Lewis base-catalyzed reactions of cyclopropenones: novel synthesis of mono- or multi-substituted allenic esters†
Yuan-Liang
Yang
,
Zhen
Zhang
,
Xiao-Nan
Zhang
,
De
Wang
,
Yin
Wei
* and
Min
Shi
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: weiyin@sioc.ac.cn; mshi@sioc.ac.cn; Fax: +86-21-64166128
First published on 25th October 2013
Abstract
The reactions of cyclopropenones with nucleophiles (H2O or methanol) could be catalyzed by nitrogen-containing Lewis bases or phosphorus-containing Lewis bases, affording the corresponding mono- or multi-substituted allenic esters in moderate to excellent yields.
The first reports on synthesis of cyclopropenones were presented by Breslow et al.1 and Volpin and co-workers2 in 1959, respectively. Recent extensive investigations have established the utility of these molecules as building blocks for the construction of biologically active compounds and important structural motifs in materials science.3 Cyclopropenone is an amphiphilic molecule, which reacts readily with both nucleophilic and electrophilic reagents; thus, they can be utilized in a wide range of organic reactions.4 Hamada and Takizawa have reported the reaction of 2,3-diphenylcycloprop-2-enone 1 with methanol in the presence of PPh3 to afford (E)-methyl 2,3-diphenylacrylate in good yield.5 Inspired by Hamada and Takizaka's work and as a part of our continuing interest on Lewis bases mediated reactions,6 we are extremely interested in the Lewis base-mediated/catalyzed chemical processes of cyclopropenones, since cyclopropenones being all-carbon 1,3-dipolar equivalents can easily react with nitrogen-containing,7 or phosphorus-containing nucleophiles.8 To our delight, we found a novel Lewis base-catalyzed reaction for synthesis of allenic esters that are efficient synthons used widely in organic synthesis.9,10 Although a variety of methods for preparation of allenes have been developed,11 there are rare reports on the synthesis of allenic esters using Lewis bases so far. The relevant studies were base-promoted alkyne isomerisation reported by Huang and Tan,12 and Lewis base-catalyzed intramolecular 1,4-addition of conjugated enynes reported by Tang.13 The established method presented here provides another novel way to access mono- or multi-substituted allenic esters.
Initial examination was carried out by using (3-oxo-2-phenylcycloprop-1-enyl)methyl acetate 2a (for the preparation of 2, see Table S in the ESI†) and a nucleophile (H2O or MeOH) as substrates in the presence of DABCO. After initial optimization, DABCO in THF was found to be a suitable system for this reaction (see Table S1 in the ESI†). Furthermore, various Lewis bases and nucleophiles were optimized. The results are summarized in Table 1 and we found that the corresponding 2-phenylbuta-2,3-dienoic acid 3a was produced in 50% yield in the presence of DABCO (20 mol%) and a nucleophile (H2O) (1.0 equiv.) (Table 1, entry 1). Under the same reaction conditions, we found that other Lewis bases such as DMAP (p-N,N-dimethylaminopyridine), PPh3, and tris(3,5-bis(trifluoromethyl)phenyl)phosphine could not catalyze this reaction (Table 1, entries 2–4). Considering that methanol is more nucleophilic than water, we chose methanol as a nucleophile to examine the reaction outcome again. The corresponding product 3a was obtained in higher yield in the presence of methanol. We further identified that 10.0 equiv. of MeOH relative to 2a was the best ratio, affording 3a in 95% yield (Table 1, entries 5–8). Other nitrogen-containing Lewis bases (DBU, Et3N, DMAP) could not promote this reaction (Table 1, entries 9–11). In the absence of DABCO, the reaction did not occur. The phosphorus-containing Lewis base PPh3 could also catalyze this reaction, giving 3a in 82% yield (Table 1, entry 14). Decreasing the catalyst loading to 10 mol% slightly reduced the yield of 3a (Table 1, entry 13). Inorganic bases (K2CO3 and CH3ONa) did not catalyze this reaction under the standard conditions (Table 1, entries 15 and 16). Thus, finally, we established the optimal reaction conditions: 20 mol% of DABCO as the catalyst in THF at room temperature, for the synthesis of compound 3a.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | NuH (equiv.) | Yielda (%) |
| a Isolated yield. Unless otherwise specified, to a mixture of 2a (0.10 mmol) and catalyst (0.02 mmol) in THF (1.0 mL) under an Ar atmosphere at 0 °C NuH was added, then the resulting mixture was stirred at room temperature for 3 days. b The reaction was conducted at room temperature for 5 hours and 4 Å MS was added. c The catalyst loading was 10 mol%. d 0.1 mmol of inorganic base was used. | ||||
| 1 | DABCO | THF | H2O (1.0) | 3a′ (50) |
| 2 | DMAP | THF | H2O (1.0) | N.R |
| 3 | PPh3 | THF | H2O (1.0) | N.R |
| 4 | P(3,5-(F3C)2CeH3)3 | THF | H2O (1.0) | N.R |
| 5b | DABCO | THF | MeOH (1.0) | 3a (78) |
| 6b | DABCO | THF | MeOH (2.0) | 3a (86) |
| 7b | DABCO | THF | MeOH (5.0) | 3a (89) |
| 8b | DABCO | THF | MeOH (10.0) | 3a (95) |
| 9b | DBU | THF | MeOH (10.0) | N.R |
| 10b | Et3N | THF | MeOH (10.0) | N.R |
| 11b | DMAP | THF | MeOH (10.0) | N.D |
| 12b | — | THF | MeOH (10.0) | N.R |
| 13b,c | DABCO | THF | MeOH (10.0) | 3a (89) |
| 14b | PPh3 | THF | MeOH (10.0) | 3a (82) |
| 15b,d | K2CO3 | THF | MeOH (10.0) | N.D |
| 16b,d | CH3ONa | THF | MeOH (10.0) | N.D |
Having identified the optimal reaction conditions, we next set out to examine the scope and limitations with various cyclopropenone derivatives 2 and MeOH. The results are summarized in Table 2. A variety of cyclopropenone derivatives 2 having either electron-donating or -withdrawing groups as substituents on the 2-, 3-, 4-positions of a benzene ring underwent the reactions smoothly, affording the corresponding products 3 in moderate to high yields (54–96%) (Table 2, entries 1–11). If R1 was 2-CO2MeC6H4, the reaction did not take place at room temperature, and was carried out at 50 °C for 30 minutes, furnishing the corresponding product 3k in 54% yield (Table 2, entry 10). When R1 was a 2-naphthyl group, the reaction also proceeded efficiently to give the corresponding product 3m in 78% yield (Table 2, entry 12). However, if R1 was a benzyl group, the reaction could not take place, presumably due to the electronic effect (Table 2, entry 13). We also investigated the reaction of substrate 2ab with an Ms protecting group, and the reaction also proceeded very well to provide 3a in 57% yield. During these investigations, we also found that the electronic effect of substituents on the benzene ring had an impact on the reaction rate. If R1 was bearing an electron-donating group, the reaction rate was slower than that of those bearing an electron-withdrawing group (Table 2, entries 1, 8, 11 vs. entries 2–7, 9, 10). The structure of 3 has been unambiguously determined by X-ray diffraction of the analogue 3j. The ORTEP drawing and its CIF data are provided in the ESI.†14
|
|
|||
|---|---|---|---|
| Entry | R1/R2 | Time | Yielda (%) |
| a Isolated yield. b The reaction was carried out at 50 °C. | |||
| 1 | 4-MeOC6H4/Ac (2b) | 8 h | 3b (93) |
| 2 | 4-NO2C6H4/Ac (2c) | 5 min | 3c (94) |
| 3 | 4-tBuC6H4/Ac (2d) | 36 h | 3d (91) |
| 4 | 4-ClC6H4/Ac (2e) | 2.5 h | 3e (96) |
| 5 | 4-BrC6H4/Ac (2f) | 2 h | 3f (83) |
| 6 | 3-FC6H4/Ac (2g) | 3 h | 3g (90) |
| 7 | 3-BrC6H4/Ac (2h) | 1 h | 3h (89) |
| 8 | 3-MeC6H4/Ac (2i) | 7 h | 3i (90) |
| 9 | 2-CNC6H4/Ac (2j) | 30 min | 3j (90) |
| 10b | 2-CO2MeC6H4/Ac (2k) | 30 min | 3k (54) |
| 11 | 2-MeOC6H4/Ac (2l) | 24 h | 3l (93) |
| 12 | 1-Naphthyl/Ac (2m) | 24 h | 3m (78) |
| 13 | Bn/Ac (2n) | 48 h | N.D |
| 14 | C6H5/Ms (2ab) | 72 h | 3a (57) |
We continued to examine the substrate generality of the reaction using various mono- or disubstituted cyclopropenone derivatives 2 with MeOH. Unfortunately, DABCO could not catalyze these reactions due to its lower nucleophilicity. Thus, we switched to more nucleophilic phosphorus-containing Lewis bases (PPh3 or PBu3). To our delight, the reactions proceeded smoothly, giving the desired products in good yields, and the results are presented in Table 3. The corresponding products 4 were obtained in moderate to high yields (57–92%). When R1 = Ph and R3 = H, R2 having no substituent on the benzene ring or having electron-donating or -withdrawing groups as substituents on the 2-, 3-, 4-positions of the benzene ring, the reactions could proceed smoothly, giving the corresponding products 4a–4b and 4f–4h in high yields (Table 3, entries 1 and 2, and 6–8). When R2 was a naphthyl or more sterically hindered phenanthryl group, the reactions also proceeded well (Table 3, entries 3–5). When 2x was used as the substrate (R1 = 4-ClC6H4, R2 = Me and R3 = H), the reaction also proceeded smoothly, affording 4i in 90% yield (Table 3, entry 9). When R1 = C6H5 and R2, R3 = (CH2)5, the reaction was conducted at room temperature for 3 days in the presence of 20 mol% PBu3, providing 4j in 79% yield (Table 3, entry 10). Finally, as for substrate 2z (R1 = C6H5 and R2 = CH3, R3 = 4-ClC6H4), the reaction was catalyzed by 20 mol% PPh3, furnishing the corresponding product 4i in 57% yield. We observed that, in general, if R3 was not a hydrogen atom, the reaction would proceed slowly, presumably due to the steric effect (Table 3, entries 1–9 vs. entries 10 and 11).
|
|
|||
|---|---|---|---|
| Entry | R1/R2/R3 | Time | Yielda (%) |
| a Isolated yield. b The reaction was catalyzed by 20 mol% PBu3. c The catalyst loading was 20 mol%. | |||
| 1 | C6H5/C6H5/H (2p) | 30 min | 4a (92) |
| 2 | C6H5/4-ClC6H4/H (2q) | 30 min | 4b (93) |
| 3 | C6H5/2-naphthyl/H (2r) | 60 min | 4c (87) |
| 4 | C6H5/1-naphthyl/H (2s) | 2 h | 4d (80) |
| 5 | C6H5/9-phenanthryl/H (2t) | 60 min | 4e (95) |
| 6 | C6H5/3-MeC6H4/H (2u) | 45 min | 4f (87) |
| 7 | C6H5/2-BrC6H4/H (2v) | 30 min | 4g (91) |
| 8 | C6H5/2,4-Cl2C6H3/H (2w) | 15 min | 4h (83) |
| 9 | 4-ClC6H4/Me/H (2x) | 30 min | 4i (90) |
| 10b | C6H5/(CH2)5 (2y) | 3 d | 4j (79) |
| 11c | C6H5/CH3/4-ClC6H4 (2z) | 3 d | 4k (57) |
We have screened various chiral monophosines or bifunctional phosphines as well as chiral multifunctional phosphines, such as phosphine-amide type catalysts, phosphine-thiourea/urea type catalysts, for their asymmetric variant. The preliminary results show that the chiral multifunctional phosphine-thiourea catalyst LB1 was fairly effective in the reaction of cyclopropenone 2q with MeOH in toluene at room temperature, giving the corresponding product (S)-4c in 80% isolated yield with 57% ee (Scheme 1, eqn (1)). Employing the substrate 2r containing a naphthyl group improved the ee value to 71% in 70% isolated yield (Scheme 1, eqn (2)). The exploration of more effective chiral phosphines and further tests for determining the substrate generality are underway.
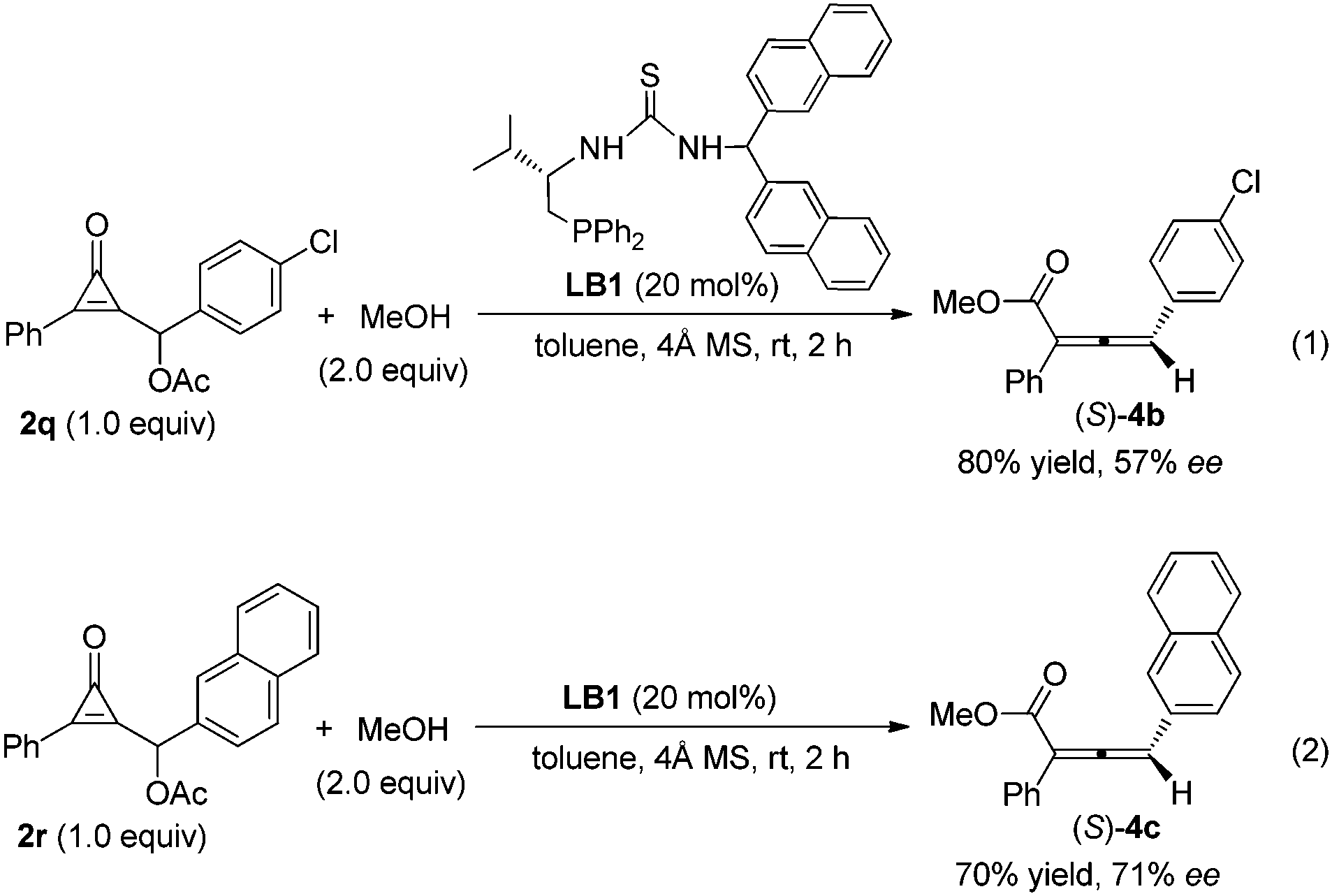 | ||
| Scheme 1 Asymmetric reaction of cyclopropenone with MeOH catalyzed by chiral phosphine catalyst LB1. | ||
A plausible mechanism for this reaction is outlined in Scheme 2. The transformation is believed to proceed via conjugate addition of Lewis base (LB) to cyclopropenone, affording zwitterionic intermediate A.15 Subsequently, methanol is deprotonated to give intermediate B. Then the three-membered ring is opened and the acetate anion is released, leading to intermediate C. The acetate anion grabs a proton of intermediate C to give intermediate D, which upon loss of the Lewis base catalyst gives rise to the final product 3. Alternatively, the conjugate addition of LB to cyclopropenone produces a cyclopropene enolate A′, which undergoes ring opening to give a ketene intermediate B′. Subsequently, the ketene intermediate E reacts with methanol to afford the final product. A more detailed investigation on the reaction mechanism will be reported in due course.
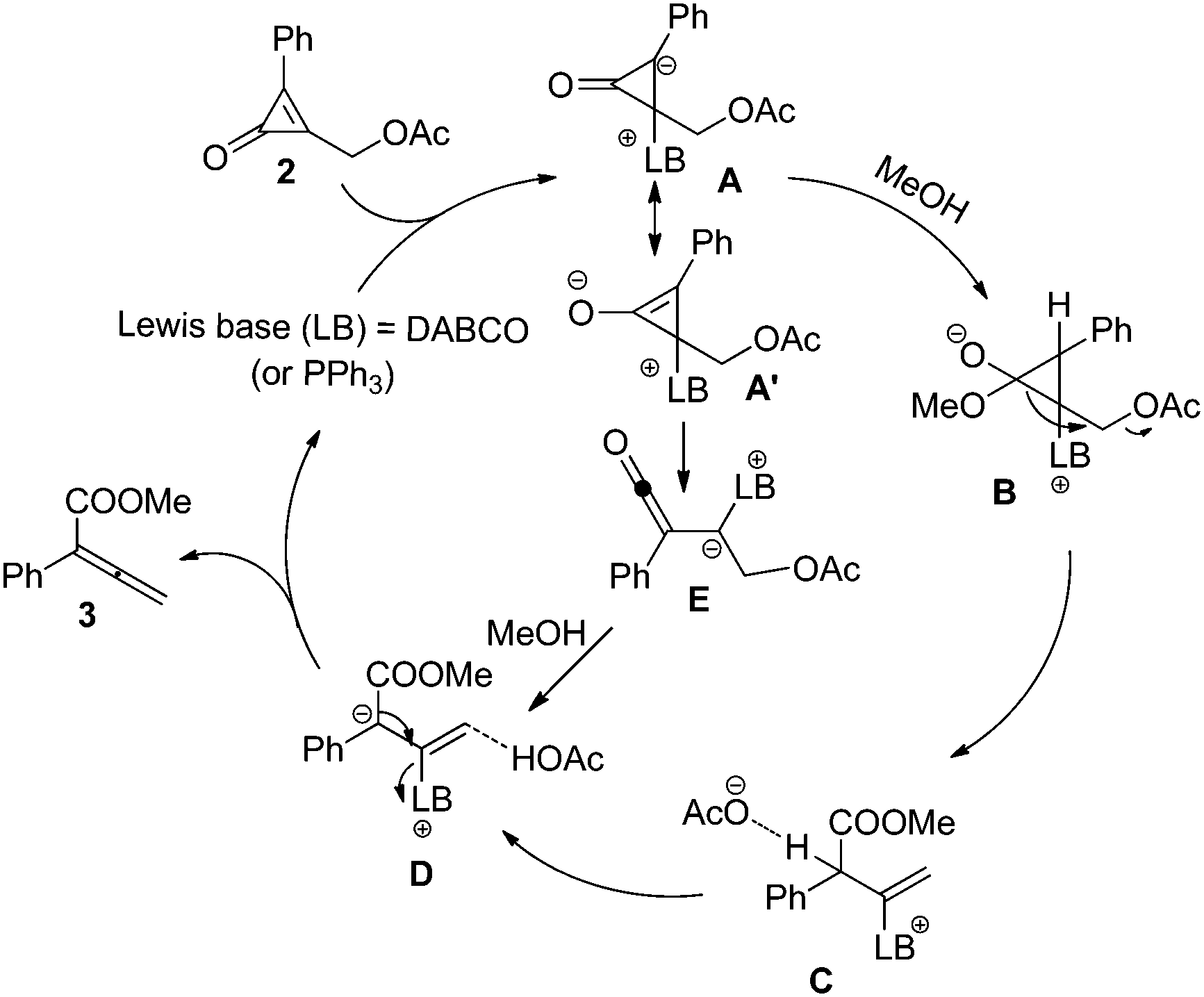 | ||
| Scheme 2 Plausible reaction mechanism. | ||
In summary, we have investigated novel and interesting reactions of cyclopropenones with alcoholic nucleophiles catalyzed by different Lewis bases, affording the corresponding allenic esters in moderate to high yields under mild conditions. This is the first example on the Lewis base-catalyzed formation of allenic esters from cyclopropenones. Based on this unprecedented method, axially chiral allenic esters have also been furnished in moderate yield and ee in the presence of multifunctional chiral phosphine LB1. A plausible reaction mechanism has also been proposed. Further studies on the mechanistic details of this catalytic system along with the reaction properties of the other cyclopropenones are currently underway in our laboratory.
We thank the Shanghai Municipal Committee of Science and Technology (11JC1402600), the National Basic Research Program of China (973)-2009CB825300, and the National Natural Science Foundation of China for financial support (21072206, 20472096, 21102166, 21372241, 21302203, 21372250, 20672127, 21121062 and 20732008).
Notes and references
- R. Breslow, R. R. Haynie and J. Mirra, J. Am. Chem. Soc., 1959, 81, 247 CrossRef CAS.
- D. N. Kursanov, M. E. Volpin and Y. D. Koreshkov, Izv. Akad. Nauk SSSR, Ser. Khim., 1959, 560 Search PubMed.
- (a) H. Tokuyama, M. Isaka, E. Nakamura and Y. Morinaka, J. Antibiot., 1992, 45, 1148 CrossRef CAS; (b) R. Ando and Y. Morinaka, J. Am. Chem. Soc., 1993, 115, 1174 CrossRef CAS; (c) R. Ando, T. Sakaki, Y. Morinaka, C. Takahashi, Y. Tamao, N. Yoshii, S. Katayama, K.-I. Saito, H. Tokuyama, M. Isaka and E. Nakamura, Bioorg. Med. Chem., 1999, 7, 571 CrossRef CAS.
- For reviews: (a) K. T. Potts and J. S Baum, Chem. Rev., 1974, 74, 189 CrossRef CAS; (b) T. Eicher and J. L. Weber, Top. Curr. Chem., 1975, 57, 1 CrossRef CAS; (c) K. Komatsu and T. Kitagawa, Chem. Rev., 2003, 103, 1371 CrossRef CAS PubMed.
- A. Hamada and T. Takizawa, Chem. Pharm. Bull., 1975, 23, 2933 CrossRef CAS.
- (a) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed; (b) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed.
- (a) A. Kascheres and D. Marchi, J. Org. Chem., 1975, 40, 2985 CrossRef CAS; (b) Y. Tamura, H. Matsushima and M. Ikeda, Tetrahedron, 1975, 32, 431 CrossRef; (c) K. N. Hemming, M. V. R. Khan, V. Kondakal, A. Pitard, M. I. R. Qamar and C. Rice, Org. Lett., 2012, 14, 126 CrossRef CAS PubMed.
- (a) A. Hamada and T. Takizawa, Chem. Pharm. Bull., 1975, 23, 2933 CrossRef CAS; (b) M. Alajarvín, C. López-Leonardo, Á. Álvarez-García, P. Llamas-Lorente, P. Sánchez-Andrada, J. Berná, A. Pastor, D. G. Baustista and P. Jones, Chem.–Eur. J., 2010, 16, 3728 CrossRef PubMed.
- For selected reviews about allene chemistry, see: (a) S. Patai, The Chemistry of Ketenes, Allenes and Related Compounds, Wiley, New York, 1980 Search PubMed; (b) S. R. Landor, The Chemistry of the Allenes, Academic Press, London, 1982 Search PubMed; (c) R. W. Bates and V. Satcharoen, Chem. Soc. Rev., 2002, 31, 12 RSC; (d) R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067 CrossRef CAS PubMed; (e) Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (f) A. Barbero and F. J. Pulido, Synthesis, 2004, 779 CAS; (g) S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed; (h) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed.
- (a) D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 13646 CrossRef PubMed; (b) S. Ma, H. Ren and Q. Wei, J. Am. Chem. Soc., 2003, 125, 4817 CrossRef CAS PubMed; (c) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426 CrossRef CAS PubMed; (d) S. Ma and Z. Gu, J. Am. Chem. Soc., 2006, 128, 4942 CrossRef CAS PubMed; (e) Y. S. Tran and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12632 CrossRef CAS PubMed; (f) C. E. Henry and O. Kwon, Org. Lett., 2007, 9, 3069 CrossRef CAS PubMed; (g) Y. Li, H. Zou, J. Gong, J. Xiang, T. Luo, J. Quan, G. Wang and Z. Yang, Org. Lett., 2007, 9, 4057 CrossRef CAS PubMed; (h) C. Zhou, C. Fu and S. Ma, Angew. Chem., Int. Ed., 2007, 46, 4379 CrossRef CAS PubMed; (i) Z. Lu, S. Zheng, X. Zhang and X. Lu, Org. Lett., 2008, 10, 3267 CrossRef CAS PubMed; (j) B. J. Cowen, L. B. Saun-ders and S. J. Miller, J. Am. Chem. Soc., 2009, 131, 6105 CrossRef CAS PubMed; (k) X. Guan, Y. Wei and M. Shi, J. Org. Chem., 2009, 74, 6343 CrossRef CAS PubMed; (l) Q.-M. Zhang, L. Yang and X.-F. Tong, J. Am. Chem. Soc., 2010, 132, 2550 CrossRef CAS PubMed; (m) G. Chai, S. Wu, C. Fu and S. Ma, J. Am. Chem. Soc., 2011, 133, 3740 CrossRef CAS PubMed; (n) X.-N. Zhang, H.-P. Deng, L. Huang, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 8664 RSC.
- For recent reviews on the syntheses of allenes, see (a) K. M. Brummond and J. E. DeForrest, Synthesis, 2007, 795 CrossRef CAS PubMed; (b) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC.
- H. Lu, D. Leow, K.-W. Huang and C.-H. Tan, J. Am. Chem. Soc., 2009, 131, 7212 CrossRef PubMed.
- (a) W. Zhang, H. Xu, H. Xu and W. Tang, J. Am. Chem. Soc., 2009, 131, 3832 CrossRef CAS PubMed; (b) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed.
- The crystal data of compound 3j have been deposited in CCDC with number 917097.
- For theoretical investigations on the relative stabilities of other possible zwitterionic intermediates, see ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data of new compounds. CCDC 917097. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc46470a |
| This journal is © The Royal Society of Chemistry 2014 |