
Fractionation of an ECM hydrogel into structural and soluble components reveals distinctive roles in regulating macrophage behavior
P. F.
Slivka
a,
C. L.
Dearth
ab,
T. J.
Keane
ac,
F. W.
Meng
a,
C. J.
Medberry
ac,
R. T.
Riggio
ad,
J. E.
Reing
a and
S. F.
Badylak
*abc
aMcGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA 15219, USA. E-mail: badylaks@upmc.edu
bDepartment of Surgery, University of Pittsburgh, Pittsburgh, PA 15219, USA
cDepartment of Bioengineering, University of Pittsburgh, Pittsburgh, PA 15219, USA
dSanford School of Medicine, University of South Dakota, Vermillion, SD 57069, USA
First published on 7th August 2014
Abstract
Extracellular matrix (ECM) derived from mammalian tissues has been utilized to repair damaged or missing tissue and improve healing outcomes. More recently, processing of ECM into hydrogels has expanded the use of these materials to include platforms for 3-dimensional cell culture as well as injectable therapeutics that can be delivered by minimally invasive techniques and fill irregularly shaped cavities. At the cellular level, ECM hydrogels initiate a multifaceted host response that includes recruitment of endogenous stem/progenitor cells, regional angiogenesis, and modulation of the innate immune response. Unfortunately, little is known about the components of the hydrogel that drive these responses. We hypothesized that different components of ECM hydrogels could play distinctive roles in stem cell and macrophage behavior. Utilizing a well-characterized ECM hydrogel derived from urinary bladder matrix (UBM), we separated the soluble and structural components of UBM hydrogel and characterized their biological activity. Perivascular stem cells migrated toward and reduced their proliferation in response to both structural and soluble components of UBM hydrogel. Both components also altered macrophage behavior but with different fingerprints. Soluble components increased phagocytosis with an IL-1RAhigh, TNFαlow, IL-1βlow, uPAlow secretion profile. Structural components decreased phagocytosis with a PGE2high, PGF2αhigh, TNFαlow, IL-1βlow, uPAlow, MMP2low, MMP9low, secretion profile. The biologic activity of the soluble components was mediated by Notch and PI3K/Akt signaling, while the biologic activity of the structural components was mediated by integrins and MEK/ERK signaling. Collectively, these findings demonstrate that soluble and structural components of ECM hydrogels contribute to the host response but through different mechanisms.
Introduction
Mammalian ECM derived from a variety of tissue sources has been extensively utilized as a surgical mesh material for many clinical applications including hernia repair,1,2 rotator cuff repair,3,4 breast reconstruction,5,6 and musculotendinous reinforcement.7 When properly prepared and implanted these 2-D sheets of ECM have been shown to function as an inductive template for the repair and reconstruction of damaged or missing tissues.8,9In vivo preclinical studies have established a temporal sequence for ECM mediated constructive remodeling beginning with a transient leukocyte response followed by a dense mononuclear cell infiltration into the ECM scaffold. Gradually the implanted scaffold is replaced with new ECM as well as small pockets of site appropriate, vascularized, and innervated tissue.9–12 The pathophysiology of ECM scaffold remodeling has been partially characterized and includes the recruitment of endogenous stem/progenitor cells13,14 and modulation of the innate immune response towards a regulatory (M2/Th2) phenotype.15–17 Macrophages that surround and infiltrate ECM scaffolds express a number of M2 markers including CD206 and CD163.16,18 Moreover, the presence of these M2 markers appears to be a strong predictor of a constructive host remodeling response.15While 2-D, flat sheets of ECM have shown clinical utility, the constraint to a sheet form has limited the potential applications for these materials. More recently, ECM scaffolds have been processed into different forms including powders, putties, and hydrogels.19–21 Of these alternative forms, hydrogels provide the most options as they can be cast into 3-D shapes for cell culture and tissue engineering applications or injected directly into host tissue.20,22,23 These injected hydrogels can fill complex cavities and retain the shape of that cavity once polymerized.24 Generation of hydrogels from ECM requires ECM to be solubilized, which is readily accomplished with pepsin digestion. Pepsin digestion has been extensively utilized to prepare collagen both for research and medical purposes and is an established industry standard.25,26 Importantly, pepsin solubilized ECM hydrogels have been shown to elicit a similar host response to the 2-D surgical mesh form of ECM.22,23
At a molecular level, ECM hydrogels consist of both structural and soluble components that contain unique molecular fingerprints. The structural components of ECM hydrogels include a number of partially digested proteoglycans and proteins such as collagens, elastin, laminin, fibronectin, hyaluronan, and heparan. The soluble components of the scaffold consist of cryptic peptide fragments generated from partial proteolysis of scaffold resident growth factors (e.g. bFGF, VEGF, IGF, TGFβ) and matricellular proteins (e.g. SPARC, tenascin, osteopontin, thrombospondin) in addition to any matricryptic peptides derived from structural components.27–29 A number of studies on stem cells and macrophages have identified different signaling cascades that are initiated by these ECM components, including mitogen-activated protein kinases (MAPKs), phosphoinositide-3 kinase (PI3K), and Wingless/Integrase 1 (Wnt), among others.30–32 Activation of these signaling cascades can originate from a number of cell surface receptors. Many structural and soluble components of ECM interact with integrins through conserved cell adhesion sequences such as RGD and VGVAPG.28,33,34 Other receptors for ECM components include growth factor receptors (FGFRs, VEGFRs, etc.), scavenger receptors (Stabilin-1, LOX-1), and toll-like receptors (TLRs).35–38 Despite these advances, no study has directly compared the structural and soluble components of ECM hydrogels to determine the unique cellular behaviors and molecular events they mediate.
The objective of the present study was to determine the biological activity of the soluble and structural components of an ECM hydrogel. The well-characterized hydrogel of the biologic scaffold referred to as urinary bladder matrix (UBM) was utilized as a model system.20,22 The structural and soluble components of UBM hydrogel were fractionated and tested for their ability to modulate stem cell and macrophage behavior—two critical cell types in the host response to ECM. The components of UBM hydrogel were tested for their ability to modulate the chemotactic and proliferative activity of human perivascular stem cells (PSCs). The components were also tested for their ability to modulate macrophage behavior by examining their effects on the phagocytic ability and secretion profile of THP1 human macrophages. Finally, a subset of signaling cascades and cell surface receptors involved in modulating the macrophage secretion profile were determined.
Materials and methods
Reagents
All chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. All cell culture media and reagents were purchased from Life Technologies (Carlsbad, CA) unless otherwise specified. All chemicals used were reagent grade or better.Urinary bladder matrix preparation
Porcine urinary bladders were acquired from market weight pigs (110–130 kg) as a byproduct of routine commercial production. The extracellular matrix from this tissue referred to as UBM was prepared as previously described.39 Briefly, the tunica serosa, tunica muscularis externa, tunica submucosa, and most of the tunica muscularis mucosa were mechanically removed and the luminal urothelial cells of the tunica mucosa were dissociated by rinsing in sterile water. The remaining tissue consisted of the basement membrane, the subjacent tunica propria of the tunica mucosa, and any resident cells in those layers. The matrix was decellularized by agitation in 0.1% peracetic acid with 4% ethanol for 2 hours at high speed followed by extensive rinsing with phosphate-buffered saline (PBS) and sterile water. Decellularization was verified using 4′-6-diamidino-2-phenylindole (DAPI, Fisher Scientific, Waltham, MA) nuclear staining and quantification of remnant DNA.40 The UBM was then lyophilized into a dry sheet and either milled into particulates using a Wiley Mill with a #60 mesh screen.19Pepsin mediated ECM solubilization
UBM was enzymatically digested as previously described20 with pepsin by mixing lyophilized, powdered UBM (10 mg mL−1) and pepsin (1 mg mL−1) in 0.01 M HCl (pH 2.0). This solution was stirred at room temperature for 48 hours. After stirring, the UBM slurry was neutralized to a pH of 7.4 in 1× PBS (137 mM NaCl, 2.7 mM KCl, 12 mM Phosphate, Fisher Scientific, Waltham, MA) to inactivate the pepsin and prepare the material for cell culture assays. A solution of pepsin (1 mg mL−1) in 0.01 M HCl, treated in the same fashion as the UBM sample, served as the control condition for all experiments. For experiments containing dose responses the pepsin control was diluted appropriately to match the concentration that was present in enzymatically digested UBM. All materials were stored at −80 °C until use.Fractionation of digested UBM
Neutralized UBM digest was incubated at 37 °C to induce gelation. The UBM hydrogel was then centrifuged at 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 30 minutes to compress the insoluble, structural components of the scaffold into a pellet, leaving a clear supernatant above the pellet (Fig. 1A). The gel pellet containing the structural components was collected and resuspended to the starting volume in 1× PBS. Due to the insolubility of the gel pellet, the gel pellet suspension was vigorously pipetted through a 10 μL pipet tip and forced through a 300 μM filter to homogenize the material as much as possible. UBM hydrogel, homogenized in the same fashion served as the control in all experiments. Homogenized suspensions were stored at −80 °C until use. The clear supernatant containing the soluble components was removed and lyophilized to dryness. The dried supernatant was rehydrated in 10% of its original volume with sterile water to drive the PBS concentration from 1× to 10×. The increase in PBS concentration salts out any remaining structural components and is a common step in the preparation of collagen.41,42 The rehydrated soluble components were centrifuged at 20000g to clarify the solution. The supernatant from this final spin was removed, diluted to the starting volume, and stored at −80 °C until use. Dilution to the starting volume for both fractionated components allowed direct comparison of the biological activity of the fractions using the same dilution factor for all materials.
000g for 30 minutes to compress the insoluble, structural components of the scaffold into a pellet, leaving a clear supernatant above the pellet (Fig. 1A). The gel pellet containing the structural components was collected and resuspended to the starting volume in 1× PBS. Due to the insolubility of the gel pellet, the gel pellet suspension was vigorously pipetted through a 10 μL pipet tip and forced through a 300 μM filter to homogenize the material as much as possible. UBM hydrogel, homogenized in the same fashion served as the control in all experiments. Homogenized suspensions were stored at −80 °C until use. The clear supernatant containing the soluble components was removed and lyophilized to dryness. The dried supernatant was rehydrated in 10% of its original volume with sterile water to drive the PBS concentration from 1× to 10×. The increase in PBS concentration salts out any remaining structural components and is a common step in the preparation of collagen.41,42 The rehydrated soluble components were centrifuged at 20000g to clarify the solution. The supernatant from this final spin was removed, diluted to the starting volume, and stored at −80 °C until use. Dilution to the starting volume for both fractionated components allowed direct comparison of the biological activity of the fractions using the same dilution factor for all materials.
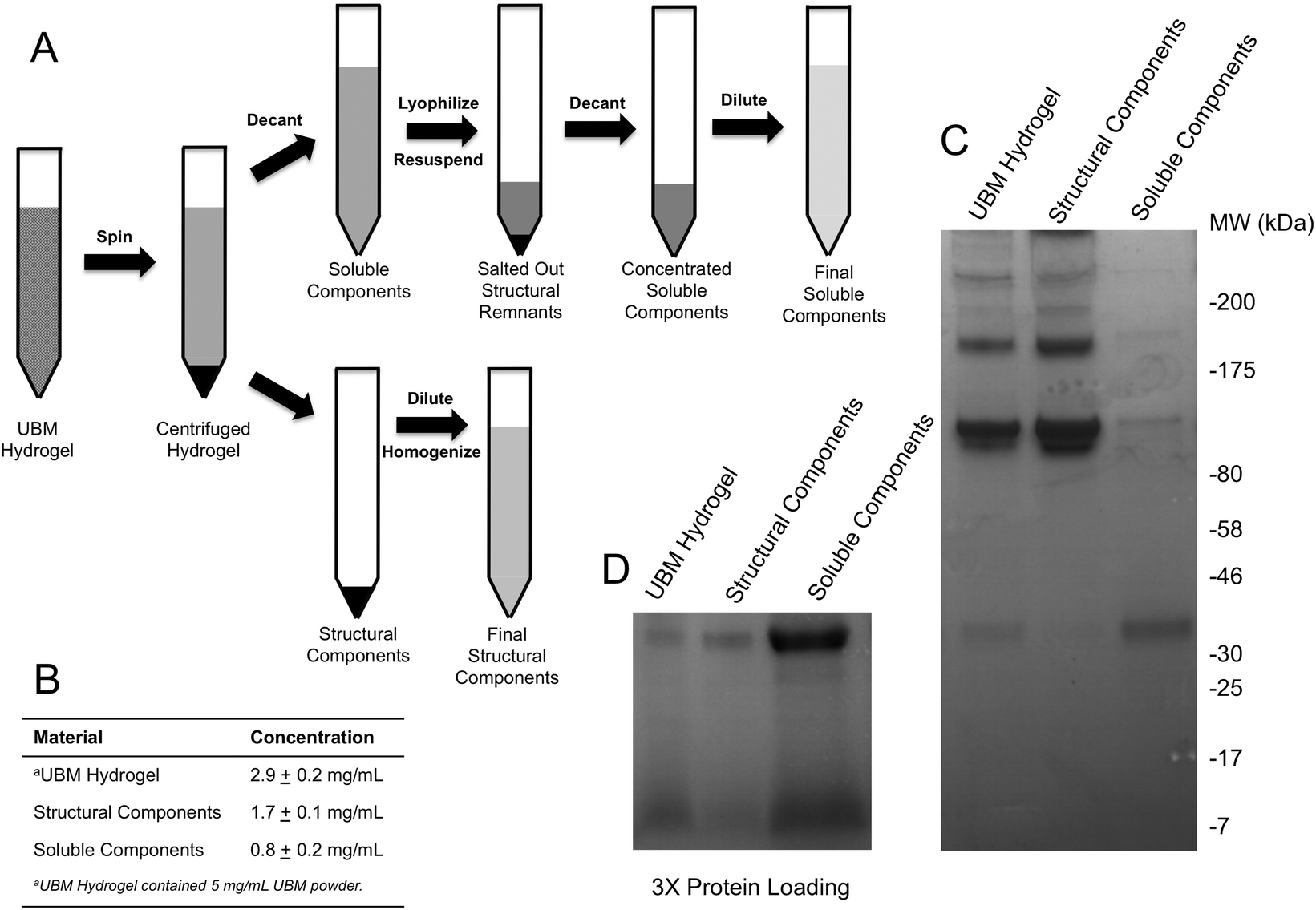 | ||
| Fig. 1 Fractionation of UBM into soluble and structural fractions. (A) Fractions were generated by forming a UBM hydrogel and centrifuging the structural components into a pellet. The liquid supernatant was removed, dried, and finally rehydrated in 10% of the original volume to increase the salt concentration and salt out any remaining structural components. Both the structural and soluble components isolated in this procedure were diluted to their original volume to allow for direct comparison of their activity. (B) The protein content of the various fractions determined by the BCA assay is shown. (C) Representative SDS PAGE gel with equal protein loading for UBM Digest, Structural Components and Soluble Components. (D) Low molecular weight bands are shown with 3 times additional protein loading. | ||
SDS PAGE and protein quantification
UBM hydrogel, structural, and soluble components were diluted 1:1 in 2× Laemmli sample buffer (Bio-Rad, Hercules, CA) and boiled at 95 °C for 8 minutes. Samples were diluted and tested for protein concentration using the Pierce BCA Protein Assay (Thermo Fisher, Waltham, MA) according to the manufacturer's instructions. Absorbances were measured at the appropriate wavelength using a Molecular Devices SpectraMax M2 plate reader (Silicon Valley, CA) and concentrations were approximated using a bovine serum albumin (BSA) standard curve. Either 15 μg (Fig. 1C) or 45 μg (Fig. 1D) of each sample—based on protein concentration—was resolved on 4–20% SDS PAGE gels (Bio-Rad, Hercules, CA) and stained with Coomassie Blue R-250 (Fisher Scientific, Waltham, MA). Gels were imaged using the Protein Simple Red Imager (Protein Simple, Santa Clara, CA).
Cell culture
THP-1 human monocytes were obtained from the American Tissue Culture Collection (ATCC, Manassas, VA) and maintained in RPMI, 10% FBS, 1% penicillin/streptomycin, 2 mM L-glutamine, and 1 mM sodium pyruvate in a humidified atmosphere at 37 °C with 5% CO2. For TNFα, PGE2, PGF2α, IL-1β, MMP2, MMP9, uPA, and IL-1RA experiments, 500000 THP-1 cells per well were plated with 320 nM phorbol 12-myristate 13-acetate (PMA) for 24 hours to induce differentiation into macrophages. Adherent macrophages were washed in PBS and placed in fresh media, followed by a 24 hour incubation in fresh media to acquiesce. THP1 macrophages differentiated with PMA and rested have been shown to exhibit nearly indistinguishable activity from human peripheral blood macrophages.43–45 Human PSCs were isolated from blood vessels in fetal muscle according to the methods outlined by Crisan et al.46–48 and maintained as reported.
Chemotaxis assay
Chemotaxis assays were performed in standard chemotaxis chambers with 8 μm filters (Neuro Probe, Gaithersburg, MD) coated with rat-tail collagen (BD Biosciences, San Jose, CA) as described previously.49 Briefly, PSCs were grown to 80–90% confluence before overnight incubation in DMEM with 0.5% heat inactivated FBS. Cells were trypsinized and resuspended in plain DMEM and 30000 cells were loaded onto the top well of the chemotaxis chamber which was separated by the filter from the lower well containing the treatment condition (580–5.8 μg mL−1 UBM hydrogel, 160–1.6 μg mL−1 soluble components, and 340–3.4 μg mL−1 structural components). Homogenization of the structural components as well as the large pore size ensured that the largely insoluble material could still diffuse through the filters. Chambers were place in a humidified atmosphere at 37 °C with 5% CO2 for 3 hours. Migrated cells were stained with DAPI (Fisher Scientific, Waltham, MA), imaged using a Zeiss Axio-Observer Z.1 microscope (Oberkochen, Germany) with 10× objective and quantified with ImageJ (NIH, Bethesda, MD). Digestion enzyme only control yielded 163 ± 35 migrated cells. This baseline value was normalized to zero and changes in chemotaxis for all treatment groups were expressed as a fold change to this control.
Proliferation assay
Human PSCs were cultured at a concentration of 5000 cells per well in a 96 well plate for 24 hours. Following 24 hours, 580–5.8 μg mL−1 UBM hydrogel, 160–1.6 μg mL−1 soluble components, or 340–3.4 μg mL−1 structural components were added to the wells along with 5-bromo-2′-deoxyuridine (BrdU; final concentration: 10 μM). Treated cells were incubated for 18 hours. The cells were then fixed with 95% methanol for 15 minutes and washed thoroughly with PBS. Cells were then incubated in 2 M HCl for 30 minutes at 37 °C and washed repeatedly with PBS. Following these washes, the fixed cells were incubated in (0.01% Triton-X100, 0.01% Tween-20, 2% horse serum) blocking buffer for 1 hour at room temperature. Cells were then incubated in primary mouse anti-BrdU (1:1000, DSHB, G3G4-c, Iowa City, Iowa) overnight at 4 °C. A donkey anti-mouse secondary antibody (1:300 Alexa Fluor 488, Life Technologies, Carlsbad, CA) was incubated for 45 minutes at room temperature. The cells were washed thoroughly with PBS and counterstained with DAPI. Whole well images were taken at 10× magnification using the mosaic function on the Zeiss Axio-Observer Z.1 microscope (Oberkochen, Germany). Cells positive for BrdU were then quantified using Cell Profiler50 and MATLAB (MathWorks, Natick, MA). Digestion enzyme only control contained 52 ± 4 BrdU+ DAPI+ nuclei per field. This baseline value was normalized to zero and changes in proliferation for all treatment groups were expressed as a percent decrease from this control.
Phagocytosis assay
Macrophages prepared as described in the Cell Culture section were treated with UBM hydrogel (290 μg mL−1) or fractionated components (80 μg mL−1 soluble, 170 μg mL−1 structural) for 48 hours. After treatment cells were washed extensively with 1× PBS and placed in fresh media containing Fluoresbrite Latex Beads (1.0 μM) and incubated for 1 hour. Cells were then washed with 1× PBS and fixed with 4% PFA for 15 minutes. After fixation cells were washed in 1× PBS and treated with Accutase for 10 minutes to lift the cells. Lifted cells were transferred to Eppendorf tubes and washed in 1× PBS. Phagocytosis was quantified using a BD FACSAria II flow cytometer to count the number of phagocytosing and non-phagocytosing cells. Between 10000 and 20000 events were counted for each treatment. Three replicate treatments were completed for each experiment and data were expressed as a percentage of phagocytosing cells.
Quantification of TNFα, PGE2, PGF2α, IL-1β, IL1-RA, MMP2, MMP9, uPA, IL-10, VEGF, and bFGF by THP-1 macrophages
Macrophages prepared as described in the Cell Culture section were treated with UBM hydrogel (290 μg mL−1) or fractionated components (80 μg mL−1 soluble, 170 μg mL−1 structural) for 48 (TNFα, IL-1β, IL-1RA, MMP2, MMP9, and uPA), or 72 (PGE2 and PGF2α) hours. For TNFα and IL-1β challenge experiments, post-treatments with lipopolysaccharide (LPS, 100 ng mL−1) were carried out for two hours. For inhibitory experiments, the specified inhibitors at the specified concentration were added to cells 1 hour prior to treatment with structural or soluble components of UBM. Culture supernatants were centrifuged at 20000g to pellet the cells and debris from the biologic scaffold treatments. Supernatants were harvested and frozen at −80 °C until the day of assay. The growth factor content of UBM and UBM hydrogel were determined using the method of Reing et al.51 Commercially available ELISA kits were used to determine the absolute quantities of TNFα (BD Bioscience, San Jose, CA), PGE2 (Enzo, Farmingdale, NY), PGF2α (Enzo, Farmingdale, NY), IL-1β (BD Bioscience, San Jose, CA), IL1-RA (R&D Systems, Minneapolis, MN), MMP2 (RayBiotech, Norcross, GA), MMP9 (RayBiotech, Norcross, GA), and uPA (RayBiotech, Norcross, GA), VEGF IL1-RA (R&D Systems, Minneapolis, MN), bFGF IL1-RA (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. Detection limits for the kits were as follows: TNFα 70 ± 3 pg mL−1, PGE2 35 ± 2 pg mL−1, PGF2α 3 ± 1 pg mL−1, IL-1β 1.7 ± 0.4 pg mL−1, IL1-RA 12 ± 3 pg mL−1, MMP2 0.41 ± 0.02 ng mL−1, MMP9 103 ± 15 pg mL−1, uPA 67 ± 4 pg mL−1, bFGF 27 ± 9 pg mL−1, and VEGF 12 ± 8 pg mL−1.
Statistics
Where appropriate, a one-way or two-way analysis of variance (ANOVA) was performed to determine significant differences with Student–Newman–Keuls post hoc testing (P ≤ 0.05). Data and error bars are reported as mean ± standard deviation unless otherwise specified.Results
Overview
The objective of this study was to determine the biological activity of the structural and soluble components of UBM hydrogels in order to better understand their role in host remodeling outcomes. To accomplish this objective, UBM hydrogels were fractionated into their soluble and structural components by centrifugation and salt precipitation. The effects of these fractions on stem cells and macrophages—two cell types that are critical to host remodeling—were investigated. The effects of structural and soluble components on stem cells were investigated by determining changes in isolated human PSC chemotaxis and proliferation. The effects of structural and soluble components on macrophages were investigated by examining changes in the secretion profile and phagocytic ability of THP1 human macrophages. Cell signaling cascades and cell surface receptors involved in regulating the secretion profile of macrophages were also determined with small molecule inhibitors.Fractionation of UBM digest
UBM hydrogels were fractionated into soluble and structural components using the scheme described in Fig. 1A. Acid digestion of ECM scaffolds solubilizes the scaffold, releases embedded soluble proteins and generates matricryptic peptides and sites.20,22,23 Neutralized UBM digest (pH 7.4) formed a relatively stiff gel at 37 °C.20,22 Centrifuging the gel at 25000g yielded a gel pellet containing the insoluble, structural scaffold components and a clear liquid supernatant. The supernatant was further refined by lyophilization and reconstitution in 10% of the original liquid volume to drive the PBS concentration to 10× (1.4 M NaCl, 27 mM KCl, 0.119 M Phosphate), which salted out any remaining structural components.25,41,42 A quick spin to remove any debris yielded a concentrated soluble component fraction of UBM hydrogel. Due to the limited yield of structural components present during the salting out step, the material could not be recovered and was thus discarded. As evidence of the removal of structural proteins, the soluble fraction could no longer form a hydrogel. In contrast, the gel pellet—containing the structural components—was completely insoluble at neutral pH and necessitated homogenization by repeated pipetting and pressing through a filter for use in cell culture assays. Homogenization of the structural components did not solubilize the material as centrifugation pelleted the material and the supernatant had no detectable protein by the BCA assay. Both the structural and soluble components were diluted to the original sample volume. The protein content was approximated with the BCA assay. The protein content of the parent material (UBM hydrogel) was 2.9 ± 0.2 mg mL−1 protein, representing 58% of the total UBM material by mass (Fig. 1B). The soluble fraction contained 0.8 ± 0.2 mg mL−1 protein and the insoluble scaffold contained 1.7 ± 0.1 mg mL−1 protein indicating that ∼85% of the protein content was retained during the fractionation process. The ∼15% reduction from the theoretical yield most likely resulted from the material discarded during the salting out step or the inaccuracy of estimating collagen rich protein samples in colorimetric protein assays.52 SDS PAGE analysis of the fractions showed that the structural fraction contained enriched quantities of high molecular weight (HMW) components (>80 kDa) compared to UBM hydrogel while the soluble fraction contained only very faint quantities of HMW proteins (Fig. 1C). The low molecular weight (LMW) end of the gel (<46 kDa) showed that the soluble fraction contained significantly enriched quantities of LMW species compared to the other materials (Fig. 1D). The multi-banding pattern demonstrated by UBM hydrogel and the structural fraction is consistent with the protein signature of collagen. A Picrosirius red stain of the structural fraction corroborated this result (data not shown).
Effect of fractions on PSCs
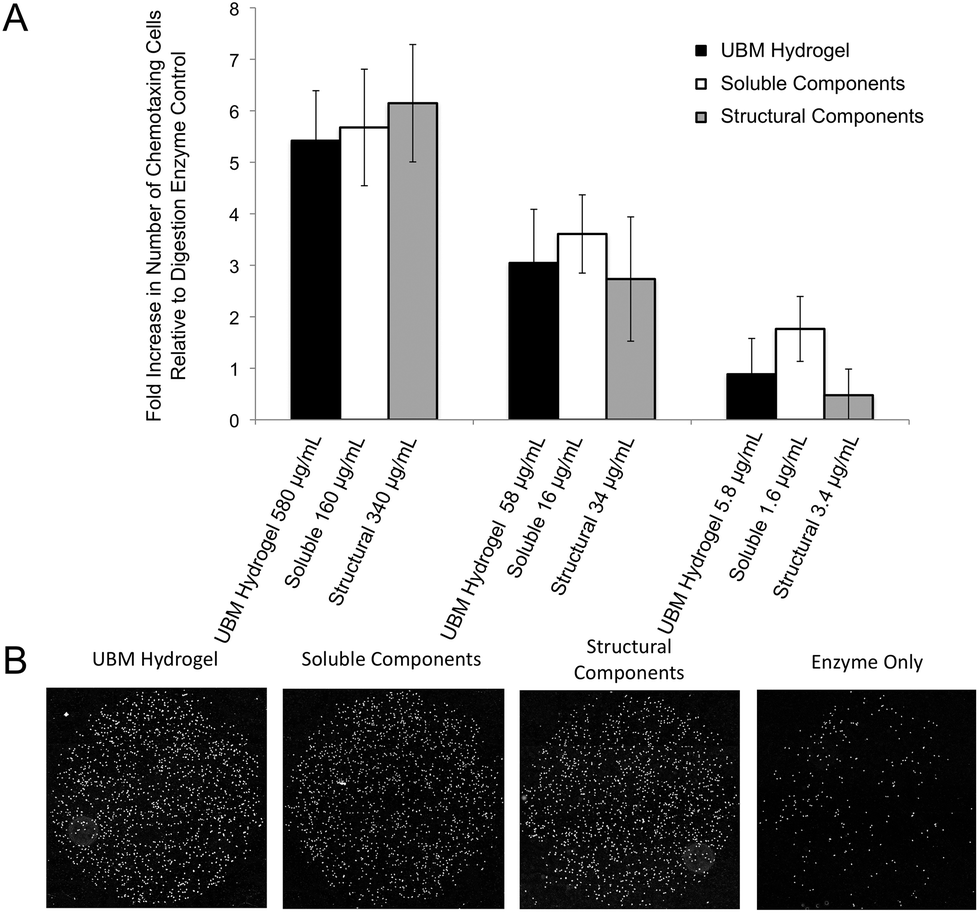 | ||
| Fig. 2 Chemotaxis of perivascular stem cells towards UBM hydrogel and its soluble and structural components. (A) PSCs were serum starved overnight, loaded into Boyden chemotaxis chambers, and allowed to migrate for 3 hours through an 8 μM collagen-coated, polycarbonate filter towards the materials shown above. Data are expressed as a fold-increase in the number of chemotaxing cells compared to a digestion enzyme only control (163 ± 35 cells), which was normalized to zero. Error bars indicate the standard error in the measurement for three experiments with four replicates per experiment. No significant differences were found between different materials at a given concentration. (B) Representative 10× magnification mosaic images from one well of migrated cells for each material at the highest concentration tested as well as the digestion enzyme only control are shown. Nuclei are shown in white. More nuclei indicate a stronger recruitment effect. | ||
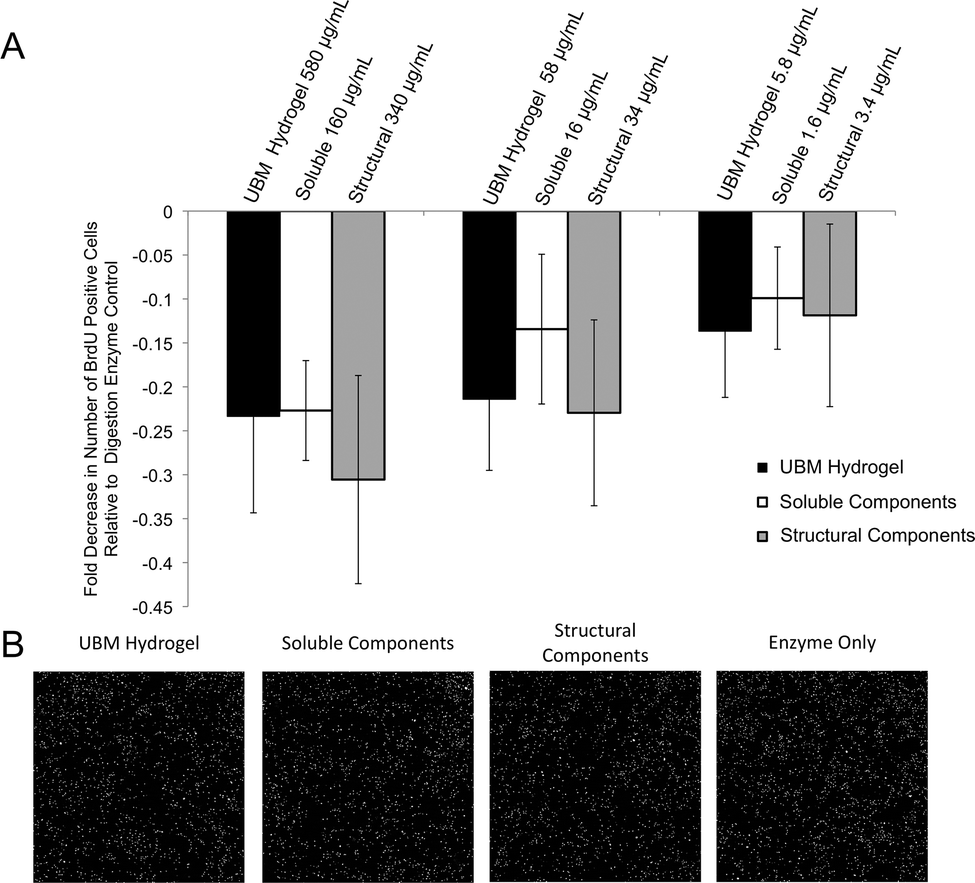 | ||
| Fig. 3 Changes in proliferation of perivascular stem cells after exposure to UBM hydrogel and its soluble and structural components. (A) PSCs were exposed to UBM or its fractionated components along with BrdU to measure changes in proliferation. The number of co-labeling (DAPI+BrdU+) nuclei was determined and the fold changes in co-labeling nuclei against a digestion enzyme only control (52 ± 4, normalized to zero) are shown. Error bars indicate the standard error in the measurement for three unique experiments with three replicates per experiment. No significant differences were found between materials at any of the concentrations tested. (B) Representative 10× magnification images of BrdU+ nuclei from PSCs treated with UBM hydrogel, its components, or digestion enzyme only control at the highest concentration tested for each condition. Greater numbers of DAPI+ nuclei indicate more proliferation. | ||
Effect of fractions on macrophage behavior
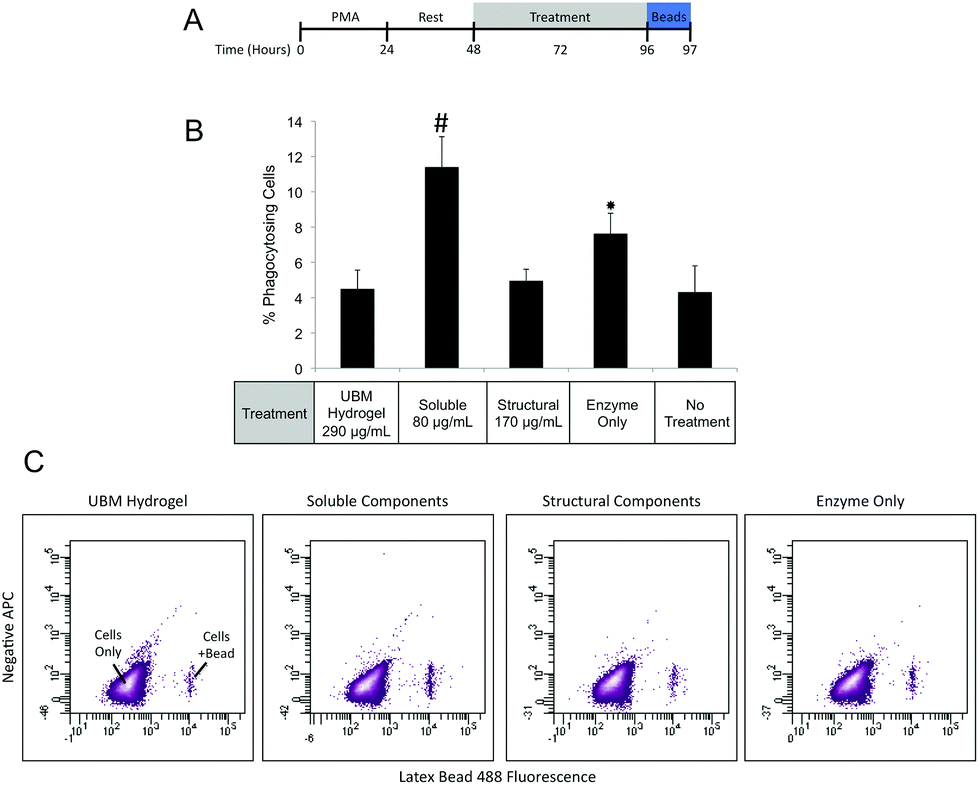 | ||
| Fig. 4 Macrophage phagocytosis of latex beads in response to UBM and its fractionated components. (A) THP-1 human monocytes were differentiated to macrophages with PMA and rested. Macrophages were treated with the specified conditions for 48 hours and exposed to latex beads. (B) The percentage of phagocytosing cells determined by flow cytometry are shown. Error bars indicate the standard deviation for six unique replicates. (#)Denotes significant increases from all other groups. (*)Denotes significant increases from UBM hydrogel, structural components, and no treatment groups. (C) Representative dot plots of macrophages phagocytosing latex beads after treatment with UBM hydrogel, soluble, or structural components and digestion enzyme only control. | ||
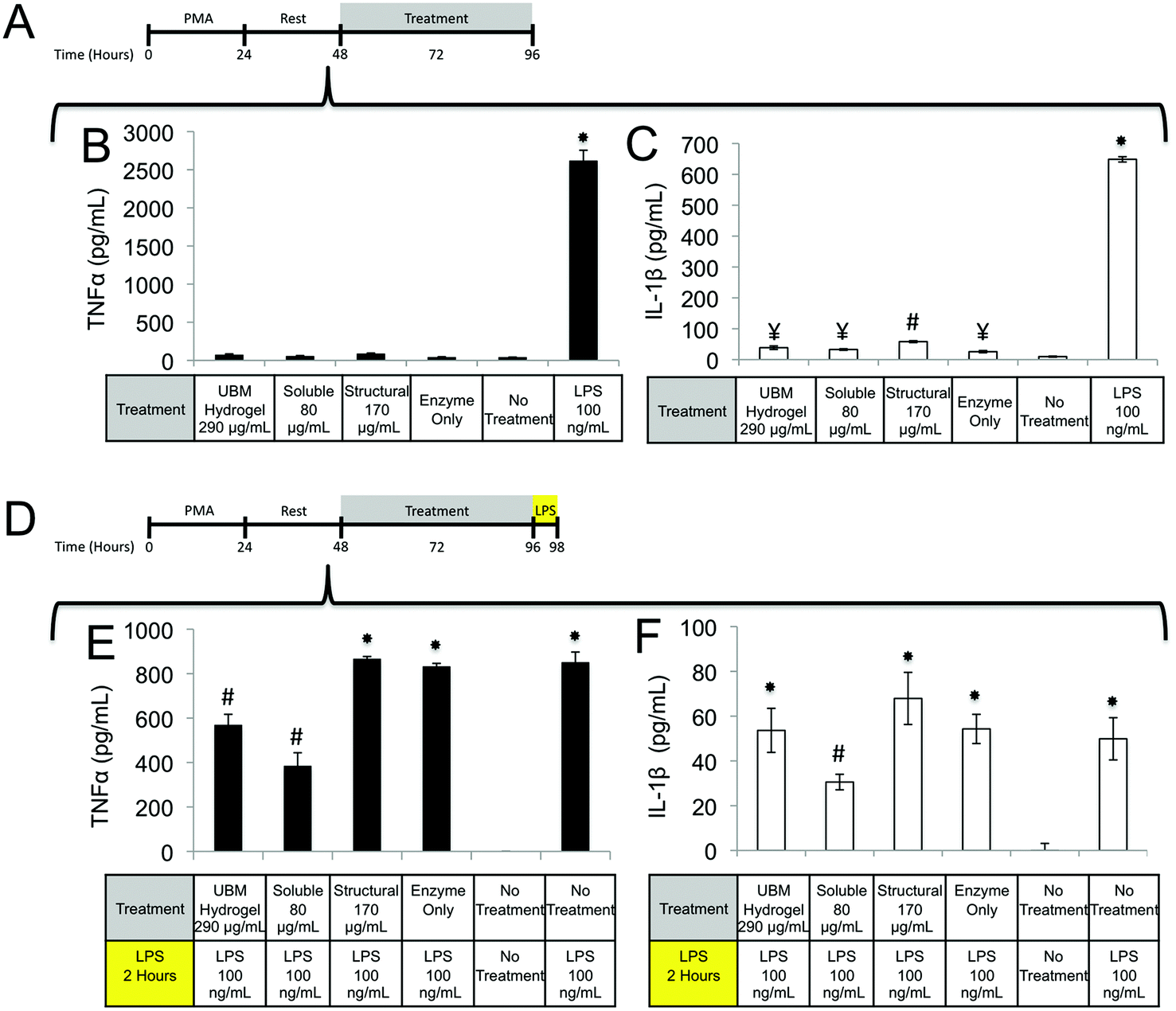 | ||
| Fig. 5 TNFα and IL-1β secretion after treatment with UBM and its fractionated components. (A) THP-1 human monocytes were differentiated to macrophages with PMA, rested, and treated with the specified conditions for 48 hours. (B, C) After treatment the concentration of TNFα (B) and IL-1β (C) in culture supernatants was determined using commercially available ELISA kits. (*)Denotes significant increases from all other groups. (¥)Denotes significant increases from no treatment control only. (#)Denotes significant increases from all other groups except LPS. (D) Macrophages treated in the same way as (B) and (C) were challenged with LPS (100 ng mL−1) to determine if treatment with the materials could prevent inflammation. (E, F) The concentration of TNFα (E) and IL-1β (F) in culture supernatants was once again determined using commercially available ELISA kits. (*)Denotes significant increases from unmarked and # marked groups. (#)Denotes significant increases from no treatment control. The data presented here represent a total of three replicates with each replicate representing duplicate samples. | ||
One possible mechanism for this suppression of inflammatory molecules is the secretion of anti-inflammatory mediators such as the IL-1 receptor antagonist (IL1-RA) and IL-10.58 To investigate if IL1-RA was secreted, macrophages were exposed to UBM hydrogel and its components for 48 hours (Fig. 6A). Treatment with the soluble components increased IL-1RA production 2.4 fold from enzyme only control while UBM and the structural components only induced a 0.3 fold increase (Fig. 6B). Secretion of the anti-inflammatory mediator, IL-10 was also examined. However, no increases in IL-10 secretion were observed, even with an additional 24 hours of incubation (data not shown). It is possible that IL-10 secretion is more acutely upregulated and the chosen timepoint is missing this transient spike. However, at the 48–72 hour time point IL-10 does not appear to be a major player.
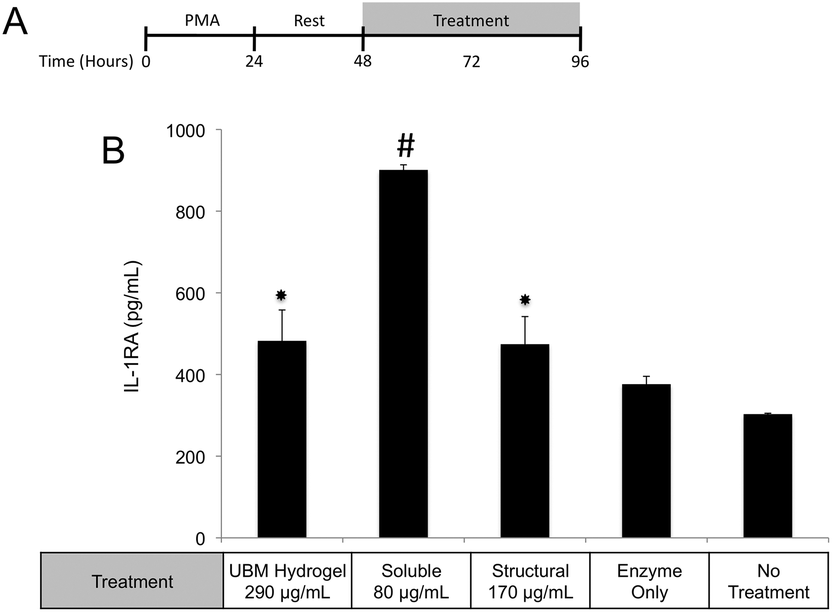 | ||
| Fig. 6 Effect of UBM and its fractionated components on IL1-RA secretion. (A) THP-1 human monocytes were differentiated to macrophages with PMA, rested and treated with UBM, soluble, or structural components for 48 hours. (B) IL-1RA in culture supernatants was measured using a commercially available ELISA kit. The data presented here represent a total of three replicates with each replicate representing duplicate samples. (#)Denotes significant increases from all other groups. (*)Denotes significant increases from no treatment and enzyme only control. | ||
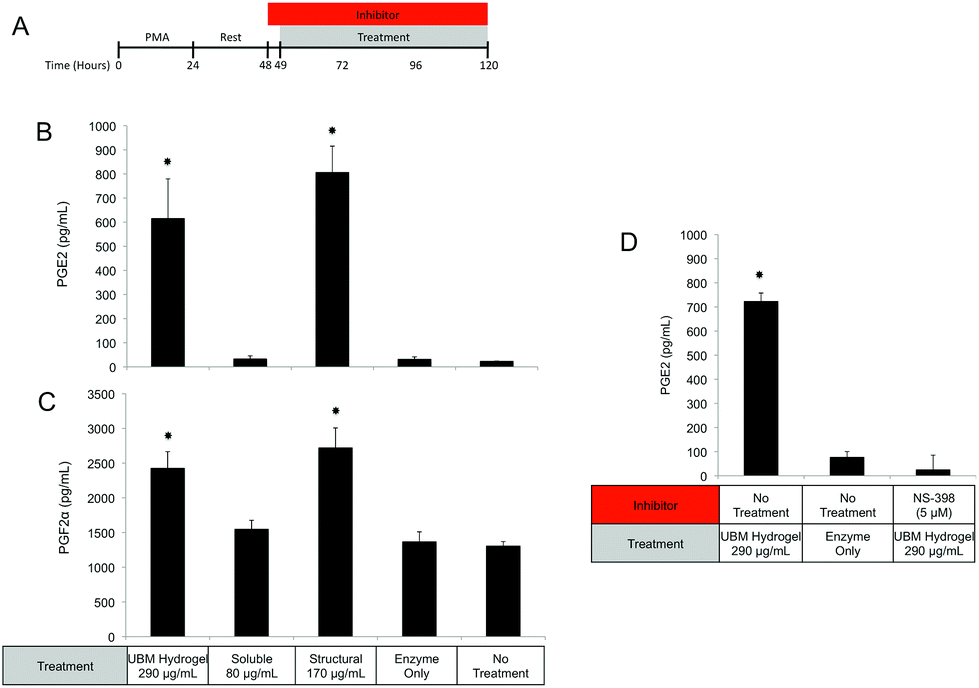 | ||
| Fig. 7 PGE2 and PGF2α secretion after treatment with UBM and its fractionated components. (A) THP-1 human monocytes were differentiated to macrophages with PMA, rested, and treated with the specified conditions for 72 hours. (B, C) The concentrations of PGE2 (B) and PGF2α (C) in culture supernatants were determined using commercially available ELISA kits. (D) The requirement of COX2 in prostaglandin production was investigated by co-administering the COX2 inhibitor, NS-398 along with UBM hydrogel. Significant increases above no treatment, enzyme only control, and soluble components are shown (*). The data presented here represent a total of three replicates with each replicate representing duplicate samples. | ||
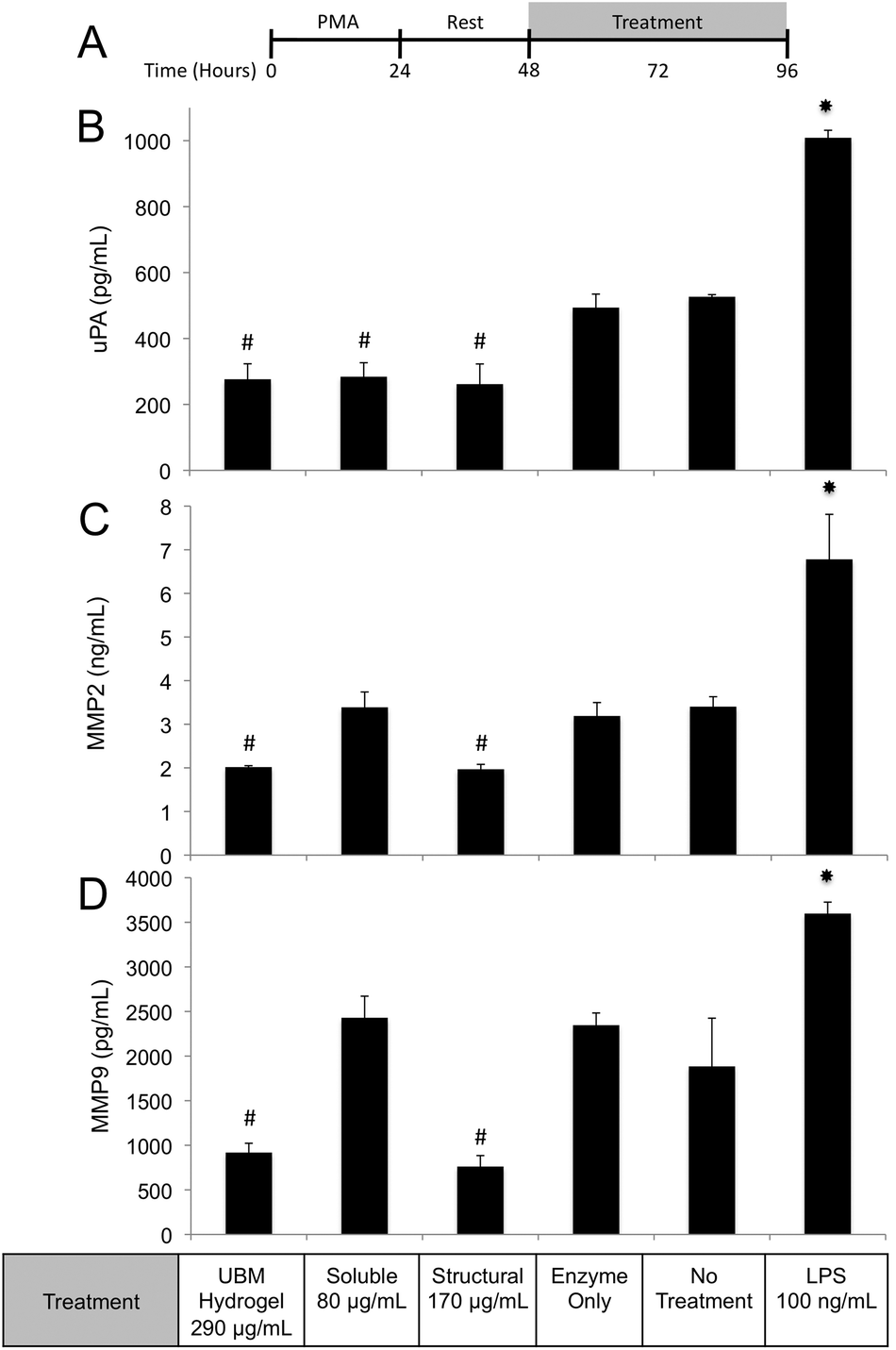 | ||
| Fig. 8 MMP2, MMP9, and uPA secretion after treatment with UBM and its fractionated components. (A) THP-1 human monocytes were differentiated to macrophages with PMA, rested, and treated with the specified conditions for 48 hours. (B, C, D) The concentrations of uPA (B), MMP2 (C) and MMP9 (D) in culture supernatants were determined using commercially available ELISA kits. Significant decreases (#) and increases (*) from no treatment and enzyme only control are shown. The data presented here represent a total of two replicates with each replicate representing three samples. | ||
Interrogation of intracellular signaling cascades and cell surface receptors
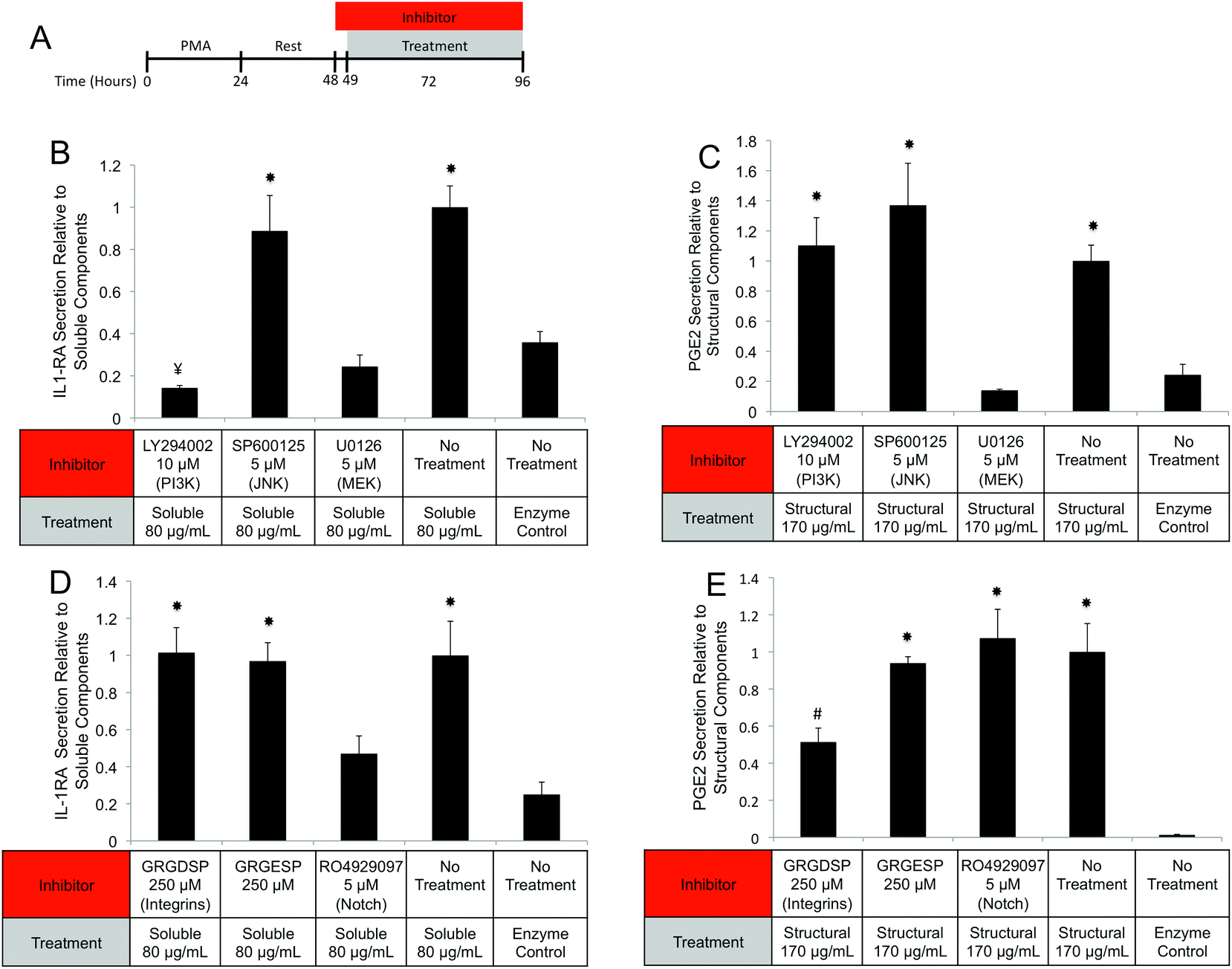 | ||
| Fig. 9 Interrogation of UBM component triggered signaling cascades and receptors. (A) THP-1 human monocytes were differentiated to macrophages with PMA and rested. Macrophages were then pretreated with inhibitors for 1 hour prior to treatment with fractionated components and co-treatments were carried out for 47 hours. The concentrations of IL-1RA and PGE2 in culture supernatants were determined using commercially available ELISA kits. (B, C) Signaling cascade inhibitors for PI3K (LY294002), JNK (SP600125), and MEK (U0126) were tested for their ability to prevent soluble component driven, IL-1RA secretion (B) or structural component driven PGE2 secretion (C). (D, E) Receptor inhibitors for integrins (RGD-active, RGE-negative control) and Notch (RO4929097) were also tested in both assays. (#)Denotes significant increases from no treatment and enzyme only control. (¥)Denotes significant decreases from no treatment and enzyme only control. (*)Denotes significant increases from unmarked, ¥, or # marked groups. The data presented here represent a total of two replicates with each replicate representing three samples. | ||
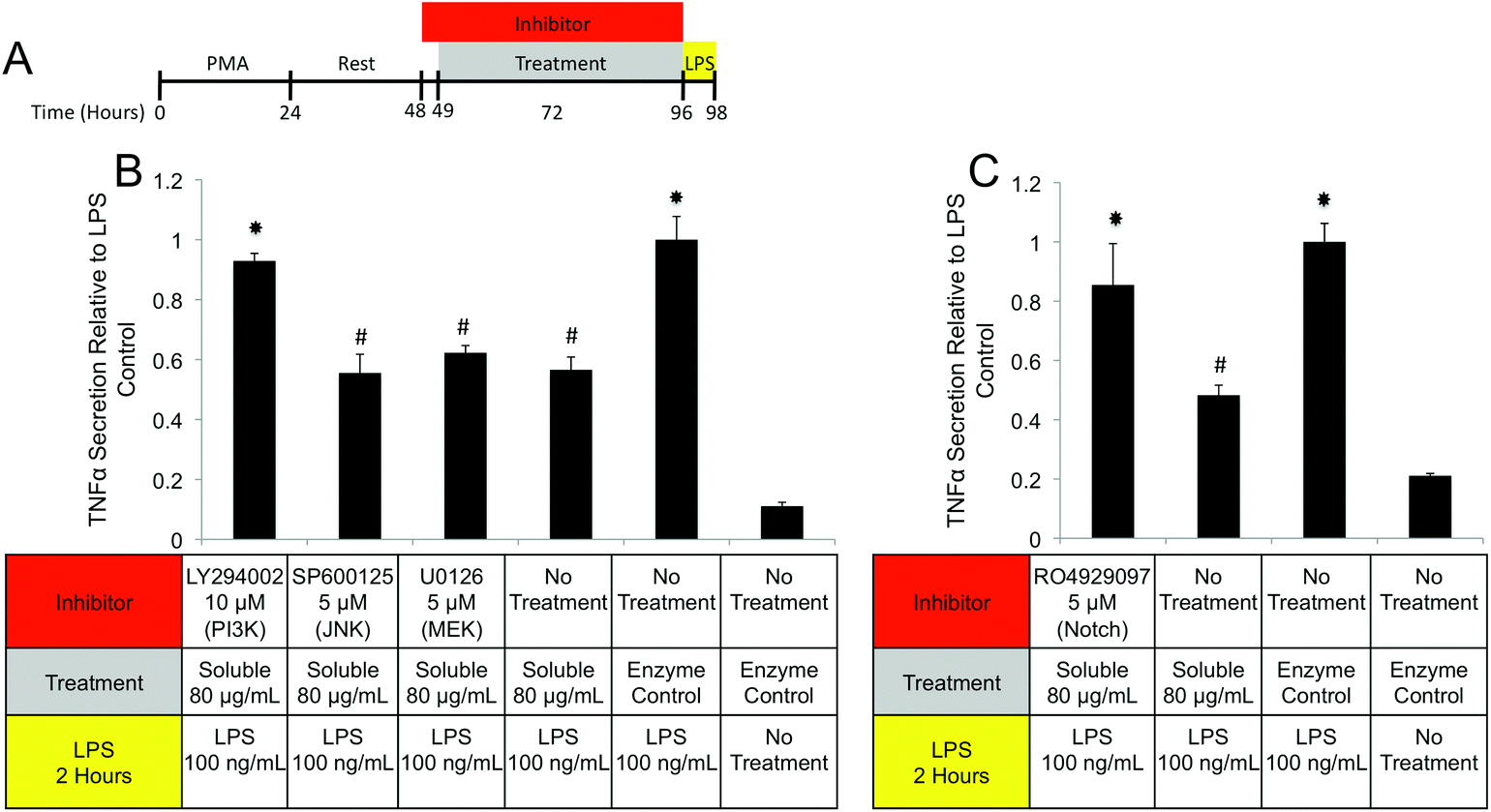 | ||
| Fig. 10 Interrogation of soluble component triggered signaling cascades and receptors. (A) THP-1 human monocytes were differentiated to macrophages with PMA and rested. Macrophages were then pretreated with inhibitors for 1 hour prior to treatment with soluble components and co-treatments were carried out for 47 hours. Cells were challenged with LPS for 2 hours and the concentration of TNFα in culture supernatants was determined using a commercially available ELISA kit. Data are expressed as relative changes from a sample treated with the specified inhibitor for 48 hours plus 2 hours of LPS challenge. (B) Signaling cascade inhibitors for PI3K (LY294002), JNK (SP600125), and MEK (U0126) were tested for their ability to prevent soluble component suppression of TNFα. (C) A Notch inhibitor (RO4929097) was also tested. (#)Denotes significant increases from no treatment control. (*)Denotes significant increases from unmarked and # marked groups. The data presented here represent a total of two replicates with each replicate representing three samples. | ||
Since MEK/ERK signaling has been shown to be highly integrin regulated, the importance of integrin activation in mediating either structural component driven PGE2 secretion or soluble component driven IL-1RA secretion was investigated.69,70 Integrin signaling was suppressed using an RGD peptide (GRGDSP). RGD peptide has been shown to inhibit integrin signaling at high doses (100–300 μM) while a point mutant RGE peptide (GRGESP) has been shown to have minimal effects.71,72 RGD peptide inhibited PGE2 production by 49% (Fig. 9E) but did not inhibit IL-1RA production (Fig. 9D). The control RGE peptide had no effect on either secreted factor.
PI3K/Akt signaling is initiated through a variety of mechanisms, including Notch activation.73 To investigate the role of Notch in mediating IL-1RA secretion and TNFα suppression, the γ-secretase inhibitor RO4929097 was utilized.74 Addition of the inhibitor reduced IL-1RA secretion by 58% (Fig. 9D) and completely prevented TNFα suppression (Fig. 10C). Given the reported toxicity of RO4929097 at high doses, the inhibitor was also tested on structural component driven PGE2 secretion. Importantly, no inhibition was observed suggesting that the toxic affects of the molecule were not prevailing at the utilized dosage (Fig. 9E).
Discussion
While significant progress has been made in the development and implementation of ECM based hydrogels, very little is known about the molecular components of these materials that facilitate the tissue remodeling response. The data presented herein suggest that both the structural components and the soluble components of the hydrogel contribute to the tissue remodeling response by stimulating stem cell chemotaxis, a decrease in stem cell proliferation, and the development of an anti-inflammatory, pro-remodeling response in macrophages. While chemotaxis and proliferation are common features of both the soluble and structural components of the hydrogel, macrophage behavior is differentially regulated with structural components contributing to the production of prostaglandins and suppression of MMPs while soluble components contribute to the suppression of inflammatory mediators as well as an increase in phagocytosis. These distinct macrophage behaviors are mediated by different signaling events with structural components relying upon integrins and MEK/ERK signaling while soluble components utilize Notch and PI3K signaling.The process of fractionation described in this study provides a simple way to fractionate soluble components of ECM hydrogels from structural. However, simply separating these two components only covers part of the story. A variety of soluble components of ECM including matricellular proteins and growth factors bind to the various structural molecules of ECM. Moreover, many of these structurally bound soluble components exhibit significant increases in potency (>100 fold) when structurally bound.33,35,38 It is unclear if the fractionation scheme described herein cleanly separates all soluble and structural molecules or if some soluble molecules are retained by the structural fraction. Attempts to quantify soluble molecules in UBM hydrogel were unsuccessful as two of the major structurally-bound growth factors present in whole UBM, VEGF (0.56 ± 0.03 ng g−1 whole UBM) and bFGF (12.6 ± 0.3 ng g−1 whole UBM) could not be detected in UBM hydrogel. It is likely that the combination of acid and pepsin digestion denatures and proteolyses soluble components leaving behind only cryptic peptides for fractionation. However, in the absence of a metric for quantifying retained soluble components, the present study cannot rule out the possibility that the biologic activity of the structural fraction originates from bound soluble molecules.
Another limitation of the scheme described here is the presentation of the structural fraction as a homogenized slurry. Manipulation of ECM ultrastructure during decellularization has been shown to reduce or eliminate some aspects of ECM bioactivity.40,75 Despite this drawback, many studies have shown that highly processed forms of ECM, including the hydrogel form used in this study retain bioactivity.22,76 Highly purified structural components of ECM (e.g. collagen, heparan, hyaluronan, etc.) also retain bioactivity in the absence of a native ECM ultrastructure.64,77,78 Based on these previous studies, the findings presented herein are relevant to understanding the biological activity of different components of ECM hydrogels. However, the extrapolation of these results to native ECM should only be done cautiously and with an understanding that native ECM structure and mechanics is compromised by fractionation.
Previous studies have established that directed migration of a variety of different endogenous stem and progenitor cells (including PSCs) towards implanted biologic scaffolds is a significant component of the remodeling response.14,79 Once embedded in the scaffold, these cells can either proliferate or slow their proliferation and differentiate to form vasculature, nerves, or other site appropriate tissues.13,30,53 The equivalent biologic activity of the structural and soluble components in mediating PSC chemotaxis and proliferation is consistent with the currently proposed paradigm of ECM scaffold mediated constructive remodeling.28,29,33,38,80 It is well accepted that both matricryptic peptides released from full-length structural ECM proteins during proteolysis as well as matricryptic sites unmasked on the scaffold itself, can drive cellular chemotaxis and proliferation as well as other behaviors such as cell shape and adhesion.28,33,80 While pepsin is undoubtedly a non-physiologically relevant enzyme, generation of bioactive cryptic peptides and sites appears to be a broad phenomenon as many different enzymes and methods (e.g. mechanical strain) have been shown to generate bioactive matricryptins.20,28,49,81 In the context of ECM hydrogel driven tissue remodeling, the biologic activity in both fractions could help control both the intensity and the duration of the host response. Studies on hydrogels have shown that embedded soluble factors diffuse from hydrogels in a controlled fashion that is governed by a variety of hydrogel properties.82,83 In remodeling ECM hydrogels, the soluble components of the hydrogel could diffuse into nearby tissues providing a prolonged burst to drive stem cell recruitment and proliferation. Subsequent to the initial burst, the structural components could provide a longer-term chemotactic and proliferative signal to continue driving the host response. This temporally controlled release could help explain why ECM hydrogels provide a more robust remodeling response than collagen hydrogels alone that may not contain the diversity and quantity of soluble components.84
Regulation of the immune response by ECM scaffolds and hydrogels has been shown to be a determinant factor in the ultimate host remodeling outcome.9,54,55 Studies have shown that the immune response to ECM is tightly regulated both spatially and temporally, and inhibiting the response can lead to poor outcomes.85,86 ECM hydrogels have been proposed to bias macrophages towards an anti-inflammatory, M2 phenotype both in vitro and in vivo.9,15,18 The biologic activity of the structural and soluble components of UBM hydrogel on THP1 macrophages described in this study supports and extends this notion. The suppression of TNFα and IL-1β secretion as well as the upregulation of IL-1RA by the soluble components of UBM hydrogel is consistent with the biologic activity of M2 cytokines (IL-4, IL-10) on macrophages.56,57 Moreover, the THP1 macrophages utilized in this study have been well documented as an inflammatory cell line secreting significantly larger quantities of M1 factors (TNFα, IL-1β) than human peripheral blood mononuclear cells.43–45 In this context, the suppression of inflammation described here is quite striking. Structural components of UBM hydrogel also exhibit M2-like activity by down regulating MMPs and releasing prostaglandins. MMP production has been associated with a classic M1 inflammatory response, while prostaglandin production has been shown to be a necessary component in the development of an M2 phenotype.87–89 Importantly, prostaglandin deficient macrophages do not become fully M2 polarized.88 Moreover, from a tissue remodeling perspective, the secretion of prostaglandins has been shown to alter cell proliferation, phagocytic behavior, and new matrix deposition.60,61 The biologic activity of the structural components is also consistent with previous studies, which have shown that cell shape, growth factor potency, and gene regulation are modulated or enhanced if cells are in contact with structural ECM components (collagen, fibronectin, laminin, etc.).28,33,35 Balancing the prostaglandin promoting activity of the structural components with the immuno-suppressive activity of the soluble components may be important as many disease models involving inflammation and hyperalgesia have shown that low levels of prostaglandins are beneficial while high levels are deleterious.90–93 The increase in phagocytosis mediated by the soluble components of UBM is another example of the balancing act between the structural and soluble components. Increases in phagocytosis are a common feature of M1 macrophages.43 However, M2 macrophages under the correct conditions can also upregulate phagocytosis.94 A recent review on macrophage phenotypes in tissue repair proposed that ECM remodeling could involve several sub-phenotypes (M1, M2a, M2b, M2c) of M1 and M2- macrophages working in concert and adjusting their sub-phenotype in response to environmental cues.59 Separating the biological activity of UBM hydrogel into soluble and structural components may help coordinate the spatial availability of macrophage subphenotypes described in the aforementioned review.
The cell signaling cascades and surface receptors identified in this study also support the notion of a balance between M1 and M2 macrophage phenotypes. Notch has been shown to play a key role in the development of an M2 phenotype.95 In the context of this study, Notch activation could suppress the expression of the PI3K negative regulator PTEN, leading to an overall increase in PI3K signaling.73 PI3K has been well documented in the promotion of an M2 phenotype as well as cell survival and angiogenesis.96,97 Unfortunately, it is unclear if UBM hydrogel contains Notch ligands (along with other cellular debris) as a consequence of decellularization or if the hydrogel stimulates Notch ligand expression on macrophages. Cell–cell contacts between neighboring macrophages cannot be ruled out due to the high seeding densities of macrophages utilized in this study. The role of Integrin activation and MEK/ERK signaling in macrophage activation is less clear. LPS mediated MEK/ERK signaling has been shown to promote a classic M1 pro-inflammatory response.89 However, separate studies have shown that IL-4 induction of T-cells utilizes MEK/ERK signaling to crosstalk with the JAK1/STAT6 pathway, driving an M2 phenotype.98 Unfortunately, studies into STAT6 activation mediated by the structural components of UBM hydrogel did not show any significant quantities of phospho-STAT6 (data not shown). Once again, these results suggest that a variety of macrophage subphenotypes are generated by the structural and soluble components. In addition, these studies underscore the importance of analyzing a variety of surface and secretory markers to describe macrophage phenotype rather than restricting analysis to one or two markers. Further studies are required to identify a minimum subset of markers to quantitatively describe the macrophage population during ECM mediated constructive remodeling. Determination of this subset may provide a framework to enhance the constructive remodeling response to ECM hydrogels by augmenting macrophage phenotype and function.
Conclusions
The structural and soluble components of ECM hydrogels distinctly influence macrophage behavior. Both components promote chemotaxis and changes in proliferation of stem cells as well as changes in macrophage behavior. In the context of macrophage behavior, these materials initiate distinctive cellular responses with structural components driving prostaglandin upregulation and MMP2, MMP9, uPA downregulation. Soluble components promote phagocytosis and suppress the production of pro-inflammatory mediators. Structural components mediate their effects from MEK/ERK signaling while soluble components mediate their effects through PI3K/Akt signaling. These studies provide a framework for a more detailed characterization of the immunomodulatory effects of ECM hydrogels.Acknowledgements
Partial funding for this study was provided by Vertex Pharmaceuticals Incorporated (Cambridge, MA). Partial funding was also provided by grant NIH R03 AG043606. The authors would like to thank Scott Johnson for preparation of UBM, and Lynda Guzik along with McGowan Institute Flow Cytometry Core. (TJK) This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under grant no. DGE-1247842. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the National Science Foundation.References
- N. A. Kissane and K. M. Itani, Plast. Reconstr. Surg., 2012, 130, 194S–202S CrossRef CAS PubMed.
- W. W. Hope, D. Griner, A. Adams, W. B. Hooks and T. V. Clancy, Plast. Surg. Int., 2012, 2012, 918345 Search PubMed.
- E. T. Ricchetti, A. Aurora, J. P. Iannotti and K. A. Derwin, J. Shoulder Elbow Surg., 2012, 21, 251–265 CrossRef PubMed.
- K. A. Derwin, S. F. Badylak, S. P. Steinmann and J. P. Iannotti, J. Shoulder Elbow Surg., 2010, 19, 467–476 CrossRef PubMed.
- M. M. Mofid, M. S. Meininger and M. S. Lacey, Eur. J. Plast. Surg., 2012, 35, 717–722 CrossRef PubMed.
- A. Cheng and M. Saint-Cyr, Clin. Plast. Surg., 2012, 39, 167–175 CrossRef PubMed.
- V. J. Mase Jr, J. R. Hsu, S. E. Wolf, J. C. Wenke, D. G. Baer, J. Owens, S. F. Badylak and T. J. Walters, Orthopedics, 2010, 33, 511 Search PubMed.
- B. M. Sicari, S. A. Johnson, B. F. Siu, P. M. Crapo, K. A. Daly, H. Jiang, C. J. Medberry, S. Tottey, N. J. Turner and S. F. Badylak, Biomaterials, 2012, 33, 5524–5533 CrossRef CAS PubMed.
- B. N. Brown and S. F. Badylak, Acta Biomater., 2012, 9, 4948–4955 CrossRef PubMed.
- V. Agrawal, B. N. Brown, A. J. Beattie, T. W. Gilbert and S. F. Badylak, J. Tissue Eng. Regen. Med., 2009, 3, 590–600 CrossRef CAS PubMed.
- N. J. Turner, J. S. Badylak, D. J. Weber and S. F. Badylak, J. Surg. Res., 2011, 176, 490–502 CrossRef PubMed.
- N. J. Turner, A. J. Yates Jr., D. J. Weber, I. R. Qureshi, D. B. Stolz, T. W. Gilbert and S. F. Badylak, Tissue Eng., Part A, 2010, 16, 3309–3317 CrossRef CAS PubMed.
- E. Vorotnikova, D. McIntosh, A. Dewilde, J. Zhang, J. E. Reing, L. Zhang, K. Cordero, K. Bedelbaeva, D. Gourevitch, E. Heber-Katz, S. F. Badylak and S. J. Braunhut, Matrix Biol., 2010, 29, 690–700 CrossRef CAS PubMed.
- A. J. Beattie, T. W. Gilbert, J. P. Guyot, A. J. Yates and S. F. Badylak, Tissue Eng., Part A, 2009, 15, 1119–1125 CrossRef CAS PubMed.
- B. N. Brown, R. Londono, S. Tottey, L. Zhang, K. A. Kukla, M. T. Wolf, K. A. Daly, J. E. Reing and S. F. Badylak, Acta Biomater., 2012, 8, 978–987 CrossRef CAS PubMed.
- B. N. Brown, J. E. Valentin, A. M. Stewart-Akers, G. P. McCabe and S. F. Badylak, Biomaterials, 2009, 30, 1482–1491 CrossRef CAS PubMed.
- J. E. Valentin, A. M. Stewart-Akers, T. W. Gilbert and S. F. Badylak, Tissue Eng., Part A, 2009, 15, 1687–1694 CrossRef CAS PubMed.
- T. J. Keane, R. Londono, R. M. Carey, C. A. Carruthers, J. E. Reing, C. L. Dearth, A. D'Amore, C. J. Medberry and S. F. Badylak, Biomaterials, 2013, 34, 6729–6737 CrossRef CAS PubMed.
- T. W. Gilbert, D. B. Stolz, F. Biancaniello, A. Simmons-Byrd and S. F. Badylak, Biomaterials, 2005, 26, 1431–1435 CrossRef CAS PubMed.
- D. O. Freytes, J. Martin, S. S. Velankar, A. S. Lee and S. F. Badylak, Biomaterials, 2008, 29, 1630–1637 CrossRef CAS PubMed.
- B. P. Hung, E. K. Salter, J. Temple, G. S. Mundinger, E. N. Brown, P. Brazio, E. D. Rodriguez and W. L. Grayson, Cells Tissues Organs, 2013, 198, 87–98 CrossRef CAS PubMed.
- M. T. Wolf, K. A. Daly, E. P. Brennan-Pierce, S. A. Johnson, C. A. Carruthers, A. D'Amore, S. P. Nagarkar, S. S. Velankar and S. F. Badylak, Biomaterials, 2012, 33, 7028–7038 CrossRef CAS PubMed.
- C. J. Medberry, P. M. Crapo, B. F. Siu, C. A. Carruthers, M. T. Wolf, S. P. Nagarkar, V. Agrawal, K. E. Jones, J. Kelly, S. A. Johnson, S. S. Velankar, S. C. Watkins, M. Modo and S. F. Badylak, Biomaterials, 2012, 34, 1033–1040 CrossRef PubMed.
- E. Bible, F. Dell'Acqua, B. Solanky, A. Balducci, P. M. Crapo, S. F. Badylak, E. T. Ahrens and M. Modo, Biomaterials, 2012, 33, 2858–2871 CrossRef CAS PubMed.
- K. Lampiaho, A. Kari, T. Hollmen, J. Pikkarainen and E. Kulonen, Biochem. J., 1967, 104, 21P CAS.
- C. A. Galea, B. P. Dalrymple, R. Kuypers and R. Blakeley, Protein Sci., 2000, 9, 1947–1959 CrossRef CAS PubMed.
- E. H. Sage and P. Bornstein, J. Biol. Chem., 1991, 266, 14831–14834 CAS.
- G. E. Davis, K. J. Bayless, M. J. Davis and G. A. Meininger, Am. J. Pathol., 2000, 156, 1489–1498 CrossRef CAS.
- P. Bornstein and E. H. Sage, Curr. Opin. Cell Biol., 2002, 14, 608–616 CrossRef CAS.
- S. Tottey, M. Corselli, E. M. Jeffries, R. Londono, B. Peault and S. F. Badylak, Tissue Eng., Part A, 2010, 17, 37–44 CrossRef PubMed.
- D. Finlay, V. Healy, F. Furlong, F. C. O'Connell, N. K. Keon and F. Martin, Cell Death Differ., 2000, 7, 302–313 CrossRef CAS PubMed.
- L. Hernandez, K. J. Roux, E. S. Wong, L. C. Mounkes, R. Mutalif, R. Navasankari, B. Rai, S. Cool, J. W. Jeong, H. Wang, H. S. Lee, S. Kozlov, M. Grunert, T. Keeble, C. M. Jones, M. D. Meta, S. G. Young, I. O. Daar, B. Burke, A. O. Perantoni and C. L. Stewart, Dev. Cell, 2010, 19, 413–425 CrossRef CAS PubMed.
- N. J. Boudreau and P. L. Jones, Biochem. J., 1999, 339(Pt 3), 481–488 CrossRef CAS.
- G. E. Davis, Biochem. Biophys. Res. Commun., 1992, 182, 1025–1031 CrossRef CAS.
- G. S. Schultz and A. Wysocki, Wound Repair Regen., 2009, 17, 153–162 CrossRef PubMed.
- J. Kzhyshkowska, G. Workman, M. Cardo-Vila, W. Arap, R. Pasqualini, A. Gratchev, L. Krusell, S. Goerdt and E. H. Sage, J. Immunol., 2006, 176, 5825–5832 CrossRef CAS.
- A. Mammoto, T. Mammoto, M. Kanapathipillai, C. Wing Yung, E. Jiang, A. Jiang, K. Lofgren, E. P. Gee and D. E. Ingber, Nat. Commun., 2013, 4, 1759 CrossRef PubMed.
- G. E. Davis, J. Mol. Cell. Cardiol., 2010, 48, 454–460 CrossRef CAS PubMed.
- D. O. Freytes, R. S. Tullius and S. F. Badylak, J. Biomed. Mater. Res., Part B, 2006, 78, 327–333 CrossRef PubMed.
- P. M. Crapo, T. W. Gilbert and S. F. Badylak, Biomaterials, 2011, 32, 3233–3243 CrossRef CAS PubMed.
- Z. Deyl and M. Adam, J. Chromatogr., 1989, 488, 161–197 CrossRef CAS.
- Z. Deyl, I. Miksik and A. Eckhardt, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2003, 790, 245–275 CrossRef CAS.
- M. Daigneault, J. A. Preston, H. M. Marriott, M. K. Whyte and D. H. Dockrell, PLoS One, 2010, 5, e8668 Search PubMed.
- S. H. Kwon, S. A. Ju, J. H. Kang, C. S. Kim, H. M. Yoo and R. Yu, Nutr. Res. Pract., 2008, 2, 134–137 CrossRef CAS PubMed.
- N. Shanmugam, Y. S. Kim, L. Lanting and R. Natarajan, J. Biol. Chem., 2003, 278, 34834–34844 CrossRef CAS PubMed.
- M. Crisan, B. Deasy, M. Gavina, B. Zheng, J. Huard, L. Lazzari and B. Peault, Methods Cell Biol., 2008, 86, 295–309 CAS.
- M. Crisan, J. Huard, B. Zheng, B. Sun, S. Yap, A. Logar, J. P. Giacobino, L. Casteilla and B. Peault, Curr. Protoc. Stem Cell Biol., 2008 Search PubMed , Chapter 2, Unit 2B 2 1–2B 2 13.
- M. Crisan, S. Yap, L. Casteilla, C. W. Chen, M. Corselli, T. S. Park, G. Andriolo, B. Sun, B. Zheng, L. Zhang, C. Norotte, P. N. Teng, J. Traas, R. Schugar, B. M. Deasy, S. Badylak, H. J. Buhring, J. P. Giacobino, L. Lazzari, J. Huard and B. Peault, Cell Stem Cell, 2008, 3, 301–313 CrossRef CAS PubMed.
- J. E. Reing, L. Zhang, J. Myers-Irvin, K. E. Cordero, D. O. Freytes, E. Heber-Katz, K. Bedelbaeva, D. McIntosh, A. Dewilde, S. J. Braunhut and S. F. Badylak, Tissue Eng., Part A, 2009, 15, 605–614 CrossRef CAS PubMed.
- A. E. Carpenter, T. R. Jones, M. R. Lamprecht, C. Clarke, I. H. Kang, O. Friman, D. A. Guertin, J. H. Chang, R. A. Lindquist, J. Moffat, P. Golland and D. M. Sabatini, Genome Biol., 2006, 7, R100 CrossRef PubMed.
- J. E. Reing, B. N. Brown, K. A. Daly, J. M. Freund, T. W. Gilbert, S. X. Hsiong, A. Huber, K. E. Kullas, S. Tottey, M. T. Wolf and S. F. Badylak, Biomaterials, 2010, 31, 8626–8633 CrossRef CAS PubMed.
- C. V. Sapan, R. L. Lundblad and N. C. Price, Biotechnol. Appl. Biochem., 1999, 29(Pt 2), 99–108 CAS.
- P. M. Crapo, S. Tottey, P. F. Slivka and S. F. Badylak, Tissue Eng., Part A, 2013, 313–323 Search PubMed.
- B. Chazaud, Immunobiology, 2014, 219, 172–178 CrossRef CAS PubMed.
- B. Deng, M. Wehling-Henricks, S. A. Villalta, Y. Wang and J. G. Tidball, J. Immunol., 2012, 189, 3669–3680 CrossRef CAS PubMed.
- P. H. Hart, R. L. Cooper and J. J. Finlay-Jones, Immunology, 1991, 72, 344–349 CAS.
- P. Wang, P. Wu, M. I. Siegel, R. W. Egan and M. M. Billah, J. Biol. Chem., 1995, 270, 9558–9563 CrossRef CAS PubMed.
- W. F. Fearon and D. T. Fearon, Circulation, 2008, 117, 2577–2579 CrossRef PubMed.
- M. L. Novak and T. J. Koh, J. Leukoc. Biol., 2013, 93, 875–881 CrossRef CAS PubMed.
- S. Rakoff-Nahoum and R. Medzhitov, J. Clin. Invest., 2007, 117, 83–86 CrossRef CAS PubMed.
- D. Van Ly, J. K. Burgess, T. G. Brock, T. H. Lee, J. L. Black and B. G. Oliver, Am. J. Physiol. Lung Cell. Mol. Physiol., 2012, 303, L239–L250 CrossRef CAS PubMed.
- M. S. Pepper, Arterioscler., Thromb., Vasc. Biol., 2001, 21, 1104–1117 CrossRef CAS.
- K. M. Khan, L. R. Howe and D. J. Falcone, J. Biol. Chem., 2004, 279, 22039–22046 CrossRef CAS PubMed.
- B. Fallica, J. S. Maffei, S. Villa, G. Makin and M. Zaman, PLoS One, 2012, 7, e48024 CAS.
- S. M. Maira, Mol. Cancer Ther., 2011, 10, 2016 CrossRef CAS PubMed.
- S. M. Maira, P. Finan and C. Garcia-Echeverria, Curr. Top. Microbiol. Immunol., 2010, 347, 209–239 CAS.
- K. Assi, R. Pillai, A. Gomez-Munoz, D. Owen and B. Salh, Immunology, 2006, 118, 112–121 CrossRef CAS PubMed.
- J. V. Duncia, J. B. Santella 3rd, C. A. Higley, W. J. Pitts, J. Wityak, W. E. Frietze, F. W. Rankin, J. H. Sun, R. A. Earl, A. C. Tabaka, C. A. Teleha, K. F. Blom, M. F. Favata, E. J. Manos, A. J. Daulerio, D. A. Stradley, K. Horiuchi, R. A. Copeland, P. A. Scherle, J. M. Trzaskos, R. L. Magolda, G. L. Trainor, R. R. Wexler, F. W. Hobbs and R. E. Olson, Bioorg. Med. Chem. Lett., 1998, 8, 2839–2844 CrossRef CAS.
- A. E. Aplin, S. A. Stewart, R. K. Assoian and R. L. Juliano, J. Cell Biol., 2001, 153, 273–282 CrossRef CAS.
- D. G. Stupack and D. A. Cheresh, J. Cell Sci., 2002, 115, 3729–3738 CrossRef CAS.
- R. B. Basani, G. D'Andrea, N. Mitra, G. Vilaire, M. Richberg, M. A. Kowalska, J. S. Bennett and M. Poncz, J. Biol. Chem., 2001, 276, 13975–13981 CAS.
- K. R. Gehlsen, W. S. Argraves, M. D. Pierschbacher and E. Ruoslahti, J. Cell Biol., 1988, 106, 925–930 CrossRef CAS.
- W. Bailis and W. S. Pear, Blood, 2012, 120, 1349–1350 CrossRef CAS PubMed.
- C. Groth and M. E. Fortini, Semin. Cell Dev. Biol., 2012, 23, 465–472 CrossRef CAS PubMed.
- D. M. Faulk, C. A. Carruthers, H. J. Warner, C. R. Kramer, J. E. Reing, L. Zhang, A. D'Amore and S. F. Badylak, Acta Biomater., 2014, 10, 183–193 CrossRef CAS PubMed.
- S. B. Seif-Naraghi, J. M. Singelyn, M. A. Salvatore, K. G. Osborn, J. J. Wang, U. Sampat, O. L. Kwan, G. M. Strachan, J. Wong, P. J. Schup-Magoffin, R. L. Braden, K. Bartels, J. A. DeQuach, M. Preul, A. M. Kinsey, A. N. DeMaria, N. Dib and K. L. Christman, Sci. Transl. Med., 2013, 5, 173ra125 Search PubMed.
- S. Franz, F. Allenstein, J. Kajahn, I. Forstreuter, V. Hintze, S. Moller and J. C. Simon, Acta Biomater., 2013, 9, 5621–5629 CrossRef CAS PubMed.
- J. Kajahn, S. Franz, E. Rueckert, I. Forstreuter, V. Hintze, S. Moeller and J. C. Simon, Biomatter, 2012, 2, 226–236 CrossRef PubMed.
- F. Li, W. Li, S. Johnson, D. Ingram, M. Yoder and S. Badylak, Endothelium, 2004, 11, 199–206 CrossRef CAS PubMed.
- V. Agrawal, J. Kelly, S. Tottey, K. A. Daly, S. A. Johnson, B. F. Siu, J. Reing and S. F. Badylak, Tissue Eng., Part A, 2011, 17, 3033–3044 CrossRef CAS PubMed.
- A. E. Carson and T. H. Barker, Regen. Med., 2009, 4, 593–600 CrossRef CAS PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- R. Censi, P. Di Martino, T. Vermonden and W. E. Hennink, J. Controlled Release, 2012, 161, 680–692 CrossRef CAS PubMed.
- M. J. Sawkins, W. Bowen, P. Dhadda, H. Markides, L. E. Sidney, A. J. Taylor, F. R. Rose, S. F. Badylak, K. M. Shakesheff and L. J. White, Acta Biomater., 2013, 9, 7865–7873 CrossRef CAS PubMed.
- J. G. Tidball, Compr. Physiol., 2011, 1, 2029–2062 Search PubMed.
- J. G. Tidball, Am. J. Physiol. Regul., Integr. Comp. Physiol., 2005, 288, R345–R353 CrossRef CAS PubMed.
- Y. Nakanishi, M. Nakatsuji, H. Seno, S. Ishizu, R. Akitake-Kawano, K. Kanda, T. Ueo, H. Komekado, M. Kawada, M. Minami and T. Chiba, Carcinogenesis, 2011, 32, 1333–1339 CrossRef CAS PubMed.
- Y. R. Na, Y. N. Yoon, D. I. Son and S. H. Seok, PLoS One, 2013, 8, e63451 CAS.
- W. C. Huang, G. B. Sala-Newby, A. Susana, J. L. Johnson and A. C. Newby, PLoS One, 2012, 7, e42507 CAS.
- N. E. Lane, J. Rheumatol. Suppl., 1997, 49, 20–24 CAS.
- W. Shen, V. Prisk, Y. Li, W. Foster and J. Huard, J. Appl. Physiol., 2006, 101, 1215–1221 CrossRef CAS PubMed.
- C. Mo, S. Romero-Suarez, L. Bonewald, M. Johnson and M. Brotto, Recent Pat. Biotechnol., 2012, 6, 223–229 CrossRef CAS.
- Y. Kharraz, J. Guerra, C. J. Mann, A. L. Serrano and P. Munoz-Canoves, Mediators Inflamm., 2013, 2013, 491497 CrossRef PubMed.
- A. Coste, M. Dubourdeau, M. D. Linas, S. Cassaing, J. C. Lepert, P. Balard, S. Chalmeton, J. Bernad, C. Orfila, J. P. Seguela and B. Pipy, Immunity, 2003, 19, 329–339 CrossRef CAS.
- Y. C. Wang, F. He, F. Feng, X. W. Liu, G. Y. Dong, H. Y. Qin, X. B. Hu, M. H. Zheng, L. Liang, L. Feng, Y. M. Liang and H. Han, Cancer Res., 2010, 70, 4840–4849 CrossRef CAS PubMed.
- T. Lawrence and G. Natoli, Nat. Rev. Immunol., 2011, 11, 750–761 CrossRef CAS PubMed.
- J. Ma, H. Sawai, N. Ochi, Y. Matsuo, D. Xu, A. Yasuda, H. Takahashi, T. Wakasugi and H. Takeyama, Mol. Cell. Biochem., 2009, 331, 161–171 CrossRef CAS PubMed.
- E. Y. So, J. Oh, J. Y. Jang, J. H. Kim and C. E. Lee, Mol. Immunol., 2007, 44, 3416–3426 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |