X. G. Jiang, J. Controlled Release, 2009, 134, 55–61 CrossRef CAS PubMed.
- S. Fokong, B. Theek, Z. J. Wu, P. Koczera, L. Appold, S. Jorge, U. Resch-Genger, M. van Zandvoort, G. Storm, F. Kiessling and T. Lammers, J. Controlled Release, 2012, 163, 75–81 CrossRef CAS PubMed.
- Y. H. Loo, C. L. Grigsby, Y. J. Yamanaka, M. K. Chellappan, X. Jiang, H. Q. Mao and K. W. Leong, J. Controlled Release, 2012, 160, 48–56 CrossRef CAS PubMed.
- J. X. Ding, C. S. Xiao, Y. C. Li, Y. L. Cheng, N. N. Wang, C. L. He, X. L. Zhuang, X. J. Zhu and X. S. Chen, J. Controlled Release, 2013, 169, 193–203 CrossRef CAS PubMed.
- X. Zeng, W. Tao, L. Mei, L. Huang, C. Tan and S. S. Feng, Biomaterials, 2013, 34, 6058–6067 CrossRef CAS PubMed.
- H. Claesson, E. Malmstrom, M. Johansson and A. Hult, Polymer, 2002, 43, 3511–3518 CrossRef CAS.
- M. M. Yallapu, B. K. Gupta, M. Jaggi and S. C. Chauhan, J. Colloid Interface Sci., 2010, 351, 19–29 CrossRef CAS PubMed.
- E. Csiszar, E. Fekete, A. Toth, E. Bandi, B. Koczka and I. Sajo, Carbohydr. Polym., 2013, 94, 927–933 CrossRef CAS PubMed.
- H. Maeda, Proc. Jpn. Acad. Ser. B: Phys. Biol. Sci., 2012, 88, 53–71 CrossRef CAS.
- C. N. Zhang, W. Wang, T. Liu, Y. K. Wu, H. Guo, P. Wang, Q. Tian, Y. M. Wang and Z. Yuan, Biomaterials, 2012, 33, 2187–2196 CrossRef CAS PubMed.
- A. Vollrath, D. Pretzel, C. Pietsch, I. Perevyazko, S. Schubert, G. M. Pavlov and U. S. Schubert, Macromol. Rapid Commun., 2012, 33, 1791–1797 CrossRef CAS PubMed.
- X. Duan and Y. Li, Small, 2013, 9, 1521–1532 CrossRef CAS PubMed.
- Y. Liu, M. K. Shipton, J. Ryan, E. D. Kaufman, S. Franzen and D. L. Feldheim, Anal. Chem., 2007, 79, 2221–2229 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |