Increasing the sustainability of membrane processes through cascade approach and solvent recovery—pharmaceutical purification case study
Jeong F.
Kim
,
György
Székely
,
Irina B.
Valtcheva
and
Andrew G.
Livingston
*
Department of Chemical Engineering and Technology, Imperial College London, Exhibition Road, London, SW7 2AZ, UK. E-mail: a.livingston@imperial.ac.uk
First published on 26th September 2013
Abstract
Membrane processes suffer limitations such as low product yield and high solvent consumption, hindering their widespread application in the pharmaceutical and fine chemicals industries. In the present work, the authors propose an efficient purification methodology employing a two-stage cascade configuration coupled to an adsorptive solvent recovery unit, which addresses the two limitations. The process has been validated on purification of active pharmaceutical ingredient (API) from genotoxic impurity (GTI) using organic solvent nanofiltration (OSN). The model system selected for study comprises roxithromycin macrolide antibiotic (Roxi) with 4-dimethylaminopyridine (DMAP) and ethyl tosylate (EtTS) as API and GTIs, respectively. By implementing a two-stage cascade configuration for membrane diafiltration, the process yield was increased from 58% to 95% while maintaining less than 5 ppm GTI in the final solution. Through this yield enhancement, the membrane process has been “revamped” from an unfeasible process to a highly competitive unit operation when compared to other traditional processes. The advantage of size exclusion membranes over other separation techniques has been illustrated by the simultaneous removal of two GTIs from different chemical classes. In addition, a solvent recovery step has been assessed using charcoal as a non-selective adsorbent, and it has been shown that pure solvent can be recovered from the permeate. Considering the costs of solvent, charcoal, and waste disposal, it was concluded that 70% solvent recovery is the cost-optimum point. Conventional single-stage diafiltration (SSD) and two-stage diafiltration (TSD) configurations were compared in terms of green metrics such as cost, mass and solvent intensity, and energy consumption. It was calculated that implementation of TSD, depending on the batch scale, can achieve up to 92% cost saving while reducing the mass and solvent intensity up to 73%. In addition, the advantage of adsorptive solvent recovery has been assessed revealing up to 96% energy reduction compared to distillation and a 70% reduction of CO2 footprint.
1. Introduction
Developing sustainable and green processes has become an important priority for the pharmaceutical and fine chemical industries in response to the growing concerns of regulatory authorities with respect to environmental impact.1,2 At the same time, the removal of genotoxic impurities (GTIs) from active pharmaceutical ingredient (API) post-reaction mixtures has gained increasing attention due to the adverse health effects of GTIs. Strict pharmaceutical regulations call for impurity levels as low as few ppm.3 It is well known that downstream separation processes account for as much as 80% of the total manufacturing costs.4,5 With profit margins growing thin, there is a huge drive for reducing both the cost and environmental impact via process intensification (PI). PI refers to a set of innovative principles in process and equipment design to achieve significant benefits in process efficiency, operating expenses, quality, and waste.6 As an effective PI tool, membrane engineering has been recognized as one of the core technologies that provides green process engineering.7Conventional separation unit operations for API purification such as recrystallization, flash chromatography, and adsorption, are thoroughly described in the literature. Recently emerging technologies, such as organic solvent nanofiltration (OSN)8,9 and molecularly imprinted polymers,10,11 as well as their synergistic combination,12 have been suggested for GTI removal.
Although nanofiltration (NF) membranes have been applied widely in water softening and wastewater treatment, polymeric membranes have suffered instability and degradation, aging, and plasticization in organic solvents and chemical compounds.13 OSN membranes now offer much higher chemical resistance to organic solvents and also to extreme pH conditions.14 It is also possible to tailor the membrane separation performance to suit specific separation requirements.15 In addition, OSN has a unique advantage in that it can perform molecular separations at ambient temperature.16 This advantage allows many thermally sensitive compounds (APIs and catalysts) to be purified under mild conditions. It is a proven technology and there are already many reported lab-scale and pilot-plant scale applications in solvent recovery,17,18 catalyst recycle,19 peptide and oligonucleotide synthesis,20 API purification,8 and many others. A recent review on OSN describes the general aspects of the technology in detail.16 Considering the many advantages of OSN, it has promising potential to develop much greener and economical processes in the pharmaceutical and fine chemicals industries. In fact, membrane processes have been chosen among the top 10 green engineering research areas by the ACS GCI Pharmaceutical Roundtable,2 and several green metrics using membranes, such as productivity-to-size (PS) ratio and modularity, have been proposed to facilitate compilation of data and comparison.6,7,21 For example, it has been shown that membranes can offer a substantial increase in reaction conversion with reduced catalyst loading, while reducing the plant size for hydrogen production.22 Also, membranes have been successfully applied to crystallization processes to improve control and efficiency.23
Despite these promising potentials, membrane processes have been facing difficulties in acceptance by the pharmaceutical industry, difficulties such as: (1) difficult performance prediction resulting in extensive membrane screening to find a suitable membrane;24 (2) ineffectively compiled literature on performance and applicability compared with the traditional operations;7 (3) insufficient separation performance13 resulting in unacceptable loss of product yield and/or purity; and (4) large solvent consumption.25 Among the aforementioned reasons, the latter two are the main reasons limiting the OSN-based membrane processes from wide implementation in pharmaceutical manufacturing.
OSN membranes separate solutes in the nanofiltration range between 100–2000 g mol−1 which lies between reverse osmosis (dense film) and ultrafiltration range (microporous). In this region, the separation mechanism has not been completely elucidated as of yet. There is a strong correlation between the size of the solute and the corresponding rejection26 where larger solutes get rejected more than small solutes. But the size parameter alone does not give satisfactory prediction of membrane performances, and other factors such as solute charge, solvent-membrane and solvent–solute interactions have been shown to affect the transport.27–29 However, as a rule of thumb, larger compounds generally exhibit higher rejection than smaller ones.
In order to fractionate solutes with high yield and purity, close to 100% rejection of one of the solutes is necessary.30 However, it is often difficult to obtain sharp enough separation to fractionate two or more solutes. In addition, to fractionate solutes using membranes, a substantial amount of solvent is required to achieve a desired purity.
In order to become a versatile process and be widely applicable, membranes must offer more flexibility during the process development stage. For most membrane process development, screening for the “right” membrane is the bottleneck stage, as membrane performances are not easily predictable and each application calls for specific separation requirements. Hence, a wide range of membranes needs to be screened. This screening step is not only time-consuming, but also it is often difficult to achieve competitive separation compared to other conventional processes such as recrystallization and flash chromatography. Therefore, it is vital to reduce or eliminate the initial membrane screening stage as more than 30% of the cost of bringing a product to market results from the process development.31
Many membrane groups have dedicated their efforts to developing membranes with higher permeability and sharper molecular separation, using for example polymers of intrinsic microporosity (PIM),32 mixed-matrix membranes (MMM),33 and graphene membranes.34 An interesting work by Vankelecom et al. to use high throughput method has shown a systematic approach to achieve such goals.35 Although the membrane permeabilities have enhanced significantly over time, the separation power of membranes has not improved as much. This lack of improvement in separation performance has a sound theoretical basis, as the Bowen & Welfoot pore-flow transport model36,37 predicts poor separation capacity even with uniform pore sizes as shown in Fig. 1.
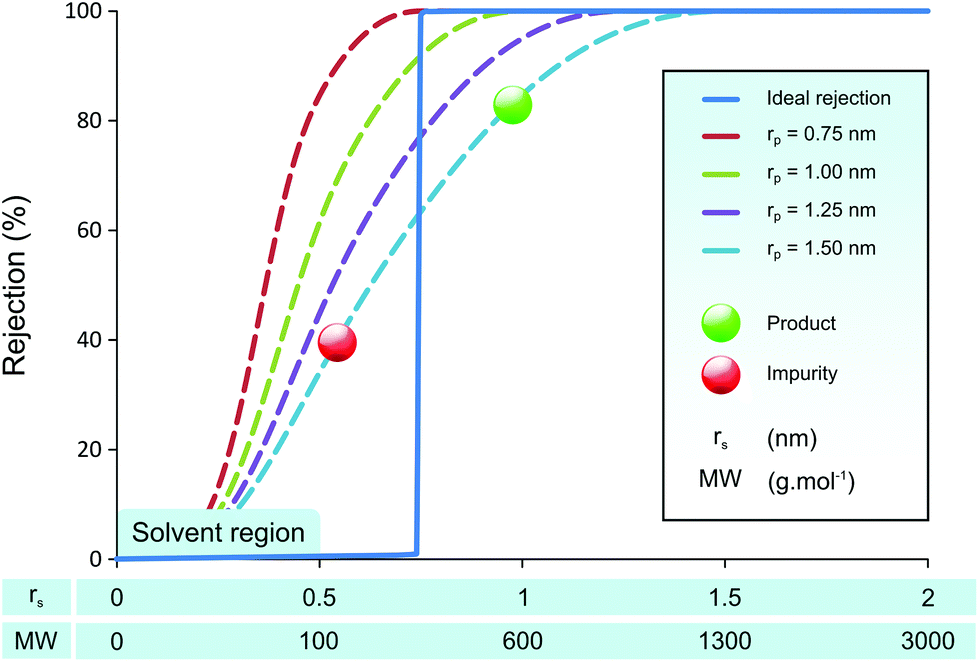 | ||
| Fig. 1 Simulated membrane rejection plotted against molecular size and approximate molecular weight for membranes with uniform pore sizes (rp). Even with perfectly uniform pore size of 1.5 nm, it is difficult to separate the product from impurity efficiently. | ||
Fig. 1 illustrates predicted membrane rejection profiles of uniform pore size membranes in the nanofiltration range. An “ideal” rejection curve is also shown for comparison. It can be seen that even with perfectly controlled pore size, it is difficult to get a clear separation of API (product) and GTIs (impurities). As the pore size of the membrane increases, the rejection curves are not shifted up the x-axis retaining their shape, rather they “smear” towards higher molecular weights. The smeared rejection profile, instead of ideal rejection, is the effect of hindered transport.38 The simulated rejection profiles shown here are typical to those observed in nanofiltration membranes.15,24 When one attempts a product purification using membranes with the rejection profile shown in Fig. 1, the obtained yield is often unfeasible because the product rejection is too low.30 The effect of product rejection on the yield is discussed in further detail in the Results and discussion section.
An alternative to finding the “right” membrane for each application is to approach the separation challenge from an engineering perspective. We seek to achieve a desired outcome by reconfiguring the membrane process, i.e. employing membrane cascades, with accessible membranes already in hand, reducing the initial screening stage time. The potential and advantages of membrane cascades have long been recognized; however, to date, several implementation challenges such as control difficulties39 have presented hurdles and few experimental data have been reported.40–42
Recently, our research group has proposed a new membrane cascade configuration30 that overcomes the previous implementation challenges. Hereby, we report on the potential of an OSN membrane cascade via an industrial application: removal of GTIs from APIs. Employing a two-stage membrane cascade configuration over single-stage diafiltration leads to a significant increase in API yield without compromising its purity. In addition, the initial membrane screening time is much reduced, or even eliminated, as the process offers much more versatility and buffers membrane performance.
Furthermore, the present work also addresses the second major challenge of membrane purification, its extensive solvent consumption. In order to comply with the high standards of API quality assurance, ultra-low GTI levels have to be achieved calling for a substantial amount of solvent. In order to minimize the solvent consumption – i.e. decrease the environmental impact and increase sustainability – an adsorption unit featuring charcoal was integrated in the process. Adsorption isotherms of the charcoal-GTI system have been studied in order to identify the isotherm behavior and optimal permeate-to-charcoal ratio for solvent recovery.
To critically assess the proposed configuration, the process economics as well as conventional green metrics such as mass and solvent intensity were evaluated. In addition, the energy consumption and carbon footprint of adsorption and distillation unit operations have been compared in detail. The authors believe the simplicity and versatility of this process configuration will boost membrane applications in industries where membranes were formerly not competitive enough in terms of economics and sustainability.
2. Results and discussion
To carry out a fractionation of solutes using membranes, typically fractionating impurities from a product, a constant volume diafiltration (CVD) process is usually employed. The CVD process works in a semi-batch manner where a feed solution is placed in contact with a membrane under pressure, and impurities are continuously removed from the system to yield pure product, or vice versa (see Experimental section). As the purification proceeds, the system volume is kept constant by feeding pure solvent into the system to make up the volume loss due to permeation, hence the term constant volume diafiltration. The system is typically characterized by the volume of solvent permeated per system volume (number of diavolumes) which is used as a time-like parameter (see Appendix A for process modeling). The molecular weights of the GTIs (<200 Da) and APIs (>500 Da) fall in the nanofiltration MW range, respectively.8 Hence, GTIs get pushed through the membrane as the APIs are retained, making OSN diafiltration a promising platform for API purification.The unique capacity of membranes to separate primarily based on molecular size is of particular advantage in API purification, since having two different classes of GTIs present in a crude API mixture is common8 and with conventional technology often requires more than one unit operation to remove them separately. In this work, we exploit this advantage by removing two different chemical classes of GTIs (DMAP – arylamine, and EtTS – arylsulfonate)55 from API (roxythromycin) simultaneously in a single membrane diafiltration (Fig. 2).
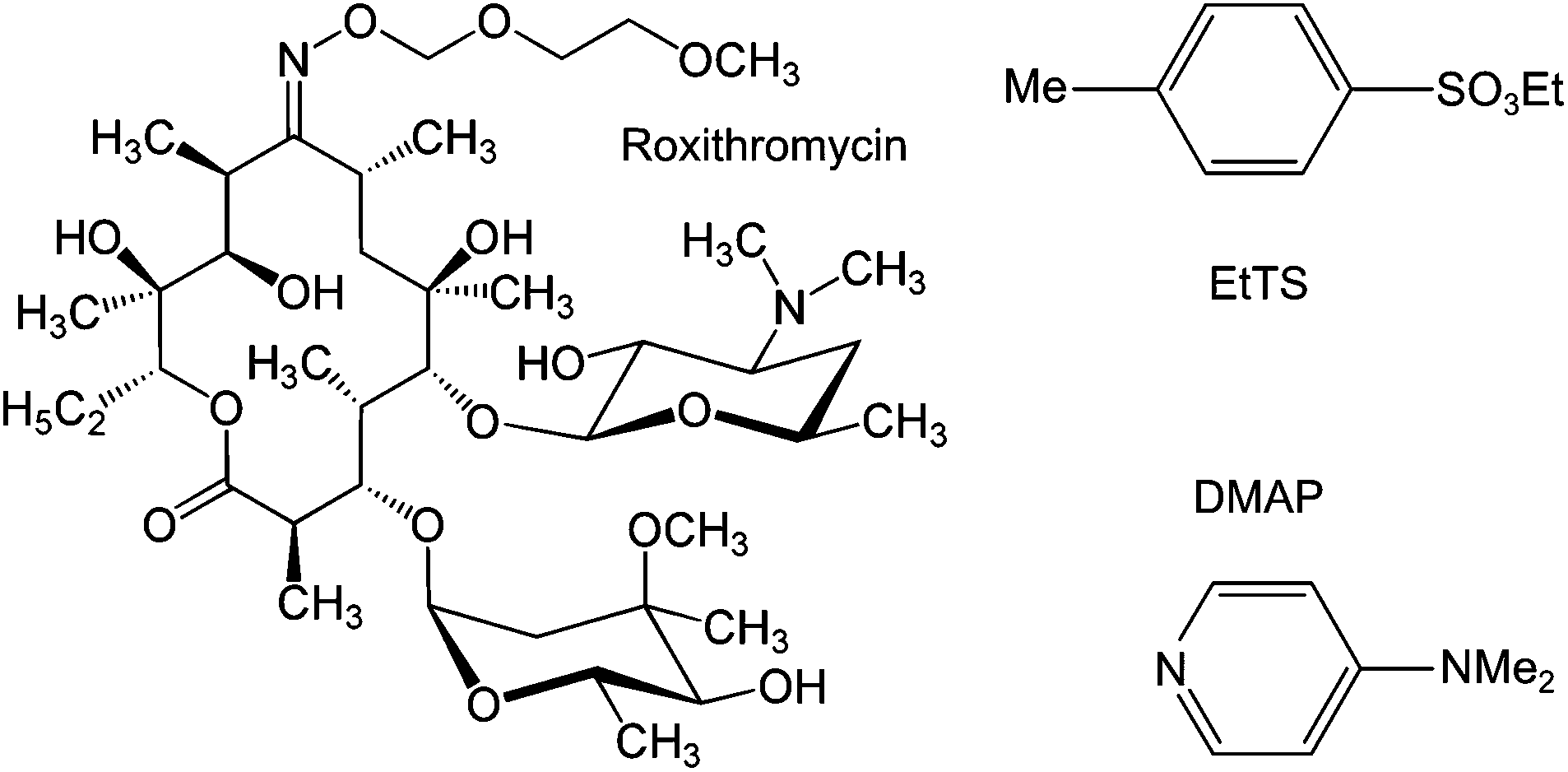 | ||
| Fig. 2 Chemical structure of the model compounds: roxythromycin (Roxi) API, ethyl tosylate (EtTS) and 4-dimethylaminopyridine (DMAP) GTI impurities. | ||
To develop the process, a suitable membrane was first selected and tested at different transmembrane pressures, followed by single stage diafiltration and two stage cascade diafiltration experiments. Also, the effectiveness of charcoal with the tested GTIs was assessed. To test the process performance, practical industry concentrations were employed: 10 kg m−3 for API and 1 kg m−3 (1000 ppm) for each GTI.8 All processes have been repeated twice and the error bars on the figures show the corresponding standard deviations. The recovered solvent was considered pure if the GTI levels were below their limit-of-detection (LOD).
2.1. Membrane selection and single stage diafiltration (SSD)
To determine a suitable membrane for API purification, a polybenzimidazole (PBI) OSN membrane was tested against the compounds of interest dissolved in methanol. However, as the solute rejections are functions of flux,43 the rejections were measured at different transmembrane pressures. The solute rejections and fluxes obtained at various pressures are shown in Table 1.| Pressure (bar) | R Roxi | R DMAP | R EtTS | Fluxa (dm3 m−2 h−1) |
|---|---|---|---|---|
| (%) | ||||
| a All measurements were made at 21 ± 0.5 °C. The average values of two membranes are reported with error bars indicating one standard deviation. | ||||
| 5 | 93.5 ± 0.3 | 10.5 ± 0.7 | 14.9 ± 0.2 | 14.0 ± 2.1 |
| 10 | 95.2 ± 0.2 | 14.3 ± 3.2 | 19.2 ± 4.7 | 24.7 ± 4.2 |
| 15 | 94.9 ± 0.2 | 15.7 ± 2.3 | 20.6 ± 3.9 | 39.7 ± 6.2 |
Table 1 shows that the selected membrane shows wide difference in rejection between the API and GTIs, where API is highly retained by the membrane while GTIs easily pass through the membrane. It can also be seen that the change of API rejection with respect to the pressure is insignificant; however, the rejection of GTIs is dependent on the transmembrane pressure, up to 35% difference. Normally, 94% API rejections shown in Table 1 are insufficient to obtain high yield after purification and further membrane screening would be necessary. However, to demonstrate the advantage of the proposed configuration and to show that excessive membrane screening period is not required, the membrane was selected to test the impact of cascade configuration. The single stage diafiltration (SSD) data obtained are presented in Fig. 3.
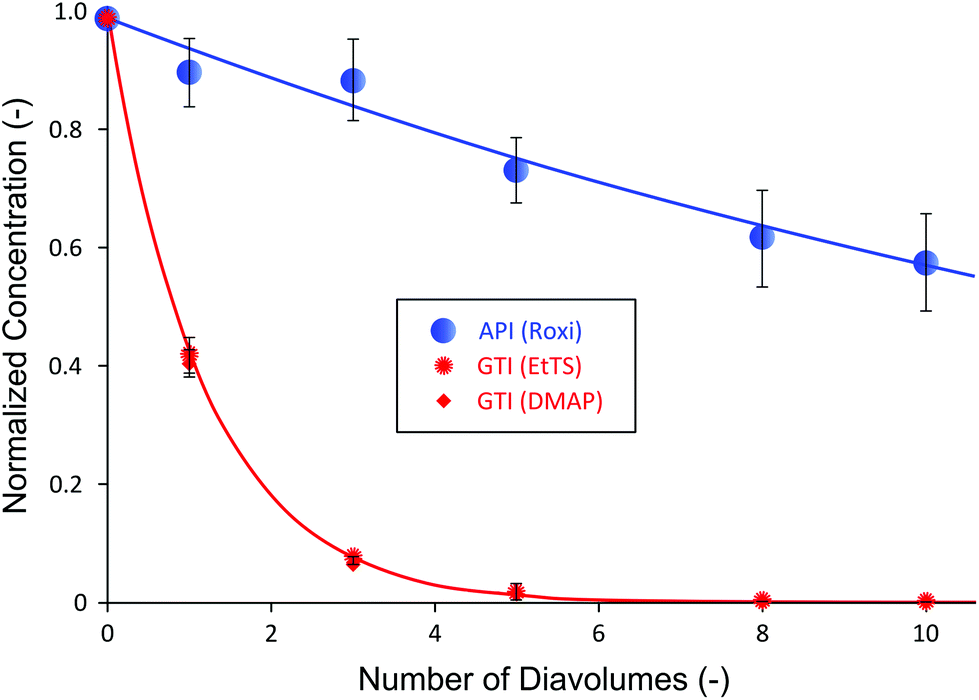 | ||
| Fig. 3 Single stage diafiltration (SSD) normalized concentration profile using PBI membranes. Both GTIs were removed at virtually the same rate. Less than 5 ppm GTI purity has been achieved after 10 diavolumes with final yield of 58%. | ||
Fig. 3 summarizes the SSD normalized concentration profile of the system using PBI membranes. The mass balance of all species fell within 4–6%. The concentration profiles of API and GTIs closely matched to model predictions simulated using rejection values from Table 1 (see Appendix A for process modeling). It can be seen that even with the moderately-high rejection of 94%, the yield of API after 10 diafiltration volumes was only 58%. Given that APIs are high-value products, such a loss is unacceptable in industry. Apart from the process yield, both GTIs showed similar rejections due to having similar molecular weights (200 and 122 g mol−1, respectively). As expected, they were removed at nearly the same rate and the final GTI concentration decreased to below 5 ppm, satisfying the typical target level of 10 ppm.25,44 Although the yield was low, the advantage of membrane processes over affinity-based purification technologies (e.g. resin, chromatography) was clearly demonstrated in this system: GTIs from different chemical classes can be removed simultaneously; usually affinity-based purifications would require specific selective agents for each chemical class of impurities.10,45 Nevertheless, the low API yield results in an unfeasible manufacturing process in terms of economics and sustainability. To address this low yield challenge, one might undertake a cumbersome screening period to find tighter membranes. However, this leads to higher GTI rejection as well, calling for even higher number of diavolumes. In addition, apart from limited availability of different types of commercial OSN membranes, it is often difficult to find the right membranes to meet the process demand. As an alternative to overcome such a challenge, a two-stage diafiltration (TSD) configuration has been assessed.
2.2. Two stage diafiltration (TSD) and comparison to single stage diafiltration (SSD)
It has been shown that to achieve a high yield using the cascade configuration, the recycle ratio,30 which is defined as the retentate (FR2) from the second stage over the feed into the second stage (FP1) (see Experimental section), must be controlled carefully. The difference in yield can be up to two-fold depending on the chosen recycle ratio. Efficient control of the membrane cascade is the key for a high yield, as poorly controlled systems can even show worse performance than the single stage performance.40,42,46 A mathematical model describing the TSD process can simulate and predict the process performance (see Appendix A). After a thorough predictive simulation based on the rejection data in Table 1, an optimum operating condition has been chosen and implemented (see Experimental section). The results from TSD are shown in Fig. 4. The mass balance for all species closed within 1–5%.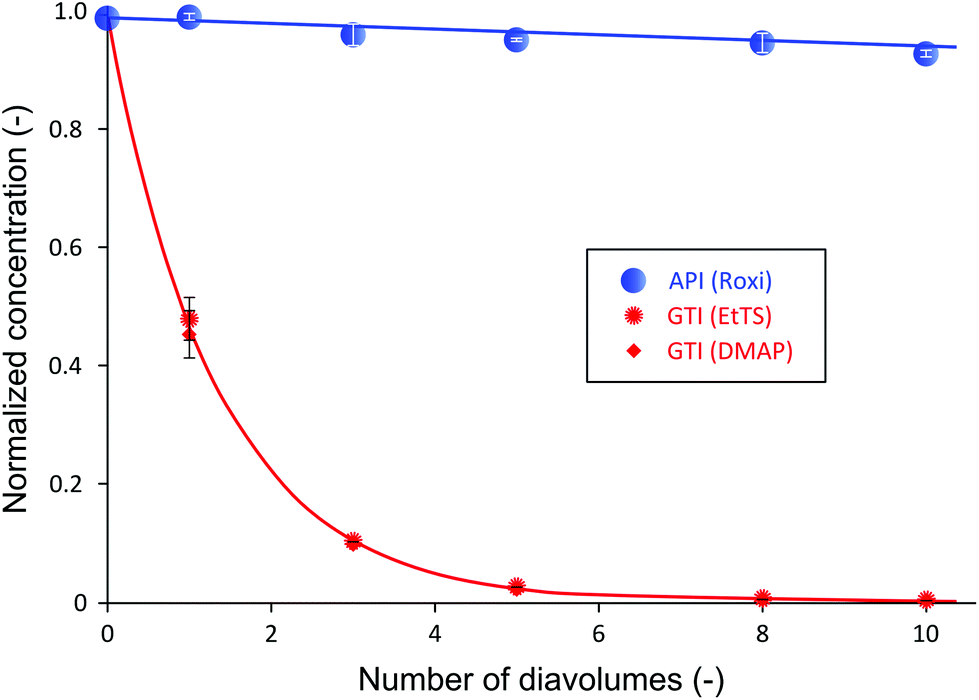 | ||
| Fig. 4 Two-stage diafiltration (TSD) normalized concentration profile using PBI membranes. Recycle ratio (rC) was set at a value of 0.5. GTI concentrations less than 5 ppm were again achieved after 10 diavolumes with the final API yield of 95%, a substantial improvement from SSD yield of 58%. | ||
Fig. 4 illustrates a drastic improvement in final yield of API, from 58% to 95%, without compromising the final purity. By introducing the second stage and setting the recycle ratio at a value of 0.5 (rC = 0.5), the actual loss of API through the membrane was considerably reduced, as predicted by the process model (Appendix A). The overall rejection of API (combined rejection of two stages) has increased from 94% in the SSD, to 99.5% in the TSD. On the other hand, although the overall rejections of GTIs have increased from 15% (SSD) to 25% (TSD), the rate of GTI permeation through the membrane remained high in accordance with the model prediction. Hence, the permeation of 10 diavolumes – the same as performed SSD – allowed the target impurity level of 10 ppm to be met. More precisely, GTI levels below 5 ppm have again been achieved, proving the two-stage membrane cascade configuration.
It has been demonstrated that the proposed membrane cascade configuration efficiently resolves the separation limitation of the single stage membrane processes. Although a set of non-commercial PBI membranes were used for this study, commercially available polyimide membranes have also shown excellent API purification performances,8 which can be applied to the proposed system. In addition, it should be stressed that the proposed process is not limited to polymeric membranes and to API purification applications but it can be applied with other types of membranes (ceramic, carbon membranes, etc.) and to other applications (catalyst recovery, membrane reactors, refinery industry, etc.). The essence of the proposed cascade configuration lies in its simplicity and versatility. Also, the process is very straightforward to implement and control.30 Furthermore, the system can be easily modeled, where the only required input parameters are the membrane permeability and solute rejections (Appendix A). For example, depending on the product rejection, the process yield improvement in moving from SSD to TSD can be predicted as shown in Fig. 5.
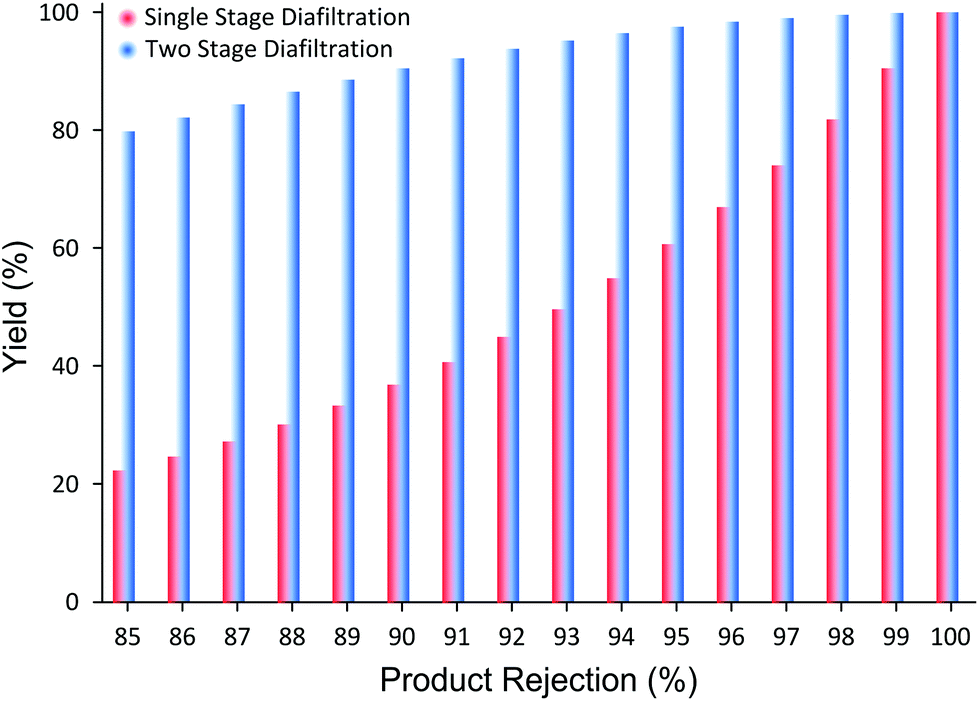 | ||
| Fig. 5 Predicted yield improvement after 10 diavolumes for different product rejection. The TSD configuration achieves significant yield improvement, overcoming the membrane limitation inherent in the SSD for rejection less than 99%. As long as the product rejection is above 90%, the process promises high product yield, making the process competitive to other traditional unit operations. | ||
Fig. 5 clearly illustrates the potential yield improvement through implementing the TSD system proposed in this work. For instance, when the membrane rejection of a product is 85%, SSD achieves only 22% yield after 10 diavolumes. However, when the TSD configuration is applied, the yield improves up to 80%, close to 4× improvement. Although the increase in yield is substantial, the yield of 80% is still unsatisfactory for high-value products. However, when the product rejection is 98%, the yield improves from 81% to 99.6% after 10 diavolumes. The implication of such yield improvement should not be understated. This difference in yield, undoubtedly, transforms the membrane process from “unfeasible” status to an economically sustainable and viable process, and makes it highly competitive to other conventional processes. Hence, the TSD configuration provides substantial improvement relative to the SSD configuration. In addition, as long as the rejections of the impurities are less than 50%, it can be shown that the final product purity will not be compromised.30 It is worth pointing out that most GTIs fall below 50% rejection on commonly available nanofiltration membranes,8 making the TSD configuration a competitive platform for API purification.
It is possible to extend the system to three or four-stage configuration which would increase the yield even further, and the model can also be generalized. However, the rejection increase of impurities mostly outweighs the yield gain and the system requires even higher diavolumes. Hence, the additional capital cost as well as the operation complexity must be carefully assessed before extending the cascade system.
With the yield challenge resolved, the process opens up new opportunities for integration with other unit operations. As the final permeate waste stream from the system mostly contains GTIs, the possibility of solvent recycle via adsorption can be exploited. Hence, the use of activated charcoal for solvent recovery has been assessed by adsorption of impurities.
2.3. Solvent recovery – adsorption isotherm and feasibility assessment
Activated charcoal is a widely used adsorbent with high adsorption capacity due to its high surface area.47 It has been applied to API purification as part of an API recrystallization process, however the selectivity between the API and GTI was poor, resulting in relatively high API loss.25 Combining membranes with different adsorbents has been reported previously12,19 and it was concluded that the major impediment to success was the API loss from the membrane stage. For the TSD configuration, the high GTI uptake of activated charcoal at low concentrations can be exploited for solvent recovery, as the permeate from the TSD system mostly contains only GTIs with negligible API. In order to determine the optimal permeate to charcoal ratio, the activated charcoal was first characterized using a conventional isotherm experiment. Adsorption isotherms are typically described using the following equation,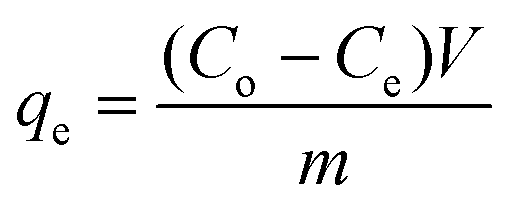 | (1) |
The adsorption isotherms obtained for both GTIs are summarized in Fig. 6.
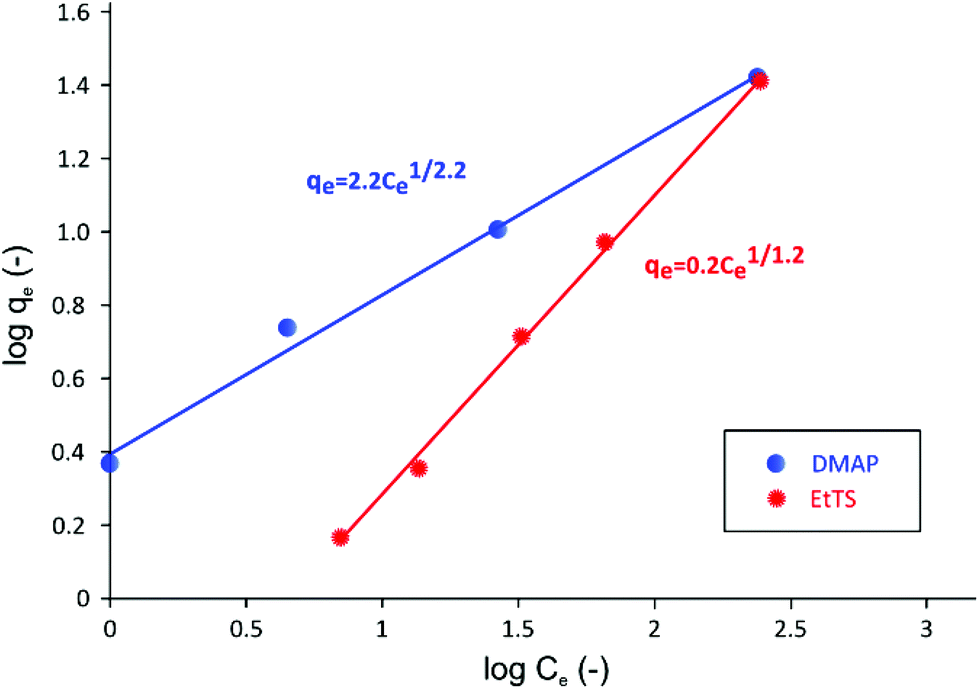 | ||
| Fig. 6 Log–log plot of adsorption isotherm of GTIs on charcoal in methanol at 21 °C. The Freundlich isotherm describes the adsorption behavior of two GTIs well. | ||
As it can be seen in Fig. 6, when isotherm data are plotted in log qevs. log Ce scale, a linear relationship can be obtained, indicating that the system follows Freundlich isotherm48 described using the following equation,
| qe = KFCe1/n | (2) |
The Freundlich isotherm has been specifically developed to describe adsorption onto charcoal, where stronger binding sites are occupied first, followed by exponential decrease in adsorption energy until all sites are occupied.49 It can be seen in Fig. 6 that the KF value for EtTS is lower than that of DMAP, showing that the adsorption strength for EtTS is weaker than that of DMAP.
At this point, two important remarks in regard to the solvent recovery experiment must be stated clearly. First, the competition for adsorption sites between the two GTIs has not been taken into account because the objective was complete GTI adsorption, and the kinetics of adsorption was outside the scope of this study. Hence, a simple Freundlich model was sufficient to describe the system. Secondly, the actual solvent recovery experiment was carried out in a batch-wise mode, rather than continuous, to assess the feasibility of adsorptive solvent recovery. Each discreet diavolume from TSD experiments was collected separately and the necessary amount of charcoal was added and stirred until all GTIs were adsorbed to a level below the limit-of-detection (LOD) (please see Experimental section for calculation).
The permeate concentration of each diavolume and the corresponding amount of charcoal required (cumulative) are shown in Fig. 7A, and the efficacy of a typical charcoal adsorption is presented in Fig. 7B.
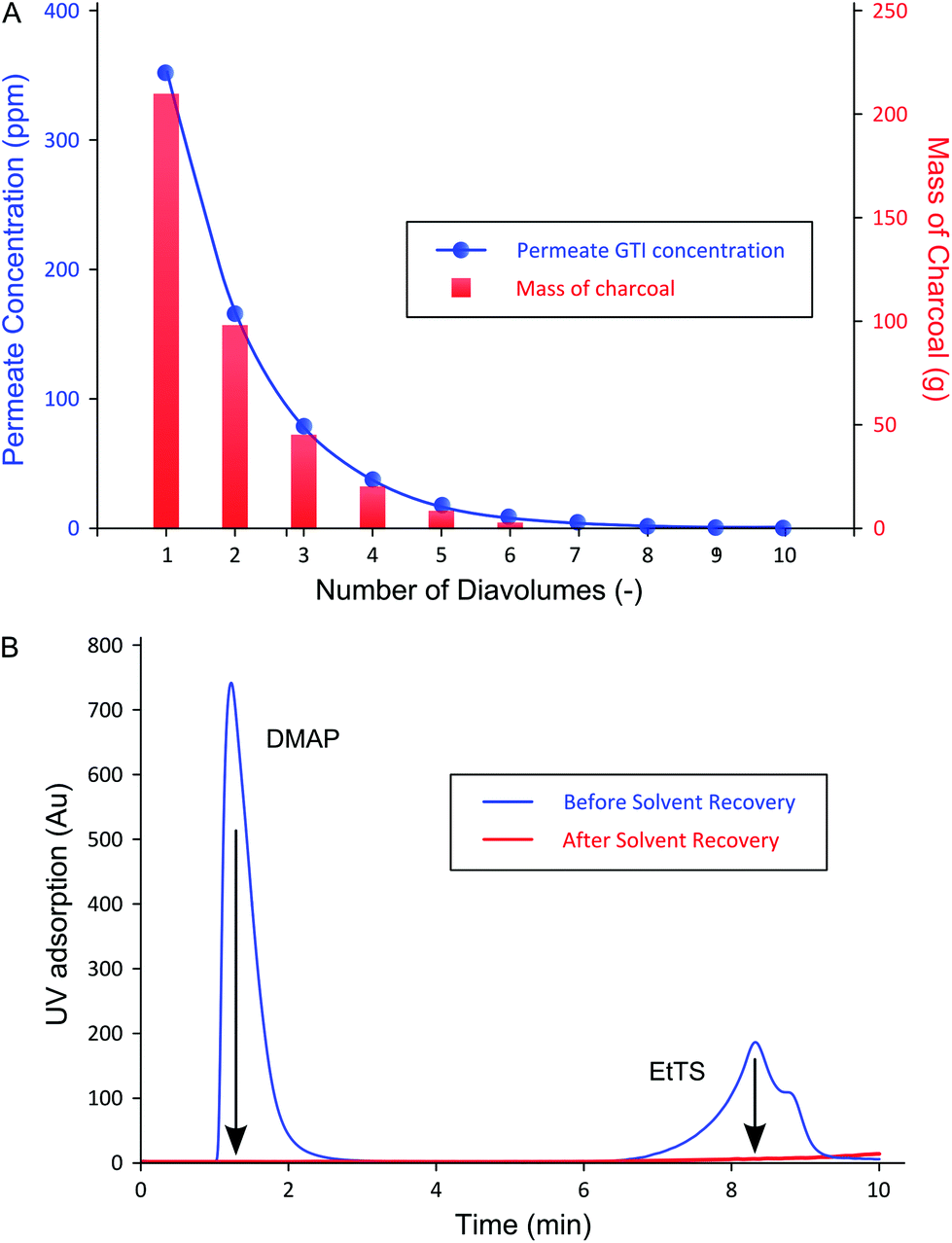 | ||
| Fig. 7 (A) GTI permeate concentration at each diavolume before adsorption and the respective amount of charcoal used to extract pure solvent. Approximately 90% of GTI permeate in the first three diavolume, requiring over 70% of the overall charcoal used. (B) A typical UV-HPLC chromatogram of crude permeate (1st diavolume data shown) and recovered solvent, which shows no remaining impurity (CGTI < LOD). Arrows show the extent to which initial permeate GTI content is reduced via solvent recovery. | ||
Fig. 7B shows that the GTI adsorption to charcoal allowed the recovery of the permeate by yielding pure solvent with concentrations below LOD for both GTIs measured by UV-HPLC, i.e. 1 and 3 ppm for DMAP and EtTS, respectively. In addition, no API was detected in the supernatant after the GTI adsorption. The purity of the recovered solvent allows its reuse in the manufacturing process.
As expected from the TSD diafiltration data (Fig. 4), it can be seen in Fig. 7A that approximately 90% of the GTIs permeate through the membrane in the first three diavolumes. After the third diavolume, the remaining 10% of the GTIs permeate at much slower rate. Fig. 7A also shows that over 70% of the required charcoal is used in the first three volumes, and it can also be seen that recycling the last diavolume takes the least amount of charcoal. This trend clearly suggests a potential for optimization. Hence, an economic analysis has been carried out in terms of cost of solvent (0.36 £ kg−1), charcoal (6 £ kg−1), and waste disposal (0.42 £ kg−1), to optimize the solvent recovery amount, as shown in Fig. 8. The used charcoal and untreated solvent were treated as solid and solvent waste, respectively.
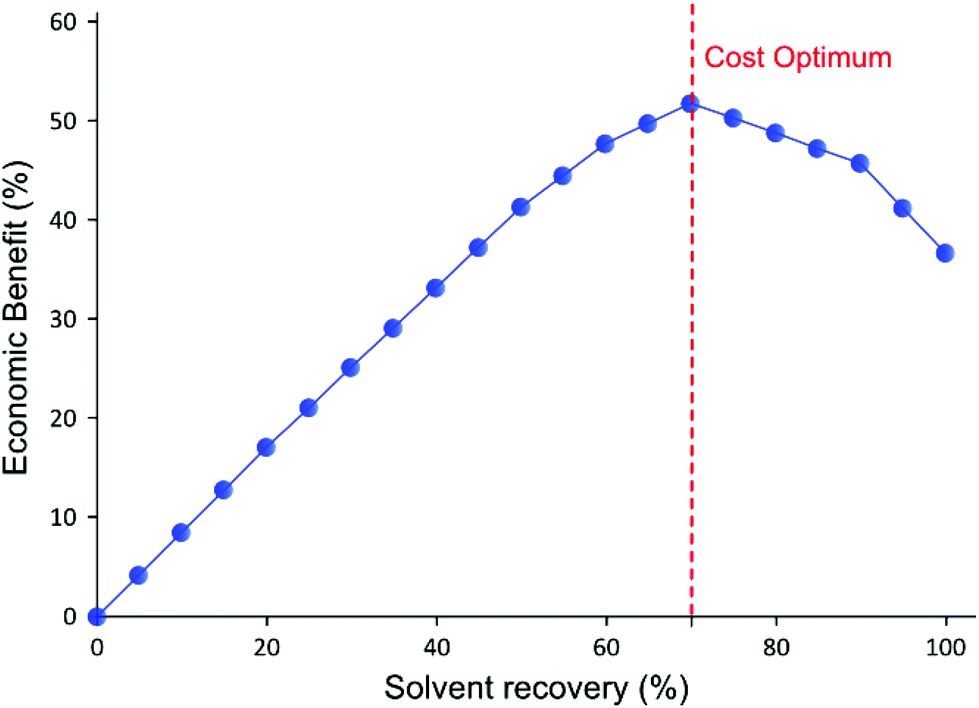 | ||
| Fig. 8 Calculated economic benefit from solvent recovery plotted versus percentage of solvent recovered curve shows an optimum for solvent recovery. The calculation is based on the cost of solvent, charcoal, and waste disposal. | ||
For the purpose of calculation, it was assumed that the last permeate diavolume was recovered first and gradually progressed towards the first permeate diavolume. Hence, 10% solvent recovery means the last 10% of the permeate has been recycled, and so on. It can be seen in Fig. 8 that the relative benefit from solvent recovery continuously increases until the maximum value at 70% solvent recovery. Then, the relative benefit decreases from the maximum value to recover the last 30% of the solvent, representing the first three diavolumes. From this point, the cost of charcoal and waste disposal overtakes the marginal gain obtained from recovering the solvent. Also, the amount of solid waste generated from charcoal increases rapidly for the last 30% (discussed in the next section). Nevertheless, it is more economical to recover 100% solvent than to have no solvent recovery, as the cost of charcoal is quite inexpensive. Also, the optimum could easily be affected by the cost of solvent and charcoal. Apart from this specific example, most API purification diafiltration have shown similar permeate concentration profiles8,25 in which 90% of impurities are removed in the first three diavolumes. Therefore, it is expected that the cost optimum solvent recovery point implementing an adsorptive solvent recovery unit would often lie between 70–80%. Nevertheless, it is expected that 100% solvent recovery would be preferred when additional factors, i.e. handling, capital cost, and greenness aspects, are considered.
2.4. Economic and green metrics analysis
Implementing a two-stage diafiltration process with a solvent recycle unit provides important process intensification of API purification. To assess the proposed process in more detail, the three processes described in this work, single-stage diafiltration (SSD), two-stage diafiltration (TSD), and two-stage diafiltration with solvent recovery (TSD + SR), are compared in terms of costs, mass and solvent intensity, energy consumption, and CO2 output, which are the key process metrics.50As briefly mentioned in the previous section, the cost of API varies widely from 100 £ kg−1 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 £ kg−1, depending on the strength, dose, production scale, and type of the API. While the economic gain from solvent recovery mainly depends on the scale of API production, its ability to reduce the overall environmental impact of the process is mainly related to the solvent used and the amount of solvent recycled. Hence, to make a reasonable comparison, the three processes are compared at different API prices. Furthermore, to compare implementing adsorption over distillation for solvent recovery, energy consumption and CO2 intensity have been compared at 70% solvent recovery.
000 £ kg−1, depending on the strength, dose, production scale, and type of the API. While the economic gain from solvent recovery mainly depends on the scale of API production, its ability to reduce the overall environmental impact of the process is mainly related to the solvent used and the amount of solvent recycled. Hence, to make a reasonable comparison, the three processes are compared at different API prices. Furthermore, to compare implementing adsorption over distillation for solvent recovery, energy consumption and CO2 intensity have been compared at 70% solvent recovery.
The magnitude of cost reduction at different API price is summarized in Fig. 9. The parameters taken into account for the calculation are cost of API loss, solvent, charcoal, membrane module, and waste disposal.
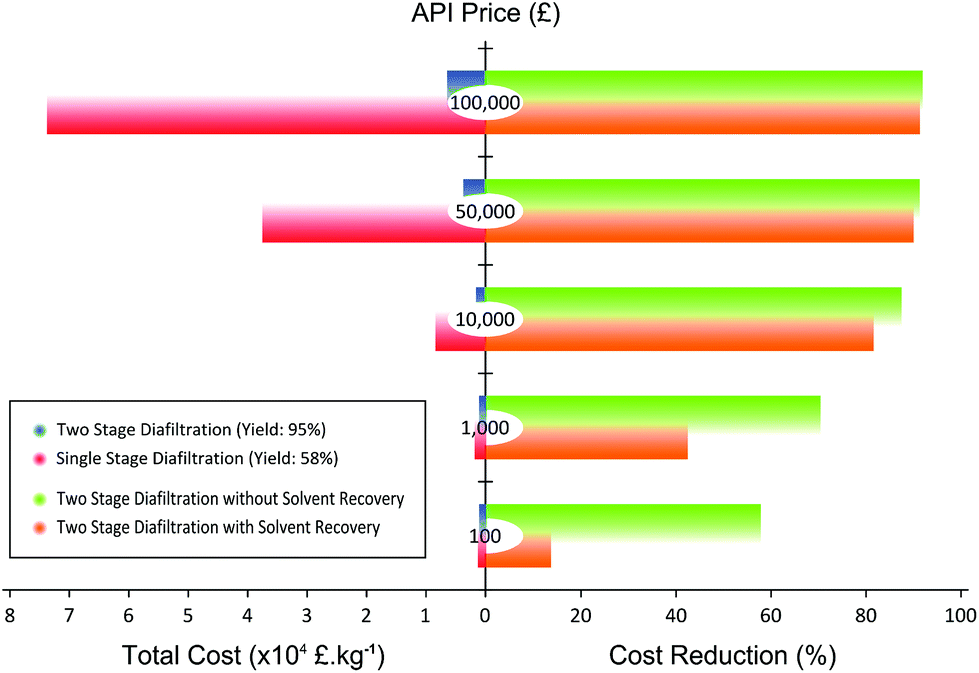 | ||
| Fig. 9 (left) Total purification cost per unit of API calculated for single-stage diafiltration (SSD) and two-stage diafiltration (TSD). The calculation includes the cost of API loss, solvent, membrane, charcoal, and waste disposal. (right) Summary of cost reduction for API purification compared to SSD by implementing TSD and TSD with 70% solvent recovery. The cost reduction is most significant when the cost of API is high. The difference between TSD and TSD + SR becomes considerable at lower API cost. Please refer to the online version to resolve the color coding of the bars. | ||
Fig. 9 clearly illustrates that the separation cost is considerably reduced when the TSD process is implemented. At an API price of 100000 £ kg−1, the cost reduction can be as high as 92%. Also, as the price of API goes down, the cost reduction also goes down. Hence, the major factor for the cost reduction comes from the API loss. As the yield of SSD is only 58% compared to the TSD yield of 95%, the loss of API is substantially reduced in TSD, improving the economics of separation. In addition, TSD + SR always compare favorably over TSD. As can be seen in Fig. 9, the gap between TSD and TSD + SR gets wider as the price of API goes down. This trend arises because the cost of solvent and waste disposal becomes comparable relative to that of API loss as the price of API goes down, and recovering 70% of the solvent contributes significantly to the process economics. In all cases, TSD + SR achieves cost saving over conventional SSD, mainly through increasing the process yield of API and reducing the solvent consumption.
Apart from the separation cost, implementation of solvent recovery brings additional advantages from the green engineering perspective. Mass intensity and solvent intensity are two of the most commonly used process metrics to describe a particular process. Due to its inherent simplicity, they are being widely applied to compare different processes.1 They are defined as,
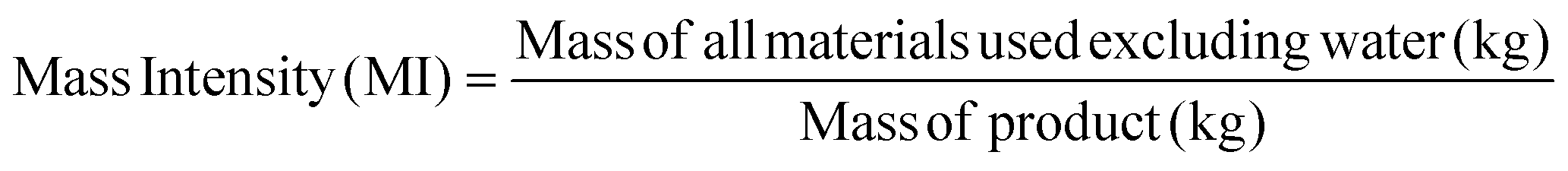 | (3) |
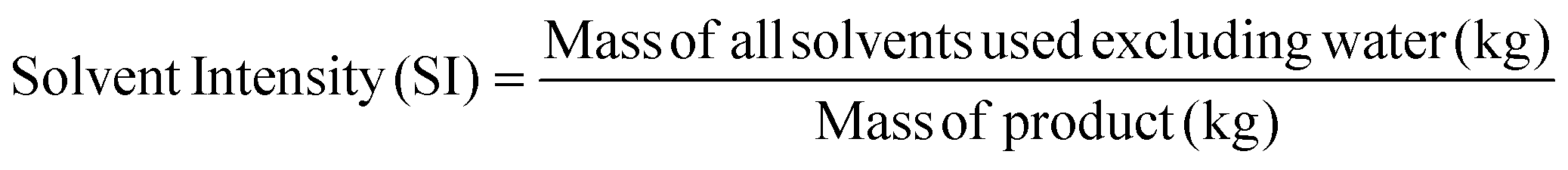 | (4) |
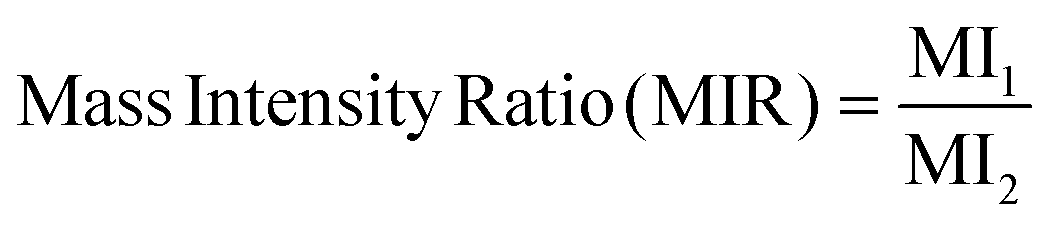 | (5) |
A solvent recovery unit achieves visible improvement in process sustainability in terms of mass intensity (MI) and solvent intensity (SI) of the process, as shown in Fig. 10.
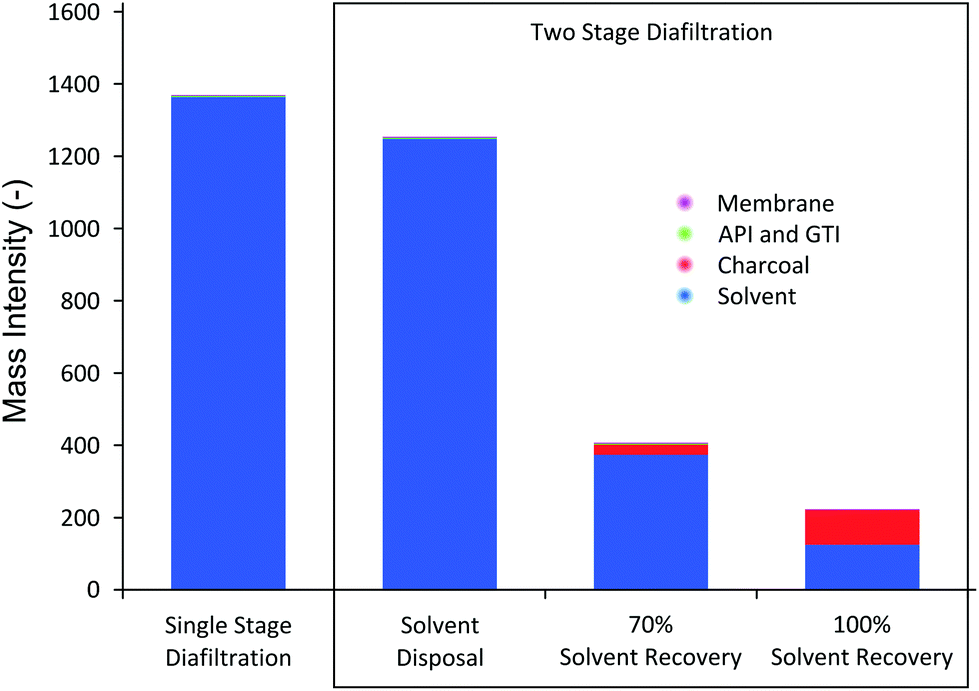 | ||
| Fig. 10 Mass intensity (MI) comparison. The majority of fractions of MI comes from solvent use. TSD achieves lower MI than SSD in all cases. | ||
The results in Fig. 10 show several interesting trends. First, compared to SSD, TSD and TSD + SR clearly achieve better MI. TSD without solvent recovery achieves 8% MI reduction, whereas TSD + 70%SR and TSD + 100%SR achieve 70% and 84% reduction in MI, respectively. It should be stressed that the overall system volume for TSD (consisting of first stage and second stage) is actually higher than SSD (consisting only of first stage), which means each given diavolume produces higher waste solvent for TSD. Nevertheless, it can be seen that the actual MI is lower because TSD achieves much higher yield, allowing lower overall solvent consumption compared to SSD. The mass intensity ratio of TSD to SSD was calculated to be 0.91, suggesting that TSD, without solvent recovery, is marginally preferred. Secondly, it can be seen in Fig. 10 that solvent consumption accounts for most of the mass intensity. As pointed out earlier, this high solvent consumption is the major challenge of membrane diafiltration processes, where for both SSD and TSD without SR, the solvent contributes over 99.9% of the calculated MI. Hence, when solvent recovery is implemented, a considerable reduction in MI can be observed, making the process greener and more sustainable. It is also apparent that the solvent fraction is lower for TSD + SR, as the mass of charcoal adsorbent contributes to the MI. The calculated mass intensity ratios of TSD + 70%SR and TSD + 100%SR were 0.29 and 0.16, respectively, approximately 4 times improvement. Lastly, the gain from recovering the last 30% of the solvent is relatively marginal as the solid waste becomes significant to the amount of solvent recovered. Nevertheless, the amount of solvent recovery depends on the type of application and hence a reasonable balance should be struck between the solvent recovery, charcoal consumption, and process operation.
MI values obtained in this study correlate well with other membrane diafiltration MI values reported in the literature.8,25 Also, it is interesting to compare MI values obtained in this work to the compiled MI data from seven major pharmaceutical companies on various processes and unit operations.51 The compiled data from 2007 presented that the maximum, minimum, and median MI values of the drug manufacturing processes in development pipeline are 887, 23, and 120, respectively. In terms of MI, membrane processes (SSD and TSD) without solvent recovery are not very competitive. However, once the solvent recovery unit is implemented, the process MI falls within the range, albeit above the median value. This trend clearly indicates that in order for membrane diafiltration unit to be competitive to conventional processes from green engineering perspective, a solvent recovery unit must be implemented. However, it should be stressed that MI values can vary considerably depending on the operating concentration range. In this study, we employed an API concentration of 10 kg m−3, but it is also common to use up to 100 kg m−3, which would reduce the MI one order of magnitude.
Both the economic and environmental impacts of a solvent recovery unit depends on the solvent type as well as the recovery process itself, i.e. whether the solvent is recovered by adsorption or distillation. For instance, chlorinated solvents (i.e. DCM) or high-boiling-point solvents (i.e. DMF) would have different impacts in process sustainability and energy consumption.52,53 The current trend is to minimize chlorinated and high-boiling-point solvent consumption, and replace them with milder and nontoxic solvents;54 however, there are still many cases where such solvents are required. Hence, a comparison between distillation and an adsorption unit in terms of energy consumption and CO2 generation considering solvents used widely in chemical processes, is presented in Fig. 11.
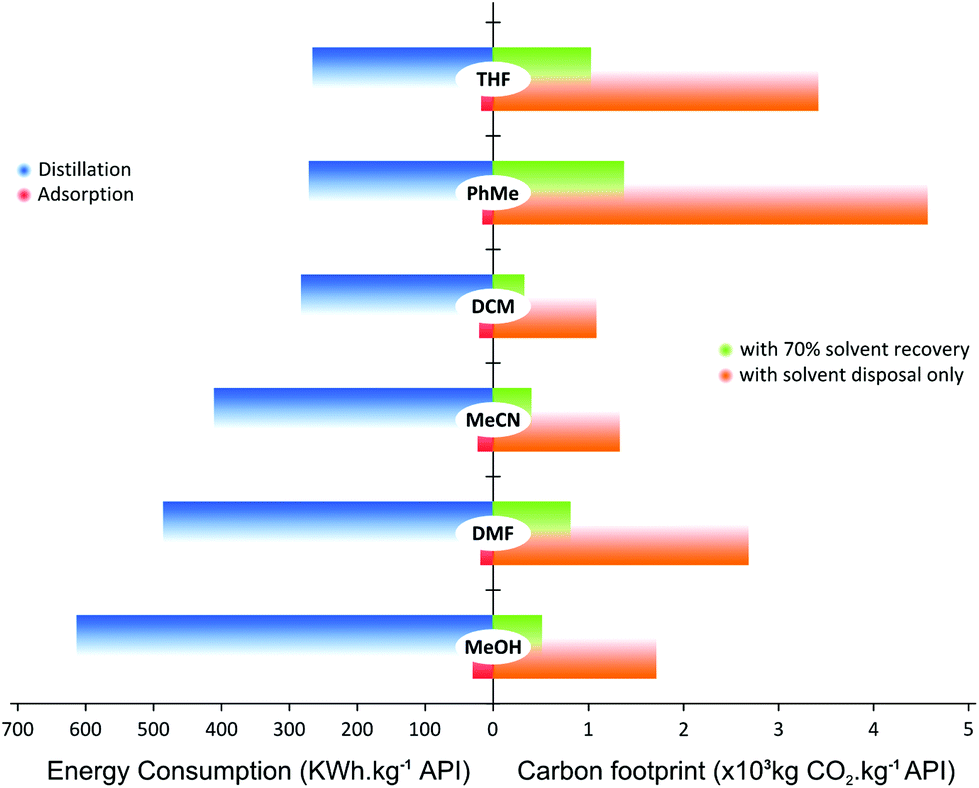 | ||
| Fig. 11 Comparison of CO2 footprint with and without solvent recovery, and of energy consumption for distillation and adsorption for solvent recovery, for commonly used organic solvents, for TSD. Please refer to the online version to resolve the color coding of the bars. | ||
Fig. 11 shows the difference in energy consumption between distillation and adsorption unit operation for TSD with 70% solvent recovery. For distillation, the calculation takes into account the solvent heat capacity, and heat of vaporization and condensation; as for the adsorption process, heat of adsorption, stirring and vacuum filtration energy have been taken into account. For comparison purposes, all calculations have been normalized to the unit of API purified. Surprisingly, the energy required to recycle MeOH is higher than that for DMF using distillation. This rather unexpected trend arises because the basis of calculation was on the same volume of solvent for the same API crude concentration (10 kg m−3). Considering the density and molecular weight of each solvent, the energy required to evaporate the same volume of different solvents does not necessarily follow the order of boiling points. On the other hand, it can be seen that the energy required for adsorption process is 92–96% lower than that from distillation.
It is interesting to compare the difference in mass of waste to be removed through solvent recovery between SSD and TSD permeate. The total waste in the permeate for SSD (total solute including API and GTI) is as high as 1.2 g waste per kg of solvent. On the contrary, the TSD configuration yields 4 times less waste in the permeate, since much less API permeates through the membrane. Such a mass gain makes adsorption processes feasible for solvent recovery. In previous studies employing conventional SSD only distillation-based solvent recovery units were investigated.25 The TSD processes coupled with an adsorptive solvent recovery unit presents a notable advantage in solvent and energy consumption.
Fig. 11 also summarizes the CO2 footprint of the process with and without solvent recycle. The calculation takes into account the amount CO2 generated from incinerating the solvents as well as the associated carbon intensity from using steam and electricity for the solvent recovery unit. The main source of CO2 comes from solvent incineration, where the carbon intensity contributions from using steam and electricity (taken as 204 and 369 kg of CO2, respectively) were less than 1% of the overall CO2 footprint. Hence, there was negligible difference in CO2 reduction between distillation and adsorption. Although the comparison was made at 70% solvent recovery, the carbon footprint can be further reduced through a higher fraction of solvent recovery. For instance, a combined approach where the first three diavolumes are recovered by distillation and the rest by adsorption, can be explored to minimize the energy consumption and CO2 footprint. Nevertheless, it is clear from Fig. 11 that recovering solvent reduces the CO2 footprint because not as much solvent is incinerated but recycled.
In summary, considering the cost (Fig. 9), mass intensity (Fig. 10), and energy consumption (Fig. 11), the overall trend leads to a clear conclusion that a solvent recovery unit is strongly preferred for membrane diafiltration processes to secure a competitive edge in comparison to other separation technologies.
3. Conclusions
In this work, previous limitations faced by membrane processes have been successfully overcome through the application of membrane cascade and solvent recovery. The proposed membrane cascade configuration was tested on a practical application: purifying an API product from two different classes of GTIs using an organic solvent nanofiltration (OSN) platform. In summary, the following conclusions can be drawn:• A two-stage membrane cascade configuration has been proposed and applied which drastically increased the process yield (58% to 95%) without compromising its final purity (below 5 ppm GTI).
• The configuration allows much higher membrane versatility, as membranes with insufficient separation performance can be used to achieve high yields. This reduces the initial membrane-screening period, which is often the bottleneck stage for membrane process development.
• Removing two classes of GTIs in a single unit operation has shown the advantage of a size exclusion membrane process over affinity-based separations.
• The TSD process achieved significant cost saving up to 92% compared to conventional single stage diafiltration. Implementing a solvent recycle unit achieved 70% and 73% reduction in mass intensity (MI) and solvent intensity (SI), respectively.
• An adsorptive solvent recovery unit consumes 92–96% less energy compared to distillation.
• A solvent recovery unit is strongly favored to make membrane diafiltration processes competitive to conventional separation techniques.
The proposed membrane process can now overcome the previous challenges such as low product yield and high solvent consumption. Through the two-stage cascade configuration, the membrane process achieves competitive yield and purity using available membranes. In addition, the process is versatile as it can be used with different types of membranes on various applications. Hence, the applicability of membrane processes in the pharmaceutical industry should be critically assessed compared to other separation techniques, with the process scale in mind, to enhance the sustainability and greenness of the industry.
4. Experimental
4.1. Materials
Acetonitrile (MeCN), N,N-dimethylacetamide (DMAc), and isopropyl alcohol (IPA), were HPLC grade and used as supplied from VWR. Celazole® S26 polybenzimidazole (PBI) solution (26 wt% polymer in DMAc containing 1.5 wt% LiCl) was purchased from PBI Performance Products Inc. Non-woven polyolefin Novatexx 2471 was from Freudenberg Filtration Technologies, Germany. Ethyl tosylate (EtTS), 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich; and PEG-400 and α,α′-dibromo-p-xylene (DBX) were purchased from Merck and VWR, respectively. Charcoal (YAO 20 × 45) and Roxithromycin samples were kindly provided by Eurocarb Ltd, UK, and Hovione FarmaCiencia SA, Portugal, respectively.4.2. Preparation of polybenzimidazole (PBI) membranes
PBI dope solutions were prepared by diluting the Celazole® S26 solution to 22 wt% with DMAc. Once homogeneous solutions had been obtained, they were cast on polyolefin non-woven support, with the casting knife height set to 250 µm. The supported film was then placed in a DI water coagulation bath. The resultant membranes were washed with IPA to ensure complete removal of water. To cross-link the polymer the membranes were immersed in a stirred 3 wt% solution of DBX in MeCN, heated at reflux for 24 hours. The membranes were then removed from the cross-linking solution and washed with IPA. Finally, the membranes were immersed in a PEG-400:IPA (1:1 v/v) impregnation bath for 4 hours and then air dried.
4.3. Membrane rejection test
All PBI membranes were screened against a test solution containing 10 kg m−3 of Roxi and 1000 ppm each of DMAP and EtTS. For each membrane a disc with an effective area of 51 cm2 was loaded into a membrane cell with six retentate side ports and one permeate port, and a volume of 0.92 dm3. A typical set-up consists of an inlet port, a retentate port fitted with a back-pressure regulator, a pressure gauge port, a thermocouple port, and a permeate port. A schematic of the system used for membrane screening and diafiltration experiments is shown in Fig. 12.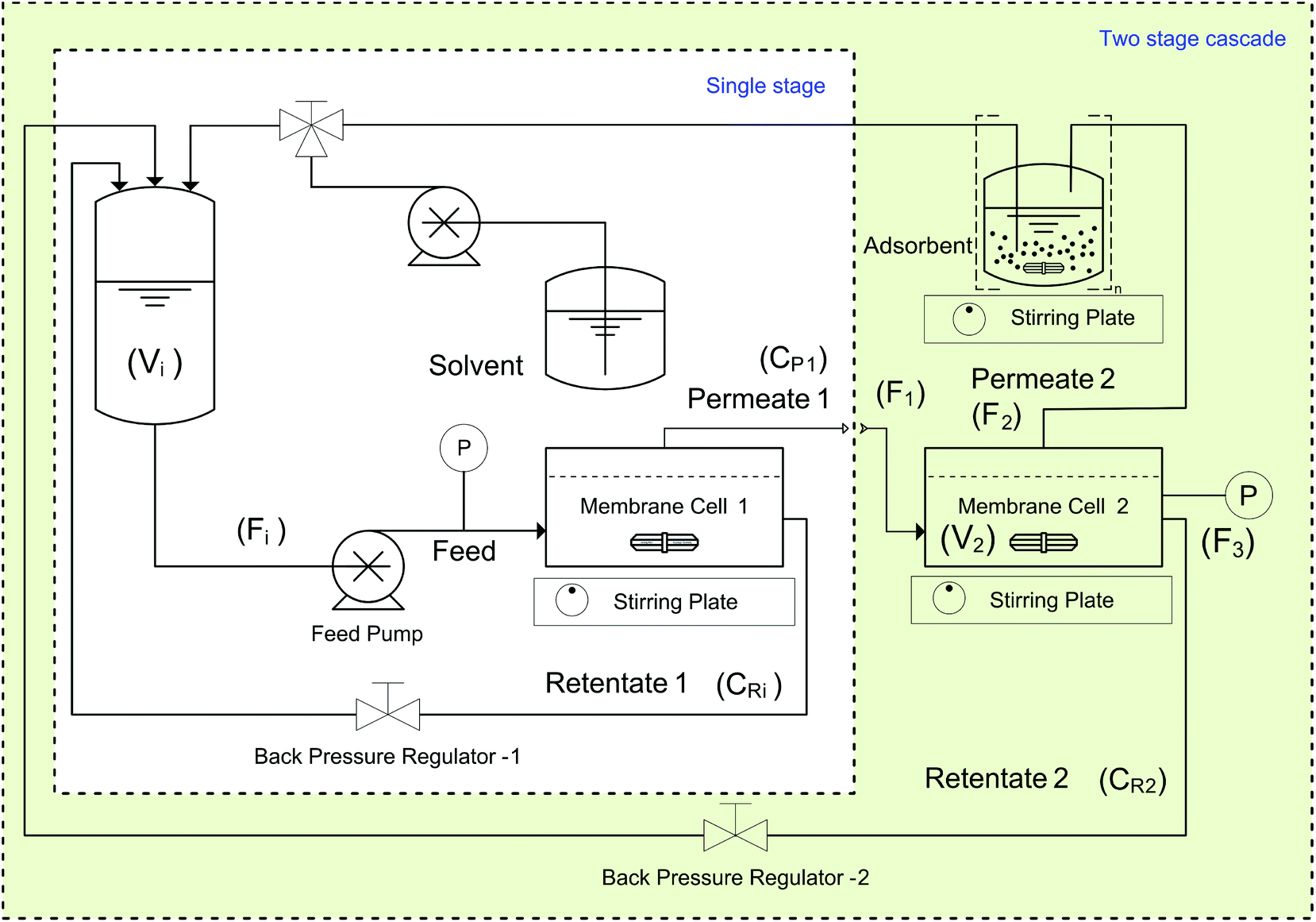 | ||
| Fig. 12 System diagrams of single-stage diafiltration and two-stage cascade diafiltration. For two-stage cascade diafiltration, the permeate from the first stage is directly connected to the feed of the second stage. The symbols in parentheses are the variables used in the process model (Appendix A). | ||
In both the single-stage and the two-stage diafiltration set-ups, a Gilson HPLC pump (Model 305) provided the flow, set at 1.8 dm3 h−1. The pressure of each cell was controlled using a back-pressure regulator, and a magnetic stirrer bar was placed inside each cell (stirred at 500 RPM) to maintain a constant hydrodynamic profile in the fluid in contact with the membrane. The feed tank volume was 0.1 dm3, and the volume of each cell plus the associated tubing was approximately 0.1 dm3.
All membranes were first washed with at least 2 dm3 of pure MeOH at 10 bar to remove all the conditioning agents and to achieve a steady-state flux. Next, a testing solution (10 kg m−3 of Roxi and 1000 ppm each of DMAP and EtTS in MeOH) was circulated at different pressures and the permeate was recycled back into the feed until a steady-state was reached. At this point, and at different times thereafter, samples of both permeate and retentate were taken for HPLC analysis to assess the membrane performance.
4.4. Single stage diafiltration
Single-stage diafiltration was performed independently with two PBI membranes using the configuration shown in Fig. 12. Initially, a test solution was circulated around the system at 5 bar. Once steady-state concentration of all the species in the retentate and permeate had been reached, a preparative single stage diafiltration was performed at 5 bar. The system was maintained at a constant volume by adding the same volume of pure solvent into the system as permeated over the same time. Permeate and retentate samples were analyzed by HPLC at different times. Mass yield of Roxi was then calculated using the following equations,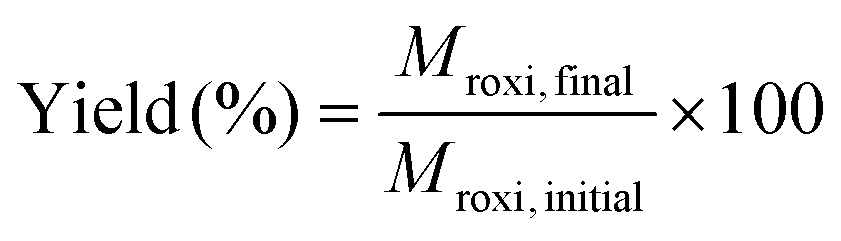 | (6) |
4.5. Two stage diafiltration
Two-stage diafiltration using the configuration shown in Fig. 12 was performed twice with a recycle ratio of 0.5 (rC = 0.5), defined in eqn (7). Without connecting the second membrane cell, the first-stage membrane was equilibrated with a testing solution, while recirculating the permeate back into the feed tank, until a steady state was achieved. Once the concentrations had become constant in the permeate and retentate, the permeate from the first stage was fed directly into the second cell (initially charged with pure solvent) as shown in Fig. 12. To control the recycle ratio (rC), the first stage and second stage transmembrane pressures were set at 15 and 5 bar, respectively.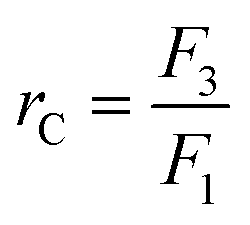 | (7) |
4.6. Charcoal isotherm determination and solvent recovery
To obtain charcoal adsorption isotherm data, 0.5 dm3 solution containing 500 ppm of each GTI (DMAP and EtTS) was first prepared. The solution was split into five 0.1 dm3 solutions and the following amount of charcoal was added: 1 g, 5 g, 10 g, 25 g, and 40 g. Each solution was stirred vigorously at 21 ± 0.5 °C and samples were taken at various times (4–20 h) to confirm equilibrium was established.The amount of charcoal required to recover pure solvent can be calculated by first equating two isotherm equations,
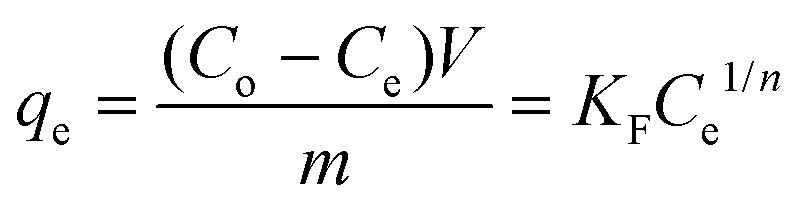 | (8) |
Solving for “m” and integrating with appropriate boundary conditions yields the amount of charcoal required:
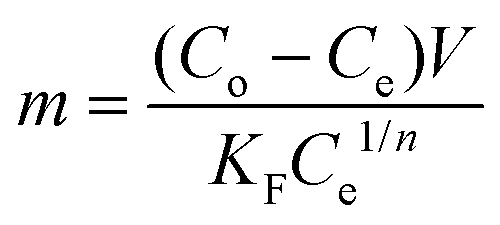 | (9) |
To recover pure solvent from the permeate, each diavolume (0.3 dm3) was collected separately and the calculated amount of activated charcoal was added. Each solution was stirred vigorously at 21 ± 0.5 °C and samples were taken at various times (4–20 h) until no more GTIs were detected in the supernatant. The recovered solvent was not re-used for the same experiment.
4.7. Analytical methods
All samples were analyzed by HPLC using an Agilent 1100 Series system equipped with an UV detector and Varian 385-LC ELSD detector. The pump flow-rate was set at 1 mL min−1, the injection volume was 30 µL, the column temperature was 30 °C, and an ACE C18 RP column was fitted. The column was eluted with a gradient of MeOH and water buffered with 5 mM diethylammonium acetate, pH 6.5. The UV wavelength was set at 260 nm, the evaporation temperature was set to 40 °C, nebulization temperature at 55 °C, and the nitrogen gas flow rate was at 1.5 SLM. Calibration curves were made for Roxi using ELSD, for DMAP and EtTS using UV.Appendix A: Process modeling
The single-stage diafiltration system, as shown in Fig. 12, can be modeled by writing a mass balance around the system. Assuming that the system operates at a constant volume and it is perfectly mixed,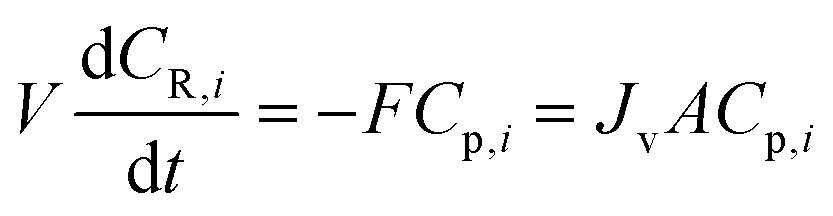 | (A1) |
Defining the observed rejection of species i as,
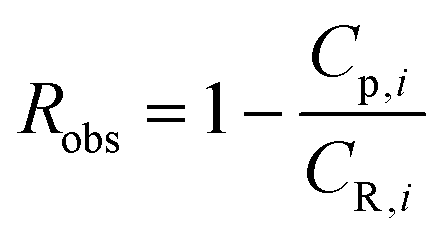 | (A2) |
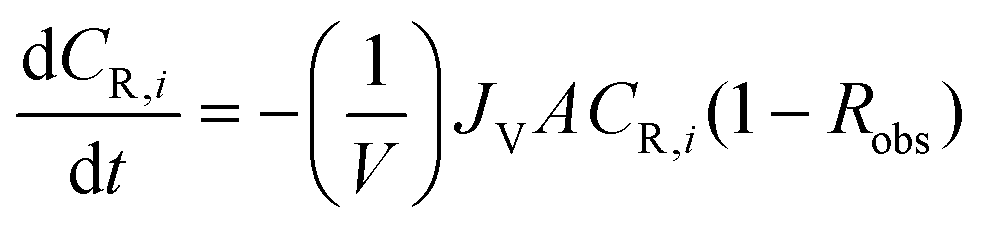 | (A3) |
This equation can be solved either numerically or analytically. When integrated analytically with appropriate boundary conditions, the following equation is obtained:
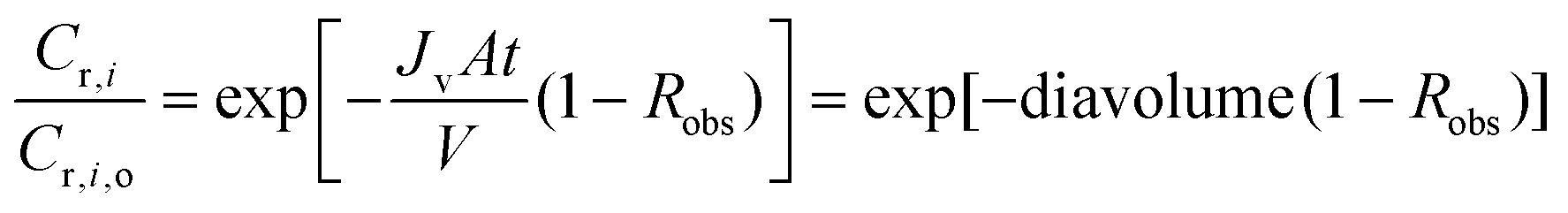 | (A4) |
A similar analysis of the two-stage diafiltration can be solved numerically using MATLAB. As the two stages are interconnected, a total of four ordinary differential equations can be written for species i and j in stages 1 and 2.
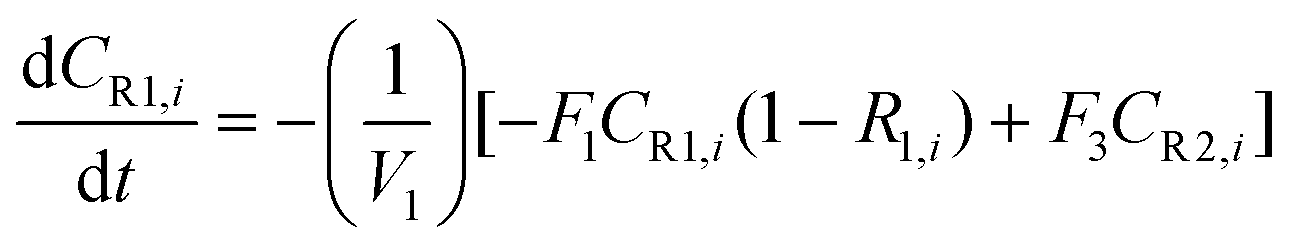 | (A5) |
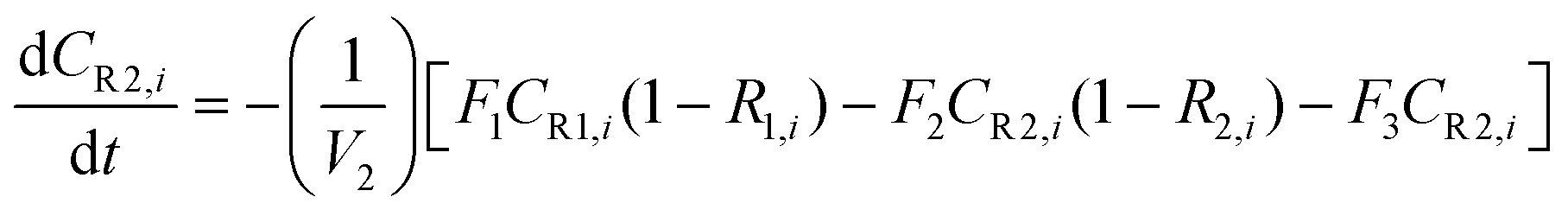 | (A6) |
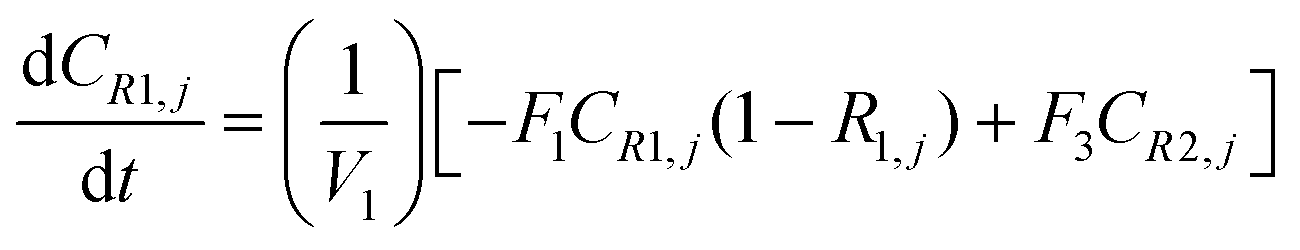 | (A7) |
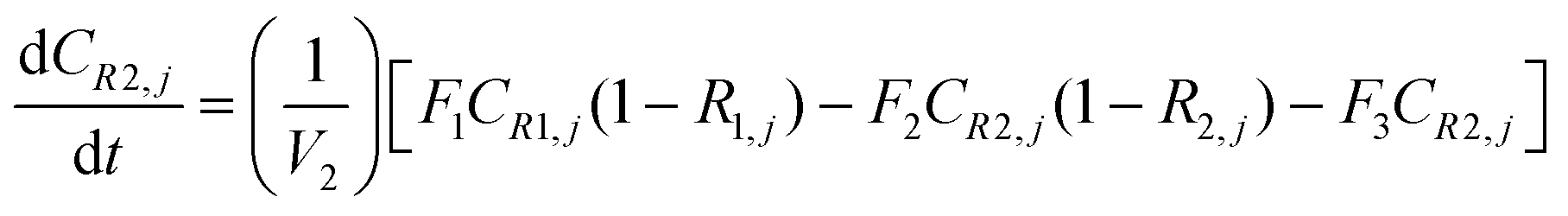 | (A8) |
Acknowledgements
J. F. Kim and I. B. Valtcheva are grateful for their PhD funding provided under the 7th Framework Program of the European Commission's Marie Curie Initiative (PITN-GA-2008-238291-MEMTIDE). G. Székely is grateful to EPSRC for his postdoctoral funding under the project EP/J014974/1 entitled Molecular Builders: Constructing Nanoporous Materials. The authors would also like to acknowledge Hovione FarmaCiencia SA, Portugal for Roxithromycin samples, and EuroCarb Ltd, UK for activated charcoal samples.References
- C. Jiménez-González, D. J. C. Constable and C. S. Ponder, Chem. Soc. Rev., 2012, 41, 1485–1498 RSC.
- C. Jiménez-González, P. Poechlauer, Q. B. Broxterman, B.-S. Yang, D. am Ende, J. Baird, C. Bertsch, R. E. Hannah, P. Dell'Orco, H. Noorman, S. Yee, R. Reintjens, A. Wells, V. Massonneau and J. Manley, Org. Process Res. Dev., 2011, 15, 900–911 CrossRef.
- Limit_of_genotoxic_impurities, EMEA, 2006.
- M. Degerman, N. Jakobsson and B. Nilsson, Chem. Eng. Technol., 2008, 31, 875–882 CrossRef CAS.
- T. Winkelnkemper and G. Schembecker, Sep. Purif. Technol., 2010, 72, 34–39 CrossRef CAS PubMed.
- E. Drioli, A. I. Stankiewicz and F. Macedonio, J. Membr. Sci., 2011, 380, 1–8 CrossRef CAS PubMed.
- E. Drioli, A. Brunetti, G. Di Profio and G. Barbieri, Green Chem., 2012, 14, 1561–1572 RSC.
- G. Székely, J. Bandarra, W. Heggie, B. Sellergren and F. C. Ferreira, J. Membr. Sci., 2011, 381, 21–33 CrossRef PubMed.
- J. Vanneste, D. Ormerod, G. Theys, D. Van Gool, B. Van Camp, S. Darvishmanesh and B. Van der Bruggen, J. Chem. Technol. Biotechnol., 2013, 88, 98–108 CrossRef CAS.
- G. Székely, E. Fritz, J. Bandarra, W. Heggie and B. Sellergren, J. Chromatogr., A, 2012, 1240, 52–58 CrossRef PubMed.
- G. Székely, J. Bandarra, W. Heggie, F. C. Ferreira and B. Sellergren, Sep. Purif. Technol., 2012, 86, 190–198 CrossRef PubMed.
- G. Székely, J. Bandarra, W. Heggie, B. Sellergren and F. C. Ferreira, Sep. Purif. Technol., 2012, 86, 79–87 CrossRef PubMed.
- B. Van der Bruggen, M. Mänttäri and M. Nyström, Sep. Purif. Technol., 2008, 63, 251–263 CrossRef CAS PubMed.
- Y. H. See Toh, F. W. Lim and A. G. Livingston, J. Membr. Sci., 2007, 301, 3–10 CrossRef PubMed.
- Y. H. See-Toh, M. Silva and A. Livingston, J. Membr. Sci., 2008, 324, 220–232 CrossRef CAS PubMed.
- P. Vandezande, L. E. M. Gevers and I. F. J. Vankelecom, Chem. Soc. Rev., 2008, 37, 365–405 RSC.
- S. Darvishmanesh, L. Firoozpour, J. Vanneste, P. Luis, J. Degrève and B. Van Der Bruggen, Green Chem., 2011, 13, 3476 RSC.
- E. M. Rundquist, C. J. Pink and A. G. Livingston, Green Chem., 2012, 14, 2197–2205 RSC.
- C. Pink, H. Wong, F. C. Ferreira and A. G. Livingston, Org. Process Res. Dev., 2008, 12, 589–595 CrossRef CAS.
- S. So, L. G. Peeva, E. W. Tate, R. J. Leatherbarrowb and A. G. Livingston, Chem. Commun., 2010, 46, 2808–2810 RSC.
- A. Criscuoli and E. Drioli, Ind. Eng. Chem. Res., 2007, 2268–2271 CrossRef CAS.
- A. Brunetti, G. Barbieri and E. Drioli, Chem. Eng. Sci., 2009, 64, 3448–3454 CrossRef CAS PubMed.
- E. Curcio, A. Criscuoli and E. Drioli, Ind. Eng. Chem. Res., 2001, 40, 2679–2684 CrossRef CAS.
- J. Stawikowska, J. F. Kim and A. G. Livingston, Chem. Eng. Sci., 2013, 97, 81–95 CrossRef CAS PubMed.
- G. Székely, M. Gil, B. Sellergren, W. Heggie and F. C. Ferreira, Green Chem., 2013, 15, 210–225 RSC.
- J. Geens, K. Boussu, C. Vandecasteele and B. Van der Bruggen, J. Membr. Sci., 2006, 281, 139–148 CrossRef CAS PubMed.
- B. Van der Bruggen, J. Schaep, D. Wilms and C. Vandecasteele, J. Membr. Sci., 1999, 156, 29–41 CrossRef CAS.
- P. Marchetti, A. Butté and A. G. Livingston, J. Membr. Sci., 2012, 415–416, 444–458 CrossRef CAS PubMed.
- S. Zeidler, U. Kätzel and P. Kreis, J. Membr. Sci., 2013, 429, 295–303 CrossRef CAS PubMed.
- J. F. Kim, A. M. Freitas da Silva, I. B. Valtcheva and A. G. Livingston, Sep. Purif. Technol., 2013, 116, 277–286 CrossRef CAS PubMed.
- P. Suresh and P. K. Basu, J. Pharm. Innovation, 2008, 3, 175–187 CrossRef PubMed.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303–307 CrossRef CAS PubMed.
- H. Vinh-Thang and S. Kaliaguine, Chem. Rev., 2013, 113, 4980–5028 CrossRef CAS PubMed.
- S. Karan, S. Samitsu, X. Peng, K. Kurashima and I. Ichinose, Science, 2012, 335, 444–447 CrossRef CAS PubMed.
- P. Vandezande, L. Gevers, J. Paul, I. Vankelecom and P. Jacobs, J. Membr. Sci., 2005, 250, 305–310 CrossRef CAS PubMed.
- W. R. Bowen and J. S. Welfoot, Chem. Eng. Sci., 2002, 57, 1121–1137 CrossRef CAS.
- W. R. Bowen and J. S. Welfoot, Chem. Eng. Sci., 2002, 57, 1393–1407 CrossRef CAS.
- W. Deen, AIChE J., 1987, 33, 1409–1425 CrossRef CAS.
- E. N. Lightfoot, T. W. Root and J. L. O'Dell, Biotechnol. Prog., 2008, 24, 599–605 CrossRef CAS PubMed.
- W. E. Siew, A. G. Livingston, C. Ates and A. Merschaert, Sep. Purif. Technol., 2013, 102, 1–14 CrossRef CAS PubMed.
- K. Mohanty and R. Ghosh, J. Membr. Sci., 2008, 307, 117–125 CrossRef CAS PubMed.
- M. Mayani, C. D. M. Filipe and R. Ghosh, J. Membr. Sci., 2010, 347, 150–158 CrossRef CAS PubMed.
- P. Aimar and R. Field, Chem. Eng. Sci., 1992, 47, 579–586 CrossRef CAS.
- R. Kecili, J. Billing, M. Leeman, D. Nivhede, B. Sellergren, A. Rees and E. Yilmaz, Sep. Purif. Technol., 2013, 103, 173–179 CrossRef CAS PubMed.
- C. Lee, R. Helmy, C. Strulson, J. Plewa, E. Kolodziej, V. Antonucci, B. Mao, C. J. Welch, Z. Ge and M. A. Al-sayah, Org. Process Res. Dev., 2010, 14, 1021–1026 CrossRef CAS.
- R. Ghosh, J. Membr. Sci., 2003, 226, 85–99 CrossRef CAS PubMed.
- G. Alberti, V. Amendola, M. Pesavento and R. Biesuz, Coord. Chem. Rev., 2012, 256, 28–45 CrossRef CAS PubMed.
- H. M. F. Freundlich, J. Phys. Chem., 1906, 57, 385–470 CAS.
- K. Y. Foo and B. H. Hameed, Chem.–Eng. J., 2010, 156, 2–10 CrossRef CAS PubMed.
- A. Lapkin and D. J. C. Constable, Green Chemistry Metrics, John Wiley & Sons, Ltd, 2009 Search PubMed.
- J. Kindervater, J. Manley and R. Henderson, ACS Green Chem. Inst., 2007 Search PubMed.
- R. Gani, C. Jiménez-González and D. J. C. Constable, Comput. Chem. Eng., 2005, 29, 1661–1676 CrossRef CAS PubMed.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. a. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- D. J. C. Constable, C. Jimenez-gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- G. Székely, B. Henriques, M. Gil, A. Ramos and C. Alvarez, J. Pharm. Biomed. Anal., 2012, 70, 251–258 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |