
Ligand capture and activation of human platelets at monolayer modified gold surfaces†
Kellie
Adamson
ab,
Elaine
Spain
a,
Una
Prendergast
a,
Robert J.
Forster
a,
Niamh
Moran
b and
Tia E.
Keyes
*a
aSchool of Chemical Sciences, Dublin City University, Dublin 9, Ireland. E-mail: kellie.adamson@dcu.ie; elaine.spain@dcu.ie; una.prendergast@dcu.ie; robert.forster@dcu.ie; tia.keyes@dcu.ie; Tel: +353 1 700 8185
bDepartment of Molecular and Cellular Therapeutics, Royal College of Surgeons in Ireland, Dublin 2, Ireland. E-mail: nmoran@rcsi.ie; Tel: +353 1 402 2153
First published on 13th August 2014
Abstract
Blood platelet adhesion is crucial in dictating haemocompatibility of medical implants and in platelet capture in diagnostics. Understanding the role of platelet activation in dictating platelet adhesion at chemically modified interfaces is important but relatively unexplored. Using scanning electron microscopy and confocal fluorescence microscopy a quantitative assessment of capture of blood platelets at self-assembled monolayers and mixed monolayers (SAMs) on gold as a function of the activation status of the platelets was conducted. Single and mixed monolayers were prepared using thiol-functionalized arginine-glycine-aspartic acid (RGD), C-Ahx-GRGDS (Ahx = aminohexanoic acid linker), thiolated poly(ethylene)glycol (PEG-COOH) and 1-octanethiol. When incubated with suspensions of resting platelets, RGD promoted platelet adhesion compared to bare or alkanethiol modified gold. Increasing the alkanethiol ratio in the deposition solution decreased the extent of platelet adhesion. Platelet adhesion increased approximately 3 fold at PEG-COO- modified surfaces compared to RGD-alone. Platelets adhered to RGD or mixed RGD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :alkane SAM surfaces were found to be captured in their resting state. In contrast, platelets captured at PEG-COO- SAM surfaces were activated by these substrates. The effect of treating platelets with the chemical activators, Mn2+ or DTT or the physiological activator, thrombin, on the capture efficiency and activation at RGD modified surfaces was also investigated. Mn2+ treated platelets presented similar adhesion to untreated platelets, while surprisingly DTT yielded a very significant decrease in platelet adhesion. And, any platelets that were captured, were in a resting state. Thrombin activated platelets were captured with similar efficiencies as untreated platelets. However, the platelets captured were fully activated. The distinction between capture of chemically and physiologically activated platelet is interesting and likely to originate from differences in the conformation of the integrin induced by each process. Finally, platelet adhesion to each surface could be reversed by incubation with a solution of linear or cyclical RGD or PEG-COO- for the RGD and PEGCOO- surfaces respectively. The specificity of platelet removal confirmed that platelet adhesion at RGD surfaces is occurring through integrin–RGD interactions.
:alkane SAM surfaces were found to be captured in their resting state. In contrast, platelets captured at PEG-COO- SAM surfaces were activated by these substrates. The effect of treating platelets with the chemical activators, Mn2+ or DTT or the physiological activator, thrombin, on the capture efficiency and activation at RGD modified surfaces was also investigated. Mn2+ treated platelets presented similar adhesion to untreated platelets, while surprisingly DTT yielded a very significant decrease in platelet adhesion. And, any platelets that were captured, were in a resting state. Thrombin activated platelets were captured with similar efficiencies as untreated platelets. However, the platelets captured were fully activated. The distinction between capture of chemically and physiologically activated platelet is interesting and likely to originate from differences in the conformation of the integrin induced by each process. Finally, platelet adhesion to each surface could be reversed by incubation with a solution of linear or cyclical RGD or PEG-COO- for the RGD and PEGCOO- surfaces respectively. The specificity of platelet removal confirmed that platelet adhesion at RGD surfaces is occurring through integrin–RGD interactions.
Introduction
In medical device or sensing applications, interfacial interactions between exogenous and bio-materials, particularly cells and proteins are crucial in dictating the efficacy and safety of a given medical component. However, controlling these interactions remains an unsolved problem which limits clinical application of many materials.1–4 For example, non-specific adsorption at implantable devices can lead to fibrosis and inflammatory complications and the need for invasive device replacement.5 In the case of blood-contacting materials, understanding and controlling, in particular, platelet interactions with the substrate is critical.6–8 Since platelet adhesion directly or through adsorbed protein on medical implants dictates haemocompatibility, and such adhesion can lead to thrombus formation.9Cell-adhesion studies have been reported across a variety of substrates including for example, glass, polystyrene and gold10–13 wherein, living cells have been shown to remodel contacting surfaces through the excretion of their own ECM proteins to provide a more ideal environment for their adhesion. In some applications this can be an advantage, for example, where re-endothelialization of an arterial wall after injury or implant is required. Self-assembled monolayers (SAMs) have been explored as a means of both promoting and preventing cell adhesion.14,15 They have the advantage of being facile to assemble and the films are usually stable and uniform.16 In applications aimed at cell capture, a particularly attractive approach is to prepare SAMS which incorporate biological ligands capable of mimicking naturally occurring biomolecular interactions.16,17 Such a strategy has been successfully applied for the integrin binding tripeptide arginine-glycine-aspartic acid (RGD), a sequence recognized by 8 out of the 24 known integrin proteins.18–20
Conversely, non-adhesive or non-fouling surfaces have exploited alkane thiols with a variety of terminal groups or poly(ethylene)glycol (PEG) to form inert surfaces which are resistant to non-specific protein or cell adhesion.21–23 While cell adhesion studies on such surfaces have been quite widely studied, blood platelet adhesion interactions with modified surfaces remain comparatively unexplored.24–30 This is in spite of their importance both in dictating the haemocompatibility of a substrate and in the case of their capture, their potential value in diagnostics. The latter in particular, is growing with increasing understanding of the role of platelets, beyond thrombosis, in a variety of disease states including inflammation and cancer.31–34
For blood platelets the RGD sequence is expected to be predominantly recognized by the integrin αIIbβ3. What distinguishes platelets from other studies on cell adhesion by RGD is the capacity of platelets to undergo activation and gross morphological change, this physiological process occurs in the early stages of thrombus formation. We were interested to see if RGD mediated both capture and activation at RGD surfaces and if activation was affected by the presence of a co-adsorbate.
RGD is one of two peptide sequences through which integrin αIIbβ3 recognizes its primary ligand fibrinogen.35,36 The RGD sequence is believed to bind principally at residues 109(118)–171(133) on the β3 chain of the integrin and has been widely applied as a therapeutic target in inhibiting thrombosis.37,38 However, whereas fibrinogen binding to blood platelets requires that they are first in an activated state, the binding of peptidic RGD does not demand this prerequisite.39,40 However, there is evidence that small molecule RGD binding can induce conformational change in αIIbβ3 and lead to platelet activation. For example, pre-incubating resting αIIbβ3 with an RGD peptide and then removing the peptide ligand through dialysis leaves αIIbβ3 in a high-affinity state for fibrinogen.41 Furthermore, integrin–RGD binding at interfaces can be affected by the activation status of the integrin.42 For example, integrin binding to polyacrylonitrile beads conjugated with RGD were found to be sensitive to the activation status of the integrin depending on the spacer length between RGD and the beads.43 A Gly-Gly-Gly linker between the beads and RGD peptide presented selective binding of high-affinity integrins, while longer linkers resulted in non-discriminative binding. Whilst for whole platelets RGD peptide ligand mediated binding to both resting and activated platelets.44,45
This contribution addresses the effect of surface chemistry on platelet adhesion, morphology and activation status across surfaces designed to be attractive or repellent to platelets. SAMs on gold prepared with thiolated RGD peptide sequence intended to capture platelet, were compared with hydrophilic anionic thiolated polyethylene glycol and hydrophobic thiolated alkane substrates intended to reduce platelet adhesion. The effect of mixing such monolayers on platelet adhesion and morphology are quantitatively compared. In particular, we were interested to see if changing the surface chemistry promoted the platelet from the resting to activated state, affected adhesion to the RGD surfaces. And, whether, platelets nominally activated through treatment with Mn2+ or DTT, or with the physiological activation via thrombin, showed inherent differences in adhesion as such distinctions might form the basis of potential screening and also yield insights into the status of the integrin at the platelet surface on such activation.46–52
Confocal fluorescence microscopy and scanning electron microscopy (SEM) were used to study platelet adhesion and spreading across the modified gold surfaces as a function of platelets activation status. CD62P (α-granule) and phalloidin (actin) staining was carried out to assess platelet activation status from the extent of α-granule and actin organization of platelets adhered to the various surfaces. Results suggest that the activation status and actin content of platelets differed depending on the surface chemistry employed. Surprisingly, platelets activated via chemical stimuli (Mn2+ or DTT) presented dramatically reduced platelet capture by RGD surfaces, whereas thrombin activated platelets showed high levels of platelet capture comparable to untreated platelets captured by RGD surfaces, but thrombin treated platelets were captured fully activated adhesion.
Materials and methods
Materials
Silicon wafers coated with a 525 μm thick layer of gold over a 50 Å titanium adhesion layer were purchased from Amsbio, USA. Aqueous gold plating solution was purchased from Technic Ink, UK. 1-Octanethiol (C8H18S, 95%), ethanol (99%), 38% paraformaldehyde (PFA), tetramethylrhodamine B isothiocyanate (TRITC)-phalloidin (λex 540 nm), thrombin from human plasma (100 U ml−1) and fluoroshield mounting media were purchased from Sigma Aldrich, Ireland. C-Ahx-GRGDS (C26H47N10O11S) peptide was purchased from Celtek Peptides, USA. Eptifibatide, (N6-(aminoiminomethyl)-N2-(3-mercapto-1-oxopropyl-L-lysylglycyl-L-αaspartyl-L-tryptophanyl-L-prolyl cysteinamide, cyclic (1–6)-disulfide), was purchased from Peptides International, USA. Peptide structure and purity was manufacturer guaranteed, presented in ESI (Fig. S1†). SH-PEG4-COOH (C11H22O6S) was purchased from Quanta Biodesign Ltd, USA. Phycoerythrin (PE) labelled mouse anti-human CD62P (λex 488 nm) was purchased from BD Biosciences, USA.Fabrication of planar gold surfaces
The gold modified silicon wafer was washed thoroughly with water and acetone before use. Teflon tape was then wrapped around half of the gold to produce a 1 cm2 final film surface area. Electrolyte solutions were degassed with nitrogen for 30 minutes prior to deposition. A gold film of 100 ± 30 nm in thickness was electrochemically deposited using from aqueous gold plating solution by applying a potential of −1 V (scan rate 100 mV s−1) versus an Ag/AgCl (sat. KCl) electrode using a Model 660 CH Instrument electrochemical workstation.Surface modification: thiolated RGD, alkane or PEG
Three thiolated species were used in this study; the platelet integrin recognizing peptide (SH)C-Ahx-GRGDS (C26H46N10O11 SH), 1-octanethiol (C8H17SH) or SH-PEG4-COOH (C11H21O6 SH). In each case, the gold substrate was immersed in a 1 μM ethanol solution of thiolated C-Ahx-GRGDS, alkane or PEG for 24 hours at room temperature. Alternatively, mixed monolayers were prepared by immersing the substrates in ethanolic solutions of RGD:PEG or RGD:alkane at ratios of 1:1, 1:5 or 1:10 for 24 hours at room temperature. The RGD concentration for SAM surface formation was maintained at 1 μM in the case of the mixed monolayers. Accepting that the ratio of components in the mixed SAMs are unlikely to match the deposition solutions, for simplicity when discussing the SAMs below we describe them in terms of their deposition solution ratios.
Electrochemical analysis of modified surfaces
Electrochemical desorption of the thiols at the chemically modified gold surfaces was carried out with the modified gold electrode in contact with a 0.1 M solution of sulphuric acid (H2SO4). Cyclic voltammetry of both bare gold and gold functionalized with RGD, alkane or PEG were carried out, cycled between −1.4 V and 1.5 V using a scan rate of 0.1 (V s−1) on the CH Instrument Model 660 work station.Preparation of washed human platelets
The use of human blood was approved by the Royal College of Surgeons in Ireland Research Ethics Committee and was collected from donors at the RCSI with their informed consent. Human blood was drawn into a syringe containing acid citrate dextrose (ACD) buffer to prevent coagulation. Modified HEPES platelet buffer (6 mM glucose dextrose D, 0.13 M NaCl, 9 mM NaHCO3, 10 mM sodium citrate, 10 mM HEPES, 3 mM KCl, 0.81 mM KH2PO4, 0.9 mM MgCl2·6H2O) was prepared and kept at room temperature. The pH was adjusted to 7.36 with acid-citrate dextrose (ACD) buffer. 5 ml of blood was transferred to a 15 ml tube and centrifuged at 150g for 10 minutes at room temperature to separate the plasma from the red and white blood cells. The PRP (Platelet Rich Plasma) upper phase was transferred to a 50 ml tube. Prostaglandin (PGE1, 1 μM concentration) was added to prevent platelet activation. The platelets were collected by centrifugation at 720g for 10 minutes. Using 500 μl platelet buffer the platelet layer was carefully removed and placed in a 15 ml tube. Platelet counts were measured using a Sysmex XE-2100 haematology analyser (Sysmex KX Series, SYSMEX UK LTD). Platelets were allowed sit for 1 hour and 1.8 mM CaCl2 was added to the platelets immediately before use.Platelet capture to modified planar gold surfaces
The platelets were diluted to 30 (±2) × 103 μl−1 platelet concentration in platelet buffer (6 mM glucose dextrose D, 0.13 M NaCl, 9 mM NaHCO3, 10 mM sodium citrate, 10 mM HEPES, 3 mM KCl, 0.81 mM KH2PO4, 0.9 mM MgCl2·6H2O). The substrate surface area, platelet volume, platelet concentration, incubation time and temperature were identical for all modified surfaces: 1 cm2, 150 μl volume of 30 (±2) × 103 μl−1 washed platelets (4.5 (±3) × 106 total platelets) incubated for 45 minutes at 37 °C respectively. It is important to note that all studies were reproducible and are representative of N = 3.Untreated platelets were incubated with each modified gold substrate at 37 °C for 45 minutes. For platelet integrin activation studies, platelets were activated according to literature methods.46,53–57 Platelets were incubated with 1 mM of Mn2+ or DTT either 15 minutes or for 2 hours at room temperature or 1 U ml−1 thrombin for 10 minutes at room temperature prior to their incubation with the modified surfaces at 37 °C for 45 minutes. To investigate reversibility of platelet adhesion, after platelets had bound to the RGD-alone modified surfaces, bound platelets were incubated with a 1 mM final concentration of RGD, PEG or the cyclic RGD peptide drug, eptifibatide for 15 minutes at 37 °C. All surfaces were washed at least three times with PBS, pH 7.4 prior to their preparation for SEM or confocal fluorescence microscopy analysis.
Platelet dehydration
Bound platelets were fixed with 2.5% glutaraldehyde for 3 hours at room temperature. To dehydrate the adherent platelets for SEM imaging, samples were incubated in 5%, 10%, 20%, 40%, 60%, 80% and 100% acetone for 15–20 minutes at a time. Samples were allowed to air dry for 2 hours before sputter coating.Scanning electron microscopy analysis
The dehydrated platelet samples were mounted onto carbon surfaces adhered to aluminium SEM stubs. Gold films were sputter coated over the platelet bound arrays using argon flow, at pressures between 3 and 4 × 10−1 mBar for 2 minutes at a voltage of approximately 40 mA resulting in a gold layer of approximately 30 nm thickness. Gold coated samples were imaged using a Hitachi S3400n SEM Tungsten system instrument. All images were collected under identical conditions at 5.00 kV accelerating voltage unless otherwise stated.Confocal fluorescence microscopy analysis
Surface bound platelets were incubated with phycoerythrin (PE) labelled CD62P (PE-CD62P, 2 mg ml−1 stock, 1/100 dilution) for 15 minutes at 37 °C. Following incubation, CD62P stained platelets were fixed with 3.8% PFA solution for 10 minutes at room temperature for confocal studies. For tetramethyl rhodamine-5-(and 6)-isothiocyanate-phalloidin staining (TRITC-phalloidin, 2 mg ml−1 stock), PFA fixed platelets were permeabilized with 0.01% triton solution for 30 minutes at room temperature. TRITC-phalloidin (1/100 dilution) was added to the surface bound platelets and incubated for 30 minutes at room temperature. All confocal samples were mounted using fluoroshield mounting media. Luminescence images were recorded on a Zeiss LSM510 Meta confocal microscope using a 64× oil immersion objective lens (NA 1.4) unless otherwise stated. A 488 nm argon ion laser was used for PE-CD62P and 540 nm HeNe laser excitation used for TRITC-phalloidin imaging. All images for quantitative platelet adhesion measurements covered an area of 200 × 200 μm. Quantitative platelet adhesion measurements, measuring the number of platelets cm−2 were carried out using ImageJ software (http://rsb.info.nih.gov/ij/).Results and discussion
Characterization of the modified gold surfaces
The SAMs were prepared on gold as it is widely used in medical devices and sensors due to its chemical stability, bio-inertness, high conductivity and its well characterized gold–thiol chemistry.58–63 The gold substrates employed comprised electrochemically deposited gold ∼100 nm thick on a silicon wafer substrate. The substrate was characterized prior to self-assembly of the monolayer using SEM and atomic force microscopy (AFM) (Fig. S2†), and was found to have an average root mean square (RMS) roughness of ∼12 ± 0.3 nm. The effect of surface modification on surface hydrophilicity was assessed by contact angle goniometry (Fig. S3†). The bare gold surface was modestly hydrophobic reflected in a static water contact angle of 61 ± 1°. Following 1-octanethiol SAM formation, the water contact angle increased to 92 ± 2°. This value is consistent with previous contact angles reported for tightly packed alkane thiol on gold.64 SH-PEG4-COOH and C-Ahx-GRGDS SAMs rendered the substrate hydrophilic presenting water contact angles of 21 ± 3° and 14 ± 0.9° respectively. In contrast, mixed deposition SAM solutions of thiolated RGD:alkane with ratios of 1:1, 1:5 and 1:10 exhibited water contact angles of 85 ± 5°, 95 ± 2° and 99 ± 3° respectively. The difference between RGD-alone on gold, with their ostensibly hydrophilic surfaces, and the mixed RGD alkane surface which is relatively much more hydrophobic may be attributed in the case of the peptide alone to a relatively disordered layer which is well hydrated. The film packing is tighter in the case of the alkane thiol containing films making them relatively more hydrophobic. This is consistent with the changes to the electroactive area of the gold with these SAMs described below. In contrast, the RGD:PEG SAM deposited at 1:1, 1:5 and 1:10 ratios were all hydrophilic with contact angles of 12 ± 1° and 12 ± 0.4° and 14 ± 1° respectively.
Voltammetry was performed to confirm SAM formation on the planar gold surfaces (Fig. S4†). The area under the gold oxide reduction peak at +0.7 V decreased by approximately 85% following alkane monolayer deposition corresponding to a reduction in the real electroactive area from 6.8 × 10−2 cm2 to 9.9 × 10−3 cm2 after modification. A comparable decrease was observed for the 1:1, 1:5 and 1:10 RGD:alkane ratios suggesting that a low defect density monolayer is formed irrespective of the composition once the alkane thiol is used in the deposition solution.
Effect of surface modification on human platelet adhesion and morphology
Platelets, in their resting (non-thrombotic) state maintain a smooth discoid shape, see Fig. 1(i). In contrast, activation triggers significant morphological change that can be characterized by 4 stages; dendritic (D), spread-dendritic (SD), spread (S) and fully spread (FS) and depending on the conditions may also include the formation of platelet aggregates. Fig. 1(i) and (ii) shows representative SEM images of platelets captured on the 1:1 RGD:alkane substrate at different stages of spreading. The SEM images presented in Fig. 2 demonstrate that the composition of the capture surface significantly affects the capture efficiency and the activation state of the captured platelets. This effect was investigated using confocal microscopy by staining the platelets with TRITC-phalloidin and measuring the number of adhered platelets using Image J software. Significantly, the number of captured platelets and their extent of activation measured from SEM and confocal imaging were indistinguishable indicating that preparation for SEM did not change the platelet morphology. A summary bar graph of platelet counts cm−2versus modified surface/platelet treatment is presented in Fig. 3. Percentage platelet adhesion is expressed in terms of the percentage of bound platelets compared to the 4500000 platelets incubated with the substrate.
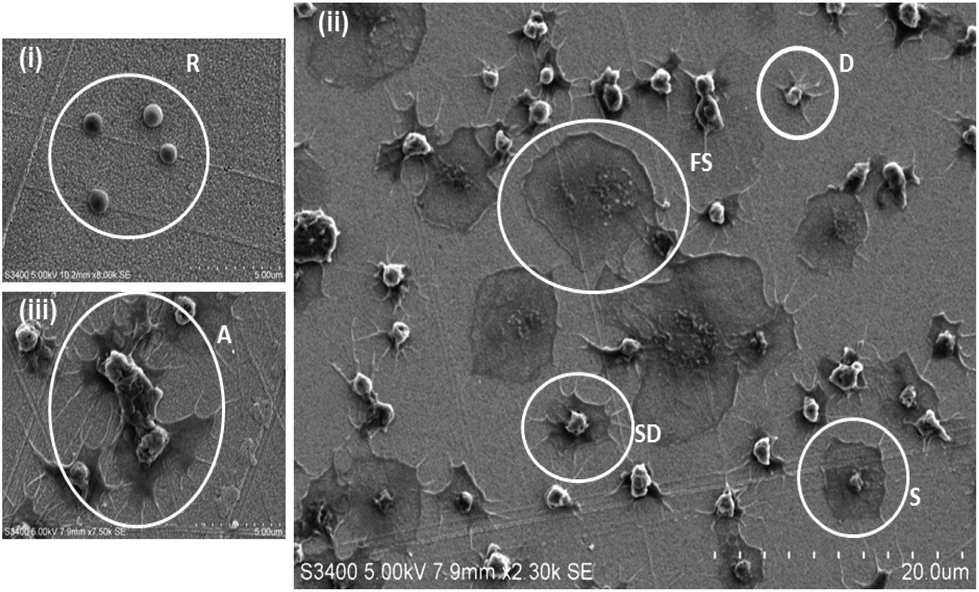 | ||
| Fig. 1 Representative SEM images of platelet adhesion to planar gold surfaces modified with a (i) hydrophobic alkane and (ii & iii) a moderately hydrophilic RGD:alkane mixed monolayer presenting the 4 stages of platelet spreading; (i) R – resting; (ii) D – dendritic; SD – spread dendritic; S – spread; FS – fully-spread and (iii) a platelet aggregate is labelled as A. 4.5 × 106 washed platelets were incubated with the self-assembled monolayer modified planar gold surfaces for 45 minutes at 37 °C. Bound platelets were dehydrated, sputter coated and imaged using 5.00 kV accelerating voltage in all cases. | ||
 | ||
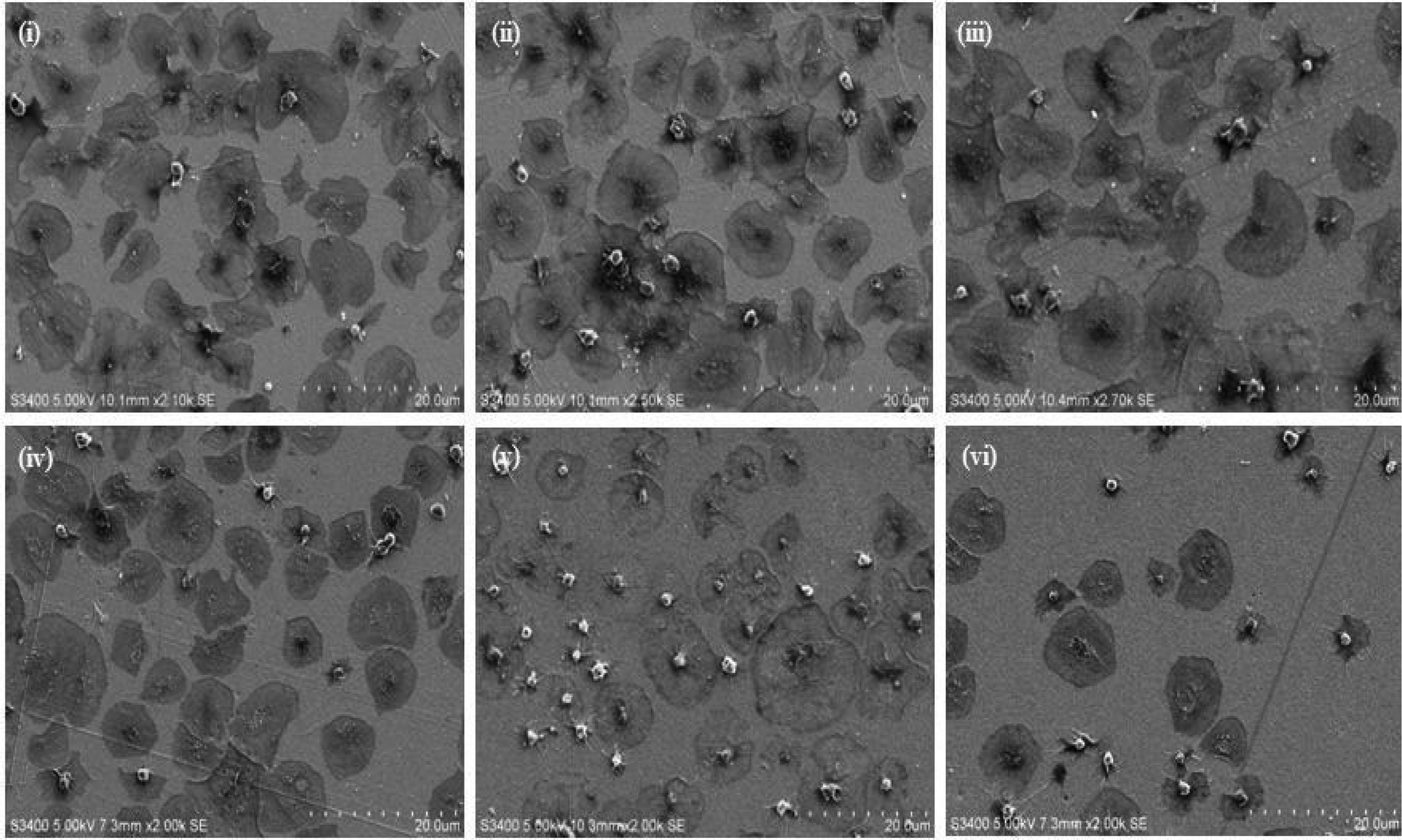
| Fig. 2 SEM images of platelets adhered to (i) an unmodified (bare) planar gold surface and gold surfaces modified with (ii) alkane alone, (iii) RGD-alone and (iv) PEG-COO-alone. (v) & (vi) Magnified images illustrating the minimal spreading of platelets bound to an (v) RGD modified planar surface and full spreading of platelets bound to a (vi) PEG-COO-modified planar surface. 4.5 × 106 platelets were incubated with the modified surfaces for 45 minutes at 37 °C. Following platelet fixation, dehydration and sputter coating, images were recorded using 5.00 kV accelerating voltage. All images are reproducible, representative over N = 3. | ||
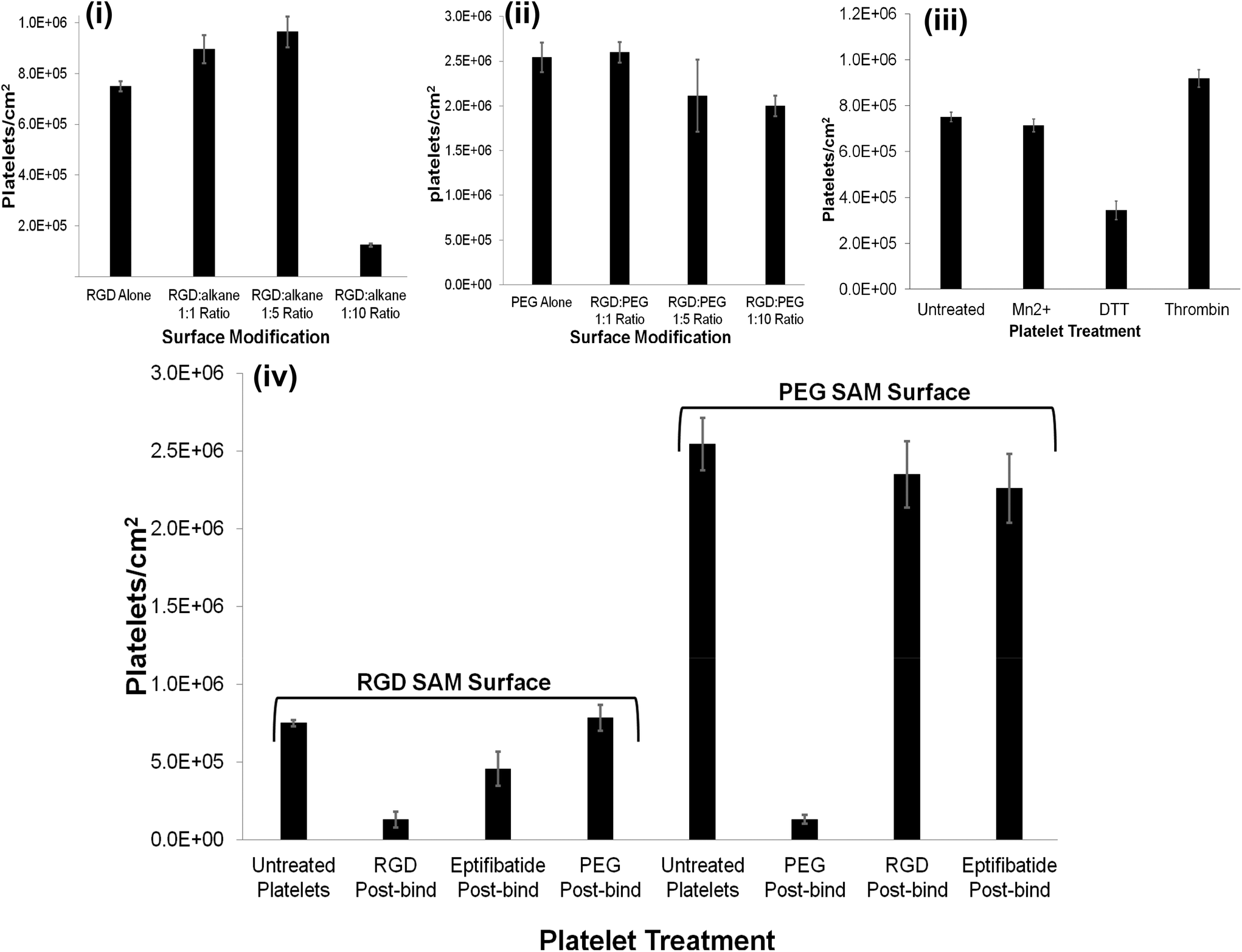 | ||
| Fig. 3 Bar graphs presenting platelet count cm−2versus surface modification: (i) RGD-alone and RGD:alkane 1:1.1:5, 1:10 ratios. (ii) PEG-COO-alone and RGD:PEG 1:1, 1:5 and 1:10 ratios. Bar graphs presenting platelet count cm−2versus platelet treatment: (iii) untreated platelets and platelets incubated with a 1 mM final concentration of Mn2+ or DTT for 2 hours or 1 U ml−1 thrombin for 10 minutes at room temperature. (iv) Untreated platelets and platelets incubated post surface binding with a 1 mM final concentration of PEG-COO-, RGD or the RGD containing drug, eptifibatide for RGD or PEG-COO- SAM surfaces. RGD peptide concentration for SAM formation was 1 μM in all ratio cases. 4.5 × 106 platelets were incubated with the modified surfaces for 45 minutes at 37 °C. Representative of N = 3. | ||
Both (i) unmodified (bare) gold and (ii) alkane thiol only controls presented minimal platelet adhesion; 21666 ± 2886 and 22500 ± 11456 platelets cm−2 (0.5(±0.1)% and 0.5(±0.3)%), respectively and any platelets that did bind were resting, reflected in their spherical shape and little or no evidence for pseudopodia formation.
The RGD-only modified gold showed enhanced platelet adhesion, with approximately 17 (±0.6)% of incident platelets retained at the surface. The morphology of the captured platelets were relatively uniform with the majority in the early stage dendritic spreading following 45 min incubation, Fig. 2(v). The relatively high numbers of bound platelets indicates the RGD is acting as a generic cell adhesion ligand.22,65–68
The extent to which RGD and alkane thiol phase separate in the monolayers is not known but Fig. 3 suggests that the capture efficiency does depend on the RGD:alkane thiol ratio. This dependence likely reflects an increase in accessibility of the individual RGD binding sites when diluted with alkane thiol.
However, for the 1:10 RGD:alkane thiol capture surface the capture efficiency is significantly lower than the RGD-alone reflecting saturation of the relatively small total number of binding sites available.
Strikingly, as shown in Fig. 3(ii), the gold surface modified with PEG-COO- alone exhibited the highest platelet adhesion at nearly 60%. In contrast to the RGD-only surface the majority of platelets bound to the PEG-COO- substrate were activated (Fig. 5(ii)) and fully spread as shown in Fig. 2(iv & vi). PEG is widely applied to surfaces to prevent adhesion of proteins or cells. However, the biocompatibility of PEG has been reported to depend greatly on PEG chain length, density and even on the method of SAM formation.59,69,70 For example, human fibroblasts exhibit higher spreading when immobilized at NH2 and COOH terminated surfaces compared to CH3 or OH surfaces, mid-range surface wettability.10 The key driver of adhesion and activation here is likely to be the carboxyl terminus on the PEG.
At physiological pH used here, this surface is expected to be anionic due to deprotonation of the carboxyl termini of the PEG thiols. Such hydrophilic, negatively charged surfaces are known to induce platelet activation. Although often cited to occur due to contact activation of factor XII, the mechanism remains the topic of discussion.71–74
As shown in Fig. 3 and 4(ii and iv), all RGD:PEG ratios showed platelet adhesion comparable to PEG-COO- alone and was accompanied full platelet spreading in all cases: (RGD:PEG 1:1 ratio, 58 ± 2%; RGD:PEG 1:5 ratio, 47 ± 9% and RGD:PEG 1:10 ratio, 45 ± 3%, respectively). This observation suggests that the anionic charge on the PEG predominantly influences adhesion. This was further confirmed by examining the effect of an RGD competitor on platelet adhesion to the PEGylated surfaces vide infra.
 | ||
| Fig. 4 SEM images of gold surfaces modified with RGD:PEG (i) 1:1, (ii) 1:5, (iii) 1:10 ratios and RGD:alkane (iv) 1:1. (v) 1:5 and (vi) 1:10 ratios. RGD peptide concentration for SAM formation was 1 μM in all ratio cases. 4.5 × 106 platelets were incubated with the modified surfaces for 45 minutes at 37 °C. Following platelet fixation, dehydration and sputter coating, images were recorded using 5.00 kV accelerating voltage. All images are reproducible, representative of N = 3. | ||
Effect of surface modification on platelet activation
Selective actin and P-selectin staining was conducted to assess the activation status of the surface bound platelets. Actin exists in two forms: monomeric globules referred to as G-actin and polymeric filaments called F-actin. F-actin is vital for the morphological changes that accompany platelet activation. F-actin content increases from approximately 40% in resting platelets to 50 to 80% in activated platelets.75 Distinct actin filament structures can be distinguished in activated platelets: filopodia, lamellipodia, a contractile ring encircling degranulating granules and thick bundles of filaments resembling stress fibres are typical structural features. Examples of such structures are presented in the ESI (Fig. S5†).76 P-selectin is a 140 kDa transmembrane glycoprotein also referred to as CD62P, which is stored in α-granules in resting platelets. CD62P is rapidly directed to the platelet surface and may be excreted upon activation and so is a useful marker for platelet activation. An example of p-selectin transport in activated platelets is presented in ESI (Fig. S6†).
Fig. 5 shows representative confocal fluorescence images of washed platelets captured at gold surfaces modified with (i) RGD-only and (ii) PEG-COO-only. Consistent with the SEM, both unmodified (bare) gold and alkane thiol modified gold showed little platelet adhesion (Fig. S7†). Platelets captured at the RGD only surface presented concentrated CD62P and phalloidin staining at the centre of the bound platelets indicating they are captured in their resting state, Fig. 5(i). In contrast, PEG-COO- adhesion presented full platelet spreading and activation with CD62P diffusion and relocation towards the periphery of the platelets, Fig. 5(ii). Correspondingly, phalloidin staining indicated significant actin reorganization in the bound platelet with the formation of actin stress fibres. In the early stages of spreading CD62P and phalloidin co-localize strongly as seen at the RGD-only surfaces but once spreading/activation occurs significant differences can be observed between both labelling agents for the PEG-COO- surfaces suggesting advanced activation. The effect of surface composition, i.e., RGD:alkane 1:1, 1:5 and 1:10 were similar results to RGD-alone (Fig. S8†) with CD62P and phalloidin staining remaining centrally located suggesting platelets are in the resting state. Equally, the surface composition does not alter the general state of activation of the platelets on PEG-COO-containing surfaces (Fig. S9†). Significant CD62P diffusion towards the periphery of the platelets was observed with phalloidin staining presenting intense longitudinal actin stress fibre formation. An RGD:PEG 1:10 ratio presented advanced CD62P relocation with a distinct increase in CD62P luminescence intensity at the platelet membrane in addition to actin stress fibre formation, all consistent with platelets in their activated state.
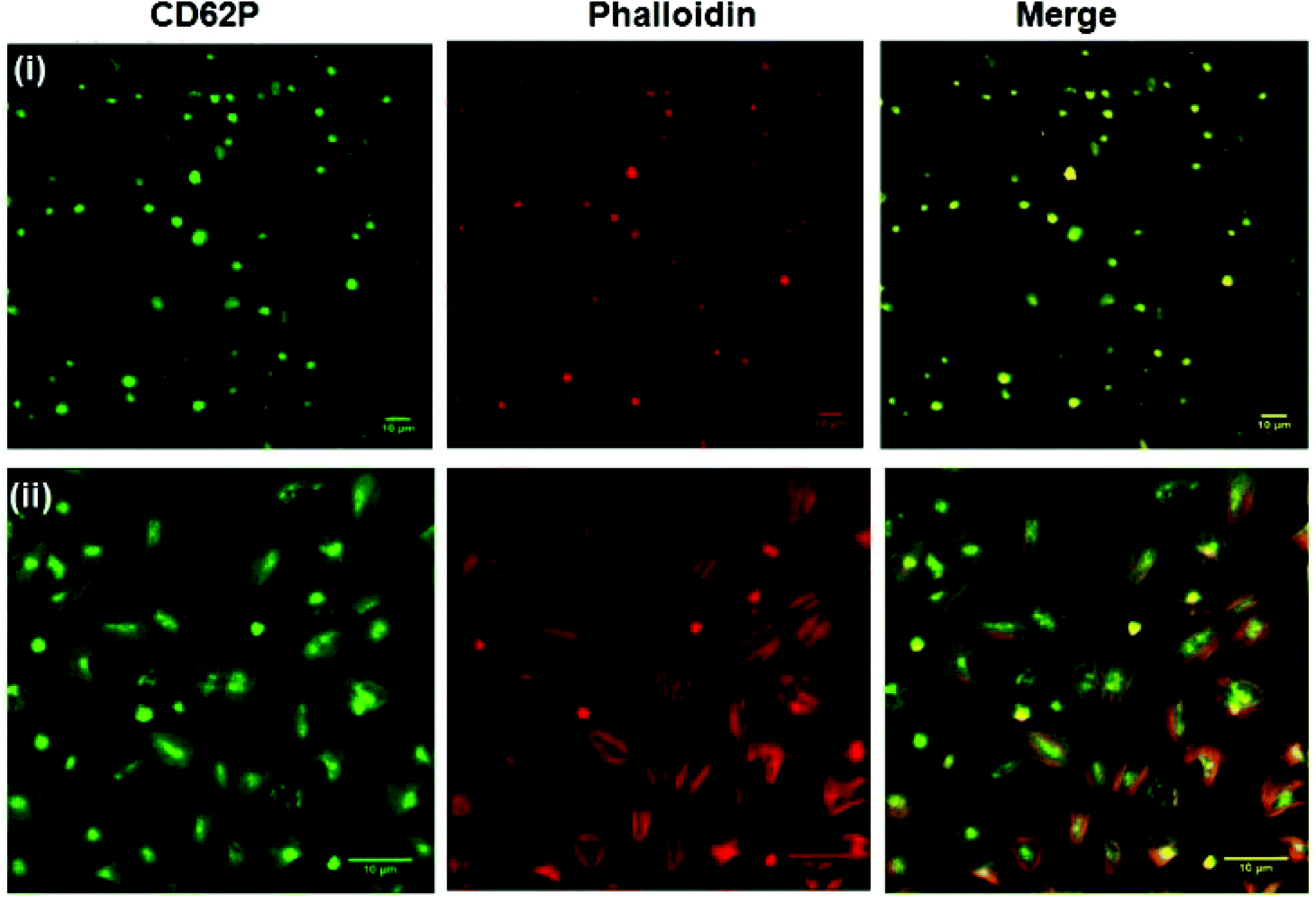 | ||
| Fig. 5 Confocal luminescence images of platelets bound to planar gold surfaces modified with (i) RGD-alone and (ii) PEG-COO- alone SAM surfaces and stained for PE-CD62P (green) and TRITC-phalloidin (red). 30 × 103 ± 2 × 103 μl−1 platelets were incubated with the modified surfaces for 45 minutes at 37 °C. Bound platelets were stained for CD62P (1/100 dilution) and phalloidin (1/100 dilution). Luminescence images were recorded using a 40× oil immersion objective lens (NA 1.4) with 488 nm argon (CD62P) and 540 nm (phalloidin) HeNe laser excitation. All images are reproducible, representative of N = 3. | ||
Integrin αIIbβ3 activation by Mn2+, DTT and whole platelet activation by thrombin yield different effects on platelet adhesion, morphology and activation status
Although “inside-out” platelet integrin αIIbβ3 activation in vivo is regulated by agonist-driven signalling, i.e. thrombin, platelet integrin αIIbβ3 can also be activated by a range of chemical stimuli including Mn2+ and DTT. These chemical stimuli have been shown to induce conformational changes in the integrin resulting in active-like states of the protein in free solution.46–49In vitro Mn2+ and DTT have been shown to increase integrin expressing cell and platelet adhesion to surfaces modified with physiological extra-cellular membrane (ECM) ligands fibrinogen and fibronectin (both contain RGD sequences).50,51 Given that RGD binding is expected to occur at the integrin RGD recognition sites whereas the PEG-COO- binding is expected to be mediated through a different mechanism, we were interested to compare the effect chemical and physiologic activation of the platelet on the recruitment of platelets to the different surfaces as a function of activator identity.Fig. 6(i) and (ii) show the effect of incubating 4.5 × 106 platelets with 1 mM Mn2+ or DTT for 2 hours at room temperature prior to their incubation with the RGD-only modified surface. 1 mM concentrations of activator were used as this is consistent with reported values required to induce integrin conformational change in the platelet.46,53–57,77 It is also important to note that shorter incubation times of 15 minutes with Mn2+ or DTT were also carried out with the same effect (Fig. S11†).
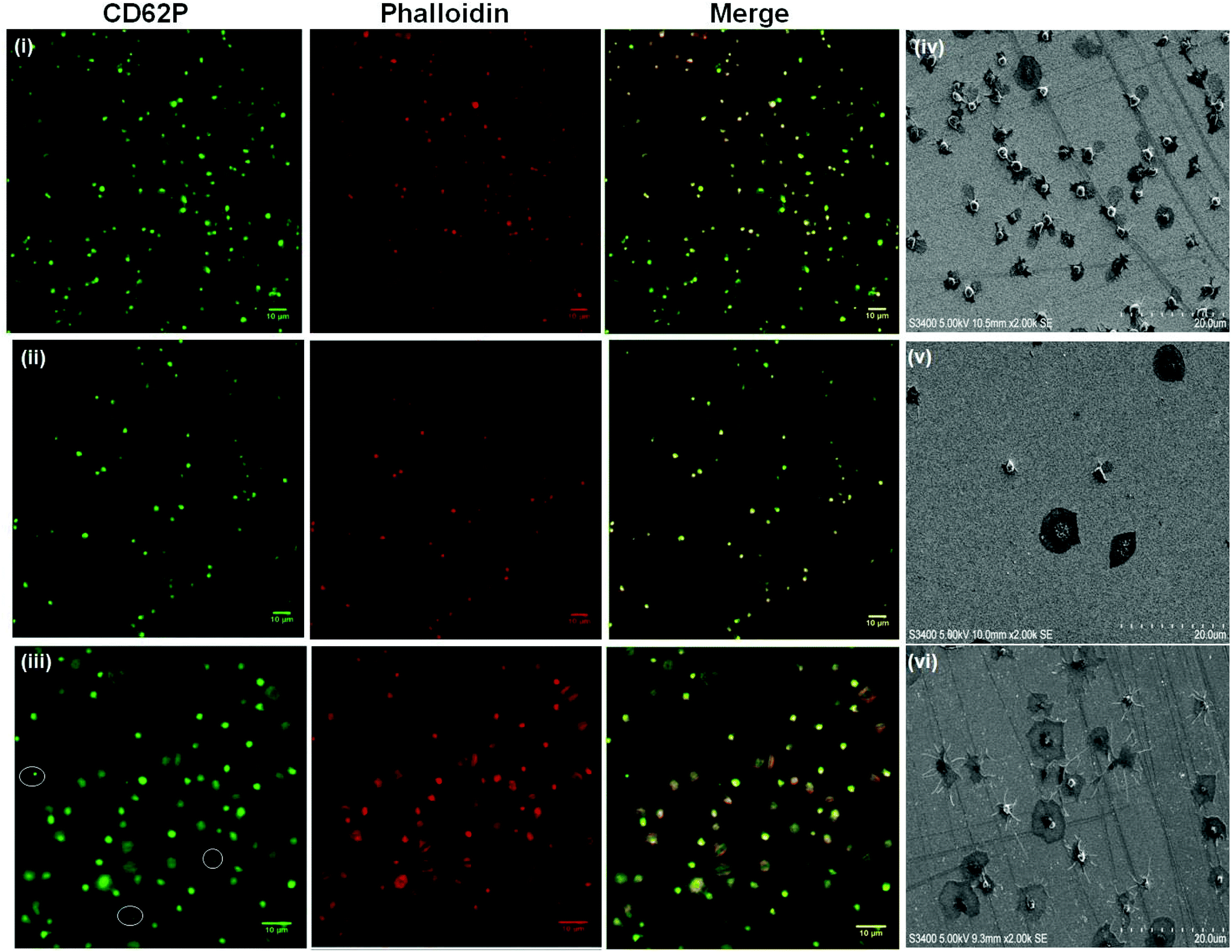 | ||
| Fig. 6 (i–iii) Confocal luminescence images of platelets treated with (i) Mn2+, (ii) DTT (1 mM final concentration, 2 hours incubation at room temperature) or (iii) thrombin (1 U ml−1 final concentration, 10 minutes incubation at room temperature) prior to incubation with planar gold surfaces modified with an RGD SAM. (iv & v) SEM images illustrating the morphology of platelets adhered to RGD SAM surfaces following treatment with a 1 mM final concentration of (iv) Mn2+, (v) DTT or (vi) thrombin under the same conditions as the confocal study. α Granule secretion is suggested following DTT and thrombin treatment, highlighted by the white circles in (iii) and suggested clearly in (vi). 4.5 × 106 platelets were incubated with the RGD SAM surfaces for 45 minutes at 37 °C. Platelet fixation, staining, mounting, dehydration and sputter coating was carried out as described. Luminescence images were recorded using a 40× oil immersion objective lens (NA 1.4) with 488 nm argon (CD62P) and 540 nm (phalloidin) HeNe laser excitation. SEM images were recorded using 5.00 kV accelerating voltage. All images are reproducible, representative of N = 3. | ||
The summary data, Fig. 3 shows, surprisingly, that Mn2+ has no effect on the platelet capture efficiency, compared with resting platelets while DTT induces a significant reduction in capture number.
The effect of Mn2+ induced integrin activation remains controversial,78 with reports suggesting Mn2+ is a highly potent effector of integrin-mediated cell adhesion at low micromolar concentrations,79,80 and that it can alter the conformation of the ligand-binding pocket.79,81 However, other investigations indicate that Mn2+ does not increase the proteins binding affinity to physiological ligands, i.e. fibronectin, and that it decreases the initial association rates of ligand with integrins resulting in only partial activation.82–84
The results presented here showed that even after an incubation time of 2 hours at room temperature, the effect of Mn2+ is weak. Mn2+ treatment does not affect the platelets ability to bind to the modified surface in this study and contrasts to previous publications reporting enhanced platelet adhesion to, for example, fibrinogen modified surfaces following Mn2+ treatment.50,51,85
DTT is known to reduce disulfide bonds within the integrins cysteine-rich domain leading to global conformational changes within both αIIb and β3 and opening of the RGD binding site.47,84,86,87 This response reportedly increases RGD mediated platelet adhesion to, for example, fibronectin modified surfaces.53,88 Rather surprisingly, Fig. 3 and 6(ii), show that in contrast to their physiological ligands, DTT treatment significantly reduced adhesion to the RGD-only surface (8 ± 0.9% adhesion compared to 17% ± 0.6) without DTT activation.
We then compared the capture of platelets that had been activated by the powerful physiological platelet activator thrombin. Thrombin, activates at least three different thrombin receptors on human platelets: G-protein coupled receptors PAR 1 and PAR 489 and the membrane glycoprotein GPIb.90 In addition, thrombin is the final enzyme in the coagulation cascade and acts to cleave the plasma protein fibrinogen into fibrin which polymerizes to form a fibrous mesh, which included with the above receptor activation, aids the platelet activation and aggregation process.91
Thrombin treatment induces the characteristic morphological change in platelets associated with activation, characterized by the loss of discoid shape and the movement of granules toward the platelet periphery.92,93 It is surprising to note that thrombin activation leads to a completely different response to DTT. As shown in Fig. 6(iii) and 6(vi) thrombin activation lead to increased adhesion (20 ± 0.8%) compared to untreated platelet adhesion (17 ± 0.6%). Full platelet spreading (Fig. S10†) was evident and activation with CD62P diffusion and relocation towards the periphery of the platelets, Fig. 6(iii) was observed. Correspondingly, Phalloidin staining indicated significant actin reorganization in the bound platelet with the formation of actin stress fibres. All consistent, with activation, as expected from thrombin activated platelets.
Overall, the lower numbers of platelets captured and their resting status after platelet treatment with DTT or Mn2+ in contrast to the number of platelets captured after thrombin treatment (increased adhesion to untreated platelets) suggests that the platelets are being activated via different processes. DTT treatment has been reported to activate only the platelet integrin αIIbβ3,46,47,94 while thrombin is known to activate the whole platelet including integrin αIIbβ3.95,96 However, in both cases it is the αIIbβ3, which is the target for the RGD surface so our results suggest that DTT, Mn2+ and thrombin all induce different conformational states within the integrin. This difference is likely to originate from steric factors affecting accessibility of the RGD binding pocket of the integrin as a result of different extents of conformational change induced by each activator.46,53,94,97 The influence of such steric effects is likely to be exacerbated by the fact that the RGD ligands are immobilised within a relatively close packed monolayer which will reduce RGD accessibility compared to other surfaces, particularly those based on the protein receptors which contain two or three binding sites per integrin.46,47,98,99
Reversibility of platelet adhesion to chemically modified surfaces
The reversibility of platelet adhesion was investigated by incubating the platelet decorated surfaces with a 1 mM solution of C-Ahx-GRGDS, eptifibatide or PEG-COO- for 15 minutes at 37 °C, presented in Fig. 3. Incubation with RGD removed approximately 83% of the bound platelets from the RGD modified surface indicating is occurring through integrin recognition of the surface confined RGD sequence. Consistent with this result, incubation with Eptifibatide, a cyclical RGD containing drug and effective integrin αIIbβ3 binding inhibitor removed approximately 39% of the bound platelets, whereas, PEG-COO- failed to remove the bound platelets from the surface at all, Fig. 3 (Fig. S12 to S14†).In contrast, incubating platelets bound to a PEG-COO- modified surface with either RGD or eptifibatide had little effect on the bound platelets removing only 7% and 11% of the platelets adhered to the surface respectively. While, incubating the platelets bound to the PEG-COO- modified surface with 1 mM PEG-COO- in PBS, pH 7.4 solution, removed approximately 95% of the platelets from the surface, Fig. 3 (Fig. S12 to S14†).
Therefore platelet adhesion to both RGD and PEG-COO- modified substrates is easily reversed by introducing an appropriate competitive agent. Reversal of cell capture at modified surfaces by competitive exposure to RGD ligand has been shown for cells previously100 but this is, we believe, the first time it has been demonstrated for platelets.
Conclusions
Understanding and controlling platelet adhesion to surfaces is important across a range of domains including implants and medical devices, while intact platelet capture in which the activation status of the platelet is controlled may be useful in diagnostics. Self-assembled monolayers of C-Ahx-GRGDS peptide on planar gold surfaces exhibit a strong affinity for resting human platelets and the captured platelets retain their resting state or are at very early stage spreading. Co-adsorption of the RGD with a second thiol had a significant impact on platelet capture. Alkane thiol alone was found to effectively inhibit platelet adhesion while carboxyl terminated PEG strongly promoted not just platelet adhesion but platelet spreading and activation. This is attributed to the anionic charge at the carboxyl terminus of the PEG used in this study. And, the presence of PEG-COO- tended to dominate the behaviour of PEG/RGD surfaces whereas RGD/alkane surfaces tended to undergo RGD like capture. Interestingly, “activating” the platelets with Mn2+ had little effect on adhesion of these platelets to the RGD modified substrates. And, surprisingly, DTT treatment of the platelets significantly inhibited binding, and the small number of platelets that bound were resting, suggesting that platelets that were activated by DTT were not capable of binding the surface immobilized RGD. Remarkably, in contrast, treatment of the platelets with physiological activator thrombin, both increased platelet adhesion at the RGD surfaces and all platelets that were captured were fully activated. The origin of these differences is likely to be steric, arising from differences in the conformation of the platelet integrin αIIbβ3 induced by each activation approach.Potentially usefully, platelet adhesion to the RGD SAMs was reversible. Incubation of the bound platelets with either linear or cyclical RGD eliminated the bound platelets (83% and 39% elimination respectively) from the surface within 15 minutes, regardless of their extent of spreading/activation. Platelet adhesion to the PEG-COO- substrate was by contrast irreversible with either linear or cyclic RGD (7% and 11% elimination respectively).
Overall, the ability to reversibly control platelet adhesion and to selectively capture platelets in a predominantly resting or activated state has value across both diagnostics and in understanding and controlling haemocompatibility of implantable materials. Future work will focus on building greater physiological relevance, evaluating these modified surfaces under bio-relevant flow conditions.
Competing interest
The authors declare no competing financial interest.Acknowledgements
This material is based upon work supported by the Health Research Board funded Scholarship program located in the Royal College of Surgeons in Ireland (Award no. PHD/2007/11). This material is based upon work also supported by the Science Foundation Ireland under grant no. [10/IN.1/B3025] and the National Biophotonics and Imaging Platform, Ireland, and funded by the Irish Government's Programme for Research in Third Level Institutions, Cycle 4, Ireland's EU Structural Funds Programmes 2007–2013. UP, RJF and TEK gratefully acknowledge EUFP7 Interreg programme under the Celtic Alliance for Nanohealth.Notes and references
- M. Schuler, G. R. Owen, D. W. Hamilton, M. de Wild, M. Textor and D. M. Brunette, et al. , Biomaterials, 2006, 27, 4003–4015 CrossRef CAS PubMed.
- G. Voskerician, M. S. Shive, R. S. Shawgo, H. Recum, J. M. Anderson and M. J. Cima, et al. , Biomaterials, 2003, 24(11), 1959–1967 Search PubMed.
- J. Song, Q. Cheng, S. Zhu and R. Stevens, Biomed. Microdevices, 2002, 4(3), 213–221 Search PubMed.
- T. Haruyama, Adv. Drug Delivery Rev., 2003, 55(3), 393–401 Search PubMed.
- Y. Onuki, U. Bhardwaj, F. Papadimitrakopoulos and D. J. Burgess, J. Diabetes Sci. Technol., 2008, 2, 1003–1015 Search PubMed.
- K. B. Neeves, A. A. Onasoga and A. R. Wufsus, Curr. Opin. Hematol., 2013, 20(5), 417–423 CrossRef CAS PubMed.
- A. G. Harvey, E. W. Will and A. Bayat, Expert Rev. Med. Devices, 2013, 10(2), 257–267 Search PubMed.
- M. B. Gorbet and M. V. Sefton, Biomaterials, 2004, 25, 5681–5703 Search PubMed.
- J. L. Harding and M. M. Reynolds, Trends Biotechnol., 2014, 32(3), 140–146 CrossRef CAS PubMed.
- T. Groth, L. Zhen-Mei, M. Niepel, D. Peschel, K. Kirchhof, G. Altankov, et al., Chemical and Physical Modifications of Biomaterial Surfaces to Control Adhesion of Cells, in Advances in Regenerative Medicine: Role of Nanotechnology, and Engineering Principles, ed. V. P. Shastri et al., Springer, 2010, ch. 13, pp. 253–284 DOI:10.1007/978-90-481-8790-4_13.
- A. Yamamoto, S. Mishima, N. Maruyama and M. Sumita, J. Biomed. Mater. Res., 2000, 50(2), 114–124 Search PubMed.
- F. Pi, P. Dillard, L. Limozin, A. Charrier and K. Sengupta, Nano Lett., 2013, 13(7), 3372–3378 Search PubMed.
- M. Mrksich, Acta Biomater., 2009, 5, 832–841 CrossRef CAS PubMed.
- E. Ostuni, G. M. Whitesides, D. E. Ingber and C. S. Chen, Methods Mol. Biol., 2009, 522, 183–194 Search PubMed.
- N. Faucheux, R. Schweiss, K. Lützow, C. Werner and T. Groth, Biomaterials, 2004, 25(14), 2721–2730 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
- L. Gonzalez-Macia, A. Morrin, M. R. Smyth and A. J. Killard, Analyst, 2010, 135, 845–867 RSC.
- M. Barczyk, S. Carracedo and D. Gullberg, Integrins, Cell Tissue Res., 2010, 339(1), 269–280 CrossRef CAS PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24(24), 4385–4415 CrossRef CAS.
- D. Pallarola, A. Bochen, H. Boehm, F. Rechenmacher, T. R. Sobahi and J. P. Spatz, et al. , Adv. Funct. Mater., 2014, 24, 943–956 Search PubMed.
- S. Bozzini, P. Petrini, M. C. Tanzi, C. R. Arciola, S. Tosatti and L. Visai, Int. J. Artif. Organs, 2011, 34(9), 898–907 CrossRef CAS PubMed.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao and B. Geiger, et al. , Biomaterials, 2007, 28, 4608–4618 Search PubMed.
- B. Zhu, T. Eurell, R. Gunawan and D. Leckband, J. Biomed. Mater. Res., 2001, 56(3), 406–416 Search PubMed.
- Y. Yang, Y. K. Lai, Q. Q. Zhang, K. Wu, L. H. Zhang and C. J. Lin, et al. , Colloids Surf., B, 2010, 79, 309–313 CrossRef CAS PubMed.
- D. Li, Q. Zheng, Y. Wang and H. Chen, Polym. Chem., 2014, 5(14), 14–24 Search PubMed.
- S. L. Goodman, S. L. Cooper and R. M. Albrecht, J. Biomed. Mater. Res., 1993, 27(5), 683–695 Search PubMed.
- S. P. Massia and J. A. Hubbell, J. Cell Biol., 1991, 114, 1089–1100 Search PubMed.
- K. Kakinoki, N. Yui and T. Yamaoka, J. Biomater. Appl., 2013, 28(4), 544–551 CrossRef PubMed.
- M. Hulander, A. Lundgren, L. Faxälv, T. L. Lindahl, A. Palmquist and M. Berglin, et al. , Colloids Surf., B, 2013, 110, 261–269 CrossRef CAS PubMed.
- B. Sivaraman and R. A. Latour, Biomaterials, 2010, 31(5), 832–839 CrossRef CAS PubMed.
- A. R. Rechner, Hamostaseologie, 2011, 31(2), 79–87 Search PubMed.
- L. Basabe-Desmonts, G. Meade and D. Kenny, Expert Rev. Mol. Diagn., 2010, 10(7), 869–874 CrossRef CAS PubMed.
- M. Dovizio, S. Alberti, P. Guillem-Llobat and P. Patrignani, Basic Clin. Pharmacol. Toxicol., 2014, 114(1), 118–127 CrossRef CAS PubMed.
- A. McNicol and S. J. Israels, Cardiovasc. Hematol. Disord.: Drug Targets, 2008, 8, 99–117 CrossRef CAS.
- E. F. Plow, T. A. Haas, L. Zhang, J. Loftus and J. W. Smith, J. Biol. Chem., 2000, 275, 21785–21788 CrossRef CAS PubMed.
- T. K. Gartner and B. S. Bennett, J. Biol. Chem., 1985, 260, 11891–11894 CAS.
- S. A. Santoro and W. J. Lawing Jr., Cell, 1987, 48, 867–973 CrossRef CAS.
- S. L. Goodman and M. Picard, Trends Pharmacol. Sci., 2012, 33(7), 405–412 Search PubMed.
- M. A. McLane, M. A. Kowalska, L. Silver, S. J. Shattil and S. Niewiarowski, Biochemistry, 1994, 301, 429–436 CAS.
- R. A. Bednar, S. Gaul Lee, T. G. Hamill, M. S. Egbertson, J. A. Shafer and G. A. Hartman, et al. , J. Pharmacol. Exp. Ther., 1998, 285, 1317–1326 Search PubMed.
- X. Du, E. F. Plow, A. L. Frelinger III, T. E. O'Toole, J. Loftus and M. H. Ginsberg, Cell, 1991, 65, 409–416 Search PubMed.
- A. Bonnefov, Q. Liu, C. Legrand and M. M. Frojmovic, Biophys. J., 2000, 78(6), 2834–2843 Search PubMed.
- J. H. Beer, K. T. Springer and B. S. Coller, Blood, 1992, 79(1), 117–128 CAS.
- A. S. Gupta, G. Huang, B. J. Lestini, S. Sagnella, K. Kottke-Marchant and R. E. Marchant, Thromb. Haemostasis, 2005, 93, 106–114 Search PubMed.
- S. A. Mousa, J. M. Bozarth, U. P. Naik and A. Slee, Br. J. Pharmacol., 2001, 133(3), 331–336 Search PubMed.
- R. I. Litvinov, C. Nagaswami, G. Vilaire, H. Shuman, J. S. Bennett and J. W. Weisal, Blood, 2004, 104, 3979–3985 CrossRef CAS PubMed.
- B. Yan and J. W. Smith, Biochemistry, 2001, 40, 8861–8867 CrossRef CAS PubMed.
- S. Tiwari, J. A. Askari, M. J. Humphries and N. J. Bulleid, J. Cell Sci., 2011, 124, 1672–1680 Search PubMed.
- J. Takagi, B. M. Petre, T. Walz and T. Springer, Cell, 2002, 110, 599–611 CrossRef CAS.
- G. P. Tuszynski and M. A. Kowalska, J. Clin. Invest., 1991, 87, 1387–1394 CrossRef CAS PubMed.
- H. Ni, A. Li, N. Simonsen and J. A. Wilkins, J. Biol. Chem., 1998, 273(14), 7981–7987 CrossRef CAS PubMed.
- M. J. Santos-Martínéz, A. Prina-Mello, C. Medina and M. W. Radomski, Analyst, 2011, 136, 5120–5126 RSC.
- H. Ni, A. Li, N. Simonsen and J. A. Wilkins, J. Biol. Chem., 1998, 273(14), 7981–7987 Search PubMed.
- R. I. Litvinov, A. Mekler, H. Shuman, J. S. Bennett, V. Barsegov and J. W. Weisel, J. Biol. Chem., 2012, 287(42), 35275–35285 CrossRef CAS PubMed.
- D. E. Macintyre and J. L. Gordon, Biochem. Soc. Trans., 1974,(2), 873–875 CAS.
- M. B. Zucker and N. C. Masiello, Thromb. Haemostasis, 1984, 51, 119–124 CAS.
- A. Margaritis, R. Priora, S. Frosali, D. Di Giuseppe, D. Summa and L. Coppo, et al. , Pharmacol. Res., 2011, 62, 77–84 CrossRef PubMed.
- E. Pensa, E. Cortés, G. Corthey, P. Carro, C. Vericat and M. H. Fonticelli, et al. , Acc. Chem. Res., 2012, 45(8), 1183–1192 CrossRef CAS PubMed.
- S. Kakinoki, N. Yui and T. Yamaoka, J. Biomat. Mater., 2012, 1–8 Search PubMed.
- B. T. Houseman and M. Mrksich, Biomaterials, 2001, 22(9), 943–955 CrossRef CAS.
- M. Mrksich, Chem. Soc. Rev., 2000, 29, 267–273 RSC.
- C. J. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef PubMed.
- B. R. Caudhry, J. D. E. T. Wilton-Ely, A. B. Tabor and D. J. Caruana, Phys. Chem. Chem. Phys., 2010, 12, 9996–9998 Search PubMed.
- C. D. Bain and G. Whitesides, Formation of Monolayers by the coadsorption of Thiols on Gold - Variation in the Length of the Alkyl Chain, J. Am. Chem. Soc., 1989, 111, 7164–7175 CrossRef CAS.
- M. Ebara, M. Yamato, T. Aoyagi, A. Kikuchi, K. Sakai and T. Okano, Adv. Mater., 2008, 9999, 1–5 Search PubMed.
- S. P. Massia and J. A. Hubbell, J. Cell Biol., 1991, 114, 1089–1100 CrossRef CAS.
- A. Kafi, W. A. El-Said, T.-H. Kim and J.-W. Choi, Biomaterials, 2012, 33(3), 731–739 CrossRef PubMed.
- S. E. D'Souza, M. H. Ginsberg and E. F. Plow, Trends Biochem. Sci., 1991, 16, 246–250 CrossRef.
- Y. Tanaka, Y. Matsuo, T. Komiya, Y. Tsutsumi, H. Doi and T. Yoneyama, et al. , J. Biomed. Mater. Res., Part A, 2010, 92(1), 350–358 Search PubMed.
- L. Perlin, S. MacNeil and S. Rimmer, Soft Matter, 2008, 4, 2331–2349 Search PubMed.
- Y. Tanaka, Y. Matsuo, T. Komiya, Y. Tsutsumi, H. Doi and T. Yoneyama, et al. , J. Biomed. Mater. Res., Part A, 2010, 92(1), 350–358 CrossRef PubMed.
- L. Perlin, S. MacNeil and S. Rimmer, Soft Matter, 2008, 4, 2331–2349 RSC.
- C. Sperling, M. Fischer, M. F. Maitz and C. Werner, Biomaterials, 2009, 30, 4447–4456 CrossRef CAS PubMed.
- E. A. Vogler and C. A. Siedlecki, Biomaterials, 2009, 30(10), 1857–1869 Search PubMed.
- P. Moix, Thrombocitopenias, Elsevier, 2nd edn, 2001 Search PubMed.
- E. L. Bearer, Cell Motil. Cytoskeleton, 1995, 30(1), 60–66 Search PubMed.
- M. Shimaoka and J. Takagi, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 485–516 CrossRef CAS PubMed.
- F. Ye, C. Kim and M. H. Ginsberg, Blood, 2012, 119, 26–33 CrossRef CAS PubMed.
- I. Dransfield, C. Cabanas, J. Barrett and N. Hog, J. Cell Biol., 1992, 116(6), 1527–1535 Search PubMed.
- J. Smith and D. Cheresh, J. Biol. Chem., 1991, 266, 11429–11432 CAS.
- A. Masumoto and M. E. Hemler, J. Biol. Chem., 1993, 268, 228–234 CAS.
- J. L. Sechler, S. A. Corbett and J. E. Schwarzbauer, Mol. Biol. Cell, 1997, 8, 2563–2573 Search PubMed.
- T. Kamata, M. Handa, Y. Sato, Y. Ikeda and S. Aiso, J. Biol. Chem., 2005, 280(26), 24775–24783 CrossRef CAS PubMed.
- J. W. Smith, R. S. Piotrowicz and D. Mathis, J. Biol. Chem., 1994, 269(2), 960–967 CAS.
- S. Urieli-Shoval, G. Shubinsky, R. P. Linke, M. Fridkin, I. Tabi and Y. Matzner, Blood, 2002, 99(4), 1224–1229 Search PubMed.
- J. J. Calvee, A. Henschen and J. Gonzalez-Rodriguez, Biochem. J., 1991, 274, 63–71 Search PubMed.
- S. O'Neill, A. Robinson, A. Deering, M. Ryan, D. J. Fitzgerald and N. Moran, J. Biol. Chem., 2000, 275, 36984–36990 Search PubMed.
- G. E. Davis and C. W. Camarillo, J. Immunol., 1993, 151(12), 7138–7150 CAS.
- G. Davi and C. Patrono, N. Engl. J. Med., 2007, 357, 2482–2494 CrossRef CAS PubMed.
- F. Adam, M. C. Guillin and M. Jandrot-Perrus, Eur. J. Biochem., 2003, 270, 2959–2970 CrossRef CAS.
- D. M. Monroe, M. Hoffman and H. R. Roberts, Arteriosclr. Thromb. Vasc. Biol., 2002, 22, 1381–1389 CAS.
- L. A. Cheryk, P. A. Gentry and F. Tablin, Comp. Haem. Int., 1997, 1, 88–94 Search PubMed.
- P. E. Stenberg, R. P. McEver, M. A. Shuman, Y. V. Jacques and D. F. Bainton, J. Cell Biol., 1985, 101(3), 880–886 Search PubMed.
- B. Z. S. Paul, G. Vilaire, S. P. Kunapuli and J. S. Bennett, J. Thromb. Haemostasis, 2003, 1(4), 814–820 CrossRef CAS.
- P. J. Sims, M. H. Ginsberg, E. F. Plow and S. J. Shattil, J. Biol. Chem., 1991, 266(12), 7345–7352 CAS.
- J. T. B. Crawley, S. Zanardelli, C. K. N. K. Chion and D. A. Lane, J. Thromb. Haemostasis, 2007, 5, 95–101 CrossRef CAS PubMed.
- K. Adamson, C. Dolan, N. Moran, R. J. Forster and T. E. Keyes, Bioconjugate Chem., 2014, 25(5), 928–944 Search PubMed.
- W. C. Kouns, B. Steiner, T. J. Kunicki, S. Moog, J. Jutzi and L. K. Jennings, et al. , Blood, 1994, 84(4), 1108–1115 Search PubMed.
- J. A. Eble, B. Beermann, H.-J. Hinz and A. Schmidt-Hedrich, J. Biol. Chem., 2001, 276, 12274–12284 CrossRef CAS PubMed.
- C. Roberts, C. S. Chen, M. Mrksich, V. Martichonok, D. E. Ingber and G. M. Whitesides, J. Am. Chem. Soc., 1998, 120, 6548–6555 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and AFM images of unmodified, electrochemically deposited planar gold surfaces; images and summary table of water contact angle measurements for all modified surfaces; electrochemical desorption spectra for all SAM surfaces; confocal luminescence reference image of phalloidin and CD62P stained activated platelets. Activation status confocal images of platelets adhered to gold alone, alkane alone, RGD:alkane and RGD:PEG 1:1, 1:5 and 1:10 ratio SAM surfaces. SEM and activation status confocal images of platelets treated prior to adhesion to RGD SAM surfaces with 1 mM DTT or Mn2+ after 15 minutes incubation at room and platelets treated post-bind with a 1 mM solution of C-Ahx-GRGDS, PEG-COO- or the cyclic RGD drug Eptifibatide. See DOI: 10.1039/c4bm00241e |
| This journal is © The Royal Society of Chemistry 2014 |