Elaboration and characterization of barley protein nanoparticles as an oral delivery system for lipophilic bioactive compounds
Jingqi
Yang
,
Ying
Zhou
and
Lingyun
Chen
*
Department of Agricultural, Food and Nutritional Science, University of Alberta, Edmonton, T6G 2P5, Canada. E-mail: lingyun.chen@ualberta.ca; Fax: +1-780-492-4265; Tel: +1-780-492-0038
First published on 27th November 2013
Abstract
This is the first report in which barley protein nanoparticles were prepared with the aim of developing a delivery system for lipophilic bioactive compounds at ambient temperature using high pressure homogenization. No organic solvents or crosslinking reagents were involved in the nanoparticle preparation. Effects of processing conditions and formulae on particle size and size distribution were investigated. Optimal nanoparticles with regular spherical shape, small size (90–150 nm) and narrow size distribution (PDI < 0.3) could be achieved at a protein weight concentration of up to 5% when the oil/protein ratio was maintained within a range of 1 to 1.5. These nanoparticles exhibited high zeta-potential (about −35 mV), high payload (51.4–54.5%) and good stability without the use of surfactants. As shown by the release test, though the bulk protein matrices of nanoparticles were degraded in the simulated gastric tract, even smaller nanoparticles were released and bioactive compounds were protected by a layer of barley protein. Then, complete release occurred in the simulated intestinal environments due to pancreatin degradation. In vitro studies showed that barley protein nanoparticles are relatively safe and could be internalized by Caco-2 cells and accumulated in the cytoplasm.
1 Introduction
Increasing scientific evidence has shown that bioactive compounds, such as polyunsaturated fatty acids, phytosterols, carotenoids and vitamins, have the potential to reduce the risk of chronic diseases and improve public health. Their efficacy depends on the dose, the preserved biological activity and the bioavailability.1 However, many lipophilic bioactive compounds exhibit low bioavailability due to insufficient residency time in the gut, or low permeability and/or low solubility within the intestinal tract, which can limit their potential activity in vivo.2 For example, of the total amount of carotenoids found in fruits and vegetables, only a small proportion is bioavailable.3 These compounds exhibit antioxidant activities which can protect cells against oxidation to reduce the risk of cardiovascular disease, cancer, and aging-related diseases. Nanoencapsulation of nutraceuticals is emerging as a promising approach for delivering health promoting substances to wide populations without harming the sensory quality of food, while providing benefits of protection and improved bioavailability. Nanoparticles can dramatically prolong the formulation residence time by decreasing the influence of intestinal clearance mechanisms and by increasing the surface area to interact with the biological support. In addition, some of them are small enough to cross the epithelial lining of the gut and are readily taken up by cells, allowing efficient delivery of active compounds to target sites in the body.4 However, the application of nanotechnology in the food industry is still limited due to the lack of effective food grade materials along with safe and economic processing.5Food grade proteins are interesting materials for nanoparticle preparation due to their excellent biocompatibility and biodegradability, as well as unique properties in forming gels, emulsions, and emulsion gels, thus offering the capacity of incorporating both hydrophilic and lipophilic bioactive molecules until release at the functional site. In spite of the promising potential, there are several formidable challenges which have to be overcome before they can be widely used as nutraceutical delivery systems. Firstly, nanoparticles prepared from protein materials involve processes such as emulsification–solvent evaporation, chemical or heat-induced crosslinking and coacervation. These normally demand heating and/or organic solvents, which cannot be applied to heat-labile bioactive compounds and may cause concerns about the potentially toxic residues.4 Secondly, feasible processing conditions for mass production of stable protein-based nanoparticles are lacking. In many cases, small molecule surfactants are required to stabilize food protein nanoparticles,6 which may result in controversies about their health risks. Moreover, due to the digestibility of food proteins in gastric environments and the high surface-to-volume ratio of nanoparticles, the incorporated nutraceuticals are normally released rapidly in the stomach, resulting in limited nutraceutical compounds that can reach small intestine where they need to be absorbed to exert health benefits. Therefore, new protein materials and feasible processing conditions are required to address the challenges.
Barley proteins are one of the most abundant and inexpensive natural food protein sources. The major fractions are hordein (about 45% w/w) and glutelin (40% to 45%). Due to their unique molecular structures and a high content of hydrophobic amino acid residues, barley proteins demonstrated good foaming,7 emulsifying,7 and film forming8 capacities. Recently, microparticles from barley protein have been developed in our group by a pre-emulsifying process followed by microfluidizing without using organic solvents or cross-linking reagents. In addition, these microparticles exhibited the ability to protect the encapsulated lipid phase in simulated gastric fluid and release them under simulated intestinal conditions.9 Therefore, it would be interesting to adapt this technology to create new nanoparticles from barley proteins for new or improved properties. This work aims to elaborate on barley protein based nanoparticles and study the impact of processing conditions on particle microscopic features including size, size distribution, morphology and surface charge. Subsequently, the in vitro release properties of the optimized barley protein nanoparticles were tested in the simulated gastro-intestinal tract using β-carotene as a model bioactive compound. Cytotoxicity and cellular uptake were also evaluated preliminarily by the Caco-2 cell model to study their biocompatibility and efficacy.
2 Materials and methods
2.1 Materials
Barley flour was milled from pearled barley grains (Falcon), which was kindly provided by Dr James Helm, Alberta Agricultural and Rural Development, Lacombe, Alberta. Barley protein was extracted from barley flour by alkaline solution according to methods previously established.10 The protein content (dry status) was 90 wt% as measured by combustion with a nitrogen analyzer (FP-428, Leco Corporation, St. Joseph, MI, USA). The canola oil used in the present work was purchased from the local supermarket. β-Carotene, pepsin (from porcine gastric mucosa, 424 U mg−1), pancreatin (from porcine pancreas), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO) and nile red were purchased from Sigma-Aldrich Canada Ltd (Oakville, ON, Canada). Alexa Fluor dyes, 4′,6-diamidino-2-phenylindole (DAPI) and mounting medium were from Life technologies (Burlington, ON, Canada). Other cell culture reagents including Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), non-essential amino acids (NEAA), HEPES solution, trypsin–EDTA and Hank's balanced salt solution (HBSS) were purchased from GIBCO (Burlington, ON, Canada). Human colorectal adenocarcinoma cell line Caco-2 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). All chemicals used in this work were of reagent grade.2.2 Nanoparticle preparation
Nanoparticles were prepared with barley protein as a coating material and canola oil as the lipid phase. β-Carotene (0.05% w/v) was added to the lipid phase as a model lipophilic bioactive compound. The oil phase was added to the aqueous protein suspension, followed by high speed homogenization (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, PowerGen, Fisher Scientific International, Inc., CA, USA) to prepare a coarse emulsion. Then, the pre-mixed emulsion was passed through a high pressure homogenizer (Nano DeBEE, Bee International, Inc., MA, USA) to form solid nanoparticles. Different processing pressures (4, 8, 12, 16 and 20 kpsi), recirculation numbers (1 to 6), protein concentrations (2, 3 and 5% w/v) and oil-to-protein ratios (0.2 to 5) were applied to prepare nanoparticles of various properties as characterized in Section 2.3. The prepared nanoparticles were stored at 4 °C with 0.025% (w/v) sodium azide until use.
000 rpm, PowerGen, Fisher Scientific International, Inc., CA, USA) to prepare a coarse emulsion. Then, the pre-mixed emulsion was passed through a high pressure homogenizer (Nano DeBEE, Bee International, Inc., MA, USA) to form solid nanoparticles. Different processing pressures (4, 8, 12, 16 and 20 kpsi), recirculation numbers (1 to 6), protein concentrations (2, 3 and 5% w/v) and oil-to-protein ratios (0.2 to 5) were applied to prepare nanoparticles of various properties as characterized in Section 2.3. The prepared nanoparticles were stored at 4 °C with 0.025% (w/v) sodium azide until use.
2.3 Nanoparticle characterization
The size, size distribution as indicated by polydispersity index (PDI), and zeta potential of the nanoparticles were measured at room temperature (23 °C) by dynamic light scattering and laser Doppler velocimetry using a Zetasizer Nano S (model ZEN 1600, Malvern Instruments Ltd, UK). The protein refractive index (RI) was set at 1.45 and dispersion medium RI was 1.33.9 The nanoparticle samples were diluted to an appropriate concentration with phosphate buffer (pH 7) to avoid multiple scattering before analysis. The data were averaged from at least three batches. The storage stability of the nanoparticles was investigated by measuring the particle size at different time intervals during storage at 4 °C in the dark. Samples were diluted one time with deionized water before storage.Methods used to quantitatively measure the surface oil percentage and encapsulation efficiency were adapted from Wang et al. with modifications.9 Pure ethanol (7 ml) was added into 3 ml nanoparticle suspension to break the particles as a major component of barley protein, hordein, is soluble in 60–70% ethanol solution.9 Then 10 ml of hexane was added and the mixture was shaken vigorously with a vortex mixer for another 1 min and allowed to stand for 1 min. These mixing and standing procedures were repeated twice. The final mixture was centrifuged at 8000g for 15 min at 20 °C. After centrifugation, 5 ml of the upper layer was transferred to a weighed tube and evaporated under nitrogen to remove the solvent. The remaining oil was weighed to the nearest 0.1 mg. The surface oil amount was measured by a similar method, but only hexane was added into the nanoparticle suspension.
The surface oil percentage was calculated by the following equation:
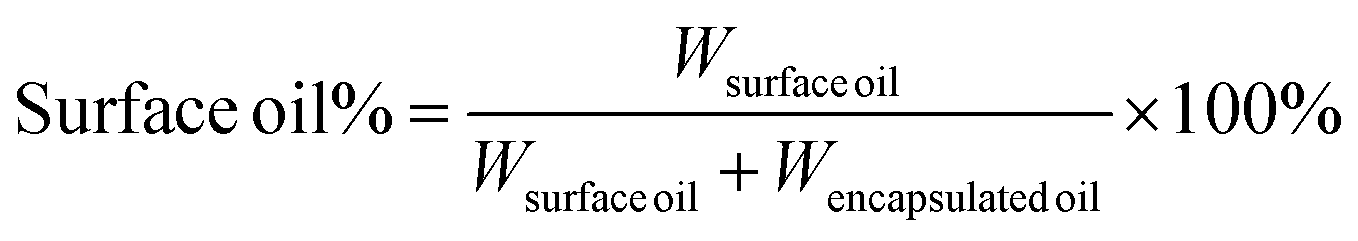 | (1) |
The encapsulation efficiency (EE) and loading capacity (LC) were calculated by the following equations:
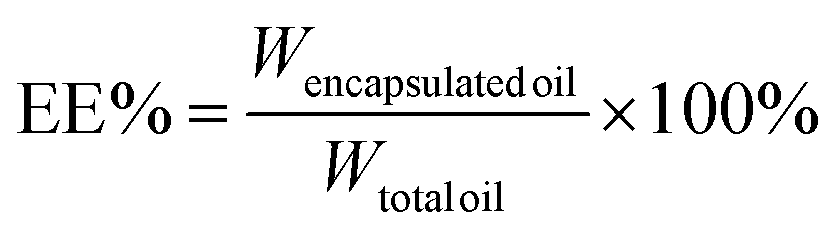 | (2) |
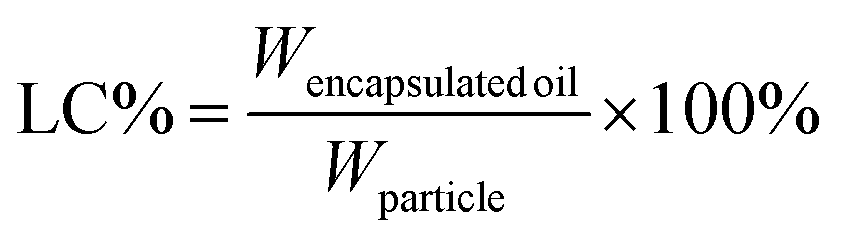 | (3) |
The morphology of the nanoparticles was observed by transmission electrical microscopy (TEM, Morgagni 268, Philips-FEI, Hillsboro, USA) at an accelerating voltage of 80 kV. Nanoparticles were negatively stained with 2% (w/v) sodium phosphotungstate (pH 7.4). One drop of the nanoparticle sample was added to a copper grid covered with nitrocellulose and kept still for 3 min. Then, a drop of sodium phosphotungstate (2%, w/v) was added to the top of the nanoparticle droplet on the grid. Excess liquid was blotted from the grid, and then samples were air dried at room temperature.
2.4 In vitro protein matrix degradation
The in vitro protein matrix degradation assays were conducted in simulated gastric fluid (SGF, pH 1.5 with 0.1% (w/v) pepsin) and simulated intestinal fluid (SIF, pH 7.4 with 1.0% (w/v) pancreatin). Changes in nanoparticle morphology after incubating in SGF and SIF were observed using the TEM and the samples were treated in the same way as above. The size change of the nanoparticles in the SGF and SIF was also monitored using the Zetasizer Nano S instrument under the same conditions as indicated above.2.5 In vitro release properties of barley protein nanoparticles
The release properties of barley protein nanoparticles were also measured in the simulated gastro-intestinal tract. Briefly, nanoparticles (containing 12 mg barley protein) were incubated in SGF at 37 °C for 1 hour. The digestion was stopped by heating the sample to 95 °C for 3 min to inactive enzymes. The released β-carotene was extracted with hexane and quantitatively determined by reading the absorbance at 450 nm with a UV-visible spectrophotometer (model V-530, Jasco, CA, USA).11After incubation in SGF for 1 hour, the pH of the mixture was adjusted to 7.5 with concentrated NaOH. Then pancreatin suspension was added to initiate the digestion and the final pancreatin concentration was 1% (w/v). The mixture was incubated at 37 °C and samples were taken at different time intervals. The digestion was stopped after 8 hours by heating the sample to 95 °C for 3 min. The degradation of nanoparticles and release of β-carotene were assessed by a lipid digestion model which was increasingly used to in vitro evaluate the lipid-based drug/nutraceutical delivery system.12 The dynamic lipid digestion was performed through the methods described by Li13 with slight modification. The released free fatty acid was titrated with 0.1 M NaOH to maintain the pH value at 7.5. The volume of NaOH added was recorded and used to calculate the concentration of free fatty acids generated during digestion. The cumulated released free fatty acid percentage was calculated using the following equations:13
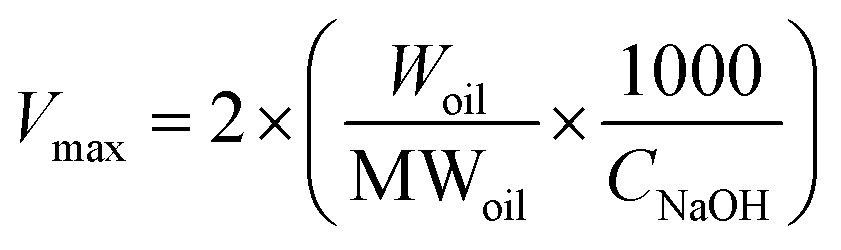 | (4) |
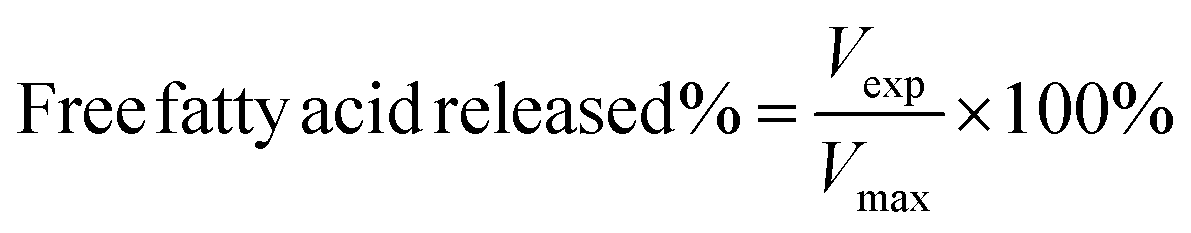 | (5) |
2.6 In vitro cytotoxicity and Caco-2 cell uptake
Caco-2 cells were grown in T-75 flasks at 37 °C in a humidified atmosphere of 5% CO2. The cells were cultured in DMEM supplemented with 20% FBS (v/v), 1% NEAA and 25 mM HEPES. The medium was changed every other day until the cells reached 80% confluence, when the cells were removed with 0.25% trypsin in 1 mM EDTA solution at 37 °C for 4–6 min and passaged with fresh medium.The cytotoxicity of the nanoparticles was examined by MTT assay. Caco-2 cells were transferred onto 96-well plates at a density of 8000 cells per well in 100 μl culture medium. The cells were grown for 24 hours to allow attachment before the experiment. Nanoparticles were added into each well to reach a final concentration of 0.5 mg ml−1, 0.25 mg ml−1 and 0.125 mg ml−1 respectively, and incubated with the cells for 6 h. 10 μl of MTT solvent (5 mg ml−1 in PBS) was then added to each well and incubated for a further 4 hours at 37 °C followed by removal of the medium from each well. 100 μl of DMSO was added to each well followed by the measurement of the absorbance at 570 nm using a microplate reader (SpectraMax, Molecular Devices, USA). The viability was expressed by the percentage of living cells with respect to the control cells.
The uptake of the nanoparticles by Caco-2 cells was studied using fluorescence-labeled nanoparticles and confocal laser scanning microscopy (CLSM). Briefly, 0.025% (w/v) nile red was dissolved in canola oil followed by centrifugation at 12000g for 10 min and the oil supernatant was used for nanoparticle preparation following the same process above. Caco-2 cells were transferred onto glass bottom microwell dishes (P35G-1.5-14-C, MatTek Corp., USA) at a density of 1 × 105 cells per dish and cultured for 5–7 days until a confluent monolayer was formed. On the day of experiment, the medium was replaced with HBSS (without phenol red) and allowed to equilibrate at 37 °C for 30 min. Following the removal of the buffer, the nanoparticle suspension in HBSS at a concentration of 0.5 mg ml−1 was added and incubated with the cells for 1, 3, and 6 h. The cells were then gently washed with PBS 3 times and fixed with 4% paraformaldehyde (w/v in PBS pH 7.2) at 37 °C for 15 min. The cell membrane and nuclei were stained with wheat germ agglutinin (WGA)–Alexa Fluor 488 conjugate and DAPI respectively and the cells were mounted with Prolong Gold Antifade Reagent. A CLSM 510 Meta (Carl Zeiss, Jena, Germany) equipped with a diode, an argon laser and a helium/neon laser, providing the excitation at 405 nm, 488 nm and 561 nm respectively, was used for observation and imaging. Images were processed with ZEN 2009LE software (Carl Zeiss MicroImaging GmbH, Germany).
2.7 Statistical analysis
All experiments were performed at least in three independent batches. Data were represented as the mean of three batches ± standard deviation. For data in figures, error bars showed standard deviations. Statistical evaluation was conducted by Student's t-test and analysis of variance (ANOVA) using SAS (SAS Institute, Inc., Cary, NC). The multiple-comparisons were evaluated by Duncan's multiple-range test. Statistical differences between samples were performed with a level of significance as p < 0.05.3 Results and discussion
3.1 Barley protein nanoparticle preparation
For globular proteins such as soy and whey protein, emulsions are normally generated after high pressure homogenization due to the fact that hydrophilic protein molecules usually form a gel-like viscoelastic thin film outside an oil droplet to stabilize it in the aqueous phase.15 Solid nanoparticles could be derived from their corresponding nanoemulsion through solidifying and/or hardening processes by adding a cross-linking reagent (e.g. glutaraldehyde and transglutaminase), or coacervating with oppositely charged polymers.16 Interestingly, solid nanoparticles were generated from barley protein stabilized coarse emulsion directly after high pressure homogenization. This phenomenon was also observed in our previous work, where barley protein formed solid microparticles during the micro-fluidization process due to the surface hydrophobic nature of their molecular structures which enabled them to adhere and completely cover the oil droplets rapidly in the pre-emulsion process. These complexes tended to strongly aggregate due to hydrophobic surface patches to form thick unruptured coatings after high pressure treatment. The morphology of the nanoparticles was shown by TEM images (Fig. 1B). The mean diameter of barley protein nanoparticles ranged from 50 to 200 nm with regular spherical shapes and smooth surfaces. Fig. 1C also showed that small oil droplets were homogeneously trapped inside the protein matrix with a honeycomb structure. The proposed nanoparticle structure is described in Fig. 1D. This type of nanoparticle structure might better carry and protect the interior dispersed phase. Our preliminary experiments indicated that processing conditions (number of passes and pressure) and particle formation solution (protein concentration, protein/oil ratio) dictated nanoparticle properties. Since the particle diameter played an important role in their physiological properties,4 in this work, the process parameters were optimized with an emphasis on the particle size and size distribution.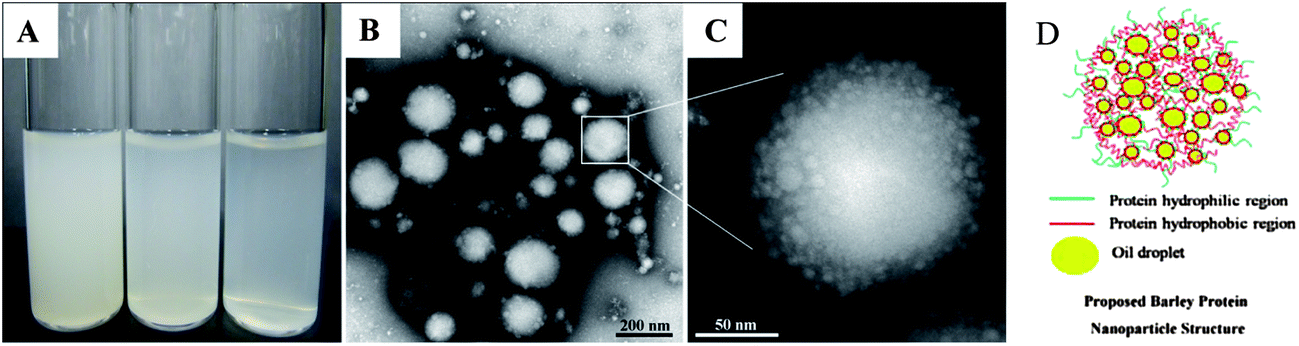 | ||
| Fig. 1 (A) Photograph of barley protein nanoparticles (from left to right, the nanoparticle concentrations are 0.2, 0.1 and 0.05 wt%); (B) TEM image of barley protein nanoparticles (2 wt% protein and 2.5% v/v oil) prepared at 16 kpsi with 14k× magnification; (C) the same sample observed with 110k× magnification; and (D) the proposed barley protein nanoparticle structure. | ||
Firstly, the particles were prepared with 2% (w/v) protein and 2.5% (v/v) oil under two selected pressures (12 and 16 kpsi) which allowed formation of well-dispersed nanoparticles with spherical shapes. Fig. 2 demonstrates that with an increase of the recirculation number from 1 to 3, diameters of particles prepared under 12 kpsi decreased from 494 to 246 nm, while particles prepared under 16 kpsi decreased from 264 to 162 nm. This result is in agreement with previous studies.18,19 Increasing recirculation time led to prolonged exposure of nanoparticles in high pressure emulsification units, which resulted in stronger energy input and a higher degree of oil droplet disruption.17 Even though a tendency to decrease the particle size was observed as the recirculation number further increased, the reductions became modest after 4 passes and leveled off gradually. The smallest mean particle size was 195 nm for 12 kpsi and 147 nm for 16 kpsi after 6 passes. Since the efficiency of particle size disruption was limited by the emulsion composition and other emulsification conditions, it was no longer efficient to minimize the particle size by adding more recirculation numbers. On the other hand, increasing the number of recirculation significantly narrowed the particle size distribution as indicated by the decreased PDI value (inset figure in Fig. 2), which was reported to have important effects on nanoparticle applications.20 PDI of the sample dropped from 0.55 to 0.33 and from 0.43 to 0.25 after 6 passes of homogenization under 12 and 16 kpsi respectively, indicating that the particle size distribution was acceptably homogenous after 6 passes, since a PDI value of less than 0.3 indicates a narrow size distribution.19 This result was expected because a longer energy input duration and a stronger dispersive effect on the oil droplets could be achieved by increasing the recirculation number.21 Thus, 6 times of recirculation was selected as the optimized condition for further experiments.
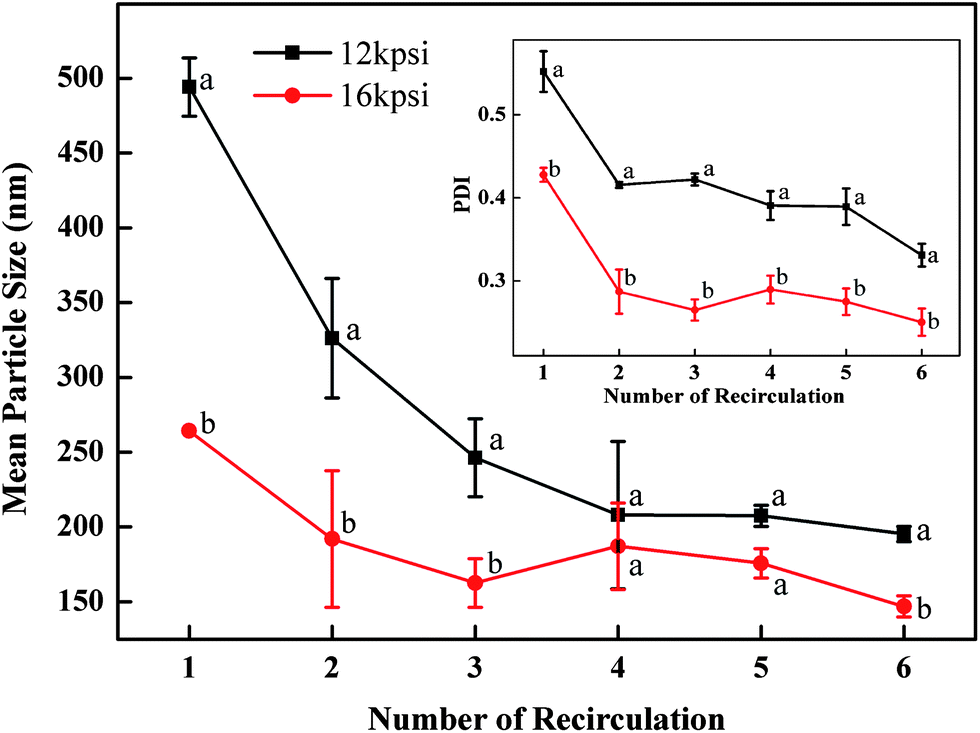 | ||
| Fig. 2 Effect of the number of recirculations on the mean particle size of samples produced with 2 wt% barley protein and 2.5% v/v oil. The inset figure shows the effect of the number of recirculations on the particle size distribution as indicated by PDI. Different letters above or below the curve indicate significant difference (p < 0.05) due to processing pressure. | ||
The influence of processing pressure on the particle size is shown in Fig. 3. Here the particles were prepared with 2 and 3% (w/v) protein and 2.5% (v/v) oil. When particles were prepared with 2% protein, the mean particle diameter dropped from 339 to 147 nm with increasing homogenizing pressure from 4 to 16 kpsi. Higher pressure input could lead to a higher degree of protein unfolding and oil droplet disruption, resulting in smaller particle sizes.18,19 In contrast, samples prepared with 3% protein had modest changes in the tested pressure range, which indicated that the emulsion formula significantly influences the particle size as well. An obvious transition point at 16 kpsi was observed for both samples. With pressure rising above 16 kpsi, particle sizes increased significantly. This ‘over-processing’ phenomenon was also observed in several emulsion systems using biopolymers as emulsifier.22 During high pressure homogenization, the particle size distribution was determined by the equilibrium between two opposite actions happening at the same time: oil droplet disruption and re-coalescence. The excessive energy input at 20 kpsi might not result in additional protein adsorption to the disrupted oil surface. Inversely, the shorter residence time of the emulsion in the emulsifying unit along with other reasons led to a higher re-coalescence rate. Furthermore, higher pressures also produced nanoparticles with narrow size distributions due to the fact that higher energy input could disrupt the large particles which survived under low pressure. The PDI values decreased from 0.4–0.6 to 0.25 for the selected two samples when pressure increased from 4 to 16 kpsi as shown in the inset of Fig. 3. A further increase of the pressure to 20 kpsi resulted in the significant increase in PDI, likely due to the re-coalescence between newly formed oil droplets.17
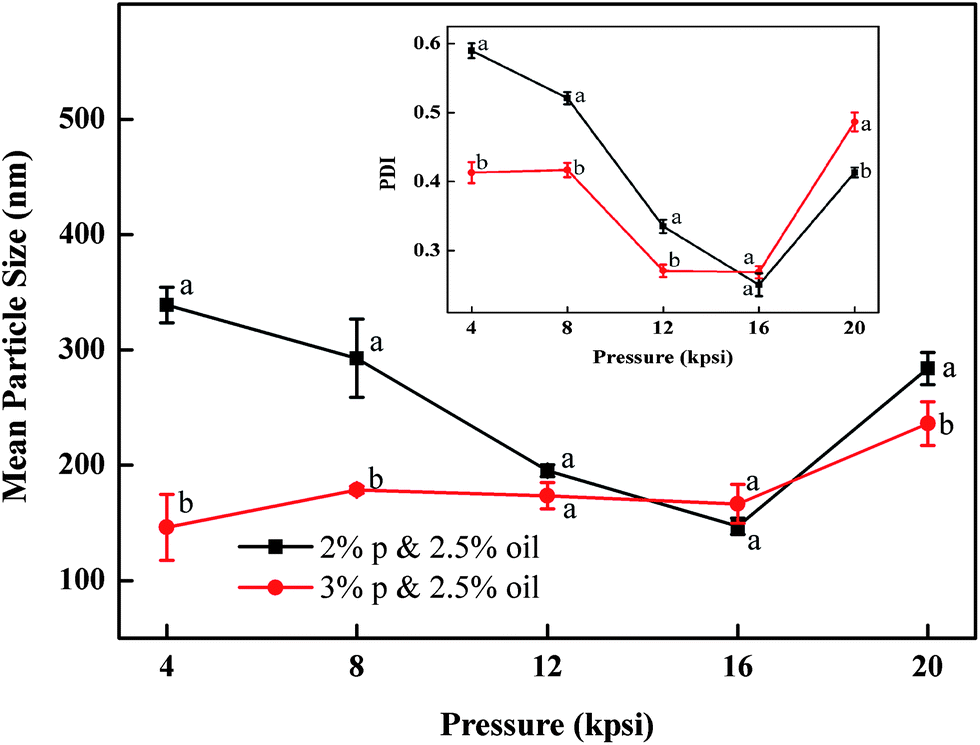 | ||
| Fig. 3 Effect of the homogenizing pressure on the mean particle size of samples produced with 2 wt% and 3 wt% barley proteins and 2.5% v/v oil by 6 passes. The inset figure shows the effect of the homogenizing pressure on the particle size distribution as indicated by PDI. Different letters above or below the curve indicate significant difference (p < 0.05) due to different formulae. | ||
| Protein concentrations | Oil contents | |||
|---|---|---|---|---|
| 1% | 2.5% | 5% | 7.5% | |
| 2% | Excessive protein precipitation | Formation of particles | Formation of particles | Particle aggregation |
| 3% | Excessive protein precipitation | Formation of particles | Formation of particles | Particle aggregation |
| 5% | Excessive protein precipitation | Excessive protein precipitated | Slightly protein precipitated | Formation of particles |
As demonstrated in Fig. 4, the mean particle diameter decreased with the increase of initial barley protein concentration. For example, with oil concentration fixed at 2.5%, the mean particle diameters were 195, 174 and 127 nm when prepared at 12 kpsi with 2%, 3% and 5% barley protein respectively. This result was expected because higher protein concentrations led to thicker coatings outside oil droplets during high pressure homogenization, which reduced the surface tension and avoided the re-coalescence between disrupted oil droplets.17 For the same reason, PDI of nanoparticle samples decreased as the initial protein concentration increased. Samples prepared with 5% protein had a PDI of 0.29 and 0.27 at 12 and 16 kpsi respectively even when the oil concentration was as high as 7.5%. On the other hand, for the same protein concentration, the particle size increased significantly with the rising oil ratio at both 12 and 16 kpsi, as shown in Fig. 4 and 5. For example, at protein concentration of 5%, the particle size increased from 92 to 179 to 202 nm when oil content was raised from 2.5 to 5 and to 7.5%, respectively. A higher ratio of the oil phase would increase the viscosity of the emulsion, and thereby more energy input was required for droplet disruption. Meanwhile, it led to higher frequency of oil droplet re-coalescence as there were not sufficient barley proteins to cover the disrupted oil droplets. The PDI of samples prepared with 3% protein at 16 kpsi increased from 0.22 to 0.44 when the oil content increased from 1% to 7.5%.
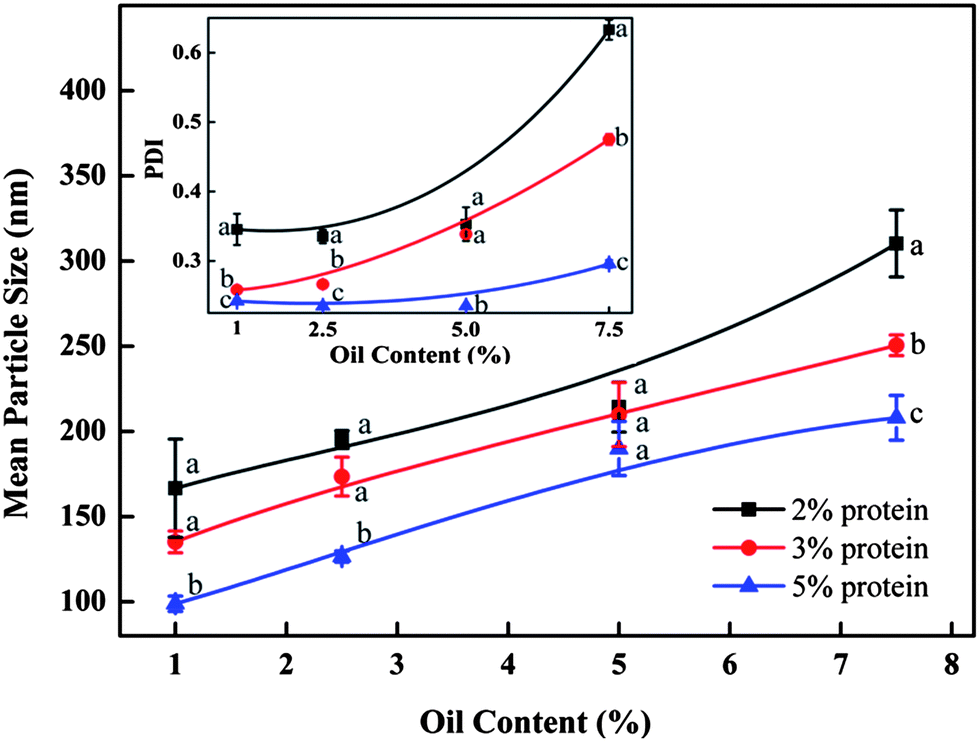 | ||
| Fig. 4 Effect of the protein concentration and oil content on the mean particle size of samples produced at 12 kpsi by 6 passes. The inset figure shows the effect of the protein concentration and oil content on the particle size distribution as indicated by PDI. Different letters above or below the curve indicate significant difference (p < 0.05) due to protein concentrations. | ||
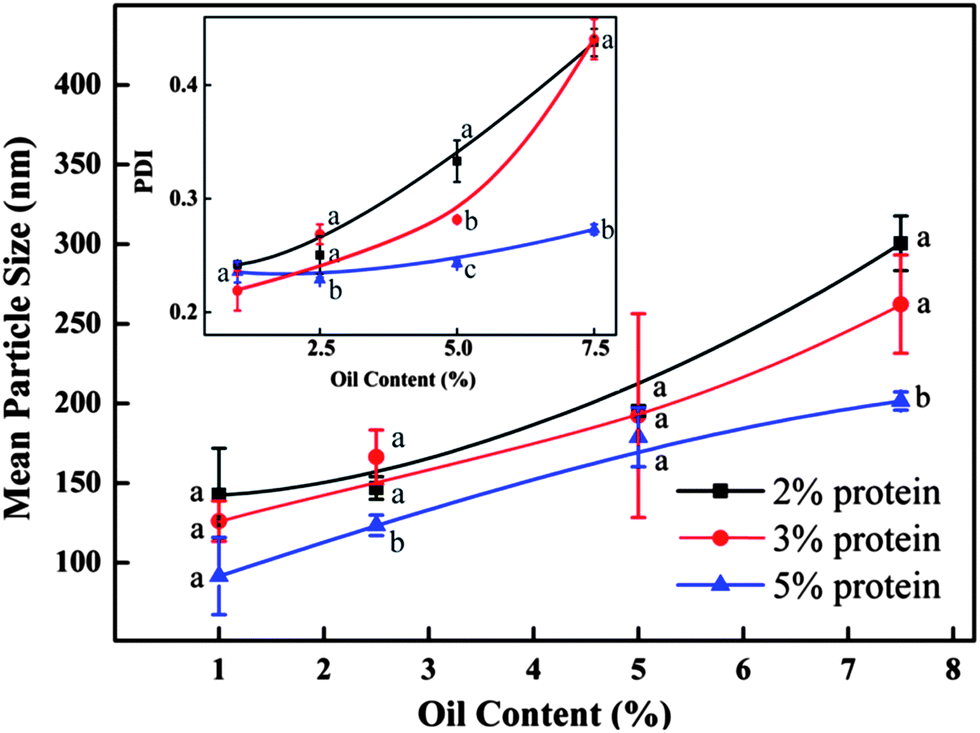 | ||
| Fig. 5 Effect of the protein concentration and oil content on the mean particle size of samples produced at 16 kpsi using 6 passes. The inset figure shows the effect of the protein concentration and oil content on the particle size distribution as indicated by PDI. Different letters above or below the curve indicate significant difference (p < 0.05) due to protein concentrations. | ||
Notably, nanoparticles with small sizes (90–150 nm) and narrow size distributions (PDI < 0.3) can be achieved with up to 5 wt% protein concentration when the oil-to-protein ratio was maintained within a range from 1 to 1.5. Zein,24 gliadin,25 soy proteins26 and milk proteins11 can only form nanoparticles at protein concentrations less than 2%. The more concentrated nanoparticles synthesised in this study were a significant improvement as mass production of nanoparticles with narrow sizes in water is a challenge facing industry and academic researchers. Samples prepared with 2% protein and 2.5–5.0% oil, 3% protein and 5% oil, as well as 5% protein and 7.5% oil, prepared at 16 kpsi and 6 passes were selected for further study due to their small size and narrow size distribution.
3.2 Nanoparticle characterization
The surface oil content, encapsulation efficiency (EE) and loading capacity (LC) of the nanoparticle samples are demonstrated in Table 2. All samples demonstrated high EE (89.2–93.5%) and LC values (51.4–54.4%) except for that prepared at 2% protein and 5% oil which only showed an EE value of 83.3% and an LC value of 46.6%. This is probably due to insufficient protein available to form a thick coating around the lipid droplets when the oil-to-protein ratio was high. The amount of surface oil is one important factor influencing the shelf life of the encapsulated oil and lipophilic bioactive compounds as it is openly exposed to the environment and has a high possibility of being oxidized or degraded. The oxidation and degradation of surface oil not only forms undesired products, but also triggers the oxidation reaction of bioactive compounds inside particles.27 It is noticed that samples prepared with higher protein concentration exhibited significantly reduced surface oil. For example the nanoparticles prepared with 5% protein and 7.5% oil had only 1.92% of total added oil attached at the particle surface although the oil-to-protein ratio is relatively high. The surface charge is another important characteristic of nanoparticles as it plays an important role in the physiological properties and stability of nanoparticles.28 The surface charge of nanoparticles is determined by the exposed side chains of the amino acid residues, the pH and the ionic strength of the environment. Barley protein nanoparticles exhibited a strong negative charge of around −35 mV in neutral aqueous solution as shown in Table 2. Negative charge is expected at neutral pH as barley protein has an isoelectric point (pI) of around 5.10 Nevertheless, it is interesting to notice that barley protein nanoparticles were much more negatively charged than many other protein nanoparticles (such as zein, gliadin and soy protein nanoparticles) which exhibited relatively low zeta-potential (−10 to −15 mV) at neutral pH.24,26,29 It could be deduced that during high pressure homogenization, barley protein reoriented its conformation with the hydrophobic region towards the oil phase and the hydrophilic region towards the water phase. Therefore, the hydrophilic amino acid residues with negative charges, such as the carboxyl group from glutamic acid, were exposed outside, rendering the nanoparticles highly negatively charged. It has been shown that nanoparticles are more stable in suspension when their zeta-potential is above ±30 mV due to the fact that the electrostatic repulsion between particles prevents them from aggregation.30| Surface oil percentage (%) | Encapsulation efficiency (%) | Loading capacity (%) | Zeta potential (mV) | |
|---|---|---|---|---|
| a Note: different letters next to the value indicate significant difference (p < 0.05) due to different formulae. | ||||
| 2% protein & 2.5% oil | 8.74 ± 1.59b | 89.19 ± 1.70ab | 46.55 ± 0.90a | −33.83 ± 0.83a |
| 2% protein & 5% oil | 8.79 ± 0.88b | 83.31 ± 1.53a | 56.12 ± 5.78a | −37.63 ± 0.75b |
| 3% protein & 5% oil | 4.80 ± 0.35ab | 92.53 ± 2.52b | 51.38 ± 1.40a | −35.47 ± 0.95a |
| 5% protein & 7.5% oil | 1.92 ± 1.30a | 93.52 ± 0.41b | 54.39 ± 0.24a | −33.67 ± 0.51a |
The stabilities of the barley protein nanoparticles were tested for 15 days during storage at 4 °C in deionized water (Fig. 6). All selected samples were rather stable during the storage period although a size increase trend was observed. The only exception was the sample prepared with 2% protein and 5% oil which underwent a more obvious size change from 196 to 289 nm due to its high surface oil content and insufficient protein coverage. The good storage stability of barley protein nanoparticles even at high protein concentrations without using surfactants was likely due to their small particle sizes, high surface charges and solid protein coatings.
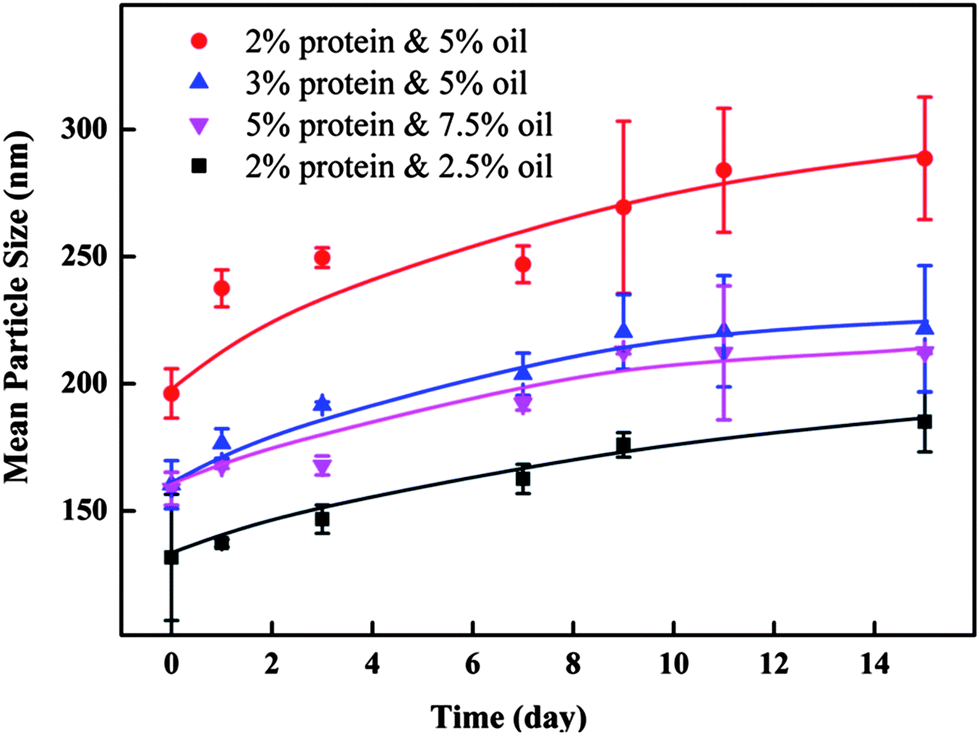 | ||
| Fig. 6 Mean particle size changes of 4 samples prepared at 16 kpsi in 15 days. | ||
3.3 In vitro release and degradation in the simulated gastro-intestinal tract
The release of incorporated ingredients from the protein matrix mainly involves three mechanisms, including diffusion from the matrix, enhanced diffusion through protein matrix swelling and liberation due to matrix degradation and erosion.31 The encapsulated lipid phase in nanoparticles is more likely to be released through the degradation of the protein matrix in the gastrointestinal (GI) tract. β-Carotene was selected as a model lipophilic bioactive compound in this study, as β-carotene has the highest pro-vitamin A activity but its low water solubility and low bioavailability have severely limited its application.3 Nanoparticles prepared with 2% protein and 2.5% oil were selected for these tests because of their optimized characteristics.The inset graph in Fig. 7 displays the cumulative release of β-carotene from barley protein nanoparticles incubated in SGF (pH 1.5 with pepsin) for 2 h. Less than 2% of the β-carotene was released in 1 h. Actually no more than 10% of the β-carotene was released from the protein matrix even after 6 hours of incubation in SGF in the presence of pepsin (data not shown). Such low release rates suggested that the barley protein nanoparticles could resist low pH and pepsin degradation, and thus provided protection for β-carotene against the harsh gastric environment. A morphology change of the nanoparticles after incubation in SGF was also observed by TEM. Interestingly, the original nanoparticles were degraded to form even smaller particles of 20–50 nm with regular spherical shapes (Fig. 8B and C). The decreased size was further supported by data from a dynamic light scattering evaluation, which showed an average particle size of 43 ± 7 nm after 1 h of incubation in SGF. This result indicates that the barley protein nanoparticle matrix was partially digested by pepsin to liberate the encapsulated oil droplets covered by a thinner but solid coating of barley protein which was indigestible by pepsin. This explained the low β-carotene release percentage in SGF. The majority of the hydrophobic amino acids on the protein chains were likely oriented towards the lipid phase, leaving the hydrophilic groups on the outside. As pepsin is most efficient in attacking peptide bonds involving hydrophobic amino acids,32 the layer of barley protein coated on the lipid surface represented a substrate that was less vulnerable to pepsin digestion. On the other hand, barley protein has a high percentage of proline (16%)10 which made degradation of the coating layer even slower as proteins with high proline content are generally more resistant to degradation by digestive enzymes in the GI tract.33 β-Carotene was susceptible to the acid environment because β-carotene can be dissociated to form carotenoid carbonations.34 Meanwhile, other common ingredients in the food matrix, such as iron, could also induce oxidation of the released β-carotene molecules, which adversely affects the bioactivity of β-carotene.35 This issue was also reported for other lipophilic bioactive compounds, such as α-tocopherol.36 Thus, maintaining the bioactive form of β-carotene until absorption is important. The resistance of barley protein nanoparticles to the stomach environment is a favorable property to improve the bioavailability of β-carotene.
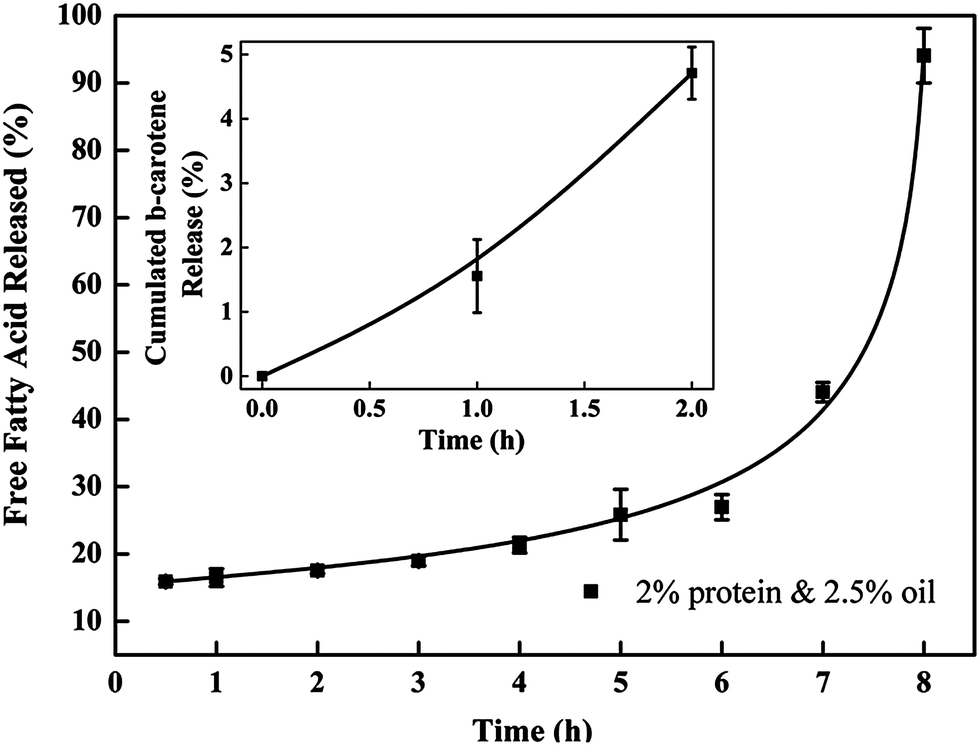 | ||
| Fig. 7 Release profile of cumulated free fatty acid from barley protein nanoparticles (2% protein and 2.5% oil in SIF for 8 hours). The inset graph shows the cumulated release percentage of β-carotene from the barley protein nanoparticles incubated in SGF for 2 h. | ||
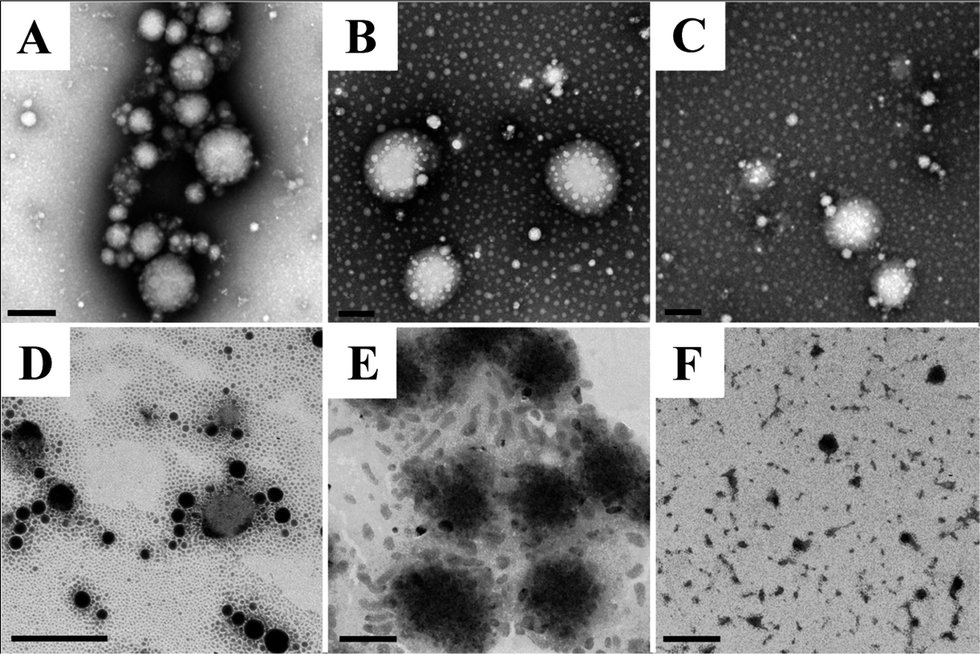 | ||
| Fig. 8 Transmission scanning microscopy micrographs of nanoparticle morphology changes during digestion in SGF and SIF for different times: (A) original nanoparticles prepared with 2% protein & 2.5% oil, (B) in SGF with pepsin for 20 min, (C) in SGF with pepsin for 1 h, (D) in SIF with pancreatin for 1 hour, (E) in SIF with pancreatin for 3 hours and (F) in SIF with pancreatin for 7 hours. All the scale bars in micrographs are equivalent to 200 nm. | ||
After incubation in SGF for 1 h, the nanoparticle samples were transferred into SIF for another 8 hours. We attempted to obtain the release profile of nanoparticles in SIF by hexane extraction. Nevertheless, the result was not consistent, which may be due to the incomplete extraction of β-carotene.11,37 Thus, the release profile of nanoparticles in SIF was measured by a method based on a pH-stat model which has been widely used in the pharmaceutical and food industries to rapidly screen lipid based formulations.12 Since the pancreatin in SIF contained appropriate concentrations of the major lipid digestive enzymes, enzymatic lipid digestion led to the generation of two free fatty acids and one monoglyceride from one triacylglcyerol molecule. Therefore, it was reasonable to quantitatively measure the release of the lipids by analyzing liberated free fatty acids in SIF media. Meanwhile, this result also indirectly demonstrated the degradation behavior of the protein coating, since the ability of lipase to come in close proximity to lipid molecules governs the rate of lipid digestion. Although measuring the liberated free fatty acids cannot directly show the release profile of the encapsulated lipophilic bioactive compounds, this in vitro digestion result in combination with TEM (showing the morphology changes) and a Caco-2 cell model (showing the nanoparticle uptake) can provide valuable information with respect to the nanoparticle degradation and release behavior in the simulated GI tract. As indicated in Fig. 7, in the first 7 hours of incubation, the accumulative release increased from ∼5–40%, then >90% free fatty acids were detected in the release media at 8 hours, indicating that ∼50% of the free fatty acids was liberated in the last hour of incubation. The degradation behavior of the above liberated smaller nanoparticles was also observed with TEM in SIF (Fig. 8D–F). After 1 hour of incubation in SIF, the liberated nanoparticles (mainly 20–50 nm) were further digested into nano-size lipid droplets, indicating that the barley protein solid coating was further degraded in SIF. Meanwhile, there was no evidence of oil droplets aggregating or coalescing at this stage, which was expected since they were covered by a thin layer of protein or protein hydrolysates. The mean particle size of samples incubated in SIF for another hour was 47 ± 1 nm. Then, nano-scaled structures were observed when the sample was incubated in SIF for 3 hours (Fig. 8E). These colloidal structures could be vesicles, mixed micelles and micelles which were able to internalize the undigested lipid molecules and β-carotene inside.12,38 Studies reported that β-carotene is likely to be absorbed through the small intestine in the form of mixed micelles and/or vesicles.3 Therefore, barley protein nanoparticles have the potential to improve the adsorption of β-carotene in small intestine. After incubation in SIF for 7 hours, the nanoparticles were further degraded into small irregular fragments. Nanoparticles with 3% protein & 5% oil and 5% protein & 7.5% oil had similar release profiles as the one showed above (data not shown).
3.4 Cytotoxicity and cell uptake
Both the cytotoxicity and the cell uptake of barley protein based nanoparticles were evaluated on a Caco-2 intestinal cell line which was derived from a human adenocarcinoma cell line. When grown on plastic dishes or filters, the confluent monolayer formed by Caco-2 cells exhibits tight junction complexes and possesses similar morphological, functional and electrical properties to human small intestinal cells. Thus, Caco-2 cells have been widely used as an in vitro model to investigate the uptake and transportation of materials through the small intestinal epithelium.39 Nanoparticles prepared with 2% protein and 2.5% oil were used in these tests.The viability of Caco-2 cells was measured by MTT assay after incubation with nanoparticles and the digested nanoparticle samples. As shown in Fig. 9, the cell viability was 88.4%, 95.3% and 92.9% for samples incubated with 0.5 mg ml−1, 0.25 mg ml−1 and 0.125 mg ml−1 nanoparticles respectively. High cell viability (about 90%) was also observed for nanoparticle samples digested by SGF for 1 hour and subsequently by SIF for 1 hour, 3 hours and 6 hours respectively. These results indicated that barley protein nanoparticles were biocompatible and had low toxicity even at high concentrations of 0.5 mg ml−1.
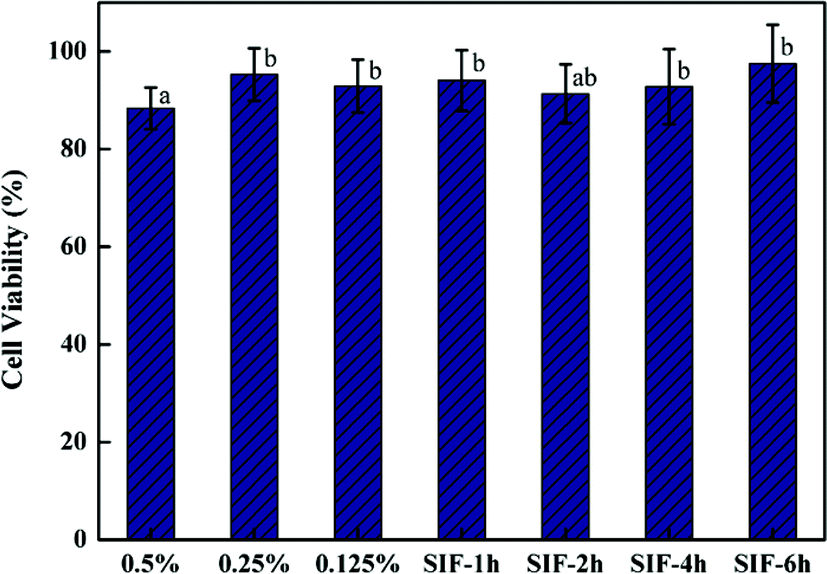 | ||
| Fig. 9 Percentage of the cell viability evaluated by MTT assay on Caco-2 cells treated with increasing concentration of barley protein nanoparticles for 6 h and with pancreatin digested nanoparticles for 1 to 6 hours respectively. Different letters above the column indicate significant difference (p < 0.05) due to the nanoparticle concentration and digestion conditions. | ||
Confocal microscopy was used to measure the amount of nanoparticles taken up by Caco-2 cells (Fig. 10). Nanoparticles were prepared with 2.5% canola oil containing 0.025% nile red which was used as a model of lipophilic bioactive compounds and is also a fluorescent stain for oil. As mentioned above, barley protein nanoparticles and their digested products were different in many physiochemical properties such as size, morphology and surface properties which governed their uptake in the small intestine.40 Therefore, intact nanoparticles and 4 selected digested products were incubated with Caco-2 cells to investigate their uptake properties. As displayed in Fig. 10A–E, red signals were clearly observed in all five samples, indicating that the encapsulated oil phase (containing nile red) was able to be effectively internalized and accumulated in the Caco-2 cell cytoplasm. Although intact nanoparticles, liberated nano-sized lipid droplets and micelles had different physiochemical properties, they all have potential to facilitate the adsorption of the encapsulated lipophilic compounds in vivo.
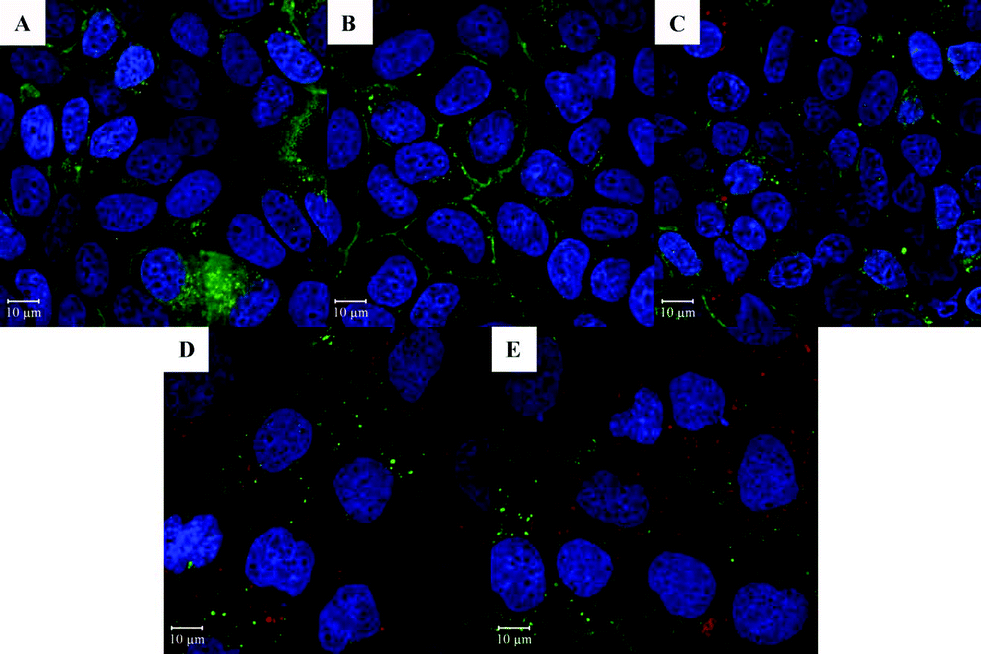 | ||
| Fig. 10 Confocal micrographs of Caco-2 cells after 6 hours incubation with (A) initial nanoparticles; (B) particles after 1 hour digestion in SGF with pepsin; (C–E) particles after another 1, 3 and 6 hours digestion in SIF with pancreatin respectively. | ||
4 Conclusions
Nanoparticles with small sizes (90–150 nm) and narrow size distributions were prepared from barley protein without the use of any organic solvents or cross-linking reagents. These nanoparticles demonstrated good storage stability in the absence of surfactants and contained a high payload (51.4–54.4%) of lipophilic nutraceutical compounds with limited surface oil. Interestingly, release experiments showed that even smaller particles (20–50 nm) were formed as a result of pepsin degradation of barley protein nanoparticle matrices. These smaller nanoparticles provided sufficient protection of the model nutrient in the SGF. Moreover, sequential lipophilic micro-domains were formed within the simulated intestinal environment, which helped stabilize water insoluble nutraceuticals in solution and may benefit their adsorption. Complete release of the model nutrients occurred after 7 hours of degradation by pancreatin. Both original barley nanoparticles and the liberated smaller ones after pepsin digestion exhibited low cytotoxicity through an in vitro study using Caco-2 cell models and they could be accumulated in the cytoplasm after being taken up into Caco-2 cells. Meanwhile, nanoparticles have a unique colloidal nature whereby their charge as well as their Brownian motion can cause the dispersion to be stable, thus they can be administered by both parenteral and non-parenteral routes, enabling wider applications. Thus, these barley protein nanoparticles have strong potential to be used as delivery systems of bioactive compounds for food, pharmaceutical and cosmetic applications. Moreover, this study provides meaningful justification for further in vivo studies to evaluate the safety and efficacy of barley protein nanoparticles as a delivery system.Acknowledgements
The authors are grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC), Alberta Crop Industry Development Fund Ltd. (ACIDF), Alberta Innovates Bio Solutions (AI Bio) and Alberta Barley Commission for financial support as well as Canada Foundation for Innovation (CFI) for equipment support. The authors are grateful to Dr Yixiang Wang who offered great help in this study. Also, the authors would like to thank Ms Arlene Oatway for her help during TEM observation.References
- H. Kaya-Celiker and K. Mallikarjunan, Food Eng. Rev., 2012, 4, 114–123 CrossRef CAS.
- L. Chen, G. E. Remondetto and M. Subirade, Trends Food Sci. Technol., 2006, 17, 272–283 CrossRef CAS PubMed.
- J. W. Erdman Jr, T. L. Bierer and E. T. Gugger, Ann. N. Y. Acad. Sci., 1993, 691, 76–85 CrossRef CAS.
- E. Acosta, Curr. Opin. Colloid Interface Sci., 2009, 14, 3–15 CrossRef CAS PubMed.
- J. Weiss, P. Takhistov and D. J. McClements, J. Food Sci., 2006, 71, R107–R116 CrossRef CAS.
- I. Ezpeleta, J. M. Irache, S. Stainmesse, C. Chabenat, J. Gueguen, Y. Popineau and A. M. Orecchioni, Int. J. Pharm., 1996, 131, 191–200 CrossRef CAS.
- J. Zhao, Z. G. Tian and L. Chen, J. Agric. Food Chem., 2010, 58, 11448–11455 CrossRef CAS PubMed.
- Y. Xia, Y. Wang and L. Chen, J. Agric. Food Chem., 2011, 59, 13221–13229 CrossRef CAS PubMed.
- R. X. Wang, Z. G. Tian and L. Chen, Int. J. Pharm., 2011, 406, 153–162 CrossRef CAS PubMed.
- C. Wang, Z. Tian, L. Chen, F. Temelli, H. Liu and Y. Wang, Cereal Chem., 2010, 87, 597–606 CrossRef CAS.
- X. Pan, P. Yao and M. Jiang, J. Colloid Interface Sci., 2007, 315, 456–463 CrossRef CAS PubMed.
- C. J. H. Porter, N. L. Trevaskis and W. N. Charman, Nat. Rev. Drug Discovery, 2007, 6, 231–248 CrossRef CAS PubMed.
- Y. Li, M. Hu and D. J. McClements, Food Chem., 2011, 126, 498–505 CrossRef CAS PubMed.
- A. Singh, B. He, J. Thompson and J. Van Gerpen, Appl. Eng. Agric., 2006, 22, 597–600 CrossRef.
- E. Dickinson, J. Chem. Soc., Faraday Trans., 1998, 94, 1657–1669 RSC.
- N. Anton, J. P. Benoit and P. Saulnier, J. Controlled Release, 2008, 128, 185–199 CrossRef CAS PubMed.
- S. M. Jafari, E. Assadpoor, Y. He and B. Bhandari, Food Hydrocolloids, 2008, 22, 1191–1202 CrossRef CAS PubMed.
- C. P. Tan and M. Nakajima, Food Chem., 2005, 92, 661–671 CrossRef CAS PubMed.
- Y. Yuan, Y. X. Gao, J. Zhao and L. Mao, Food Res. Int., 2008, 41, 61–68 CrossRef CAS PubMed.
- C. A. Guimaraes, F. Menaa, B. Menaa, J. S. Quenca-Guillen, J. D. Matos, L. P. Mercuri, A. B. Braz, F. C. Rossetti, E. R. M. Kedor-Hackmann and M. Santoro, Thermochim. Acta, 2010, 505, 73–78 CrossRef CAS PubMed.
- R. H. Muller, D. Harden and C. M. Keck, Drug Dev. Ind. Pharm., 2012, 38, 420–430 CrossRef CAS PubMed.
- S. M. Jafari, Y. He and B. Bhandari, Eur. Food Res. Technol., 2007, 225, 733–741 CrossRef CAS.
- E. Dickinson and T. van Vliet, in Food colloids, biopolymers and materials, ed. E. Dickinson and T. van Vliet, Royal Society of Chemistry, Cambridge, 2003, vol. 284, pp. 68–83 Search PubMed.
- Y. C. Luo, Z. Teng and Q. Wang, J. Agric. Food Chem., 2012, 60, 836–843 CrossRef CAS PubMed.
- M. A. Arangoa, M. A. Campanero, M. J. Renedo, G. Ponchel and J. M. Irache, Pharm. Res., 2001, 18, 1521–1527 CrossRef CAS.
- J. Zhang, L. Liang, Z. Tian, L. Chen and M. Subirade, Food Chem., 2012, 133, 390–399 CrossRef CAS PubMed.
- Y. D. Kim and C. V. Morr, J. Agric. Food Chem., 1996, 44, 1314–1320 CrossRef CAS.
- D. J. McClements and J. Rao, Crit. Rev. Food Sci. Nutr., 2011, 51, 285–330 CrossRef CAS PubMed.
- M. A. Arangoa, G. Ponchel, A. M. Orecchioni, M. J. Renedo, D. Duchene and J. M. Irache, Eur. J. Pharm. Sci., 2000, 11, 333–341 CrossRef CAS.
- L. F. Lai and H. X. Guo, Int. J. Pharm., 2011, 404, 317–323 CrossRef CAS PubMed.
- L. Chen, in Designing functional foods: measuring and controlling food structure breakdown and nutrient absorption, ed. D. J. McClements and E. A. Decker, CRC Press, Oxford, 2009, pp. 572–600 Search PubMed.
- L. Chen and M. Subirade, Biomaterials, 2005, 26, 6041–6053 CrossRef CAS PubMed.
- D. J. Simpson, Plant Sci., 2001, 161, 825–838 CrossRef CAS.
- V. V. Konovalov and L. D. Kispert, J. Chem. Soc., Perkin Trans. 2, 1999, 901–910 RSC.
- C. S. Boon, D. J. McClements, J. Weiss and E. A. De.cker, Crit. Rev. Food Sci. Nutr., 2010, 50, 515–532 CrossRef CAS PubMed.
- L. Liang, V. L. S. Line, G. E. Remondetto and M. Subirade, Int. Dairy J., 2010, 20, 176–181 CrossRef CAS PubMed.
- L. Beaulieu, L. Savoie, P. Paquin and M. Subirade, Biomacromolecules, 2002, 3, 239–248 CrossRef CAS PubMed.
- O. Hernell, J. E. Staggers and M. C. Carey, Biochemistry, 1990, 29, 2041–2056 CrossRef CAS.
- I. J. Hidalgo, T. J. Raub and R. T. Borchardt, Gastroenterology, 1989, 96, 736–749 CAS.
- T. Jung, W. Kamm, A. Breitenbach, E. Kaiserling, J. X. Xiao and T. Kissel, Eur. J. Pharm. Biopharm., 2000, 50, 147–160 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |