Bilirubin oxidase bioelectrocatalytic cathodes: the impact of hydrogen peroxide†
Ross D.
Milton
a,
Fabien
Giroud
b,
Alfred E.
Thumser
c,
Shelley D.
Minteer
b and
Robert C. T.
Slade
*a
aDepartment of Chemistry, University of Surrey, Guildford, GU2 7XH, UK. E-mail: r.slade@surrey.ac.uk
bUniversity of Utah, 315 S 1400 E, Salt Lake City, Utah 84112, USA
cDepartment of Biochemistry and Physiology, University of Surrey, Guildford, GU2 7XH, UK
First published on 29th October 2013
Abstract
Mediator-less, direct electro-catalytic reduction of oxygen to water by bilirubin oxidase (Myrothecium sp.) was obtained on anthracene-modified, multi-walled carbon nanotubes. H2O2 was found to significantly and irreversibly affect the electro-catalytic activity of bilirubin oxidase, whereas similar electrodes comprised of laccase (Trametes versicolor) were reversibly inhibited.
Multi-copper oxidases (MCOs), such as bilirubin oxidase (BOd) and laccase, are commonly incorporated within enzymatic biological fuel cells (EBFCs) as cathodic biocatalysts responsible for the 4-electron reduction of dissolved O2 to H2O.1–6 BOd and laccase both contain 3 different copper centres that are responsible for the catalysis of substrates, and they are classified as type 1 (T1), type 2 (T2) and a binuclear type 3 (T3). The T2 and T3 sites are combined in a tri-nuclear cluster (TNC).7 The single-electron oxidation of substrates takes place at the proximal T1 site, with the 4-electron reduction of O2 taking place at the TNC. Following the oxidation of substrates, 4 electrons are quickly transferred between the T1 site and the TNC (in 4 × 1-electron transfers) of MCOs via a His-Cys-His chain.7,8 It has also been reported that laccase and BOd can promote the reduction of O2 to H2O via a (albeit unfavourable) 2-electron peroxide intermediate step.9,10
In contrast to laccase (Trametes versicolor), BOd (Myrothecium sp.) is capable of reducing O2 in neutral or near-neutral pH conditions.11 The operation of a cathode in neutral pH conditions is favourable for enzymatic devices incorporating glucose-oxidising enzymes such as glucose oxidase (GOx) or glucose dehydrogenase (GDH) that commonly have neutral optimum pH's. This has implications for devices in physiological conditions, pH 7.3–7.4 is being considered to be physiological pH.3,12
The electro-catalytic reduction of O2 by MCOs can be facilitated by an electron-mediated route (mediated electron transfer, MET) or by direct electron transfer between the enzyme and electrode (DET). Common electron mediators for the oxygen reduction reaction (ORR) with BOd or laccase include ABTS‡13,14 and osmium-based redox polymers.15,16 DET of laccase has been reported at gold, graphite and carbon nanotube surfaces, where anthracene moieties induce and enhance communication between the T1 site of the MCO and the supporting electrode.2,17–21 The reduction of O2 by DET can even be observed for laccase on unmodified gold electrodes.22 Furthermore, DET by BOd has also been demonstrated on many electrode architectures,4,5,23 including by the attachment of poly-cyclic moieties to graphite electrodes.24
It is widely reported that the activity of laccase and BOd can be inhibited by halide anions, but BOd is reported to be less-sensitive to Cl− ions.11,12,25 It has been shown that the mechanisms of inhibition of MCOs by F− and Cl− differ; F− has been demonstrated to act as a non-competitive inhibitor that directly disrupts electron transfer between the T1 and TNC of MCOs whereas Cl− has been shown to behave as a competitive inhibitor (with a physiological concentration of around 150 mM), that suppresses the oxidation of substrates (and communication with mediators) at the T1 site.11,22,26,27 Thus, it has been shown that DET with the T1 site can introduce varying degrees of Cl− resistance to MCOs. F−, however, still remains a problem for DET-based cathodes.22
Work by Calvo et al. in 2010 reported the inhibition of laccase (Trametes trogii) by H2O2 during O2 reduction by MET via an osmium redox polymer.15 We have previously demonstrated the inhibition of laccase (Trametes versicolor) by H2O2 on DET-based cathodes; it was shown that GOx anodes produced significant quantities of H2O2 to effectively inhibit laccase in membrane-less glucose/O2 EBFCs.28 This could suggest that H2O2 inhibits MCOs in a similar fashion to F− (non-competitive); this may differ between MCOs.
Fig. 1 presents cyclic voltammograms of BOd cathodes immobilised on a Toray carbon paper electrodes comprised of anthracene-modified multi-walled carbon nanotubes (Ac-MWCNTs). Complete electrode preparation procedures are detailed within the ESI.† Comparison of cyclic voltammograms recorded under argon and under flowing air indicates the DET reduction of O2 by BOd, at an onset potential of approximately 0.57 V (vs. Ag/AgCl) at pH 6.5 and reaching a current density of approximately 0.28 mA cm−2 (at a scan rate of 1 mV s−1). Further potentiostatic experiments (Fig. 2 and in the ESI† in Fig. S3) under air-purging conditions provide catalytic current densities of approximately 0.36 mA cm−2 at 0.2 V (vs. Ag/AgCl). The reduction of O2via DET by these cathodes was also evaluated between pH 4.5 and 7.5 (Fig. S1 and S2A and B, ESI†), with the optimum pH found to be 6.5. The reduction of O2via DET by BOd was also investigated on hydroxylated MWCNTs (OH-MWCNTs) that serve as a precursor to the Ac-MWCNTs. Ac-MWCNTs showed improved cyclic voltammograms, suggesting that the presence of anthracene moieties can favourably orientate BOd to facilitate DET with a lower over-potential for O2 reduction. Furthermore, the reduction of O2via DET by BOd on Ac-MWCNTs gave comparable performance to laccase/Ac-MWCNTs cathodes under similar conditions and optimal pH (ESI,† Fig. S4).
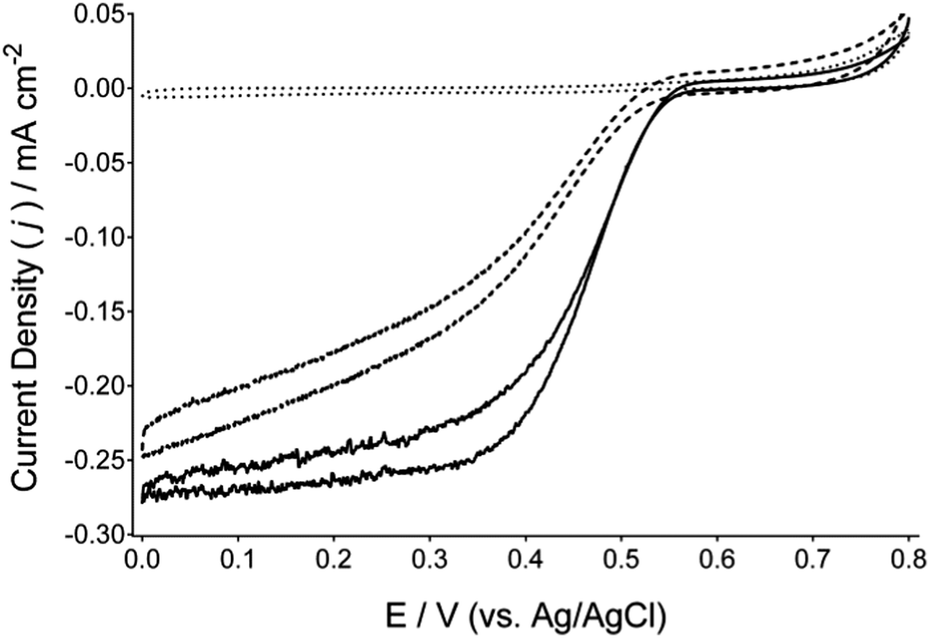 | ||
| Fig. 1 Cyclic voltammograms of BOd/Ac-MWCNTs Toray-paper electrodes under argon purged (dotted line) and air-purged (solid line) hydrodynamic conditions, at a scan rate of 1 mV s−1 (0.2 M citrate/phosphate buffer, pH = 6.5). The dashed line presents a BOd/OH-MWCNTs Toray-paper electrode (same conditions), where the OH-MWCNTs is the precursor to the Ac-MWCNTs. | ||
 | ||
| Fig. 2 Chronoamperometric trace for a (A) BOd/Ac-MWCNTs (pH 6.5) and (B) laccase/Ac-MWCNTs (pH 4.5) Toray-paper electrode in a hydrodynamic citrate/phosphate buffer solution (0.2 M), poised at 0.2 V (vs. Ag/AgCl). n.b. air and argon gasses were purged individually – air purging was terminated at t = 3600 seconds. | ||
Fig. 2A presents the chronoamperometric trace of a BOd/Ac-MWCNTs electrode with additions of air, H2O2 and catalase at differing times during the experiment. Upon the initiation of air-purging, a current response of approximately 0.36 mA cm−2 is observed, for the reduction of O2 by BOd via DET. The addition of ABTS to the electrolyte (final concentration of 0.2 mM) results in an approximate improvement of 11% of the current density, suggesting that most of the immobilised BOd undergoes DET. H2O2 was added to the electrolyte, and after 2000 seconds of exposure (t = 3000 seconds) an addition of catalase was made (final concentration of 20 μg ml−1). After subsequent intermittent periods of argon-purging (with a further addition of catalase), no significant recovery of the catalytic current for the reduction of O2 was observed for both MET and DET. This could suggest that exposure to H2O2 had denatured or strongly (perhaps irreversibly) inhibited the catalytic activity of BOd (measured up to 3000 seconds). Electrode rinsing and electrolyte replacement did not recover any lost catalytic current.
The effect of H2O2 on the laccase/Ac-MWCNTs cathodes was also investigated under similar conditions (Fig. 2B). Although a current density of approximately 0.29 mA cm−2 was observed, the addition of ABTS (0.2 mM) results in an approximate improvement of 59% of the DET current (implying not all immobilised laccase undergoes DET). A concentration of 20 mM H2O2 was found to sharply remove approximately 75% of the total catalytic current, which was recovered to 98% after the first addition of catalase (20 μg ml−1). Argon-purging confirmed enzymatic activity (and no interference from catalase). During argon cycling, the total catalytic current did not return to 98%; no air-purging was performed and therefore the dissolved O2 concentration was recovered via solution perturbation (hydrodynamic) and the decomposition of any remaining H2O2 by catalase.
The catalytic activities of these BOd/Ac-MWCNTs and laccase/Ac-MWCNTs electrodes were also tested in the presence of 150 mM Cl− and 15 mM F− (Fig. S3A and B, respectively (ESI†)). For the BOd/Ac-MWCNTs electrode, the addition of ABTS (final concentration of 0.2 mM) saw an approximate increase in 3% of the catalytic current; the addition of Cl− and F− gave decreases in total catalytic current of approximately 15% and 11%, respectively. Furthermore, argon-purging the solution removed catalytic activity. For the laccase/Ac-MWCNTs electrode, the addition of ABTS (final concentration of 0.2 mM) gave an increase of approximately 70% of the catalytic current from DET. The addition of Cl− removed the current resulting from mediated electro-catalysis, with further removal of approximately 19% of the DET current. The addition of F− completely inhibited catalytic activity of laccase. Control experiments with bovine serum albumin as a protein showed no significant response upon the addition of any of ABTS, Cl−, F− and H2O2 (data not shown). The catalytic activity of laccase (determined by chronoamperometry) in the presence of H2O2 was also studied (Fig. S5, ESI†).
Specific enzymatic activities were spectrophotometrically determined to be 12.1 ± 0.2 U mg−1 and 10.3 ± 0.2 U mg−1 for BOd (Myrothecium sp., pH 6.5) and laccase (Trametes versicolor, pH 4.5) respectively, using ABTS as the substrate. For BOd, this decreased by approximately 65% after 5 min of exposure to a 25 mM H2O2 solution. The rapid removal of H2O2 by catalase did not recover specific enzymatic activity (ESI†).
Although the laccase/Ac-MWCNTs electrodes presented within this study are sharply and significantly inhibited by H2O2, the addition of catalase can effectively restore enzymatic activity. Thus, the co-immobilisation of catalase (or other H2O2-decomposing moiety) within EBFC electrodes (albeit effective) could be replaced with “on the spot” treatments to remove H2O2. For BOd, however, it is suggested that catalase (or another H2O2-decomposing moiety) is permanently required within EBFCs that are likely to produce H2O2; this study suggests that BOd is less resistant to H2O2 than laccase and that enzymatic activity cannot be easily restored by the removal of H2O2 (albeit at relatively high concentrations). Further studies are required to determine whether the concentrations of H2O2 produced as a function of anodic side reactions (for example, by GOx) in common EBFCs, are sufficient to be detrimental to the performance of BOd-containing devices.
The authors thank Sekisui Diagnostics (UK) for their kind donation of BOd. This research was financially supported by the U.K. Engineering and Physical Sciences Research Council's SuperGen Biological Fuel Cells Consortium (EPSRC contract EP/H019480/1). The authors thank National Science Foundation Grant #105797 and the Air Force Office of Scientific Research for funding.
Notes and references
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- M. Minson, M. T. Meredith, A. Shrier, F. Giroud, D. Hickey, D. T. Glatzhofer and S. D. Minteer, J. Electrochem. Soc., 2012, 159, G166–G170 CrossRef CAS PubMed.
- M. Shao, M. N. Zafar, M. Falk, R. Ludwig, C. Sygmund, C. K. Peterbauer, D. A. Guschin, D. MacAodha, P. ÓConghaile, D. Leech, M. D. Toscano, S. Shleev, W. Schuhmann and L. Gorton, ChemPhysChem, 2013, 14, 2260–2269 CrossRef CAS PubMed.
- M. T. Meredith and S. D. Minteer, Annu. Rev. Anal. Chem., 2012, 5, 157–179 CrossRef CAS PubMed.
- E. Katz and K. MacVittie, Energy Environ. Sci., 2013, 6, 2791–2803 CAS.
- N. Mano and L. Edembe, Biosens. Bioelectron., 2013, 50, 478–485 CrossRef CAS PubMed.
- L. Quintanar, C. Stoj, A. B. Taylor, P. J. Hart, D. J. Kosman and E. I. Solomon, Acc. Chem. Res., 2007, 40, 445–452 CrossRef CAS PubMed.
- S. Shleev, J. Tkac, A. Christenson, T. Ruzgas, A. I. Yaropolov, J. W. Whittaker and L. Gorton, Biosens. Bioelectron., 2005, 20, 2517–2554 CrossRef CAS PubMed.
- S. Brocato, C. Lau and P. Atanassov, Electrochim. Acta, 2012, 61, 44–49 CrossRef CAS PubMed.
- M. R. Tarasevich, V. A. Bogdanovskaya and L. N. Kuznetsova, Russ. J. Electrochem., 2001, 37, 833–837 CrossRef CAS.
- U. Salaj-Kosla, S. Poeller, Y. Beyl, M. D. Scanlon, S. Beloshapkin, S. Shleev, W. Schuhmann and E. Magner, Electrochem. Commun., 2012, 16, 92–95 CrossRef CAS PubMed.
- X. Wang, M. Falk, R. Ortiz, H. Matsumura, J. Bobacka, R. Ludwig, M. Bergelin, L. Gorton and S. Shleev, Biosens. Bioelectron., 2012, 31, 219–225 CrossRef CAS PubMed.
- P. Jenkins, S. Tuurala, A. Vaari, M. Valkiainen, M. Smolander and D. Leech, Bioelectrochemistry, 2012, 87, 172–177 CrossRef CAS PubMed.
- K. Sadowska, K. Stolarczyk, J. F. Biernat, K. P. Roberts, J. Rogalski and R. Bilewicz, Bioelectrochemistry, 2010, 80, 73–80 CrossRef CAS PubMed.
- P. Scodeller, R. Carballo, R. Szamocki, L. Levin, F. Forchiassin and E. J. Calvo, J. Am. Chem. Soc., 2010, 132, 11132–11140 CrossRef CAS PubMed.
- J. Gallaway, I. Wheeldon, R. Rincon, P. Atanassov, S. Banta and S. C. Barton, Biosens. Bioelectron., 2008, 23, 1229–1235 CrossRef CAS PubMed.
- M. S. Thorum, C. A. Anderson, J. J. Hatch, A. S. Campbell, N. M. Marshall, S. C. Zimmerman, Y. Lu and A. A. Gewirth, J. Phys. Chem. Lett., 2010, 1, 2251–2254 CrossRef CAS PubMed.
- C. F. Blanford, R. S. Heath and F. A. Armstrong, Chem. Commun., 2007, 1710–1712 RSC.
- M. T. Meredith, M. Minson, D. Hickey, K. Artyushkova, D. T. Glatzhofer and S. D. Minteer, ACS Catal., 2011, 1, 1683–1690 CrossRef CAS.
- N. Lalaoui, K. Elouarzaki, A. L. Goff, M. Holzinger and S. Cosnier, Chem. Commun., 2013, 49, 9281–9283 RSC.
- M. Bourourou, K. Elouarzaki, N. Lalaoui, C. Agnes, A. Le Goff, M. Holzinger, A. Maaref and S. Cosnier, Chem.–Eur. J., 2013, 19, 9371–9375 CrossRef CAS PubMed.
- U. Salaj-Kosla, S. Poeller, W. Schuhmann, S. Shleev and E. Magner, Bioelectrochemistry, 2013, 91, 15–20 CrossRef CAS PubMed.
- D. Leech, P. Kavanagh and W. Schuhmann, Electrochim. Acta, 2012, 84, 223–234 CrossRef CAS PubMed.
- L. dos Santos, V. Climent, C. F. Blanford and F. A. Armstrong, Phys. Chem. Chem. Phys., 2010, 12, 13962–13974 RSC.
- J. Hirose, K. Inoue, H. Sakuragi, M. Kikkawa, M. Minakami, T. Morikawa, H. Iwamoto and K. Hiromi, Inorg. Chim. Acta, 1998, 273, 204–212 CrossRef CAS.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- C. Vaz-Dominguez, S. Campuzano, O. Rudiger, M. Pita, M. Gorbacheva, S. Shleev, V. M. Fernandez and A. L. De lacey, Biosens. Bioelectron., 2008, 24, 531–537 CrossRef CAS PubMed.
- R. D. Milton, F. Giroud, A. E. Thumser, S. D. Minteer and R. C. T. Slade, Phys. Chem. Chem. Phys., 2013, 15, 19371–19379 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S5, experimental details and procedures. See DOI: 10.1039/c3cc47689h |
| ‡ ABTS = 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid). |
| This journal is © The Royal Society of Chemistry 2014 |