 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of advanced catalyst development for Fischer–Tropsch synthesis of hydrocarbons from biomass derived syn-gas
Hessam
Jahangiri
a,
James
Bennett
*b,
Parvin
Mahjoubi
c,
Karen
Wilson
*b and
Sai
Gu
a
aCranfield University, Whittle Building, Cranfield, Bedfordshire, MK43 0AL, UK. E-mail: s.gu@cranfield.ac.uk
bEuropean Bioenergy Research Institute (EBRI), Aston University, The Aston Triangle, Birmingham, B4 7ET, UK. E-mail: k.wilson@aston.ac.uk
cSchool of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: parvin.mahjoubbi@gmail.com
First published on 9th May 2014
Abstract
Fischer–Tropsch synthesis (FTS) is a process which converts syn-gas (H2 and CO) to synthetic liquid fuels and valuable chemicals. Thermal gasification of biomass represents a convenient route to produce syn-gas from intractable materials particularly those derived from waste that are not cost effective to process for use in biocatalytic or other milder catalytic processes. The development of novel catalysts with high activity and selectivity is desirable as it leads to improved quality and value of FTS products. This review paper summarises recent developments in FT-catalyst design with regards to optimising catalyst activity and selectivity towards synthetic fuels.
 Hessam Jahangiri | Hessam Jahangiri obtained his master degree in Chemical Engineering at the University of Birmingham in 2012. He subsequently joined Cranfield University and in 2013 began work on a collaborative programme with the European Bioenergy Research Institute (EBRI) at Aston University to pursue his PhD degree with an emphasis on biomass thermal conversion to produce bio-oils coupled with upgrading the bio-oil by zeolite cracking and co-processing in existing refineries under the supervisions of Professors Sai Gu and Karen Wilson. |
 James Bennett | James Andrew Bennett obtained his Master and PhD at the University of Leicester, where he investigated the use of perfluoroalkyl moieties to allow heterogenisation of homogeneous catalysts over zirconium phosphonate supports. He then worked at the University of Birmingham, researching biogenic heterogeneous catalysts composed of transition metal nanoparticles supported on bacterial biomass, using waste sources of metals and biomass to produce “green” catalyst materials. He is currently working with Professors Karen Wilson and Adam Lee at the European Bioenergy Research Institute (EBRI) at Aston University, developing environmentally sustainable catalysts derived from Red Mud industrial waste for pyrolysis oil upgrading. |
 Parvin Mahjoubi | Parvin Mahjoubi gained her bachelor and master degrees in Chemical Engineering at Amirkabir University of Technology (Tehran Polytechnic) and University of Birmingham, respectively. |
 Karen Wilson | Karen Wilson holds a Chair of Catalysis and is Research Director of the European Bioenergy Research Institute at Aston University, where she also holds a Royal Society Industry Fellowship in collaboration with Johnson Matthey. Karen's research interests lie in the design of heterogeneous catalysts for clean chemical synthesis, particularly the design of tuneable porous materials for sustainable biofuels and chemicals production from renewable resources. Karen has a BA (Hons) in Natural Sciences from the University of Cambridge (1992), an MSc with distinction in heterogeneous catalysis from the University of Liverpool (1993) and a PhD (1996) in heterogeneous catalysis and surface science from the University of Cambridge. Following post-doctoral research at Cambridge and the University of York, Karen was appointed to her first independent academic position at York in 1999 where she stayed until 2009 after appointment to a Readership in Physical Chemistry at Cardiff University. |
 Sai Gu | Sai Gu holds a Chair of Bioenergy Technology and is the Director of Centre for Renewable and Biofuels in Cranfield University. He obtained a Ph.D from the University of Nottingham and further developed his research career at the University of Cambridge as a post-doc researcher. He is leading one of the largest energy research teams in the UK, covering a wide range of biofuel and clean energy research. His team has won numerous awards for high quality publications including the UK Scopus Young Researcher Award in Engineering and the Felix Weinberg Prize from the Institute of Physics. |
1. Introduction
Mounting concerns over dwindling petroleum oil reserves, in concert with growing governmental and public acceptance of the anthropogenic origin of rising CO2 emissions and associated climate change, is driving academic and commercial routes to utilise renewable feedstocks as sustainable sources of fuel and chemicals. The quest for such sustainable resources to meet the demands of a rapidly rising global population represents one of this century's grand challenges.1 Sustainable Catalytic Conversion of renewable substrates derived from biomass offers the most readily implemented, and low cost, solution for transportation fuels,2 and the only non-petroleum route to organic molecules for the manufacture of bulk, fine and speciality chemicals and polymers3 required to meet future societal demands.4,5 In order to be considered truly sustainable, biomass feedstocks must be derived from sources which do not compete with agricultural land use for food production, or compromise the environment e.g. via deforestation.6 Potential feedstocks include cellulosic or oil based materials derived from plant or aquatic sources, with the so-called biorefinery concept offering the co-production of fuels, chemicals and energy,7 analogous to today's petroleum refineries which deliver high volume/low value (e.g. fuels and commodity chemicals) and low volume/high value (e.g. fine/speciality chemicals) products, maximizing biomass valorisation.8Thermochemical conversion of biomass can be performed by gasification, pyrolysis or combustion,9 with gasification the most widely used commercial route to convert biomass into synthesis gas the major constituents of which are carbon monoxide (CO), hydrogen (H2) and methane (CH4). Thermal gasification of biomass represents a convenient route to produce syngas from intractable materials particularly waste.10 In 1922 Franz Fischer and Hans Tropsch developed a heterogeneously catalysed process (Fischer–Tropsch synthesis (FTS)) for the transformation of synthesis gas (syngas, CO + H2) into different hydrocarbons fractions (diesel fuels, gasoline, lower olefins, etc.) (Fig. 1).11,12 The motivation for this work was to allow nations with no natural oil reserves to produce liquid fuels for transportation from coal. Liquid fuels are preferred for use in transportation since they have higher energy density than coal but are more easily and safely stored than gas, which has the highest energy density of the three phases.
 | ||
| Fig. 1 The three stages of Fischer–Tropsch synthesis. Adapted with permission from ref. 12, Copyright © 2010, Elsevier. | ||
The commercialisation of FTS began in 1936 in Germany with >1 million tons of FTS liquid produced annually in the 1940s.13 The first coal-based construction plant was built in Sasolburgh in South Africa in 1952 due to the cheap domestic coal available in this country.13 With the need for non-fossil derived fuels there is now growing interest in the use of FTS-processes to convert such (waste) biomass derived syn-gas to liquid hydrocarbons for synthetic fuels or chemical feed-stocks.
The following exothermic reactions take place during FT synthesis, with alkanes and alkenes the desired products:12
| Alkane production: (2n + 1) H2 + nCO → CnH2n+2 + nH2O | (1) |
| Alkene production: 2nH2 + nCO → CnH2n + nH2O | (2) |
| Water-gas shift reactions: CO + H2O ↔ CO2 + H2 | (3) |
The water-gas shift (WGS) reaction also takes place over most of FT catalysts (reaction (3)) and provides a means to alter the CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 distributions. Side reactions such as those producing alcohols and undesired carbonaceous deposits via Boudouard reactions may occur (reactions (4) and (5)).
:H2 distributions. Side reactions such as those producing alcohols and undesired carbonaceous deposits via Boudouard reactions may occur (reactions (4) and (5)).
| Alcohols: 2nH2 + nCO → CnH2n+2O + (n − 1)H2O | (4) |
| Boudouard reaction: 2CO → C + CO2 | (5) |
The FTS has been developed to allow the transformation of various feed stocks into liquid products, including gas-to-liquid (GTL), coal-to-liquid (CTL) and biomass-to-liquid (BTL) technologies. In addition to natural gas, coal and biomass can be converted to syngas by partial oxidation, steam reforming or gasification processes. Moreover, different hydrocarbons may be directly produced from syngas by developing highly selective FT catalysts. Therefore, FT synthesis is viewed as a valuable process to produce super-clean fuels from syngas derived from non-petroleum resources as shown earlier in Fig. 1.12
The FT reaction is a polymerisation process, involving adsorption, chain initiation and chain growth termination. Fig. 2 illustrates the dissociative adsorption of carbon monoxide on metal atoms and carbide species formation. Insertion of adsorbed dissociated hydrogen into this carbide species produces the active CH2 intermediate which leads to the propagation step. The resulting alkyl chain desorbs from the metal after hydrogenation and β-scission, forming olefins or paraffin. Alternative mechanistic pathways have been proposed, but all of them have involved initiation, propagation and termination steps.15
 | ||
| Fig. 2 Classic mechanism pathway. Reprinted with permission from ref. 14, Copyright © 2009, Elsevier. | ||
The resulting FTS hydrocarbon products follow an arithmetical distribution identified as the Anderson–Schulz–Flory (ASF) distribution. Eqn (1) and (2) show that the chain growth probability (α) describes the weight fraction of a single product in FTS which depends on the rates of propagation and termination where Mn/n is the product weight fraction and rp and rt are rate constants of propagation and termination, respectively.15
| Mn/n = (1 − α)2α(n−1) | (1) |
| α = rp/(rp + rt) | (2) |
As can be seen in Fig. 3, according to the Anderson–Schulz–Flory distribution law, more than 50 wt% of the product mixture is C5–C22 hydrocarbons for an α-parameter of 0.7 to 0.875. At higher values of α-parameter the fraction of C5–C22 hydrocarbon products decreases sharply. Low molecular-weight, gaseous products, such as methane, are of lower commercial value than longer chain products. Methane is normally underestimated by the ASF distribution and it decreases to a few wt% at the high alpha values. Therefore to maximise the profitability of the FTS, conditions which favour high α-parameters should be used to optimise the selectivity to the more valuable C5–C22 hydrocarbon products.14
 | ||
| Fig. 3 Anderson–Schulz–Flory (ASF) values sensitivity. Reprinted with permission from ref. 14, Copyright © 2009, Elsevier. | ||
2. Key factors for FTS catalyst design
The design of heterogeneous catalysts with optimum performances for a given process requires a consideration of a combination of chemical, physical and mechanical properties. Anderson et al. has defined a “triangular concept” for catalyst design which has been adapted for FTS by Farrauto and Bartholomew, as illustrated in Fig. 4. In addition the physic-chemical properties of the active phase, specifically formulation shape, size and crystallinity of the nanoparticles are significant factors for optimisation during catalyst design.16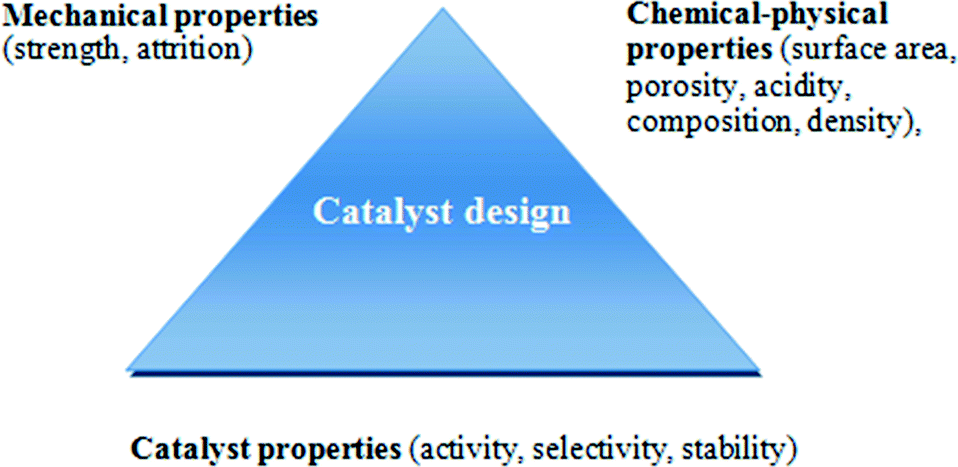 | ||
| Fig. 4 Concept of triangle of catalyst design. Reprinted with permission from ref. 16, Copyright © 2006, Wiley and Sons. | ||
2.1 Active phase identification
It has been reported that the activity of FT catalysts are dependent on H2 adsorption capacity, ability to dissociatively adsorb CO the reducibility of other metal oxide components. Based on literature reviews14,17,18 it shown that transition metals belonging to groups III–VI of the periodic table are ineffective for FTS. While, they are desirable for dissociative adsorption of CO their tendency to form highly stable oxides, means catalysts are not easily reduced under usual FTS conditions. Moreover, transition metals belonging to groups XI and XII plus Ir, Pt and Pd are favour non-dissociative CO adsorption and are not sufficiently active in FTS. Fe, Co, Ni, Ru and Os are commonly accepted to be the best catalytic materials for use FT synthesis, with occasionally Re and Rh reported to exhibit acceptable catalytic activity for FTS.Ru is one of the most active catalysts for FTS operating at low reaction temperature producing long chain hydrocarbons without the need for any promoters. However, it is very expensive and a limited world resource and therefore it is not considered a sustainable option for use in industrial processes. Nickel, while another suggested catalyst for FT process, has a high hydrogenation activity and thus undesired high selectivity to methane production. Therefore, Co and Fe are the deemed to be the best metals for application in industrial scale FTS processes.19
Fe while economically attractive and highly abundant, has a very low selectivity to paraffins, favouring the production of olefins and oxygen and unfortunately deactivates more quickly than Co based catalysts.20 Although Co is more expensive than Fe, Co has a good selectivity to paraffins, low selectivity to olefins and oxygen, and is more resistant to deactivation.
Therefore, Co is the preferred choice to create long chain paraffins, while Fe is the better selection to produce olefins. To make a more informed selection between Fe and Co, the nature of the carbon feedstock is an essential factor that must be considered. Fe has a high activity for water gas shift (WGS) and hence it is appropriate for hydrogen-poor feed stocks derived from biomass or coal.20 Fe is thus useful for syngas conversion with lower ratios of H2/CO = 0.5–2.5 obtained from biomass or coal, but it is not suitable for conversion of H2-rich syngas derived from methane. Consequently, the Fe based catalysts are better for alkene production from syn-gas, BTL and CTL technologies. On the other hand, Co provides better catalytic performance at high H2/CO ratios (i.e. 2 and above) and is therefore, a better selection where the carbon feed stock is natural gas. Due to the instability of Fe, more modification is generally required to improve activity and selectivity, with rapid catalyst deactivation still the major challenge for Fe based catalysts.21
FT processes are usually categorised based on operating conditions; typically high-temperature FT (HTFT) operates at 300–350 °C and low-temperature FT (LTFT) operates at 200–240 °C.20,22 The typical reactors designed for FT processes are fixed bed, slurry bubble column reactor, and circulating and fluidised-bed reactor,23 with fluidised bed reactors mostly applied for HTFT processes using Fe catalysts to produce C1–C15 hydrocarbon fractions. Slurry-phase and fixed-bed reactors are typically used for LTFT process with both Co and Fe catalysts to produce linear long-chain hydrocarbons (such as paraffins and waxes). Although HTFT processes are mostly used for liquid fuels production, some valuable chemicals, such as gasoline and low molecular weight olefins, can be extracted. Recent developments in FT processes are based on LTFT technologies and involve syngas with a high H2/CO ratio-generated by vapour reforming or auto-thermal reforming. In LTFT processes, Fe catalysts produce waxy materials such as paraffins and high molecular mass linear waxes, while Co catalysts provide better catalytic performance for long-chain hydrocarbons synthesis because of their high conversion, stability and high hydrocarbons productivity.7,20,22
2.2 Catalyst promoters
Effective reduction of the active phase plays an important role in optimising catalyst performance, with the addition of small quantities of promoters during the formulation of the catalyst found to significantly enhance the reducibility of Co and Fe. Furthermore, promoters improve the activity and selectivity of heterogeneous catalysts by influencing the catalyst's structural properties through varying the active phase structure, or modifying the electronic character of the active phase. Generally, metal oxides (MnO and ZnO), alkali metals (Na, K, Rb, Cs) and certain transition metals (Cu, Pd, Pt, Ru) or carbonates are applied as promoters for the Fe and Co-based FT catalysts.242.3 Catalyst support shape and size
The third crucial factor that must be considered for catalyst design is the mechanical resistance, morphology and porosity of the support. Accordingly, the selection of support which stabilises the resulting Co or Fe nanoparticles is critical in determining FT-catalyst activity and stability. Altering the support's surface structure and pore size may improve the metal dispersion, reducibility and the diffusion coefficients of reactants and products. Al2O3, SiO2, TiO2 and ZrO2 are used as catalyst supports in the FTS due to their high surface area and strong mechanical strength. Low metal-support interactions are important for catalyst activity and stability. The strong Co/Al2O3 interaction decreases reducibility and catalyst activity. On the contrary, the Co/SiO2 interaction is weak and leads to superior catalyst reducibility and activity.20,25Furthermore, periodic mesoporous silicas and carbon supports are considered as the model systems to investigate FT catalyst performance.26,27 For the production of hydrocarbons between C5 and C12, which are gasoline-range hydrocarbons (GRHs), further processes are required. Conventional FT-catalysts combined with acidic zeolite catalysts have been advanced as attractive hybrid catalysts for GRHs production.28
2.4 Nature of Co and Fe nanoparticles in FT-catalysts
The product selectivity and activity FT-catalysts in CO conversion depends not only on the nature of the catalyst but also on the reactor design and operating conditions. Several publications and reviews have studied the effect of the reactor design and operating conditions in FTS.13,22,29–32 The nature of the exposed facets and size of Co and Fe and associated activity in FTS is expected to be controlled by the nature of the support and promoters.Catalyst activity may be defined in a number of ways for the purpose of comparison: measurement of the turnover frequency (TOF) (or activity per site), conversion per catalyst volume, conversion per gram of catalyst and conversion per surface area. In the case of a supported metallic cobalt catalyst, the number of cobalt surface atoms can be measured by pulse titration of hydrogen or carbon monoxide chemisorption on the reduced catalyst.33 Identification of the active site in Fe-based catalysts by this method is however complicated by the fact that Fe is often present as a combination of different phases including iron oxide, metallic iron and iron carbides under operating conditions. The active species for iron catalysts remains the subject of debate and is often ill-defined, with some researchers claiming Fe3O4 is the active catalyst, while others claim that it is the carbide phases in FTS.32,33 Therefore, TOF values based on total metallic surface Fe are often not reliable for assessing FT-catalyst performance.33
In contrast TOF values for Co catalysts are simpler to compare, enabling insight into the most active crystal phase can be gleaned. Metallic cobalt exists as different crystal structures, with hcp (α) and fcc (β) unit cells. Crystalline cobalt is stable as a pure fcc unit cell with a diameter <20 nm.35 In FTS, the Co–hcp catalysts are found to produce much higher CO conversions than Co–fcc catalysts (Fig. 5). Furthermore, Co–hcp catalysts produce more C5+ hydrocarbons in comparison with fcc phases. The activity and selectivity of Co/SiO2 (20 wt% cobalt) catalysts as both hcp and fcc phases are shown in Table 1.34
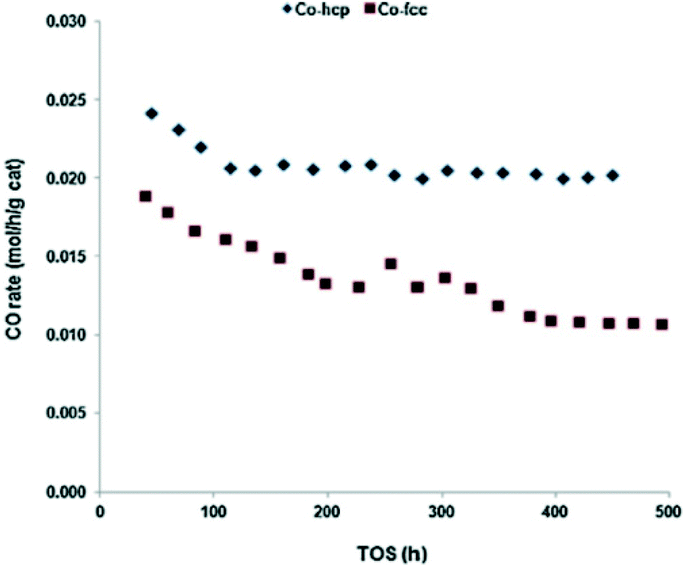 | ||
| Fig. 5 CO conversion rate as a function of time-on-stream (TOS) for Co/SiO2 (20 wt% cobalt) catalysts (Co-hcp, Co-fcc) with conditions: 200 °C, P = 19.9 bar, H2/CO = 2, syngas SV = 3. Reprinted with permission from ref. 34, Copyright © 2013, Elsevier. | ||
| Catalyst | TOS (h) | CO. conv. (%) | Selectivity (C-%) | TOF (s−1) | |
|---|---|---|---|---|---|
| CH4 | C5+ | ||||
| Co-hcp | 16 | 63.1 | 3.5 | 88.2 | 0.095 |
| 450 | 55.5 | 4.2 | 90.2 | 0.068 | |
| Co-fcc | 13 | 45.1 | 6.4 | 86.1 | 0.092 |
| 492 | 32.1 | 7.2 | 82.9 | 0.049 | |
The effect of cobalt particle size in the range of 2.6 to 27 nm has been investigated by Bezemer et al. in FTS.36 The performance of the FTS catalyst was found to be independent of cobalt particle size for diameters >8 nm. Reducing the average diameter of cobalt particles to <6 nm resulted in decreased FTS activity and selectivity when compared to particles >8 nm.
2.5 Bimetallic Fe/Co catalysts
Bimetallic catalysts also show great promise for application in FTS and their use is reviewed by Calderone et al.37 Most studies of bimetallic catalysts have focused on a combination of conventional FTS transition metals which are Co, Fe, Ru and Ni, with some reports demonstrating an improvement in catalyst activity upon combining two metals.37–41 Co and Fe are the most commonly used metals in FTS, as previously mentioned, with bimetallic catalysts generally more selective towards to C6+ hydrocarbons.37 The order of impregnation of metal over the support is also important, and Arai et al. reported that FeCo/TiO2 catalyst in which iron is added to the support first, followed by cobalt has a higher activity in FTS in comparison with pure metal catalysts.38In contrast, Duvenhage reported that while CoFe/TiO2 catalysts had superior performance to monometallic Fe/TiO2 this was inferior to pure Co/TiO2.42 However, both CoNi/TiO2 and NiFe/TiO2 are more active catalysts than pure metal analogues.37 Likewise, FeCo/silica is reported to have improved CO conversion in comparison with Co/silica, with the former producing mainly C1–C4 hydrocarbons, whereas the latter produces C5+ hydrocarbons.43 In bimetallic catalysts, the olefin/paraffin product ratio is generally found to be increased by the addition of metallic Fe,44 while the addition of Fe to Co increases alcohol production.44
In industrial-scale FTS reactors, the cost of using vast quantities of Co catalyst is a significant limiting factor. The prohibitive cost of the Co active phase can be alleviated by replacing the bulk of the material, i.e. the core of the catalyst particles, with a cheaper material, such as iron oxide. Calderone et al. prepared FTS catalysts composed of iron oxide cores coated with a shell of metallic cobalt. A range of core/shell ratios were tested, with the resulting FeCo particles supported on nanosized alumina powder at a loading of 20 wt% cobalt. The core–shell catalysts were less active than pure iron catalysts but maintained constant activity throughout the test duration, whereas the pure Fe catalyst rapidly deactivated. A cost analysis of such an approach for a range of metal oxide cores covered by a thin Co shell is shown in Fig. 6 which reveals the potential savings at different core size/particle size ratios using various metal oxide/cobalt combinations.45
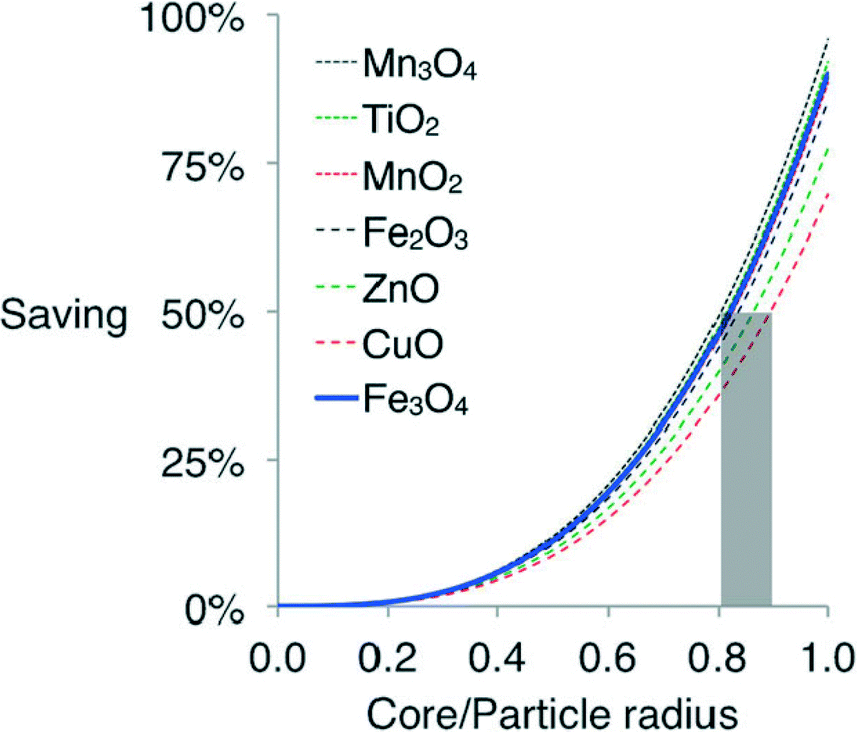 | ||
| Fig. 6 Comparative cost of spherical cobalt shells with different metal oxide cores. Reprinted with permission form ref. 45, Copyright © 2013, Wiley and Sons. | ||
3. Deactivation of cobalt catalysts
In order to extend catalyst lifetime, the deactivation mechanism of the FTS catalysts must be understood so that they may be prevented and to allow effective regeneration of the active phase. Due to the relatively high cost of the metal, the deactivation of cobalt is a very important factor in FTS development.20 The following mechanisms have been proposed for cobalt deactivation:46,47•Oxidation of active metallic Co to inactive CoOX
•Poisoning by sulphur and nitrogen present in the synthesis gas feed (especially in the CTL processes)
•Formation of Co-support compounds such as cobalt aluminates and cobalt silicates
•Sintering of Co crystallites leading to reduction of surface area
•Formation of carbon or “coking”
•Carbon-induced reconstruction of surfaces
During the last 15 years, the vast majority of the research on Co catalyst deactivation has focused on the mechanism of oxidation as the cause of reduced activity. According to the 133 papers reviewed, around 60% of researches focus on the oxidation issue for cobalt catalyst deactivation because oxidation is the main cobalt catalysts deactivation in FTS (Fig. 7).46 These factors will be considered in turn.
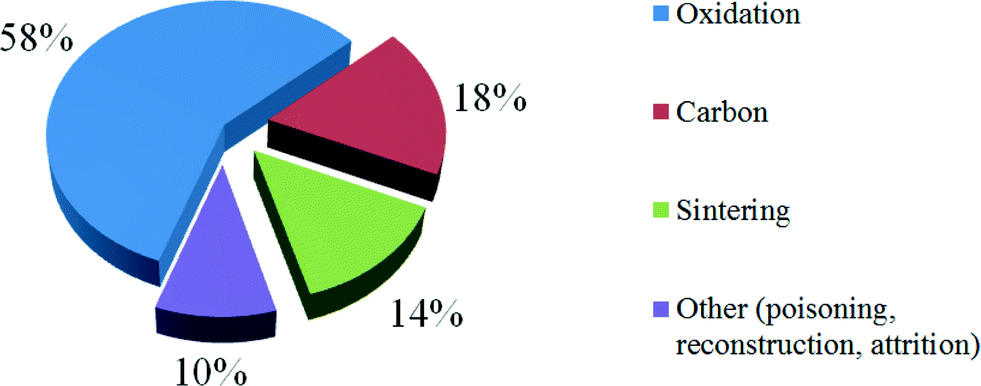 | ||
| Fig. 7 Current focus on cobalt catalyst deactivation according to research articles from 1995 to 2009 in FTS. Reprinted with permission from ref. 46, Copyright © 2010, Elsevier. | ||
3.1 Deactivation by oxidation
Oxidation of metallic Co species leads to deactivation of the catalyst and reduced reaction rates during FTS. The formation of cobalt oxide, formed by oxidation of metallic Co with water, is a possible reason for catalyst deactivation during FTS, since water is a by-product of the reaction.20,48 In reality, the oxidation of bulk metallic Co bulk Co(II)O is not feasible during FTS, as p(water) relative p(hydrogen) must be >128 at 493 K for bulk oxidation of metallic Co to be thermodynamically feasible (Fig. 8).49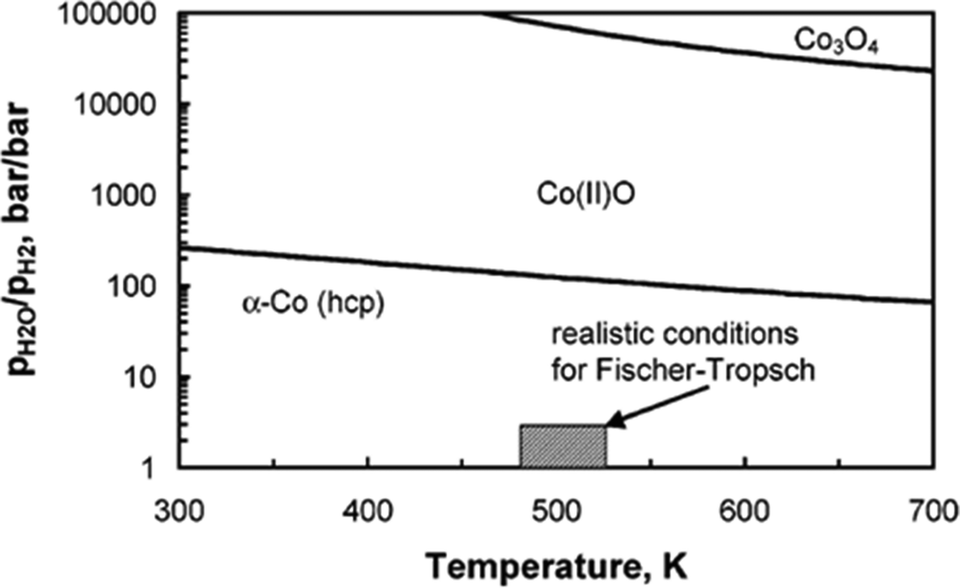 | ||
| Fig. 8 Metallic cobalt bulk stability diagram and different cobalt oxide phases as function of temperature and ratio of water partial pressure to hydrogen partial pressure. Reprinted with permission from ref. 49, Copyright © 2005, American Chemical Society. | ||
The oxidation of small cobalt crystallites or formation of an oxide shell might be possible under conditions which do not allow the formation of bulk cobalt oxide. Therefore, it can be anticipated that nano-sized crystalline material is less resistant to oxidation than the bulk crystalline materials.49 The oxidation of nano-sized metallic cobalt to cobalt oxide has been proposed as a major deactivation mechanism in FTS.48 However, in the recent years research has concluded that the oxidation of bulk or surface metallic cobalt is not a significant deactivation mechanism for supported Co catalysts with an average crystallite size of ≥2 nm in FTS.46,50
3.2 Deactivation by poisoning
The sulphur-compounds present in natural gas and coal act as poisons for FT catalysts. It has been illustrated that the presence of sulphur decreases FT catalyst activity and this decrease is proportional to the concentration of sulphur in the syngas.The CO conversion is found to decrease for Co/Al2O3 with increasing amount of sulphur in the feed (0, 10, 100, 250 and 2000 ppmw) at conditions 220 °C, 20 bar, 2 molH2 molCO−1, 5000 cm3 (STP)CO+H2 h−1 gcat−1. The selectivity of different products (methane, ethylene, C5+ and olefins) is represented in Fig. 9.51
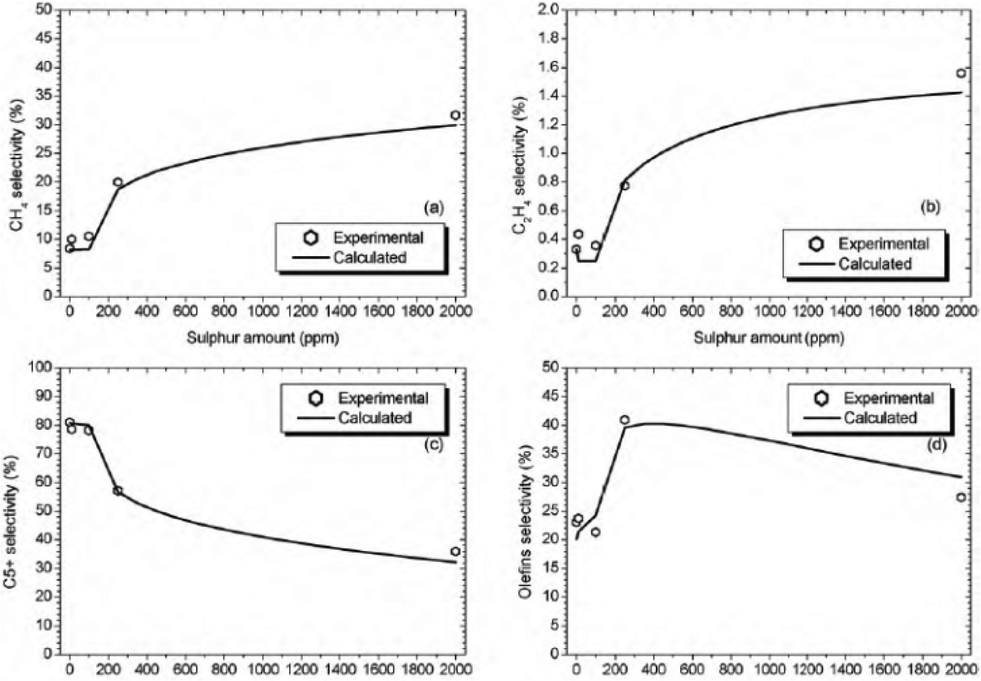 | ||
| Fig. 9 Methane (a), ethylene (b), C5+ (c) and olefins (d) selectivities with different amounts of sulphur on Co/Al2O3 under equal conditions (220 °C, 20 bar, 2 mol H2 mol−1 CO, 5000 cm3 (STP) CO + H2 h−1 gcat−1). Reprinted with permission from ref. 51, Copyright © 2010, Elsevier. | ||
The formation of C5+ hydrocarbons decreases with increasing sulphur loading and therefore the selectivity of reactions drives towards light hydrocarbons. At low amounts of sulphur (<100 ppm), CO conversion is reduced, while the product distribution (selectivity) is not changed significantly.51 Cobalt-based catalyst poisoning in FTS by nitrogen-containing compounds such as HCN and NH3 has been reported to deactivate cobalt catalysts rapidly.52,53 However this is reversible as the presence of nitrogen has less of a poisoning effect and can be removing by hydrogen treatment.51,54
3.3 Deactivation by sintering
Cobalt crystallite sintering is another possible cause of catalyst deactivation and was identified as the main cause of deactivation of a Co/SiO2 FTS catalyst.55,56 In addition, extended X-ray absorption fine structure (EXAFS) studies of a 20 wt% Co/Al2O3 catalyst showed a large increase in the degree of Co–Co metal coordination in the used catalysts relative to fresh catalyst. This indicated substantial growth of the metal particles due to sintering which led to catalyst deactivation. The addition of promoters such as Pt and Ru (0.05 wt%) was found to lead to improved activity but made catalysts more prone to deactivation by sintering.57,58Long term lifetime studies of Co FT-catalysts in a slurry bubble column reactor reveal the most severe sintering occurs over the first 20 days which caused the initial deactivation because of loss of active surface area. The degree of sintering slows after 20 days (Fig. 10).59
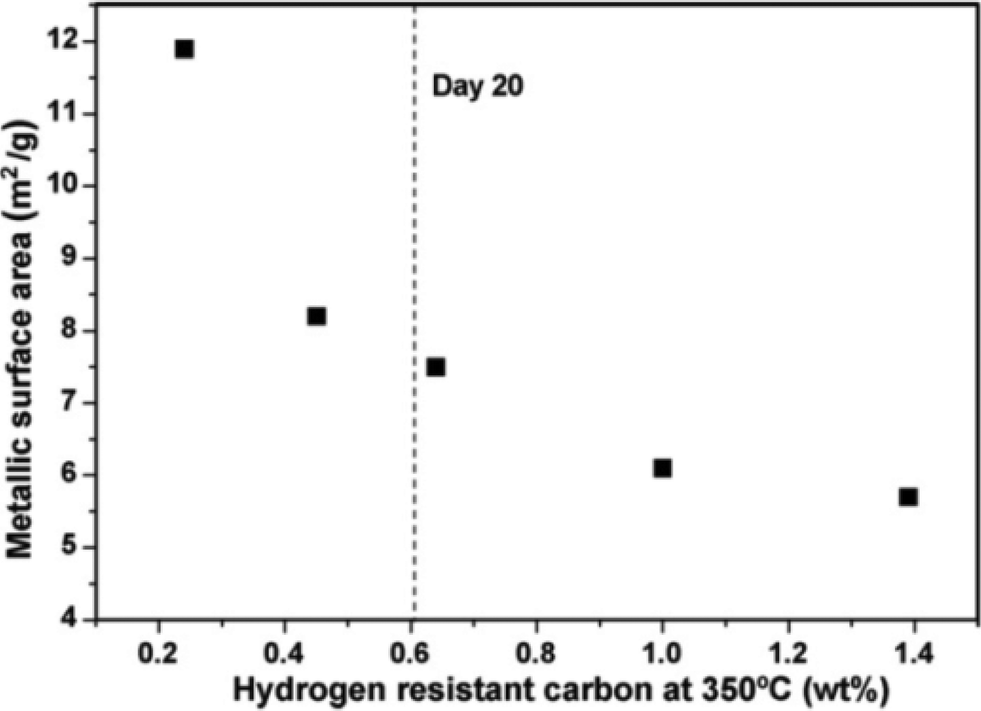 | ||
| Fig. 10 Metallic surface areas as s function of the H2 resistant polymeric carbon for Co/Pt/Al2O3 (20 wt% Co, 0.05 wt% Pt) catalyst in slurry bubble reactor. Sintering does not decrease sharply after 20 days. Reprinted with permission from ref. 59, Copyright © 2009, Elsevier. | ||
3.4 Deactivation by carbide formation
After prolonged periods of time on stream, cobalt-based FT catalysts the formation of carbides such as Co2C or Co3C have been reported. Normally, the role of carbides in deactivation of Co catalysts is minor compared to that in Fe FT-catalysts because the rate of carbon diffusion into Co to form carbides is negligible. The possibility of cobalt carbide formation is low under typical FTS conditions and all carbon is converted to hydrocarbon products. Therefore, although Co2C and Co3C are stable between temperatures of 500–800 °C, a typical operating temperature range for FTS, such phases have not been observed during FTS.21 Doping of metallic Co with La2O3 is found to promote formation of Co carbides and this is thought to be the cause of this catalyst exhibiting greater selectivity towards alcohols.60 Catalysts deactivated by carbon deposition, may be regenerated by oxidation.463.5 Effect of water
When using alumina or silica as support material, cobalt aluminate and silicate are formed in the FTS due to the presence of by-product water. Water increases the rate of metal aluminate and silicate formation from Co metallic catalysts. Li et al. reported the effect of water on 12 wt% Co/SiO2 catalyst properties in a continuous stirred tank reactor (CSTR) in FTS. It is concluded that the addition of water in the range of 5–25% (vol.%) raised CO conversion. Furthermore, slow addition of water (8 vol.%) increased the CO conversion and did not cause significant catalyst deactivation as can be seen in Fig. 11.61 According to Table 2, addition of >8 vol.% water to the catalyst caused the catalyst's deactivation. The degree of deactivation increases with increasing partial pressure of water.55,61 However, in the recent years, it has been demonstrated that the amount of cobalt aluminates formed on the catalyst is not significant to be a major deactivation pathway under typical FTS conditions (T = 230 °C, P = 20 bar).50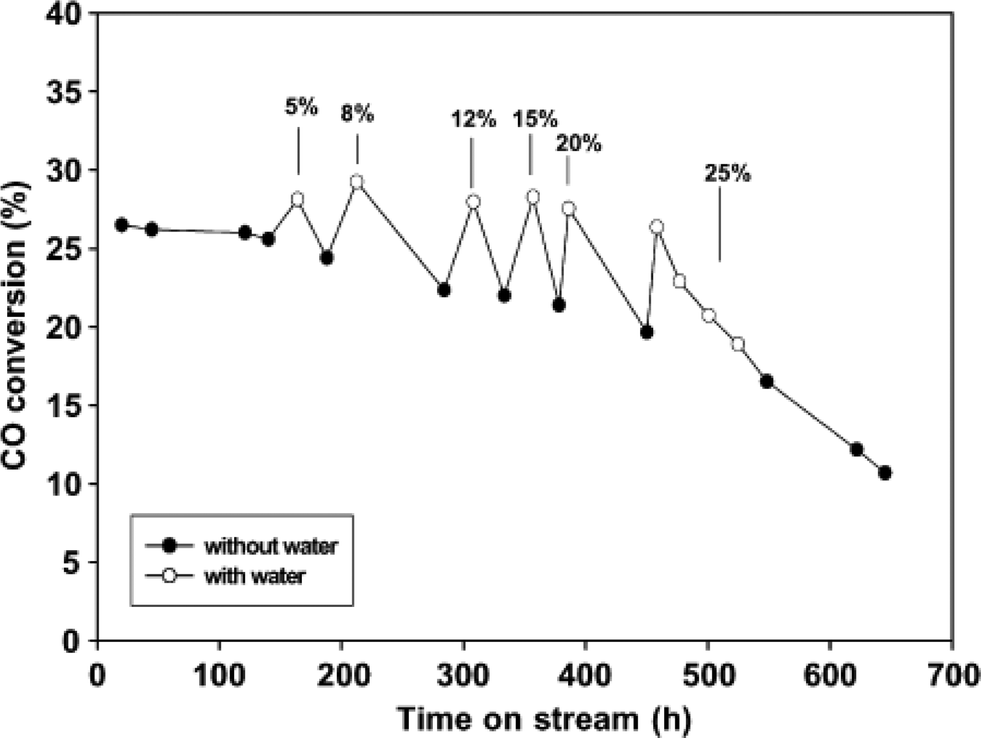 | ||
| Fig. 11 Water effect on CO conversion for 12 wt% Co/SiO2 catalyst with conditions: 210 °C, P = 20 bar, H2/CO = 2.0, SV = 8 SL gcat−1 h−1. Reprinted with permission from ref. 61, Copyright © 2002, Elsevier. | ||
| Water amount added (vol.%) | CO conversion (%) | Water partial pressure (bar) | P H2O/PH2 |
|---|---|---|---|
| 0 | 22.5 | 0.9 | 0.12 |
| 5 | 28.1 | 2.2 | 0.29 |
| 8 | 29.2 | 3.0 | 0.38 |
| 12 | 28.1 | 3.9 | 0.51 |
| 15 | 28.9 | 4.8 | 0.62 |
| 20 | 28.6 | 5.7 | 0.74 |
| 25 | 27.4 | 6.6 | 0.83 |
3.6 Deactivation by surface reconstruction
Cobalt catalysts supported on carbon nanofibre can be deactivated due to surface reconstruction. Bezemer et al. reported that a decrease of Co coordination number (Co particle size < 6 nm) led to nanoparticle reconstruction with flattening of the particles and a decrease in the catalyst activity.36 Schulz et al. found CO-induced reconstruction is an activation mechanism of bimetallic cobalt FTS catalysts.62 Strong CO chemisorption leads to segregation of the active metallic surface species and an increase in the number of active sites. Reduction with hydrogen can also be used to regenerate the optimal cobalt surface structure for FTS catalysis.46 The deactivation mechanisms for cobalt catalysts are summarised in Table 3.| Mechanism | Severity | Importance in FTS | Comment |
|---|---|---|---|
| Sulphur poisoning | High | Not important | Can be removed from syngas |
| Oxidation | None | Not important | Not observed |
| Sintering | High | Important | Can cause 30% reduce in activity |
| Carbon deposition | High | Important | Gradual largely polymeric carbon deposition with TOS |
| Surface reconstruction | Medium | Possibly important | Induced by carbon and may also play a role in genesis of the B5-type active site |
4. Deactivation of iron catalysts
Determining the phase and the nature of the active sites of metallic iron catalysts is still a huge challenge in FTS. There is a great deal of speculation about the causes of loss of catalytic activity for metallic Fe catalysts in FTS. Four main deactivation mechanisms have been defined in the literature as follows:21,63•Reoxidation
•Poisoning (mainly, sulphur application)
•Sintering
•Carbon formation
4.1 Deactivation by reoxidation
Catalyst deactivation by reoxidation is mainly caused by the presence of water in FTS. Iron-based FTS catalysts have a much shorter lifetime than cobalt catalysts because cobalt has a lower reoxidation rate than iron. Iron-based catalyst deactivation is mostly caused by high partial pressures of water leading to active phase reoxidation under LTFT conditions.63 The metal oxide reducibility increases in ascending order from Fe, Co, Ni to Ru. It has been found that for metallic iron catalysts, Fe3O4 is formed during FTS, although no oxides are found for Co, Ru and Ni.21,64 Most studies found that the active phase of iron is oxidised gradually during the FTS to form Fe3O4 which is inactive in FTS.21,65,66 Duvenhage et al. showed that catalyst deactivation, due to Fe0 oxidation by water, was greater at the end of a packed bed in a continuous flow FTS reactor. This was attributed to accumulating partial pressure of steam as the feed proceeded through the catalyst bed. Fig. 12 shows the reduced activity of catalyst at the bottom of the packed bed caused by increased water content.67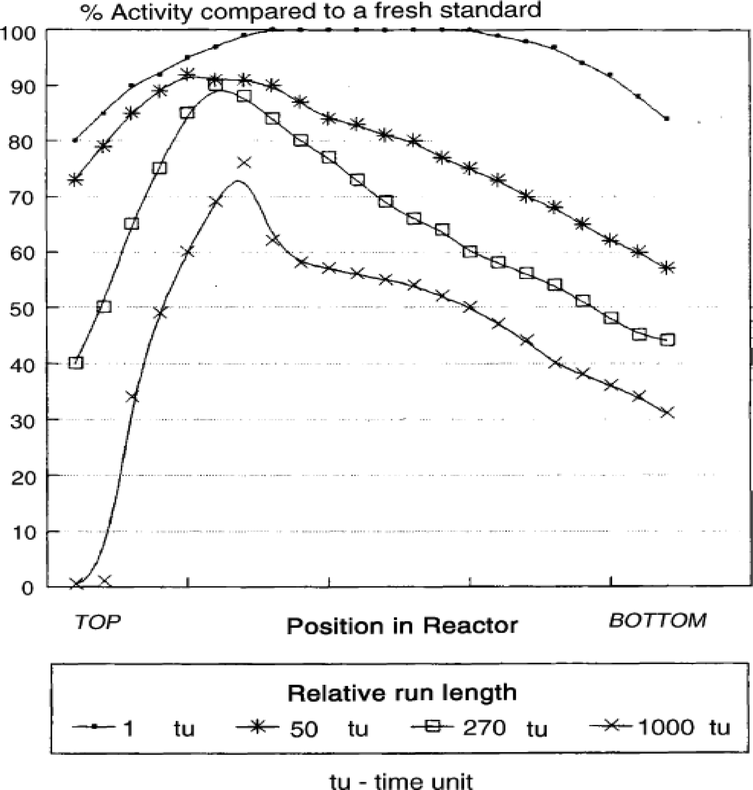 | ||
| Fig. 12 Iron relative activity profile in different time periods on line. Reprinted with permission from ref. 67, Copyright © 2005, Elsevier. | ||
It is therefore advised to choose operating conditions for FTS which minimise the partial pressure of water present. In order to reactivate the oxidised iron catalysts, they may be reduced by hydrogen treatment at high temperatures (>350 °C).21
4.2 Deactivation by poisoning
According to the literature, the well-known poisons for iron are electronegative atoms such as oxygen, bromine, chlorine and sulphur with the latter being the strongest poison.21,63,68,69 Despite this, Baoshan Wu et al. revealed that iron catalysts containing small amounts of sulphur have improved activity and selectivity to heavier hydrocarbons when used in fixed bed reactors (FBR) and continuous stirred tank slurry reactors (CTSR) for FTS.68 Furthermore, Bromfield et al. reported that adding small amounts of Na2S (500–5000 ppm) to a precipitated iron catalyst containing a mixture of different iron and iron oxide phases yields a catalyst with activity four times greater than a sulphur-free Fe catalyst (Fig. 13). Low sulphide loadings increased the olefin/paraffin ratio. However, high sulphide loadings became poisonous to the catalyst resulting in decreased CO conversion. The sulphided iron catalyst compositions and catalytic properties are compared in Table 4.69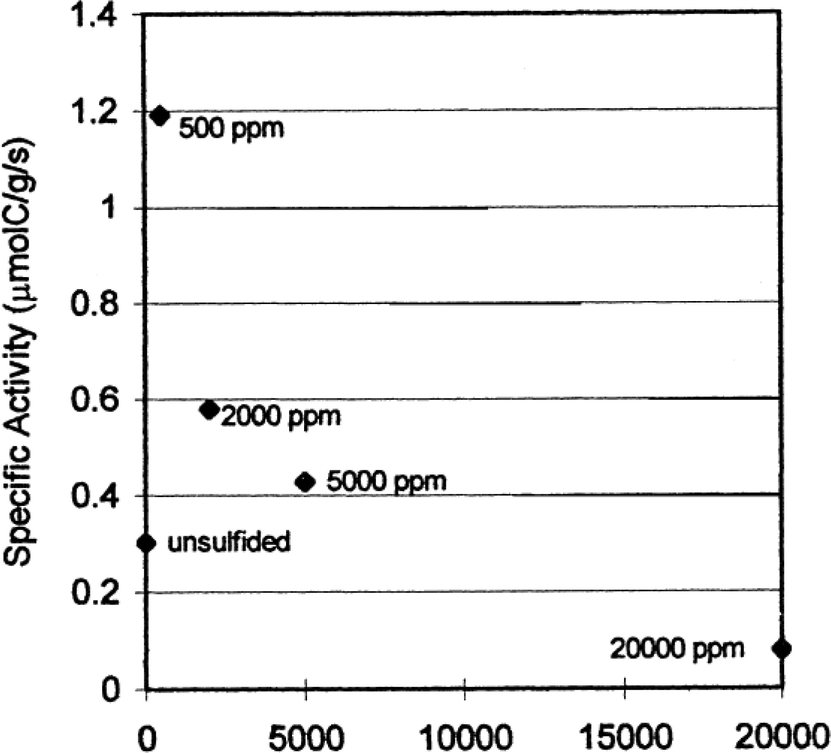 | ||
| Fig. 13 Effect of different amounts of sulphide on specific activity for iron catalyst. Reprinted with permission from ref. 69, Copyright © 1999, Elsevier. | ||
| Catalyst | Total CO conversion (%) | % CO converted to hydrocarbons (excluding CO2) | α-Value |
|---|---|---|---|
| Unsulfided | 26.5 | 22.9 | 0.80 |
| 500 ppm S | 53.7 | 49.0 | 0.78 |
| 2000 ppm S | 53.8 | 48.4 | 0.83 |
| 5000 ppm S | 48.0 | 39.8 | 0.65 |
| 20 000 ppm S | 9.95 | 6.5 | 0.53 |
4.3 Deactivation by sintering
All the Fe-based catalysts can be deactivated by sintering of the active phase. Sintering is a process by which small crystallites grow due to migration or ripening and coalescence phenomena. The rate of sintering depends on the Tamman temperature of the material and this value is defined as half of bulk melting point. Above this temperature, atoms on the surface of the crystallite become mobile and as a result nanoparticles begin to sinter.21,70 For iron, the Tamman temperature is 633 °C, which is significantly higher than typical FTS conditions of 200 to 300 °C. Sintering is believed to occur due to the heat produced during FTS, which is a strongly exothermic reaction, and thus it is possible through localised hot-spots for the iron catalyst crystallites to be exposed to much higher temperatures compared to the actual FTS reaction temperature.21,32 Therefore, for iron it is difficult to verify that reaction induced sintering is a main cause of catalyst deactivation mechanism during FTS. However, Duvenhage et al. reported Fe catalyst deactivation from high partial water pressures in the bottom portion of a fixed bed reactor caused hydrothermal sintering.674.4 Deactivation by carbon deposition
The deposition of carbonaceous material (fouling), can lead to catalyst deactivation. During the FTS reaction, insoluble carbonaceous and high molecular-weight wax compounds are formed which may reduce the activity of the catalyst and increase methane selectivity over time. In Fe catalysts, amorphous carbon is mainly deposited in LTFTS (<280 °C), while the formation of graphitic carbon, which leads to coke deposition, occurs under HTFT conditions (>280 °C). Iron carbide is actually postulated an active phase for FTS, with inter-conversion of iron carbide species leading to catalyst deactivation.65 The catalytic activities of the different iron carbide species are proposed to be fundamentally different,65 thus a hypothesised mechanism for FT-catalyst deactivation involves an transformations of active Fe phase of ε-carbide, X-carbide, ε′-carbide, or more generally FexC or metallic α-Fe to inactive or less active catalyst phases. FTS activity is maintained and even improved in the presence of nanocrystalline graphite,71 thus deactivation of Fe-based FTS catalysts by carbon would require the laydown of large, graphitic carbon overlayers.5. Effect of catalyst promoters
Promoters are additives that improve the catalytic activity and selectivity of heterogeneous catalysts by affecting the structural or electronic properties of the active phase. For FT synthesis, Co and Fe metallic catalysts are normally combined with noble metal, alkali metal or transition metal oxides and ions as promoters to improve their catalytic performance. The addition of chemical promoters is important for the optimisation of active and selective FT catalysts.245.1 Promoter effects on metallic Co catalysts
Noble metals and transition metal oxides are distinctive promoters applied for metallic cobalt catalysts. Tsubaki et al.72 investigated the effects of various noble metal promoters loaded on 10 wt% Co/SiO2, and observed that the rate of hydrogenation of CO increased in ascending order as Co/SiO2 < Pt–Co/SiO2 < Pd–Co/SiO2 < Ru–Co/SiO2 (H2/CO = 2/1, P = 1 MPa, T = 513 K, W/F = 5 gcat h mol−1). Interestingly, the addition of even a very small amount of Ru increased the degree of Co reduction and increased the H2 TOF of surface Co. However, the addition of Pt and Pd increased the degree of cobalt reduction but decreased the TOF.72 The use of Pt significantly increased the FT reaction rate and CH4 selectivity, but decreased the C5+ selectivity.73Rhenium is another commonly applied promoter for cobalt catalysts. The addition of Re raised the dispersion of Co supported on TiO2 by preventing agglomeration of CoOX particles during calcination.74 Storsaeter et al.75 demonstrated that Re doping on Co/Al2O3, Co/TiO2 and Co/SiO2 catalysts increased the rate of hydrocarbon production (per gram of catalyst) and promoted selectivity to C5+ hydrocarbons. In addition to noble metals, some metal oxides are used to promote the structural stability and catalytic performance of metallic Co catalysts in FTS.20,74 ZrO2 is one of the best metal oxide promoters to enhance the CO conversion and C5+ selectivity of a Co/SiO2 catalyst.76 The effects of ZrO2 promoters on a 20 wt% Co/Al2O3 were studied by Jongsomjit et al.77 who report ZrO2 impregnation of γ-Al2O3 improved the reducibility of Co species by preventing the formation of aluminate on Co surface. Increasing the Zr content was found to raise the olefin:paraffin ratio in C2–C17, and decrease the hydrogenation ability of ZrO2-promoted catalysts.78
Another efficient metal oxide promoter for metallic Co catalysts is manganese oxide. Morales et al.79 reported that the introduction of 2 wt% Mn into a 7.5 wt% Co/TiO2 catalyst increased the conversion of CO and the rate of FT reaction. It was also found that the addition of MnOX raised the C5+ selectivity and decreased the CH4 selectivity. The key factor in obtaining such promotion was the selection of the optimum preparation method.80,81 The olefin to paraffin ratio in C2–C8 products was increased drastically through increasing the content of Mn in Mn–Co/TiO2, and hence the addition of MnOX reduced the hydrogenation ability of Co species.79,82
5.2 Promoter effects on metallic Fe catalysts
Fe-based catalysts are distinguished from other FTS catalysts by higher CO conversion, flexibility in FTS operating conditions and increased selectivity to lower olefins.83 However, the use of promoters is necessary in order to increase the selectivity to desired products in FTS. Alkali metal ions are the most commonly applied promoters in Fe-catalysed FT synthesis.84 The alkali metal ions promote CO chemisorption and inhibit H2 chemisorption by affecting the electronic character of iron, which leads to lower rates of FTS, higher molecular weight of products, and greater olefin content.85–87Ngantsoue-Hoc et al.88 have investigated the effect of alkali metal ions on metallic iron catalyst (at 270 °C and 13 bar) in a slurry phase reactor. It was found that the addition of Na+ and K+ improves the activity of Fe in FTS and also WGS reaction, while Cs+, Rb+ and Li+ act as catalyst poisons for both FTS and WGS at low conversions of CO. The promoting or poisoning effects of the alkali metals arose from their basicities and their influence on competitive adsorption of CO and olefins.
The addition of K+ is found to enhance the catalytic performance of Mn-modified Fe catalyst and increased the CO conversion in a fixed-bed reactor.84 The addition of ~1.5 at% K+ to an iron–manganese catalyst, increased the selectivity to C5+ hydrocarbons, decreased CH4 production and prevented rapid catalyst deactivation.89 Feyzi et al.24 found that the use of 6.0 wt% K promoter in a Fe–Mn catalyst provides optimum catalytic performance for synthesis gas conversion to hydrocarbons (especially C2–C4 olefins/C2–C4 alkanes = 1.45) in a fixed-bed reactor (H2/CO = 2/1, P = 1 atm, T = 250 °C, GHSV = 1200 h−1). Moreover, the addition of noble metals (Cu and Ru) and some transition metal oxides (Al2O3, ZnO, MnO) are also reported to promote the Fe-catalysed FT reaction by a combination of enhanced their structural integrity and catalytic properties.
Yang et al.90 researched the conversion of H2 and CO over Fe-based catalysts using various promoters and found the most promising to be Mn and Ca. Manganese is a well-known promoter for Fe catalysts which increases dispersion and increases selectivity to lower alkenes. When used in a slurry reactor Fe–Mn catalysts are found to give high CO conversions and C2–C4 selectivity.91 Similar catalyst properties of Fe–Mn were observed for FTS by Liu et al. in a continuous flow reactor.92Table 5 summarises the effect of different promoters on Fe–Mn catalytic performance. Based on the given information, it can be concluded that the use of 2 wt (%) K provides the optimum catalytic performance for synthesis gas conversion to light olefins and better olefins selectivity.24
| Promoter | |||||
|---|---|---|---|---|---|
| Rb | Cs | K | Zn | Ce | |
| CO conversion (%) | 28.7 | 38.4 | 43.8 | 34.1 | 32.5 |
| Product selectivity (%) | |||||
| CH4 | 24.5 | 21.1 | 16.4 | 22.3 | 25.1 |
| C2H6 | 6.7 | 7.4 | 8.3 | 8.8 | 4.8 |
| C2H4 | 7.4 | 9.4 | 10.8 | 9.4 | 6.3 |
| C3H8 | 4.5 | 5.2 | 6.3 | 6.4 | 7.3 |
| C3H6 | 3.8 | 6.3 | 8.9 | 7.7 | 8.3 |
| C4H10 | 5.3 | 6.3 | 7.3 | 6.3 | 4.6 |
| C4H8 | 5.1 | 7.1 | 10.1 | 7.4 | 5.9 |
| CO2 | 12.2 | 10.1 | 8.3 | 10.8 | 11.4 |
| C5–C9 | 21.1 | 15.4 | 14.4 | 14.4 | 18.1 |
| C10+ | 9.4 | 11.7 | 9.2 | 6.5 | 8.2 |
| Olefins/paraffins | 0.99 | 1.21 | 1.36 | 1.14 | 1.22 |
The effect of different transition metals (Zr, Mn, Cr, Mo, Ta and V) on the catalytic performance of Fe–Cu based catalysts has also been explored, which showed that the utilisation of these transition metals promotes both CO hydrogenation and WGS activity.93 Further studies suggested that the addition of these transition metal promoters increased the iron dispersion, but did not reduce the BET surface area or reducibility of catalyst.94
According to the literature review, it has been revealed that most of these promoters function by enhancing the carburisation and reduction of Fe precursor species, but do not affect the overall activity (TOF) of the iron active site in the operating catalyst. There is a tendency for studies to combine various modifiers with different functions to decrease the selectivity to CH4 and CO2 and promote the formation of light olefins. Given the multitude of effects such promoters can have it is essential to gain improved insight into the synergistic interactions of such modifiers with Fe-based catalyst if efficient and rational design protocols are to be realised.
6. Support effects on the catalytic performance of metal catalysts
The choice of support material in FT-catalysts is of paramount importance to:95–98•Optimise the dispersion of the active phase, a high surface area support gives better dispersion and reducibility of the active phases
•Improve the heat and mass transfer in an exothermic or diffusion-limited reaction
•Stabilises the active phases against morphological change and loss of surface area during the reaction
•Sustains the mechanical strength of the catalysts
The surface area and pore structure of the support provides significant improvements to catalytic performance as it controls the morphology, dispersion and reducibility of the active phase.20,99,100 The size, distribution and shape of the pores is an important factor when there is resistance towards diffusion of reactants/products,95,101–105 thus materials, which can easily be manipulated to produce the optimum textural properties are desirable. The chemical interaction between the active phase and the support has been established as extremely important during FTS. A strong interaction affects the ease of reduction of the active phase while a weak interaction may lead to poor dispersion of the active phase.106
Most investigations of support effects in FT-catalysis have focussed on the impact of Al2O3, SiO2, TiO2, mesoporous and zeolite supports on catalytic properties of cobalt catalysts (rather than iron).57,61,106,107 The majority of the products of the FTS process are linear hydrocarbons, particularly C5+. The supports used to promote linear hydrocarbon products are typically Al2O3, SiO2 and TiO2.25 Mesoporous supports, such as MCM-41 and SBA-15 silicas are also beneficial because the active metal (Co, Fe or Ru) nanoparticles are highly dispersed over the large surface area and this can lead to desirable catalytic performance in the FTS.109–111 Carbon-based materials have also drawn much interest as an appropriate catalyst support because of their, resistance to basic and acidic conditions and their high thermal stability during FTS.27,112 In order to produce gasoline-range hydrocarbons (GRHs, C5–C12 hydrocarbons) and improve the octane number, further processing of the products after FTS may be needed, with zeolites the popular support for GRH production.28,113,114
6.1 Support effects of SiO2, Al2O3, TiO2
When using cobalt-based catalysts for FTS, the most commonly used support materials are ZrO2, Al2O3, SiO2 and TiO2. CO conversion is related to the dispersion of metallic Co over the support. The dispersion, and therefore, the average particle size affects chain growth and the selectivity to C5+ hydrocarbons and is also correlated with the ease of diffusion of materials through the support's pore network.20 Bartholomew et al. found that decreasing the Co loading on the support (or increasing dispersion) significantly reduces the catalytic activity of cobalt, with a 3 wt% Co/Al2O3 ~20 times less active than 15 wt% Co/Al2O3.The product selectivity is also a function of Co dispersion and the nature of the support, with higher selectivity to the lighter hydrocarbons and CO2 productivity observed for catalysts with high dispersion that are less easily reduced.20,108 Comparison of the selectivity and activity of Co/γ-Al2O3 to those of Co/ZrO2,115 reveals Al2O3 gives rise to a poorly reducible active phase compared to ZrO2. The later was also capable of adsorbing hydrogen via a spillover mechanism due to the strong interaction strength between the support and metal allowing higher CO conversion to be observed a low operation pressure.115
Bartholomew and Reuel studied the C5+ hydrocarbons selectivity using a Co catalyst (10 wt% cobalt loading) on different supports (H2/CO = 2, T = 225 °C and P = 1 atm).108 The study found that the C5+ hydrocarbons selectivity increased in the order: Co/MgO < Co/C < Co/A12O3 < Co/SiO2 < Co/TiO2 as shown in Table 6.108
| Catalyst (10 wt% cobalt) | Average carbon number | Weight percentage hydrocarbon selectivities | ||||
|---|---|---|---|---|---|---|
| C1 | C2–C4 | C5–C12 | C13+ | Alcohols | ||
| Co/SiO2 | 4.0 | 29 | 27 | 42 | 0.2 | 1.3 |
| Co/Al2O3 | 3.8 | 32 | 31 | 35 | 0.7 | 1.3 |
| Co/TiO2 | 5.0 | 16 | 30 | 52 | 1.7 | 1.1 |
| Co/MgO | 1.9 | 55 | 39 | 6.2 | 0 | 0.6 |
| Co/C (type UU) | 2.3 | 53 | 31 | 16 | 0 | 0 |
| Co/C (Spheron) | 2.1 | 66 | 23 | 11 | 0 | 0 |
Hydrogen chemisorption and temperature programmed reduction (TPR) may be used in order to measure the reducibility of a supported cobalt catalyst. Jacobs et al. studied about the reducibility of Co over supports such as Al2O3, TiO2, SiO2, and ZrO2 with promoters, including metal cation (K+) and noble metals (Ru, Re and Pt). The reducibility of the cobalt oxide species on different supports increases in ascending order from SiO2 to TiO2 to Al2O3.116 In order to decrease the presence of cobalt oxide, Pt and Ru are added as a catalyst promoter, with Re believed to aid cobalt oxide reduction by interaction with the support. Additionally, the noble metal promoters increase the initial activity of the catalysts by improving the reduction of smaller cobalt particles which interact strongly with the support. Contrarily, the addition of La, Zr, K and B metals which are not reducible, caused an increase in cobalt oxide reduction temperature. Zr metal was recommended to improve the dispersion of Co over Al2O3 and SiO2 supports, with increased cobalt loading producing increased average Co cluster size and reducibility of Co oxide.116 Studies by Enache et al. show that hydrogen adsorption and spillover when using ZrO2 supports increases the ease of Co oxide reduction, leading to a superior FTS catalyst with higher activity and α-parameter than for Co/Al2O3.115
In a study comparing CO conversion over metallic cobalt catalysts with different supports (TiO2, SiO2 and Al2O3), Iglesia revealed that, the turnover frequency (TOF) for CO conversion during FTS under operating conditions of pressure and temperature are 20 bar and 200 °C was not highly dependent on Co dispersion over the range 0.01–0.12.74
In a slurry phase reactor, the turn-over frequency of cobalt-based catalysts was reportedly influenced by the cobalt particle size, the average pore size, the degree of cobalt species reduction and potential for re-oxidation by water produced in situ during the FTS reaction.25 The catalytic performances of 20 wt% Co/SiO2 (CoS), Co/γ-Al2O3 (CoA) and Co/TiO2 (CoT) catalysts in slurry phase FTS are shown in Fig. 14.25
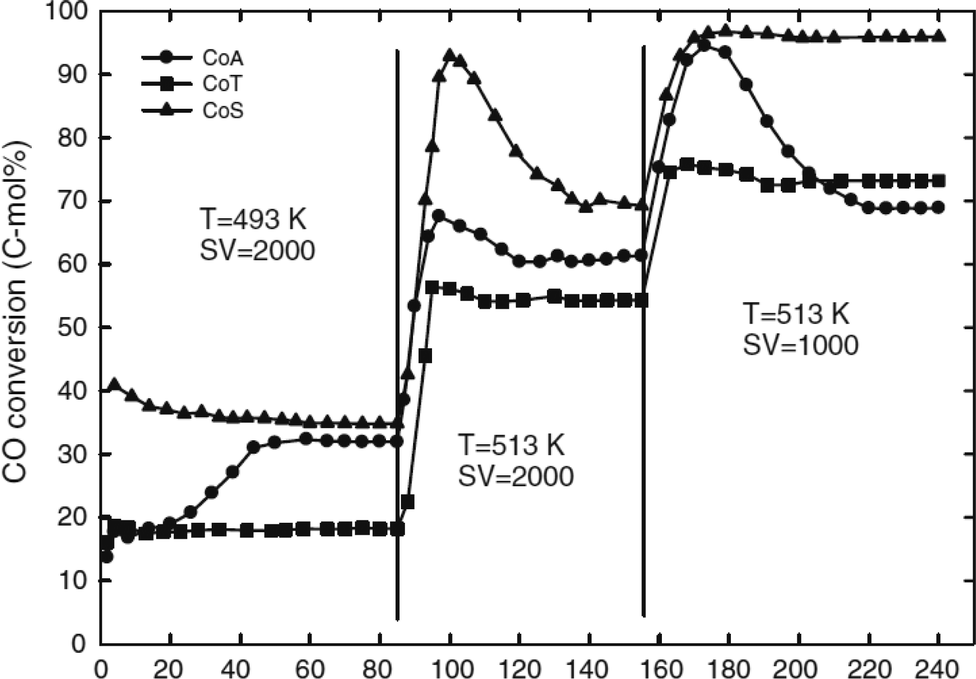 | ||
| Fig. 14 CO conversion vs. time on the stream (h) on different FTS catalysts in conditions: 220–240 °C, 20 bar, 1000–2000 L kgcat−1 h−1, H2/CO/CO2/Ar (mol%) = 57.3/28.4/9.3/5.0. Catalysts: 20 wt% Co/SiO2 (CoS), Co/γ-Al2O3 (CoA) and Co/TiO2 (CoT). Reprinted with permission from ref. 25, Copyright © 2009, Springer. | ||
The diameter of the catalyst pellet also affects the selectivity and synthesis rate during FTS, with Iglesia et al. reporting that pellet diameters > 0.36 mm lead to increased selectivity to CO2 and methane. Furthermore, larger pellet sizes (1–3 mm) increased diffusion limitations resulting in reduced rates of reaction.
Catalysts with egg-shell structures formed by having the active Co components located close to the outer surface of the support (SiO2 pellet, powder, etc.) give improved C5+ selectivity and FT reaction rates.117 20 wt% Co/SiO2 was claimed to be the preferable catalyst as it exhibited the best performance due to the high degree of cobalt species reduction. Co/γ-Al2O3 had the smallest particle size of cobalt and the highest dispersion but had poor reducibility of cobalt species due to the strong interaction between the metal and support. Co/TiO2 catalyst had a larger pore size than alumina and exhibited improved diffusion of FTS products. The CO conversions of these three catalysts are summarised in Table 7. Co/γ-Al2O3 produces small amounts of oxygenated products and exhibits low C5+ selectivity due to catalyst deactivation resulting from aggregation of catalyst and oxidation of Co metal particles by the in situ generated water during the FTS reaction.25
| Notation | CO Conversion (mol%) | TOF (×10−3 s−1) | Product selectivity (C-mol%) | |||
|---|---|---|---|---|---|---|
| C1 | C2–4 | C5+ | Olefins in C2–4 | |||
| T = 220 °C | SV = 2000 (L kgcat−1 h−1) | |||||
| CoAl | 31.9 | 12.2 | 4.3 | 5.8 | 89.9 | 55.6 |
| CoT | 18.2 | 27.9 | 1.7 | 3.1 | 95.2 | 58.6 |
| CoS | 34.8 | 20.6 | 2.1 | 4.5 | 93.4 | 56.1 |
| T = 240 °C | SV = 2000 (L kgcat−1 h−1) | |||||
| CoA | 61.3 | 23.4 | 7.5 | 10.2 | 82.3 | 42.7 |
| CoT | 54.3 | 83.1 | 5.5 | 6.1 | 88.4 | 37.0 |
| CoS | 69.2 | 40.9 | 6.9 | 10.0 | 83.1 | 41.9 |
| T = 240 °C | SV = 1000 (L kgcat−1 h−1) | |||||
| CoA | 68.8 | 13.1 | 5.8 | 7.8 | 86.4 | 44.8 |
| CoT | 73.2 | 56.0 | 9.6 | 8.6 | 81.8 | 29.5 |
| CoS | 95. 8 | 28.3 | 31.0 | 29.3 | 39.7 | 13.2 |
In conclusion, the support effects on the catalytic behaviour in FTS are significant and extremely complicated and warrant more study. It is difficult to compare the effect of supports to each other. The support's pore structure, the pore size distribution, the interaction between the active metal (precursor) and the support and the location of the active metal particles are the most important issues connected with C5+ selectivity and productivity. These factors are related to the metal's reducibility and the size of the metal particles (morphology). Furthermore, the diffusion of reagents and products can vary for supports composed of the same material but possessing different pore structures.
6.2 Mesoporous support materials
MCM-41 and SBA-15 are mesoporous silica supports which are commonly used in FTS catalyst. The strengths of these materials are their narrow well-define pore size distribution, high surface area and controllable acid and base properties. The average pore diameter of these materials is <50 nm.26,118 Periodic mesoporous silicas (MCM-41, SBA-15 and SHS) with metallic cobalt are reported as interesting catalysts for FTS exhibiting superior product yields.109,110As can be seen in Table 8, Jung et al. demonstrated that the use of 0.5 wt% Co/SHS (mesoporous silica, hollow sphere) catalyst in a fixed bed reactor had greater catalytic activity and selectivity towards C5+ hydrocarbons when compared with other supported catalysts. Generally, the performance of mesoporous silicas with cobalt-based catalysts depends on the particle size of cobalt and the structure of the mesoporous support (pore size distribution and pore diameter).110
| Sample (0.5 wt% cobalt) | Pore diameter (nm) | Mean Co particle (nm) | CO conversion (%) | CO2 selectivity (%) | Hydrocarbon selectivity (%) | ||
|---|---|---|---|---|---|---|---|
| CH4 | C5–9 | C5+ | |||||
| Co/SiO2 | 6 | 9.01 | 60.1 | 14.20 | 29.4 | 26.6 | 55.2 |
| Co/MCM-41 | 3.7 | 9.05 | 63.8 | 12.98 | 27.2 | 27.2 | 58.8 |
| Co/SHS | 12.6 | 18.3 | 75.5 | 8.95 | 19.8 | 31.0 | 70.4 |
HMS and Al-HMS are ordered mesoporous silicas with hexagonal pore structures produced via neutral templating routes with alkyl amine surfactants. Co/HMS catalysts show superior catalytic activity and C5+ selectivity than MCM-41 or Al-HMS variants because the Co/HMS catalyst has a shorter channel size and larger mesoporous diameter.118 Khodakov et al. confirmed that the pore size of periodic mesoporous silicas affected the reaction rate and selectivity of Co catalysts in FTS, with reaction rates in FTS higher for cobalt catalysts with a pore diameter of >3 nm. In addition, larger catalysts pore diameter resulted in higher selectivity towards C5+ hydrocarbons.119
Ruthenium has been used as a promoter for cobalt catalysts supported on MCM-41 and SBA-15 in FTS.120 Addition of ruthenium to silica-supported catalysts with small pore sizes (dp = 3–4 nm) led to an increase in the FTS reaction rate. However, when added to catalysts with large pore size (dp = 5–6 nm), the ruthenium had a reduced effect on the FTS reaction rates. Hong et al. reported that for supports with large pores addition of ruthenium had a diminished effect on the cobalt dispersion and cobalt reducibility.120 Co/SiO2 exhibited a distinct tendency toward the C19+ fraction hydrocarbons in FTS. However, the use of HMS as support for Co is suitable for producing diesel fraction hydrocarbons (C8 to C21) due to HMS pore structure.121 Although support effects have been widely reported for cobalt-based FTS catalysts, there is a lack of information about iron-based catalysts with mesoporous supports for the FTS in the literature.
6.3 Carbon supported catalysts
Recently, carbon supported catalysts with attractive porous structures have received a lot of attention for use in FTS. Nanoporous carbon supports have been defined as “the next generation of mesoporous materials”.122 Porous carbon is an attractive material for use as a catalytic support for FTS because it can be prepared cheaply from a wide variety of low cost precursors, it is chemically stable and biocompatible under non-oxidising conditions, it has high thermal conductivity, has good mechanical stability, easy handling and good electrical conductivity. In comparison with mesoporous silica, mesoporous carbon is more resistant to structural collapse in aqueous environments due to hydrolytic effects.122Carbon nanofibre (CNFs), carbon nanotubes (CNTs),123 carbon spheres (CSs)124 and multi-walled carbon nanotubes (MWCNTs)125 have all been studied as nanoporous carbon supports in FTS catalysis. While the use of porous carbon catalysts increases the selectivity towards heavier hydrocarbons, these catalysts are prone to deactivation since the catalytic sites sinter after prolonged use (around 125 h and between 318 °C and 418 °C).126
CNT supports have a central internal pore structure which distinguishes them from CS, CNF and CMC (carbon micro-coil) supports. The internal and external diameters of CNTs are typically between 5–12 nm and 10–25 nm, respectively.126,127 Recently, the majority of research on porous carbon supports has focused on supported metal catalysts on the external surface of CNT. Metallic cobalt catalysts have been supported on CMC, CNT and CNF supports,128 with the use of inert carbon supports reported to improve the reducibility of cobalt oxide under typical FTS conditions. However, despite claims the deposition of catalytic sites on the interior surface of the nanotubes produces more stable catalysts by minimising sintering,126 it is surprising the selective deposition of metallic nanoparticles in the nanotubes inner cavity has received little attention.126 Pt, Ru and Ir have also been shown to be promoters which enhance the catalytic properties of carbon supported cobalt catalysts.128,129
Carbon-supported iron catalysts for FTS have received much more attention than cobalt catalyst. Metallic iron supported on carbon can be used as catalyst for FTS in order to produce light olefins.130 Selectivity to light olefins of up to 60% was observed with iron nanoparticles on carbon nano-fibre supports promoted by sulphur plus sodium.131 Alkali-earth metals with strong basic properties, such as Ca, Sr and Mg, can be used as promoters to improve the catalytic performance of Fe on carbon support in the FTS process.130 The location of the iron particles on the support has an effect on the resulting catalytic properties. Chen et al. observed that 10 wt% Fe located on the interior surface of CNTs (CNT-in) had increased activity in FTS than Fe particles on the exterior surface (CNT-out) (Fig. 15). Also, the C5+ hydrocarbons selectivity for CNT-in catalyst was twice as high as the CNT-out.127 The improved activity and selectivity of the CNT-in catalyst was due to changes in the redox properties of the confined Fe and a decreased potential for Fe particle sintering.
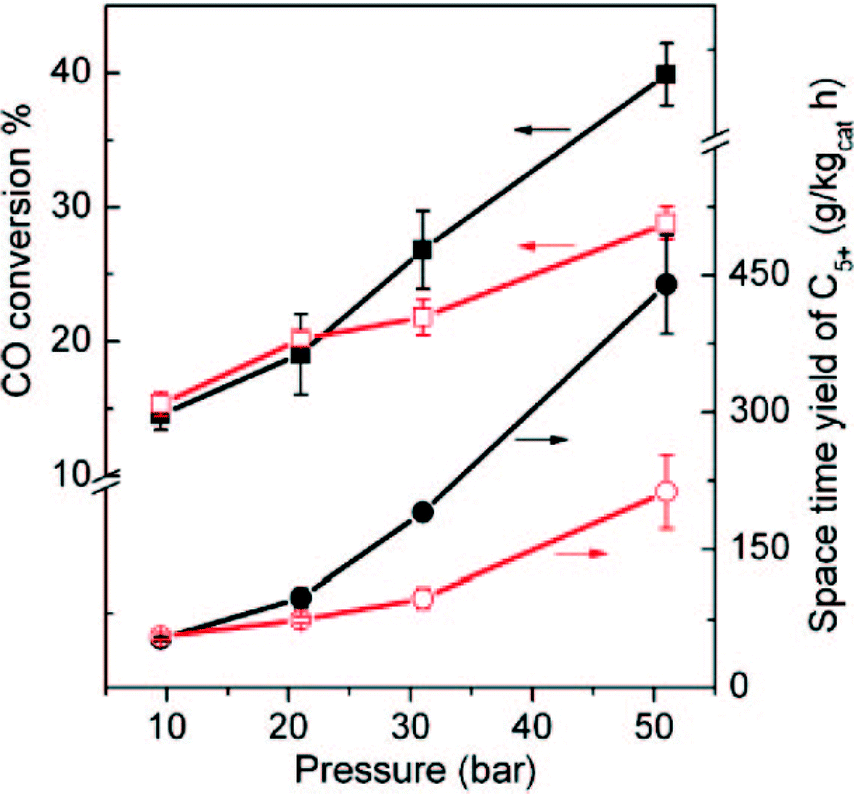 | ||
| Fig. 15 FTS conversions of 10 wt% Fe-out-CNT and Fe-in-CNT at different pressures at 270 °C. Circle symbols represent the space-time yield of C5+ hydrocarbons and square shows the CO conversion. Hollow symbols show Fe-out-CNT and filled one denote Fe-in-CNT. Reprinted with permission from ref. 127, Copyright © 2008, American Chemical Society. | ||
6.4 Zeolite supported catalysts
Zeolites possess shape selectivity and do not allow the formation of products, transition states or intermediates larger than the size of zeolite's channels or cavities. Therefore, zeolite-supported catalysts lead to formation of lighter hydrocarbons with limited chain growth in FTS. In addition, the zeolite acidity can cause isomerisation, aromatisation and secondary cracking reactions of the primary FT hydrocarbons and contribute to a wider product distribution.132It has been reported that Co-based FTS catalysts supported on mesoporous and microporous molecular sieve catalysts have superior activity and C5+ hydrocarbon selectivity in FTS when the Co metal particles are located inside the channels rather than on the exterior surface. The former materials, with active metal sites located inside the support's pore structure, could provide the essential geometric constraints to control the product distribution. Most of the studies on mesoporous molecular sieves focus on mesoporous silicas (MCM-41 and SBA-15). The majority of the publications involving microporous zeolite materials focused on Faujasite, ZSM-5 and mordenite catalysts. Selection of an appropriate mesoporous or microporous material with the desired acid–base properties is key to controlling the FT product selectivity.
Metallic cobalt-coated zeolite MCM-22 with structure of MWW was studied for FTS by Ravishankar et al.134 Zeolite MCM-22 has a high external surface area and low fraction of micropores. The selectivity of Co supported on MCM-22 catalyst was dependent on the ratio of Si/Al within the zeolite and the degree H+ exchange. Methane selectivity was strongly dependent on the degree of ion exchange of H+ by Na+. Selectivity to methane is reduced and C5+ selectivity is increased with increasing Si/Al ratio. According to the literature, Co/Na–MCM-22 (Si/Al = 200) produced higher C5+ selectivity and lower methane selectivity than Co/SiO2 under the same reaction conditions P = 12.5 bar, T = 280 °C and H2/CO = 2.117
Zeolites ITQ-2 and ITQ-6 were used as supports to prepare a 20 wt% Co FTS catalyst by Concepción et al. and compared to other zeolite and silica supports.133 These zeolite catalysts were prepared by delamination of a layered ferrierite and MCM-22 support. The structures of ITQ-2 and ITQ-6 are represented in Fig. 16. ITQ-2 has narrow sheets of 2.5 nm height with “cups” in a hexagonal array (0.7 × 0.7 nm) with both sides connected into the sheet by a double-6-membered ring (MR) window. The sheets consist of a circular 10-MR channel system with a sinusoidal pattern (Fig. 16a). The ITQ-6 external surface consists of cups surrounded by 10-MR and therefore it has a smaller diameter than the 12-MR of ITQ-2 (Fig. 16b).133 The ITQ-6 layers do not have 10-MR channels. Concepción et al. found that the Co/ITQ-6 was the most active catalyst of those tested, with a higher reaction rate in FTS compared to Co/MCM-41 and Co/SiO2 respectively (Table 9). Furthermore, Co/ITQ-6 had a high activity due to a good dispersion and high reducibility of cobalt. The cobalt particle dispersion and reducibility in Co/ITQ-2 and Co/SiO2 catalysts were approximately equal, as were the reaction rates of FTS. The Co/ITQ-6 and Co/ITQ-2 also had a higher selectivity to C5+ hydrocarbons than Co/MCM-41 and Co/SiO2. This was attributed to a higher concentration of unsaturated Co0 sites in the delaminated ITQ zeolites, as identified by FTIR characterisation.133
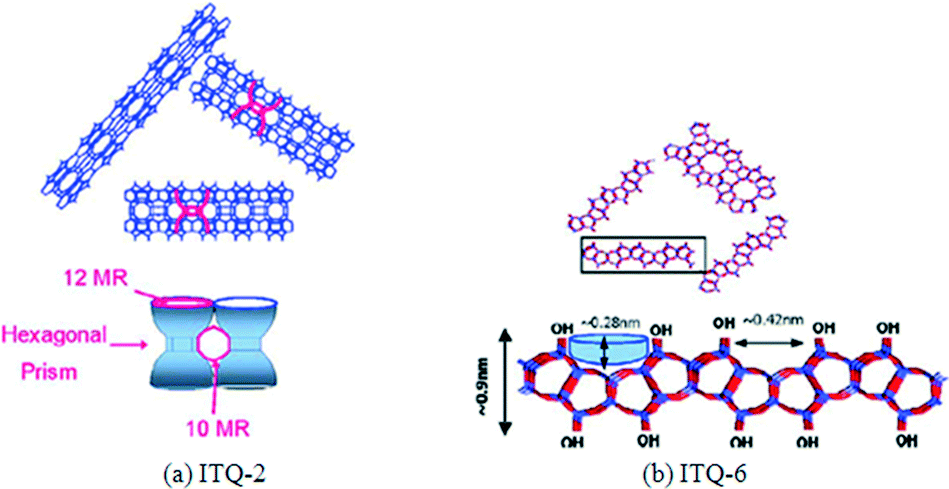 | ||
| Fig. 16 Structures illustration of the (a) ITQ-2 and (b) ITQ-6 catalysts. Reprinted with permission from ref. 133, Copyright © 2004, Elsevier. | ||
| Catalyst (20 wt% cobalt) | CO Conversion (%) | Reaction rate (10−3 s−1) | TOF (10−2 s−1) | Hydrocarbon distribution (%C) | α | ||
|---|---|---|---|---|---|---|---|
| C1 | C2–C4 | C5+ | |||||
| Co/ITQ-6 | 37.5 | 5.51 | 4.5 | 10.7 | 11.0 | 78.3 | 0.85 |
| Co/ITQ-2 | 21.9 | 3.30 | 3.4 | 13.2 | 14.2 | 72.6 | 0.83 |
| Co/MCM-41 | 24.3 | 3.80 | 7.2 | 25.6 | 29.7 | 44.7 | 0.76 |
| Co/SiO2 | 20.2 | 3.01 | 2.6 | 16.6 | 17.9 | 65.5 | 0.81 |
Other zeolites explored include LTL (Linde Type L) zeolites in the potassic form (K-LTL) which when doped with metallic iron was used as catalyst in FTS under reaction conditions of P = 20 bar, T = 543 K and H2/CO = 2 gave CO conversions of ~40%.135 Faujasite zeolites, including X- and Y-type zeolites, with a supercage of diameter 1.3 nm were proposed as potentially useful supports for controlling metal nanoparticle size (<2 nm).
However, Tang et al. observed that Co and Fe impregnated Faujasite materials were poor catalysts because reduction of metal particles inside the pores and cages was incomplete at moderate temperatures below 500 °C. The reduction of the non-noble metal cations was unsuccessful because of the strong chemical interaction between the anionic zeolite frameworks and the cationic metal-precursor.136
Bessell reported that, strongly acidic ZSM-5 catalysts are good candidates to produce gasoline products with high octane numbers.137 Metallic cobalt (10 wt%) on ZSM-5, ZSM-11, ZSM-12 and ZSM-34 supports was tested in a comparative study in order to determine the influence of the zeolite pore structure in the FTS process. As can be seen in Table 10, the CO conversion (%) of these zeolites are ranked as ZSM-34 < ZSM-5 < ZSM-11 < ZSM-12 in ascending order, while ZSM-34 catalyst has the highest selectivity to gasoline range hydrocarbons. This improved the activity was a result of increasing the pore size of the zeolite supports. The acidic strength and acid site concentration of these zeolites decreased in the order of ZSM-34 > ZSM-5 > ZSM-11 > ZSM-12. As the zeolite acidity decreased, the hydrocarbon products became lighter and less n-alkanes were produced, which indicated selectivity was governed by accessibility of the acid sites within the pore channels rather than simply the overall acidic strength.138
| Catalyst (10 wt% Co) | CO conversion (%) | Hydrocarbon selectivity (%) | |||
|---|---|---|---|---|---|
| C4–9 | C10–11 | C12–13 | C14–18 | ||
| Co/ZSM-5 | 60 | 0.34 | 0.25 | 0.29 | 0.45 |
| Co/ZSM-11 | 61 | 0.25 | 0.18 | 0.23 | 0.42 |
| Co/ZSM-12 | 79 | 0.26 | 0.19 | 0.21 | 0.40 |
| Co/ZSM-34 | 45 | 0.55 | 0.55 | 0.61 | 0.68 |
For ZSM-5 catalysts, the Si/Al ratio plays a significant role in controlling olefin selectivity and activity of FTS catalysts. Kang et al. compared Co/ZSM-5 with different Si/Al ratios for direct production of GRHs and found that ZSM-5 with low a Si/Al ratio of ~25 was good catalyst.139,140 Highest olefin selectivity in the C2–C4 range and high CO conversion was observed for 20 wt% Fe/ZSM-5 with the ratio of Si/Al = 25 (Table 11). Furthermore, by increasing the Si/Al ratio, decreases the Fe reducibility and weak acid site density, which was believed to be the cause of the decrease in olefin selectivity and CO conversion.141
| Si/Al ratio | CO conversion (%) | CO2 selectivity (%) | Selectivity in hydrocarbons (%) | O/(O + P) | ||
|---|---|---|---|---|---|---|
| C1 | C2–C4 | C5+ | ||||
| 25 | 80.7 | 37.7 | 18.3 | 24.9 | 56.8 | 39.7 |
| 40 | 78.9 | 37.1 | 17.5 | 23.7 | 58.8 | 37.1 |
| 140 | 61.6 | 29.1 | 12.6 | 16.5 | 70.9 | 27.9 |
As previously discussed, acidic zeolite supported FT-catalysts can produce high octane, gasoline range hydrocarbons. Another popular class of catalyst receiving much attention are zeolite hybrid catalysts.142 The in situ upgrading of primary FTS143 products using zeolite hybrid catalysts is an attractive approach. The zeolite hybrid catalysts consist of a conventional FTS catalyst (SiO2, Al2O3, etc.) combined with an acidic zeolite.
Zeolite hybrid catalysts with silica-supported cobalt were successfully used by Martínez et al. for cracking C13+ long chain hydrocarbons to gasoline-range branched products in FTS under conditions P = 20 bars, T = 250 °C and H2/CO = 2.142 A range of different support types, including ultrastable Y (USY), HBeta zeolite (HBeta), H mordenite (HMOR) and Zeolite Socony Mobil-5 (HZSM-5) were investigated and it was observed that the selectivity to C5–C8 branched products varied for different support types. The yield of C5–C8 products increased in the following order: USY < HBeta < HMOR < HZSM-5 which is represented in Fig. 17.142 It is reported that for the zeolite hybrid catalysts, metallic Fe is preferable to metallic Co because the optimum temperature for zeolite operation is around 275 °C, while for cobalt catalyst is around 235 °C.143 Therefore, for cobalt catalysts, increasing the temperature of the reaction increases selectivity to undesired methane and CO2 products. Co is susceptible towards sintering at higher temperatures, however in contrast the optimum temperature for iron catalysts is the same as for the zeolite (275 °C) yielding high CO conversion and low methane selectivity.143
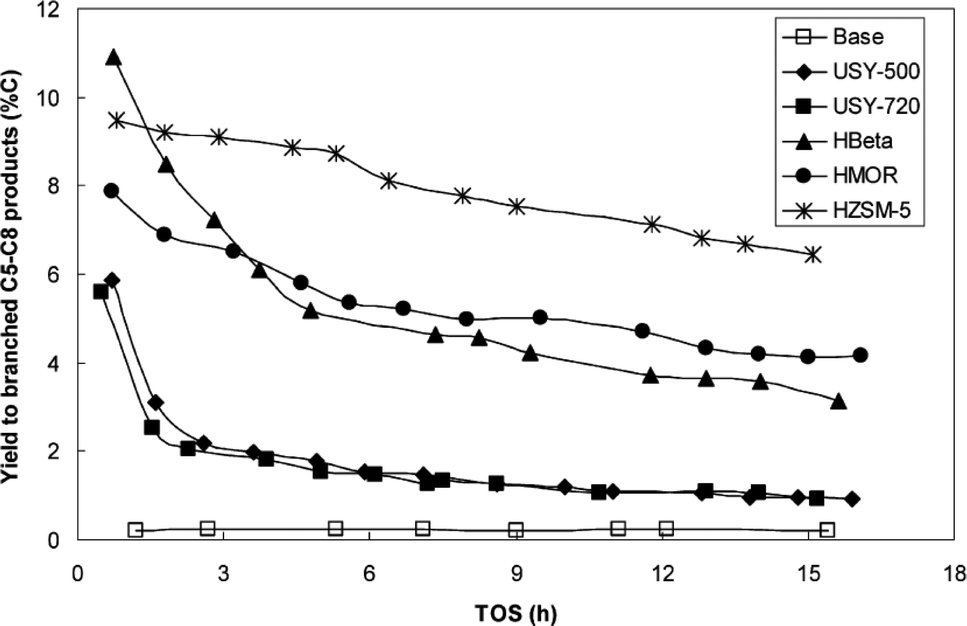 | ||
| Fig. 17 Yield to branched C5–C8 products (% C) vs. time-on-stream (TOS) for the base and hybrid catalyst in conditions: 250 °C, 20 bar, H2/CO = 2 and 13.5 lsyngas gcat−1 h−1. Reprinted with permission from ref. 142, Copyright © 2007, Elsevier. | ||
7. Conclusions, summary and outlook
The Fischer–Tropsch synthesis (FTS) is used to convert a mixture of carbon monoxide and hydrogen (syngas) derived from gasification of biomass or solid organic matter to valuable hydrocarbons which may be used as fuels and chemicals. Optimisation of the selectivity and activity of the catalysts is a crucial but challenging goal for FTS. In order to produce a wide variety of products, the factors affecting the selectivity and activity of the catalysts must be understood. This review paper has covered the FTS catalyst selectivity and activity in detail including the following components.Choice of metal
Some metals present desirable activity in the FTS. Transition metals which are in the 3rd, 4th, 5th and 6th group are not suitable catalysts for the FTS. The suitable metal catalysts for the FTS are cobalt, iron, nickel and ruthenium, with the latter being the most active catalyst. Ruthenium is prohibitively expensive for use in industrial processes. Nickel has high activity for hydrogenation and high selectivity for methane formation which is undesirable product in the FTS process. Iron and cobalt are the only two metals which are used in FTS for industrial applications. The iron metal catalyst has low selectivity to long chain paraffins, while it produces a high amount of oxygen for valuable chemicals such as alcohols and aromatics. Iron catalysts are deactivated more easily than cobalt catalysts. Furthermore, most of the catalyst deactivation for iron and cobalt is a result of catalyst oxidation. Iron is cheaper than cobalt, while cobalt has a high selectivity to form long chain paraffins and has low selectivity to olefin and oxygenation and is also more resistant to catalyst deactivation. Therefore, in order to form long chain paraffins, cobalt is the preferable catalyst, whereas to produce olefins and valuable chemicals (alcohols, aromatics), iron is the more suitable catalyst.Promoter
Metallic cobalt and iron catalysts require promoters such as alkali metals, transition metal oxides, metal ions or noble metals to provide better catalytic performance. Alkali metal ions are mainly applied as promoters for iron catalysts in FT synthesis. The addition of K+ to Fe–Mn catalysts increases the CO conversion in fixed-bed reactors. Furthermore, the addition of noble metals promotes Fe catalytic performances. The use of metal transition oxides (Zr, Mn, Cr, Mo, Ta and V oxides) for the iron catalyst can improve CO hydrogenation and WGS reaction. Noble metals and transition metal oxides are distinctive promoters applied for Co-based catalysts. Additional of a small amount of Ru increases the Co reducibility significantly. The addition of Pt promoter to cobalt catalyst increases the FT reaction rate, while decreasing the C5+ selectivity. Rhenium is another desirable promoter for cobalt catalysts which improves the selectivity to C5+ hydrocarbons. Addition of ZrO2 and MnOX to cobalt catalysts improves the CO conversion and the selectivity of C5+ hydrocarbons.Support
The choice of catalyst support is an important factor for product selectivity in the FTS because it enhances the metal dispersion, reducibility, stability, mechanical strength and facilitates heat and mass transfer. Conventional supports for producing C5+ hydrocarbons over cobalt-based catalysts are ZrO2, Al2O3, SiO2 and TiO2. The CO hydrogenation activity of these supports in descending order is Co/TiO2 > Co/SiO2 > Co/Al2O3. In pellet-form supports, a pellet diameter of more than 0.36 mm increases both methane and CO2 selectivities. In metallic cobalt catalysts, if the CO/H2 ratio decreases then C5+ selectivity decreases and lower olefins are produces.Egg-shell catalyst, in which the active cobalt particle is located near the outer surface of the pellet, enhances the FTS reaction rate and C5+ hydrocarbon selectivity. In addition, egg-shell Co/SiO2 catalyst yields the maximum C5+ selectivity in comparison with other conventional supports. Periodic mesoporous silicas supports such as MCM-41, SBA-15, MCM-22 and SHS affect the catalytic performance in the FTS. Mesoporous supports with iron and cobalt metal can produce higher CO conversion and C5+ hydrocarbon selectivity than conventional supports such as SiO2 and Al2O3. Cobalt supported upon mesoporous materials shows good catalytic performance in FTS. Hexagonal mesoporous silica (HMS) has been compared with MCM-41 and the Co/HMS has superior catalytic activity and C5+ hydrocarbon selectivity. There are reports in the literature on the utilisation of mesoporous supports for metallic iron catalysts in the FTS.
Furthermore, nano-structured carbon supports such as carbon nanotubes (CNTs), carbon nanofibers (CNFs), carbon spheres (CSs) and multi-walled carbon nanotubes (MWCNTs) have been used for the FTS process. In comparison with mesoporous silica, mesoporous carbon is more resistant to changes of the support structure in aqueous environments. Iron and cobalt metal catalysts can be placed inside the CNT channel (Fe or Co/CNTs-in) as well as on the exterior surface of the CNT support (Co/CNTs-out). It has been observed that if the metal catalyst is located inside the pores then heavier hydrocarbon selectivity is achieved. Amongst CNF, CNT, CMC and CS as carbon nanoporous supports, the CNT supports have the highest selectivity to C5+ hydrocarbons. In order to produce light olefins, Fe supported on carbonaceous materials can be used. Iron metal situated inside CNTs support, has higher C5+ hydrocarbons selectivity than when supported on the exterior surface of the CNTs.
Finally, zeolite supported catalysts have received much attention for production of gasoline range hydrocarbons. The shapes of the zeolite's cages do not allow the formation of hydrocarbons with sizes larger than the size of the channel and, therefore, it can be used to crack heavier hydrocarbons to gasoline range hydrocarbons. It has been found that Fe metal catalyst supported on Faujasite zeolite has significant C5+ hydrocarbons selectivity. The Fe/ZSM-5 catalyst with Si/Al ratio of 25 has higher CO conversion and light olefin selectivity. Additionally, the Co-based ZSM catalysts with Si/Al ratio of 25 lead to better production of gasoline range hydrocarbons compared to those of higher Si/Al ratio. Zeolite hybrid catalysts, which are zeolites coupled with conventional supported catalyst (Co/SiO2, Fe/Al2O3, etc.) are promising FTS catalysts. Co zeolite hybrid catalysts are inferior to Fe zeolite hybrid catalysts in terms of CO conversion and hydrocarbon selectivity. Further work is predicted to focus on the modification of bifunctional FT catalysts with customised product activity and selectivity which can be used for industrial applications.
Nomenclature
| ASF | Anderson–Schulz–Flory |
| BTL | Biomass to liquid |
| CNFs | Carbon nanofibers |
| CNTs | Carbon nanotubes |
| CO | Carbon monoxide |
| Co | Cobalt |
| CSs | Carbon spheres |
| CTL | Coal to liquid |
| FTS | Fischer–Tropsch synthesis |
| GHSV | Gas hourly space velocity |
| GRHs | Gasoline range hydrocarbons |
| GTL | Gas to liquid |
| HMS | Hexagonal mesoporous silica |
| HTFT | High temperature Fischer–Tropsch |
| LTFT | Low temperature Fischer–Tropsch |
| MPS | Mesoporous silica |
| MWCNTs | Multi-walled carbon nanotubes |
| TOF | Turn over frequency |
| TON | Turn over number |
| TOS | Time on stream |
| WGS | Water gas shift |
| α | Chain growth probability |
| SHS | Silica hollow sphere |
| CMC | Carbon micro-coils |
| TPR | Temperature programmed reduction |
| MCM | Mobil composition of matter |
| HMS | Hexagonal mesoporous silica |
| ZSM | Zeolite Socony Mobil |
Acknowledgements
We thank the EPSRC (EP/K000616/1 and EP/K036548/1) for financial support and the Royal Society for the award of an Industry Fellowship (KW).References
- B. Walter, J. F. Gruson and G. Monnier, Oil Gas Sci. Technol., 2008, 63, 387–393 Search PubMed
.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed
- G.-Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS PubMed
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC
- F. Danielsen, H. Beukema, N. D. Burgess, F. Parish, C. A. Brühl, P. F. Donald, D. Murdiyarso, B. Phalan, L. Reijnders, M. Struebig and E. B. Fitzherbert, Conservation Biology, 2009, 23, 348–358 CrossRef PubMed
- B. Kamm and M. Kamm, Chem. Ing. Tech., 2007, 79, 592–603 CrossRef CAS
- B. Kamm, Angew. Chem., Int. Ed., 2007, 46, 5056–5058 CrossRef CAS PubMed
- E. G. Pereira, J. N. da Silva, J. L. de Oliveira and C. S. Machado, Renewable Sustainable Energy Rev., 2012, 16, 4753–4762 CrossRef CAS PubMed
- U. Arena, Waste Manage., 2012, 32, 625–639 CrossRef CAS PubMed
- A. K. Dalai and B. H. Davis, Appl. Catal., A, 2008, 348, 1–15 CrossRef CAS PubMed
-
M. A. Fahim, T. A. Alsahhaf and A. Elkilani, in Fundamentals of Petroleum Refining, ed. M. A. Fahim, T. A. Alsahhaf and A. Elkilani, Elsevier, Amsterdam, 2010, pp. 303–324 DOI:10.1016/B978-0-444-52785-1.00012-7
- D. Leckel, Energy Fuels, 2009, 23, 2342–2358 CrossRef CAS
- C. Perego, R. Bortolo and R. Zennaro, Catal. Today, 2009, 142, 9–16 CrossRef CAS PubMed
- C. Perego, Rendiconti Lincei, 2007, 18, 305–317 CrossRef
-
R. J. F. C. H. Bartholomew, Fundamentals of Industrial Catalytic Processes, 2nd edn, 2005 Search PubMed
- O. O. James, A. M. Mesubi, T. C. Ako and S. Maity, Fuel Process. Technol., 2010, 91, 136–144 CrossRef CAS PubMed
- V. Ponec, Catal. Today, 1992, 12, 227–254 CrossRef CAS
- H. Schulz, Appl. Catal., A, 1999, 186, 3–12 CrossRef CAS
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC
- M. R. Rahimpour and H. Elekaei, Fuel Process. Technol., 2009, 90, 747–761 CrossRef CAS PubMed
- B. H. Davis, Catal. Today, 2002, 71, 249–300 CrossRef CAS
- M. Feyzi, M. Irandoust and A. A. Mirzaei, Fuel Process. Technol., 2011, 92, 1136–1143 CrossRef CAS PubMed
- J.-H. Oh, J. W. Bae, S.-J. Park, P. K. Khanna and K.-W. Jun, Catal. Lett., 2009, 130, 403–409 CrossRef CAS PubMed
- A. Y. Khodakov, Catal. Today, 2009, 144, 251–257 CrossRef CAS PubMed
- T. Fu, Y. Jiang, J. Lv and Z. Li, Fuel Process. Technol., 2013, 110, 141–149 CrossRef CAS PubMed
- Y.-P. Li, T.-J. Wang, C.-Z. Wu, H.-B. Li, X.-X. Qin and N. Tsubaki, Fuel Process. Technol., 2010, 91, 388–393 CrossRef CAS PubMed
- S. R. Deshmukh, A. L. Y. Tonkovich, K. T. Jarosch, L. Schrader, S. P. Fitzgerald, D. R. Kilanowski, J. J. Lerou and T. J. Mazanec, Ind. Eng. Chem. Res., 2010, 49, 10883–10888 CrossRef CAS
- A. P. Steynberg, M. E. Dry, B. H. Davis and B. B. Breman, Fischer–Tropsch Technology, 2004, 152, 64–195 CAS
- R. M. Malek Abbaslou, J. S. Soltan Mohammadzadeh and A. K. Dalai, Fuel Process. Technol., 2009, 90, 849–856 CrossRef CAS PubMed
- M. E. Dry, Catal. Today, 2002, 71, 227–241 CrossRef CAS
- B. H. Davis, Ind. Eng. Chem. Res., 2007, 46, 8938–8945 CrossRef CAS
- M. K. Gnanamani, G. Jacobs, W. D. Shafer and B. H. Davis, Catal. Today, 2013, 215, 13–17 CrossRef CAS PubMed
- O. Kitakami, H. Sato, Y. Shimada, F. Sato and M. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13849–13854 CrossRef CAS
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. D. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 CrossRef CAS PubMed
- V. R. Calderone, N. R. Shiju, D. C. Ferre and G. Rothenberg, Green Chem., 2011, 13, 1950–1959 RSC
- H. Arai, K. Mitsuishi and T. Seiyama, Chem. Lett., 1984, 1291–1294 CrossRef CAS
- Y. H. Bi and A. K. Dalai, Can. J. Chem. Eng., 2003, 81, 230–242 CrossRef CAS
- T. Ishihara, N. Horiuchi, T. Inoue, K. Eguchi, Y. Takita and H. Arai, J. Catal., 1992, 136, 232–241 CrossRef CAS
- T. Ishihara, N. Horiuchi, K. Eguchi and H. Arai, J. Catal., 1991, 130, 202–211 CrossRef CAS
- D. J. Duvenhage and N. J. Coville, Appl. Catal., A, 1997, 153, 43–67 CrossRef CAS
- V. A. de la Peña O'Shea, M. C. Álvarez-Galván, J. M. Campos-Martín and J. L. G. Fierro, Appl. Catal., A, 2007, 326, 65–73 CrossRef PubMed
- A. Tavasoli, M. Trepanier, R. M. M. Abbaslou, A. K. Dalai and N. Abatzoglou, Fuel Process. Technol., 2009, 90, 1486–1494 CrossRef CAS PubMed
- V. R. Calderone, N. R. Shiju, D. Curulla-Ferrè, S. Chambrey, A. Khodakov, A. Rose, J. Thiessen, A. Jess and G. Rothenberg, Angew. Chem., Int. Ed., 2013, 52, 4397–4401 CrossRef CAS PubMed
- A. M. Saib, D. J. Moodley, I. M. Ciobîcă, M. M. Hauman, B. H. Sigwebela, C. J. Weststrate, J. W. Niemantsverdriet and J. van de Loosdrecht, Catal. Today, 2010, 154, 271–282 CrossRef CAS PubMed
- A. M. Saib, A. Borgna, J. V. De Loosdrecht, P. J. Van Berge and J. W. Niemantsverdriet, J. Phys. Chem. B, 2006, 110, 8657–8664 CrossRef CAS PubMed
- J. van de Loosdrecht, B. Balzhinimaev, J. A. Dalmon, J. W. Niemantsverdriet, S. V. Tsybulya, A. M. Saib, P. J. van Berge and J. L. Visagie, Catal. Today, 2007, 123, 293–302 CrossRef CAS PubMed
- E. van Steen, M. Claeys, M. E. Dry, J. van de Loosdrecht, E. L. Viljoen and J. L. Visagie, J. Phys. Chem. B, 2005, 109, 3575–3577 CrossRef CAS PubMed
- D. J. Moodley, A. M. Saib, J. van de Loosdrecht, C. A. Welker-Nieuwoudt, B. H. Sigwebela and J. W. Niemantsverdriet, Catal. Today, 2011, 171, 192–200 CrossRef CAS PubMed
- C. G. Visconti, L. Lietti, E. Tronconi, P. Forzatti, R. Zennaro and S. Rossini, Catal. Today, 2010, 154, 202–209 CrossRef CAS PubMed
- P. J. Van Berge and E. A. Caricato, Nitrogen containing compounds for cobalt based Fischer-Tropsch catalysts as a deactivation mechanism, Oral Presentation at Catalysis Society of South Africa (CATSA), Kruger National Park, South Africa, 2000 Search PubMed.
- V. R. R. Pendyala, M. K. Gnanamani, G. Jacobs, W. Ma, W. D. Shafer and B. H. Davis, Appl. Catal., A, 2013, 468, 38–43 CrossRef CAS PubMed
- F. G. Botes, J. W. Niemantsverdriet and J. van de Loosdrecht, Catal. Today, 2013, 215, 112–120 CrossRef CAS PubMed
- G. Kiss, C. E. Kliewer, G. J. DeMartin, C. C. Culross and J. E. Baumgartner, J. Catal., 2003, 217, 127–140 CAS
- G. W. Huber, C. G. Guymon, T. L. Conrad, B. C. Stephenson and C. H. Bartholomew, Catalyst Deactivation 2001, Proceedings, 2001, 139, 423–430 CAS
- G. Jacobs, P. M. Patterson, Y. Q. Zhang, T. Das, J. L. Li and B. H. Davis, Appl. Catal., A, 2002, 233, 215–226 CrossRef CAS
- T. K. Das, G. Jacobs, P. M. Patterson, W. A. Conner, J. L. Li and B. H. Davis, Fuel, 2003, 82, 805–815 CrossRef CAS
- D. J. Moodley, J. van de Loosdrecht, A. M. Saib, M. J. Overett, A. K. Datye and J. W. Niemantsverdriet, Appl. Catal., A, 2009, 354, 102–110 CrossRef CAS PubMed
- G. Jiao, Y. Ding, H. Zhu, X. Li, J. Li, R. Lin, W. Dong, L. Gong, Y. Pei and Y. Lu, Appl. Catal., A, 2009, 364, 137–142 CrossRef CAS PubMed
- J. L. Li, G. Jacobs, T. Das, Y. Q. Zhang and B. Davis, Appl. Catal., A, 2002, 236, 67–76 CrossRef CAS
- H. Schulz, Z. Q. Nie and F. Ousmanov, Catal. Today, 2002, 71, 351–360 CrossRef CAS
- R. L. Espinoza, A. P. Steynberg, B. Jager and A. C. Vosloo, Appl. Catal., A, 1999, 186, 13–26 CrossRef CAS
-
R. B. Anderson, L. J. E. Hofer, E. M. Cohn, H. Steiner, M. Greyson and S. W. Weller, in Catalysis, ed. P. H. Emmett, Van Nostrand-Reinhold, New York, 1956, vol. 4 Search PubMed
- S. A. Eliason and C. H. Bartholomew, Appl. Catal., A, 1999, 186, 229–243 CrossRef CAS
- S. Z. Li, R. J. O'Brien, G. D. Meitzner, H. Hamdeh, B. H. Davis and E. Iglesia, Appl. Catal., A, 2001, 219, 215–222 CrossRef CAS
- D. Duvenhage and N. Coville, Appl. Catal., A, 2006, 298, 211–216 CrossRef CAS PubMed
- B. Wu, L. Bai, H. Xiang, Y.-W. Li, Z. Zhang and B. Zhong, Fuel, 2004, 83, 205–212 CrossRef CAS
- T. C. Bromfield and N. J. Coville, Appl. Catal., A, 1999, 186, 297–307 CrossRef CAS
-
S. A. Stevenson, J. A. Dumesic, R. T. K. Baker and E. Ruckenstein, Metal Support Interactions in Catalysis, Sintering, and Redispersion (Van Nostrand Reinhold Catalysis Series), New York, 1987 Search PubMed
- W.-S. Ning, N. Koizumi and M. Yamada, Catal. Commun., 2007, 8, 275–278 CrossRef CAS PubMed
- N. Tsubaki, S. L. Sun and K. Fujimoto, J. Catal., 2001, 199, 236–246 CrossRef CAS
- W. Chu, P. Chernavskii, L. Gengembre, G. Pankina, P. Fongarland and A. Khodakov, J. Catal., 2007, 252, 215–230 CrossRef CAS PubMed
- E. Iglesia, Appl. Catal., A, 1997, 161, 59–78 CrossRef CAS
- S. Storsater, O. Borg, E. Blekkan and A. Holmen, J. Catal., 2005, 231, 405–419 CrossRef PubMed
- H. P. Withers, K. F. Eliezer and J. W. Mitchell, Ind. Eng. Chem. Res., 1990, 29, 1807–1814 CrossRef CAS
- B. Jongsomjit, J. Catal., 2003, 215, 66–77 CrossRef CAS
- H. Xiong, Y. Zhang, K. Liew and J. Li, J. Mol. Catal. A: Chem., 2005, 231, 145–151 CrossRef CAS PubMed
- F. Morales, F. Degroot, O. Gijzeman, A. Mens, O. Stephan and B. Weckhuysen, J. Catal., 2005, 230, 301–308 CrossRef CAS PubMed
- F. Morales, D. Grandjean, F. M. F. de Groot, O. Stephan and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2005, 7, 568–572 RSC
- F. Morales, F. M. F. de Groot, P. Glatzel, E. Kleimenov, H. Bluhm, M. Havecker, A. Knop-Gericke and B. M. Weckhuysen, J. Phys. Chem. B, 2004, 108, 16201–16207 CrossRef CAS
- F. Morales, E. Desmit, F. Degroot, T. Visser and B. Weckhuysen, J. Catal., 2007, 246, 91–99 CrossRef CAS PubMed
- D. L. King, J. A. Cusumano and R. L. Garten, Catal. Rev.: Sci. Eng., 1981, 23, 233–263 CAS
- Y. Yang, Appl. Catal., A, 2004, 266, 181–194 CrossRef CAS PubMed
- S. Soled, E. Iglesia and R. A. Fiato, Catal. Lett., 1991, 7, 271–280 CrossRef
- A. P. Raje, R. J. O'Brien and B. H. Davis, J. Catal., 1998, 180, 36–43 CrossRef CAS
- H. Schulz and A. Zein El Deen, Fuel Process. Technol., 1977, 1, 45–56 CrossRef CAS
- W. Ngantsoue-Hoc, Y. Q. Zhang, R. J. O'Brien, M. S. Luo and B. H. Davis, Appl. Catal., A, 2002, 236, 77–89 CrossRef CAS
- N. Lohitharn and J. Goodwinjr, J. Catal., 2008, 260, 7–16 CrossRef CAS PubMed
-
C. H. Yang and A. G. Oblad, Catalytic synthesis of light olefinic hydrocarbons from CO and H/sub 2/over some iron catalysts, 1978 Search PubMed
- L. Bai, H. W. Xiang, Y. W. Li, Y. Z. Han and B. Zhong, Fuel, 2002, 81, 1577–1581 CrossRef CAS
- Y. Liu, B.-T. Teng, X.-H. Guo, Y. Li, J. Chang, L. Tian, X. Hao, Y. Wang, H.-W. Xiang, Y.-Y. Xu and Y.-W. Li, J. Mol. Catal. A: Chem., 2007, 272, 182–190 CrossRef CAS PubMed
- N. Lohitharn, J. G. Goodwin Jr and E. Lotero, J. Catal., 2008, 255, 104–113 CrossRef CAS PubMed
- N. Lohitharn and J. Goodwinjr, J. Catal., 2008, 257, 142–151 CrossRef CAS PubMed
- M. Arsalanfar, A. A. Mirzaei, H. R. Bozorgzadeh, A. Samimi and R. Ghobadi, J. Ind. Eng. Chem., 2014, 20, 1313–1323 CrossRef CAS PubMed
- L. Spadaro, F. Arena, M. Granados, M. Ojeda, J. Fierro and F. Frusteri, J. Catal., 2005, 234, 451–462 CrossRef CAS PubMed
- D. B. Bukur, X. Lang, D. Mukesh, W. H. Zimmerman, M. P. Rosynek and C. P. Li, Ind. Eng. Chem. Res., 1990, 29, 1588–1599 CrossRef CAS
- H. N. Pham, A. Viergutz, R. J. Gormley and A. K. Datye, Powder Technol., 2000, 110, 196–203 CrossRef CAS
- C. M. A. Parlett, D. W. Bruce, N. S. Hondow, A. F. Lee and K. Wilson, ACS Catal., 2011, 1, 636–640 CrossRef CAS
- G. R. Moradi, M. M. Basir, A. Taeb and A. Kiennemann, Catal. Commun., 2003, 4, 27–32 CrossRef CAS
- J.-P. Dacquin, J. Dhainaut, D. Duprez, S. Royer, A. F. Lee and K. Wilson, J. Am. Chem. Soc., 2009, 131, 12896–12897 CrossRef CAS PubMed
- J. Dhainaut, J.-P. Dacquin, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 296–303 RSC
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC
- C. Pirez, J.-M. Caderon, J.-P. Dacquin, A. F. Lee and K. Wilson, ACS Catal., 2012, 2, 1607–1614 CrossRef CAS
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and F. Villain, J. Phys. Chem. B, 2001, 105, 9805–9811 CrossRef CAS
- S. L. Soled, E. Iglesia, R. A. Fiato, J. E. Baumgartner, H. Vroman and S. Miseo, Top. Catal., 2003, 26, 101–109 CrossRef CAS
- A. Tavasoli, K. Sadagiani, F. Khorashe, A. A. Seifkordi, A. A. Rohani and A. Nakhaeipour, Fuel Process. Technol., 2008, 89, 491–498 CrossRef CAS PubMed
- R. C. Reuel and C. H. Bartholomew, J. Catal., 1984, 85, 78–88 CrossRef CAS
- J. Panpranot, J. G. Goodwin and A. Sayari, Catal. Today, 2002, 77, 269–284 CrossRef CAS
- J.-S. Jung, S. W. Kim and D. J. Moon, Catal. Today, 2012, 185, 168–174 CrossRef CAS PubMed
- I. T. Ghampson, C. Newman, L. Kong, E. Pier, K. D. Hurley, R. A. Pollock, B. R. Walsh, B. Goundie, J. Wright, M. C. Wheeler, R. W. Meulenberg, W. J. DeSisto, B. G. Frederick and R. N. Austin, Appl. Catal., A, 2010, 388, 57–67 CrossRef CAS PubMed
- R. M. Malek Abbaslou, J. Soltan and A. K. Dalai, Fuel, 2011, 90, 1139–1144 CrossRef CAS PubMed
- F. G. Botes and W. Böhringer, Appl. Catal., A, 2004, 267, 217–225 CrossRef CAS PubMed
- Y.-P. Li, T.-J. Wang, C.-Z. Wu, X.-X. Qin and N. Tsubaki, Catal. Commun., 2009, 10, 1868–1874 CrossRef CAS PubMed
- D. Enache, Appl. Catal., A, 2004, 268, 51–60 CrossRef CAS PubMed
- G. Jacobs, T. K. Das, Y. Q. Zhang, J. L. Li, G. Racoillet and B. H. Davis, Appl. Catal., A, 2002, 233, 263–281 CrossRef CAS
- E. Iglesia, S. L. Soled, J. E. Baumgartner and S. C. Reyes, J. Catal., 1995, 153, 108–122 CrossRef CAS
- D. H. Yin, W. H. Li, W. S. Yang, H. W. Xiang, Y. H. Sun, B. Zhong and S. Y. Peng, Microporous Mesoporous Mater., 2001, 47, 15–24 CrossRef CAS
- A. Y. Khodakov, A. Griboval-Constant, R. Bechara and V. L. Zholobenko, J. Catal., 2002, 206, 230–241 CrossRef CAS
- J. Hong, P. A. Chernavskii, A. Y. Khodakov and W. Chu, Catal. Today, 2009, 140, 135–141 CrossRef CAS PubMed
- E. Lira, C. M. López, F. Oropeza, M. Bartolini, J. Alvarez, M. Goldwasser, F. L. Linares, J.-F. Lamonier and M. J. Pérez Zurita, J. Mol. Catal. A: Chem., 2008, 281, 146–153 CrossRef CAS PubMed
- A. Stein, Z. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS
- H. Xiong, M. A. M. Motchelaho, M. Moyo, L. L. Jewell and N. J. Coville, Catal. Today, 2013, 214, 50–60 CrossRef CAS PubMed
- H. Xiong, M. A. M. Motchelaho, M. Moyo, L. L. Jewell and N. J. Coville, J. Catal., 2011, 278, 26–40 CrossRef CAS PubMed
- L. Guczi, G. Stefler, O. Geszti, Z. Koppany, Z. Konya, E. Molnar, M. Urban and I. Kiricsi, J. Catal., 2006, 244, 24–32 CrossRef CAS PubMed
- R. M. M. Abbaslou, A. Tavassoli, J. Soltan and A. K. Dalai, Appl. Catal., A, 2009, 367, 47–52 CrossRef CAS PubMed
- W. Chen, Z. L. Fan, X. L. Pan and X. H. Bao, J. Am. Chem. Soc., 2008, 130, 9414–9419 CrossRef CAS PubMed
- Y. Yang, K. Chiang and N. Burke, Catal. Today, 2011, 178, 197–205 CrossRef CAS PubMed
- B. Li, C. Wang, G. Yi, H. Lin and Y. Yuan, Catal. Today, 2011, 164, 74–79 CrossRef CAS PubMed
- S.-H. Kang, H. M. Koo, A. R. Kim, D.-H. Lee, J.-H. Ryu, Y. D. Yoo and J. W. Bae, Fuel Process. Technol., 2013, 109, 141–149 CrossRef CAS PubMed
- H. M. T. Galvis, J. H. Bitter, C. B. Khare, M. Ruitenbeek, A. I. Dugulan and K. P. de Jong, Science, 2012, 335, 835–838 CrossRef PubMed
- M. Dalil, M. Sohrabi and S. J. Royaee, J. Ind. Eng. Chem., 2012, 18, 690–696 CrossRef CAS PubMed
- P. Concepción, C. López, A. Martínez and V. Puntes, J. Catal., 2004, 228, 321–332 CrossRef PubMed
- R. Ravishankar, M. M. Li and A. Borgna, Catal. Today, 2005, 106, 149–153 CrossRef CAS PubMed
- M. V. Cagnoli, N. G. Gallegos, A. M. Alvarez, J. F. Bengoa, A. A. Yeramian, M. Schmal and S. G. Marchetti, Appl. Catal., A, 2002, 230, 169–176 CrossRef CAS
- Q. H. Tang, Q. H. Zhang, P. Wang, Y. Wang and H. L. Wan, Chem. Mater., 2004, 16, 1967–1976 CrossRef CAS
- S. Bessell, Appl. Catal., A, 1993, 96, 253–268 CrossRef CAS
- S. Bessell, Appl. Catal., A, 1995, 126, 235–244 CrossRef CAS
- S. H. Kang, J. H. Ryu, J. H. Kim, P. S. Prasad, J. W. Bae, J. Y. Cheon and K. W. Jun, Catal. Lett., 2011, 141, 1464–1471 CrossRef CAS
- S. Wang, Q. Yin, J. Guo, B. Ru and L. Zhu, Fuel, 2013, 108, 597–603 CrossRef CAS PubMed
- S.-H. Kang, J. W. Bae, K.-J. Woo, P. S. Sai Prasad and K.-W. Jun, Fuel Process. Technol., 2010, 91, 399–403 CrossRef CAS PubMed
- A. Martínez, J. Rollán, M. Arribas, H. Cerqueira, A. Costa and E. Saguiar, J. Catal., 2007, 249, 162–173 CrossRef PubMed
- A. N. Pour, M. Zare, S. M. Kamali Shahri, Y. Zamani and M. R. Alaei, J. Nat. Gas Sci. Eng., 2009, 1, 183–189 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2014 |