In silico design of bio-based commodity chemicals: application to itaconic acid based solvents
Laurianne
Moity
a,
Valérie
Molinier
a,
Adrien
Benazzouz
a,
René
Barone
b,
Philippe
Marion
c and
Jean-Marie
Aubry
*a
aUniversité Lille Nord de France, USTL, ENSCL, E.A. 4478 Chimie Moléculaire et Formulation, Cité Scientifique, 59652 Villeneuve d'Ascq Cedex, France. E-mail: jean-marie.aubry@univ-lille1.fr
bUniversité Paul Cézanne, ISM2, UMR 7313, Faculté des Sciences de St Jérôme, 13397 Marseille Cedex 20, France
cSolvay Recherches & Innovation, Centre de Lyon, 85 rue des Frères Perret, Saint-Fons Cedex, France
First published on 13th September 2013
Abstract
Global issues are deeply changing the face of the chemical industry. Both the feedstock – biomass vs. oil – and the chemical transformations used are now chosen to comply with a sustainable development. In this work, a computer-assisted organic synthesis program, named GRASS (GeneratoR of Agro-based Sustainable Solvents), is proposed to help in the design of sustainable solvents from biomass feedstock. A careful selection of industrially relevant chemical transformations has been performed to propose a set that allows obtaining in a few steps (1 to 3) a large number of commodity chemicals from a chosen set of bio-based building-blocks. Emphasis has been put on solvents since this type of compound is undergoing a deep renewal due to more stringent regulatory constraints, but the same approach may be extended to other commodity chemicals. This methodology has been exemplified starting from itaconic acid, a multifunctional bio-based building block. We checked a posteriori that all itaconic acid based-solvents reported in the literature do indeed belong to the set of virtual solvents generated by GRASS, along with an extended list of new derivatives that could be of potential interest.
Introduction
All industries are now facing severe environmental issues, particularly the rarefaction of oil resources coupled with a chaotic oil market and the necessity to decrease greenhouse gases emissions.1 Even though shale gas provides an opportunity to reduce oil dependence, its exploration and production have become highly controversial issues.2–4 An international commitment has arisen based on questions related to the rarefaction of oil resources and to greenhouse gases emissions, notably via the emergence of international agencies and the increase in the number of funded research programs on related topics. These unquestionable global issues find a particular echo in the chemical industry that has been asked to “lead in developing the technological dimension of a sustainable civilization” during the Rio Conference in 1992.5 This led to various initiatives, such as Responsible Care Initiative and the Responsible Care Global Charter of the Chemical Industry, and also to the establishment of the principles of Green Chemistry and Green Engineering.6–10Also driven by economical and strategic reasons, sometimes speeded up by legislative constraints, the chemical industry is progressively turning to more sustainable solutions. This implies re-thinking its production methods from a global point of view, in a “cradle-to-grave” analysis: feedstock (starting material and reagents), chemical transformations and processes, and also target final products are carefully chosen, with the help of tools such as the life cycle assessment and/or the 12 principles of green chemistry.6
Particularly, due to their large-scale use in a great number of industrial and general public applications, solvents are among the commodity chemicals that are the most affected by new regulations (REACH, VOC's or HAP's compounds). Many of the traditional organic solvents (halogenated compounds, aromatics, glycols ethers) are banned or about to be, and there is, therefore, an urgent need to find alternatives with good ESH (environment, safety and health) profiles, often called “green solvents”.11–15 Recently, we have reviewed the currently available “green solvents” and compared them to the classical organic solvents using the theoretical COnductor-like Screening MOdel for Real Solvents (COSMO-RS)16 approach.17 This study provides a virtual panorama of the green and classical solvents, which helps in the search for potential alternatives. It also sheds light on the paucity or, on the contrary, the over representation of green alternatives in some chemical families. Indeed, current green solvents are mostly aprotic dipolar (e.g. glycerol triacetate), amphiprotic (e.g. ethanol) and polar protic (e.g. glycerol carbonate) compounds while strong and weak electron pair donor bases (as alternatives to NMP, DMF for instance) are scarcely represented. There is thus a need to develop new green solvents with the prerequisite properties. The design of these new sustainable solvents should obey the principles of green chemistry in particular for the choice of starting materials, preferably from renewable feedstock,18,19 and of the economically viable and environmentally-friendly transformations.
The purpose of this work is to propose a computer-assisted tool for the valorization of bio-based building blocks into target intermediate or commodity chemicals, such as solvents, through selected chemical transformations. A representative polyfunctional building-block, itaconic acid, has been chosen as an example of a starting material to illustrate the in silico methodology developed in this work.
The development of Computer-Aided Organic Synthesis (CAOS) tools to help organic chemists is not a novel topic20–23 (and ref. within it) since, as early as 1969, Wipke and Corey proposed the first retrosynthetic program named OCSS (Organic Chemical Simulation of Synthesis),24 in which the fundamental rules of organic chemistry that are still used today (retrosynthesis, disconnection, synthons etc.) were first described.25 Since then, a number of retrosynthetic programs have been developed (such as SECS,26 SYNCHEM,27 EROS,28 SYNGEN,29 SOS,30 PASCOP,31 HOLOWIN,32 SYNSUP33), the main differences lying in the way of coding the reactions to prune the synthetic tree and programs that are either interactive or automatic.
Only few programs have been developed to be used in the forward direction, even if some can be used in both ways: CAMEO,34 PSYCHO,35,36 FORWARD,37 ROBIA,38 SOS,39,40 and NELB41 are the main ones. CAMEO and SOS are based on mechanism considerations, while PSYCHO and others are based on reaction types. These programs usually do not promote unanimity within the organic chemists’ community because organic synthesis raises many challenges of feasibility depending on various factors such as reactivity of the substrate and experimental conditions. Therefore, in the case of complex molecules (as pharmaceuticals), synthesis prediction is not straightforward and computers will seldom, if ever, succeed, as the knowledge of a specialized chemist, coupled with a careful analysis of the literature, is more relevant. However, in the case of large scale and inexpensive chemicals, molecules and transformations must be simple, inexpensive and efficient. Thus synthesis planning should be easier and is expected to be performed by a computer.
That is why we have developed in this work a program working in the forward direction that is both easy to use and flexible. It has been named GRASS (GeneratoR of Agro-based Sustainable Solvents) to underline the will to generate chemical structures that can serve as commodity chemicals, particularly solvents, obtained from bio-based building blocks through a limited number of industrially relevant and environmentally-friendly transformations. The GRASS program initially derives from the SOS program (forward direction) that underwent various modifications.40,42,43 GRASS is developed to generate all the possible products that can be formed from a selected bio-based building block and readily available co-reactants according to well-chosen transformations. The aim is to generate libraries of virtual structures (also called library combinatorial design), a concept extensively used in the pharmaceutical domain,44 but not, to the best of our knowledge, for the design of commodity chemicals. These virtual candidates can then be evaluated according to synthesis feasibility and physico-chemical criteria (boiling point, melting point, vapour pressure, solubilizing power, etc.) relevant for a given application. GRASS can thus serve as a tool that helps in designing new agro-based solvents in silico but could also be used for the design of other commodity chemicals or intermediates. The methodology has been exemplified to generate potential bio-based solvents starting from itaconic acid, an unsaturated carboxylic diacid available from biomass and of special interest because of its polyfunctionality.
Experimental section
GRASS software
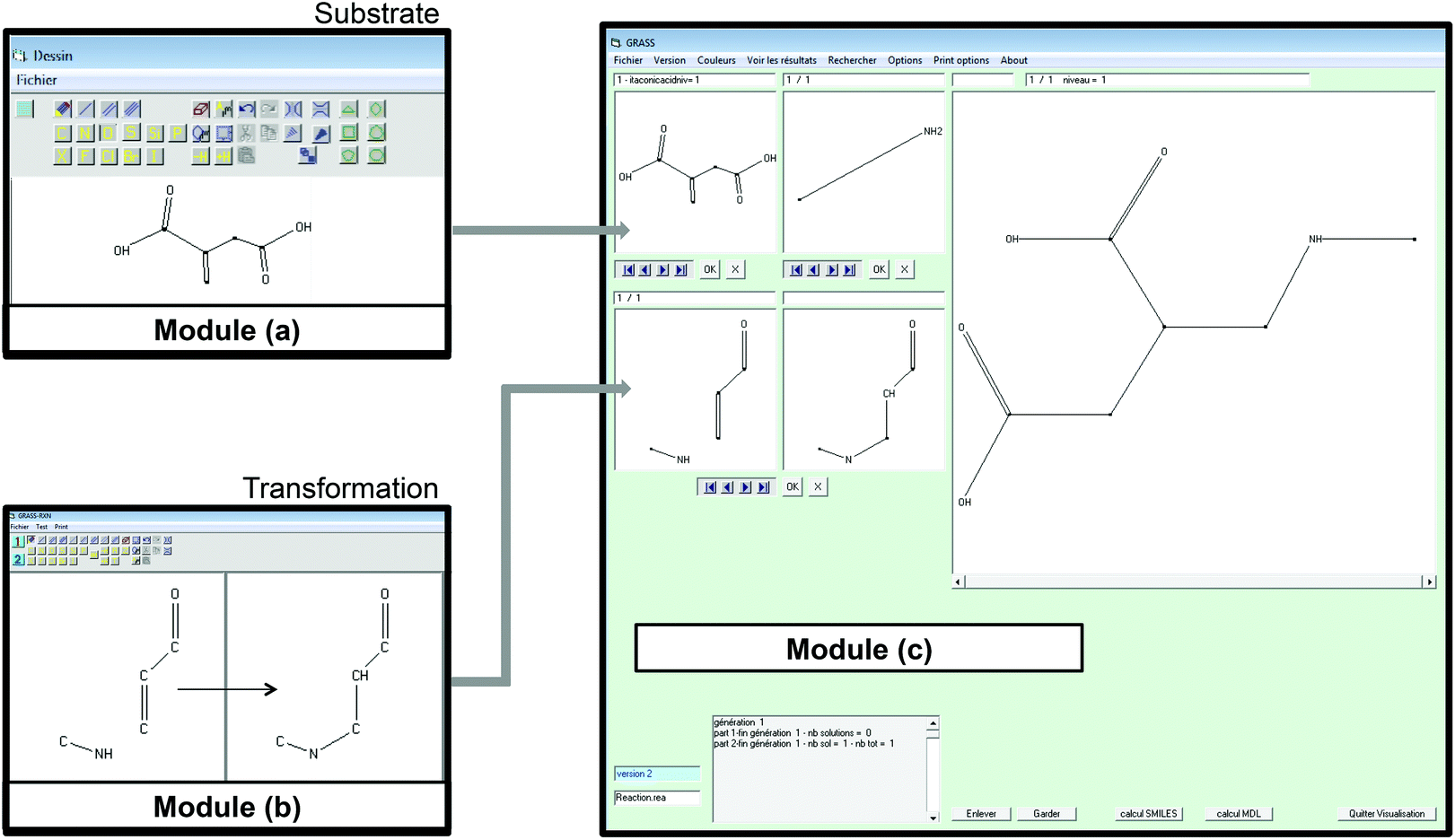 | ||
| Fig. 1 Screenshots of the graphical editors of the three modules used within GRASS. Module (a) allows drawing of the starting materials, module (b) is used to encode the selected reactions and module (c) corresponds to the combinatorial chemistry program. | ||
In module (a), substrates are drawn as 2D molecules and then saved in independent files. In module (b), reactions that take place during the process of generation are encoded via a graphical editor by drawing only the reacting functionalities. Some tests can be added, as for instance “oxidation of alcohol implies that reactant possesses a primary or secondary alcohol and not a tertiary one”. An example of reactions that can be coded via the graphical editor is shown in Fig. 1 (1,4-addition of an amine derivative). To generalize the encoded reactions, it is possible to use generic atom terms such as T that corresponds to all heteroatoms, or Z that corresponds to electro-withdrawing groups by mesomeric effect (esters, nitriles, ketones), instead of using the full description with all the involved atoms. Thus, 1,4-addition of a heteroatom on an activated carbon–carbon double bond can be encoded as:
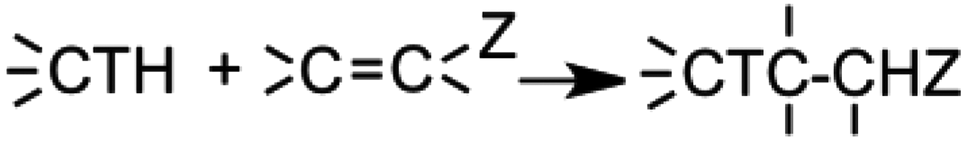
In module (c), all products that can be theoretically formed from substrates and chosen reactions are generated systematically. For instance, for the esterification of an alcohol, the combinatorial chemistry program first looks for the moieties 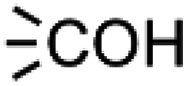 and
and 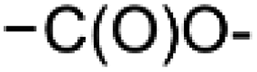 within the substrates. If these moieties are found, the program verifies that they correspond to an alcohol and to a carboxylic acid function respectively. If this step is verified, the program replaces moieties
within the substrates. If these moieties are found, the program verifies that they correspond to an alcohol and to a carboxylic acid function respectively. If this step is verified, the program replaces moieties 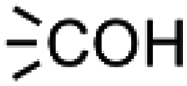 and
and 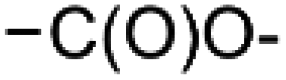 by the final moiety
by the final moiety 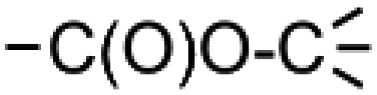 . The program repeats these procedures until all the reactions are considered and thus all the moieties have been replaced. It is important to mention that the transformations of starting substrates into products correspond to one generation. All the products generated at the first generation can then be considered as substrates and be transformed again into new compounds, called the second generation (the program only saves new structures). It is thus possible to have access to the nth generation of compounds from starting substrates, the number of generations being set by the user.
. The program repeats these procedures until all the reactions are considered and thus all the moieties have been replaced. It is important to mention that the transformations of starting substrates into products correspond to one generation. All the products generated at the first generation can then be considered as substrates and be transformed again into new compounds, called the second generation (the program only saves new structures). It is thus possible to have access to the nth generation of compounds from starting substrates, the number of generations being set by the user.
A screenshot of the man–machine interface of the combinatorial chemistry program (module (c)) is presented in Fig. 1. The two windows at the top left show the starting substrates (methylamine and itaconic acid) and the two at the bottom left show reactions. The large window on the right hand side shows virtually generated molecules. A comment window at the bottom left indicates that the analysis was performed on one generation leading to 1 structure at the first generation.
• GRASS takes into account the fact that a substrate may be a polyfunctional compound with repeated functionalities and thus generates all the possible combinations of each transformation. As an example, when the substrate has two non-equivalent carboxylic acid functions, as itaconic acid does, and when the encoded reaction is esterification, the software generates 3 compounds, which are the 2 monoesters and the one diester.
• It is possible to generate automatically 1, 2, or n generations of products and then to retrieve the synthetic pathway that has been used to virtually generate each product;
• Products virtually generated are available as SMILES or MDL files that can be used as input for next steps (such as SciFinder or Belstein bibliographic searches, or QSPR and others models predictions);
• Virtual products can be sorted out considering the number of atoms, number of rings, number of atoms in a ring, molecular weight, the presence of particular functions or chemical moieties (substructures).
Another feature that could be further implemented in GRASS is to consider the non-compatibility of all the functions of the substrate with respect to the transformation. For instance, if a substrate is composed of ketone and alkene functions, it may be difficult to carry out only the hydrogenation of the ketone without hydrogenating the alkene function.
Bibliographic search
All bibliographic searches have been carried out using the web version of SciFinder (https://scifinder.cas.org/scifinder/) as a reliable collection of substances, reactions and references. Substance information (such as CAS registry number, melting and boiling points, synthesis pathways, etc.) has been collected thanks to the “explore substance” option using the SMILES code and then the “substance details” option in which experimental data have been collected.COSMO-RS calculations
The molecular geometries are first sketched as three-dimensional structures with Arguslab (http://www.arguslab.com/, Release 2004). Conformational analysis is carried out for each structure using the COSMOconf script.45 It involves semi-empirical AM1 (program: MOPAC 7 locally modified by COSMOlogic) calculations followed by a more accurate density functional theory (DFT) treatment of the most important AM1 conformers. The Turbomole program is used to perform the DFT/COSMO geometry optimizations according to the standard quantum chemical method for COSMO-RS i.e. the DFT functional B88-PW8646 with a triple zeta valence polarized (TZVP) basis set. All further details and references regarding DFT/COSMO calculations with Turbomole can be found elsewhere.47 Subsequent to this conformational search and optimization, COSMO surfaces, “σ-profiles” and “σ-potentials” are generated via COSMOtherm (C21_0111 version).48 The relative contributions of each conformer are determined by an iterative procedure using the Boltzmann weight of the free energies of the conformers. The mathematical procedure described in ref. 17 and 49 is used to locate the virtual solvents in the previously defined classes.Results and discussion
Methodology
The methodology used to generate virtual commodity chemicals is presented schematically in Fig. 2. It is exemplified with the generation of itaconic acid-based solvents. The present section details blocks 1 to 4 that are the basis of the generation of new molecules. The software developed (1) has been named GRASS as the acronym of GeneratoR of Agro-based Sustainable Solvents. The GRASS program (1) requires three inputs: a bio-based building block, in this case itaconic acid (3) and a list of readily available co-reactants (4) that are transformed according to carefully selected transformations (2). Then, virtual products are automatically generated, which corresponds to a set of structures named 1st generation. This set can, in turn, be an input (3) to GRASS and transformed again. After 3 generations, all the sets are put together to highlight head of series of potential commodity chemicals. It is then necessary to seek among all virtual compounds those that possess appropriate properties for the intended application.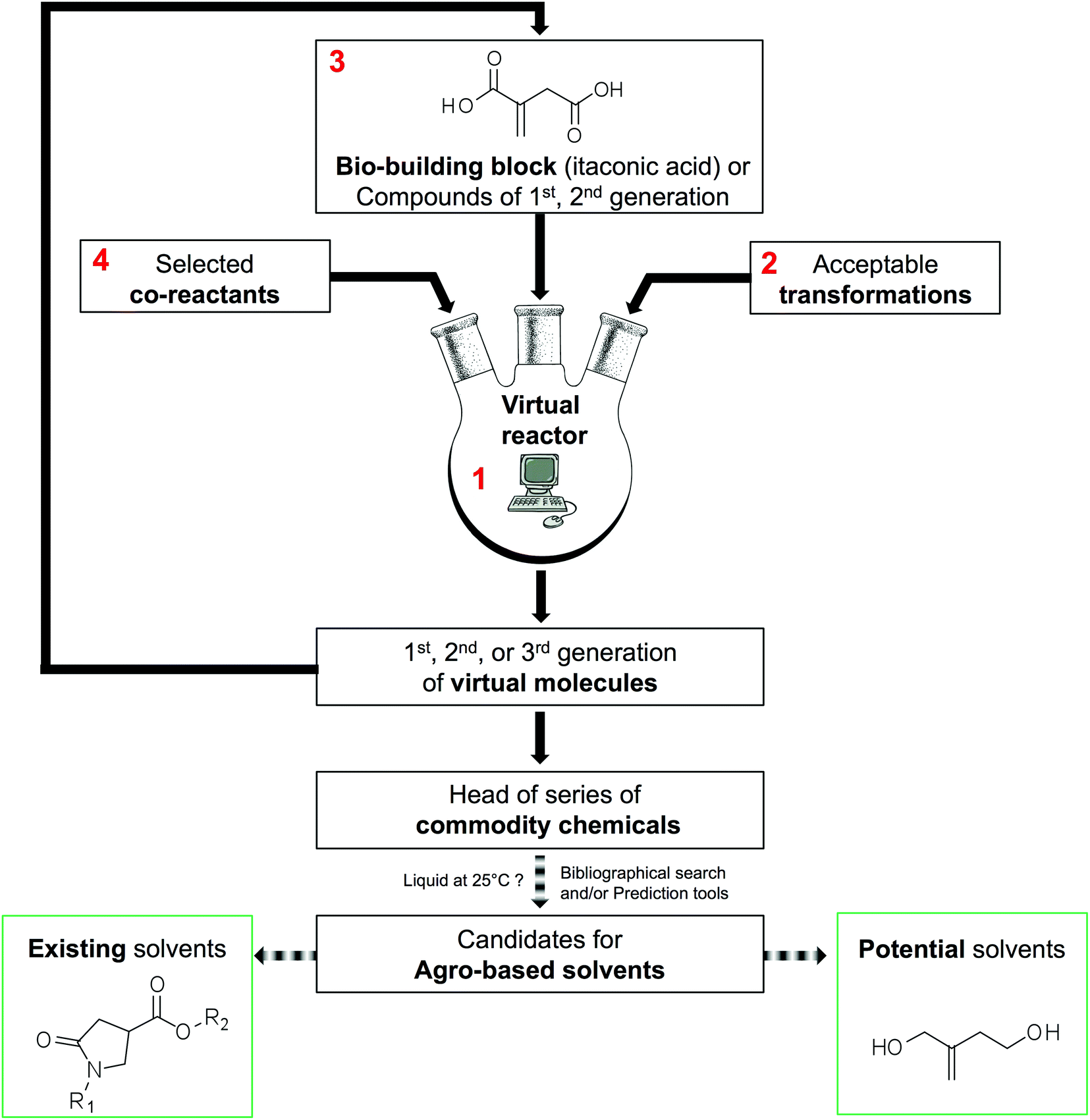 | ||
| Fig. 2 Methodology used to generate automatically virtual commodity chemicals thanks to GRASS (1), exemplification with the generation of solvents derived from itaconic acid (3) that reacts with selected co-reactants (4) according to acceptable transformations (2). After sorting the head of series that have been automatically generated through three generations, virtual agro-based solvents are split into two sets: existing solvents (validation of the methodology) and potential solvents. | ||
As we are looking for compounds that can serve as solvents, the sort criterion used is the melting temperature that must be below 25 °C so that the compound is liquid at room temperature. This sorting can be achieved by a literature survey when the generated compounds are not too numerous and already described in the literature. In contrast, when the number of candidates increases or when the compounds are not referenced in the literature, it is necessary to rely on melting point prediction models (e.g. the group contribution method of Joback and Reid50). Virtual solvents are then split into two groups: the group of known solvents that are used to test the effectiveness of the strategy and a set containing the potential solvents that might be of interest although they are not described as such in the literature.
In the last few years, the pharmaceutical industry has devoted a similar effort to analyze and classify the most frequently used reactions in medicinal chemistry. In 2005, Pfizer reviewed ca. 3000 reactions used in their research facilities since 1985 and classified them as COOH-derivative interconversion, C–N bond formation, C–O bond formation, C–C bond formation, Red-ox, salt formation and other types of reactions.51 GSK also recently classified 4800 most used reactions in 14 categories (alkylation, condensation, Pd-catalyzed coupling, etc.)52 and have outlined some of the properties that a new reaction should have in order to be used in medicinal chemistry. AstraZeneca, GSK and Pfizer joined forces to assess the processes used in pharmaceutics, from safety, environmental, and economic points of view among others.53 In 2006, they provided a complete survey of the reactions used for the preparation of drug candidate molecules54 by analyzing the syntheses of 128 compounds. This landscape of the most frequently used reactions in medicinal chemistry has already been cited more than 350 times. A similar analysis was performed in 2011 by the Cancer Research UK Drug Discovery.55 In 2007, a roundtable gathering of the main actors of the pharmaceutical industry allowed identifying the key research areas for the development of green chemistry practices.56 This brainstorming allowed analyzing the reactions currently used, regarding their frequency of use, waste generation and process hazard. The work of Vasilevich et al. in 2012 on the analysis of reactions used in Natural Products chemistry should also be pointed out, where a toolbox of the main categories of chemical reactions used is also provided.57
On the other hand, to the best of our knowledge, no similar review is available in the literature for the synthesis of commodity chemicals, which are undifferentiated relatively simple molecules produced on a massive scale and at the lowest cost. The reason is probably that the chemistry in this case is much less complicated and implies less sophisticated reactions. It has, however, to comply with constraints of (i) costs (the price of a conventional solvent rarely exceeds 5€ per kg), (ii) large-scale synthesis in a limited number of steps, (iii) availability of the raw materials, and (iv) establishment of safe and cost effective procedures with high yields and limited by-products.
To provide a short-list of acceptable transformations, we have first listed as exhaustively as possible the chemical reactions used on large scale by reviewing literature dealing with industrial chemistry of commodity chemicals58 and the preparation of the classical organic solvents.59 As most of these reactions start from basic petroleum-based substrates, they take place under specific conditions (temperature, pressure, and gas phase) that are not always suitable for the bio-based substrates that GRASS is supposed to consider.
For instance, the industrial synthesis of methanal goes through the oxidative dehydrogenation of methanol in the presence of Ag or Cu catalysts at about 700 °C,58 which are reaction conditions that will probably not be applicable to a more fragile starting substrate. The generic transformation “oxidation of alcohol to aldehyde”, however, can be envisaged since it can be performed under less stringent conditions. To enlarge the scope of transformations to be implemented in GRASS, reactions more specific to more sensitive polyfunctional synthons were considered through the work of Corma et al.19 that gives a comprehensive review of the chemical transformations of bio-based building blocks. Finally, the most used reactions in the pharmaceutical industry presented in the work of Carey et al.54 were also considered. Altogether, a list of 493 reactions was established, 224 concerning commodity chemicals, 153 more sensitive polyfunctional synthons and 116 pharmaceuticals.
This list of 493 chemical reactions was then reduced to a set of 104 generic transformations, indicating the two by two reacting starting functions and the final function created. This list is presented in Table 1 with the most representative industrial examples. This drastic set reduction is explained by the fact that some of the reviewed reactions are redundant and should be counted only once as a generic transformation. Redundancy can be linked to the same reaction being applied to various substrates: e.g. carboxylic esters formations have been reviewed several times (considering different alcohols reacting with various carboxylic acids, such as acetic acid, lauric acid, etc.) and has been considered once as the single generic esterification transformation. Various experimental conditions can also lead to redundancies within the reviewed reactions. For instance, oxidation of isopropanol into acetone can be performed following, at least, three procedures: at 250–270 °C and 25–30 bar over a special supported copper catalyst (Deutsche Texaco), at about 150 °C and atmospheric pressure in a high-boiling solvent using RANEY® nickel or copper chromite (IFP), at 90–140 °C and 3–4 bar with a small amount of H2O2 as an initiator (Shell and Du Pont).58 In that case, this oxidation has been counted once as a generic transformation from an alcohol (primary or secondary one) into a ketone. Dihydroxylation of alkene is another example of such redundancy since it can be performed according to two procedures involving either potassium permanganate or osmium oxide coupled with hydrogen peroxide. In that case, only one generic transformation of alkene into the dihydroxylated derivative in oxidative conditions has been considered.
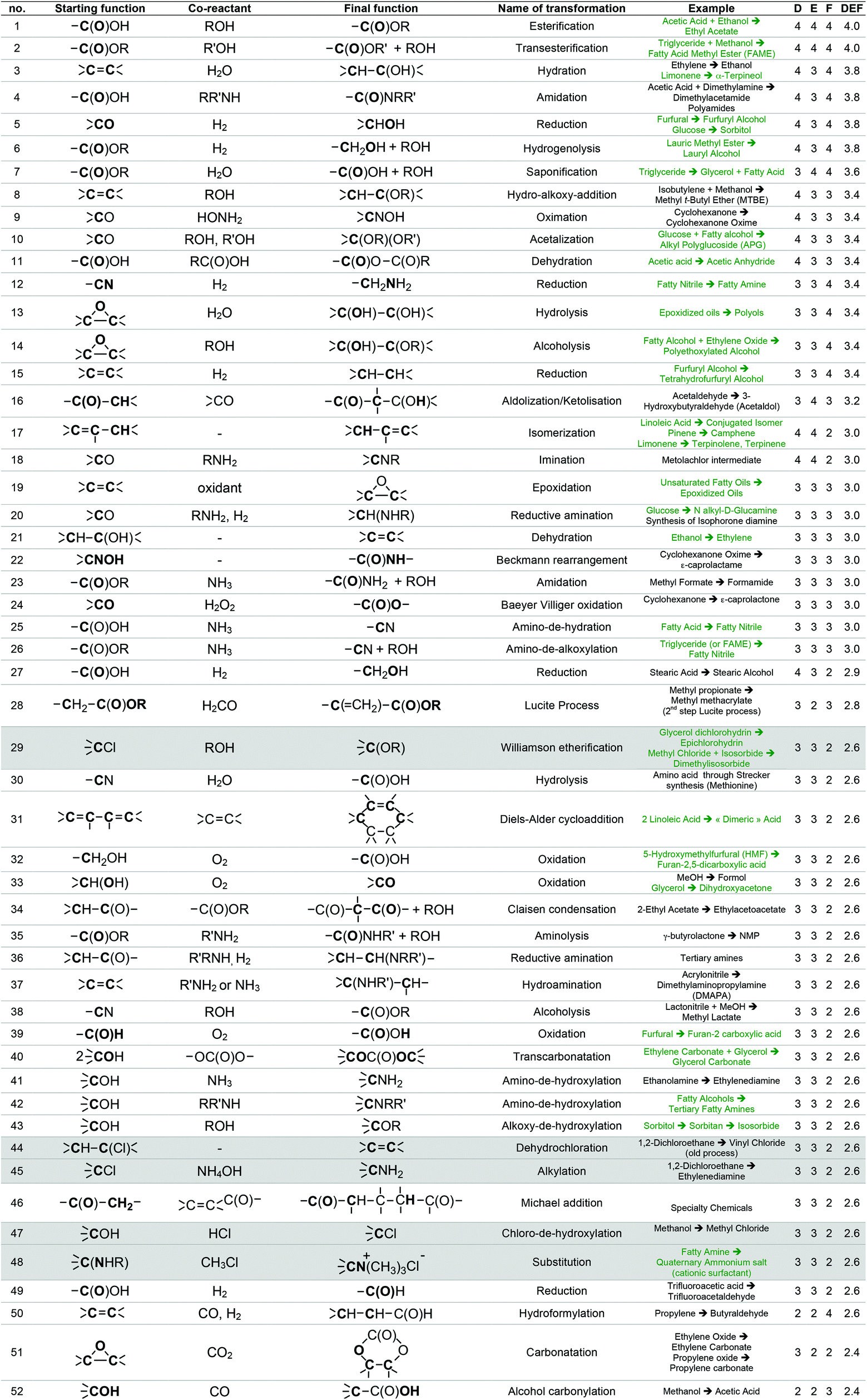
|
|
The list of 104 generic transformations was then further shortened by considering the following three criteria:
1. Durability (D), which can be evaluated by screening the 12 principles of Anastas,6 in particular the type of reagents implied (environment, safety and health profile), the atom economy60 of the transformation, the amount of waste generated (E-factor)61 and the innocuousness of the reaction.
2. Easiness (E), i.e. conditions of temperature, pressure, process required, which have to be convenient with the large-scale synthesis of the commodity chemicals GRASS is supposed to generate.
3. Frequency of use (F) at the industrial scale today, which accounts for the workability of the transformation. A transformation that is widely used at industrial scale leads, most of the time, to high volumes of consumption. For instance, esterification from a carboxylic acid and an alcohol is carried out at a world scale of more than 500 kt year−1 (synthesis of ethyl acetate, butyl acetate, butyl acrylate, etc.), transesterification of an ester with an alcohol implies several Mt year−1 in the biodiesel industry, and amide formation from a carboxylic acid and an amine is also employed at the scale of several Mt year−1 in the polyamide industry. The fact that a transformation is versatile, i.e. adaptable to a various range of substrates is taken into account within the F criterion. A non-versatile transformation, even though it is efficient for a given substrate, will be less frequently used at large scale. In such case, F criteria will be decreased.
To illustrate the durability (D) criteria, two pathways for the preparation of the diesters of ethyl-succinic and methyl-glutaric are presented in Fig. 3. For the sake of clarity, only the methyl-glutaric acid derivative is presented. These diesters are attracting interest because they are eco-friendly solvents resulting from the valorization of the dinitrile derivatives obtained as by-products of the preparation of adiponitrile, a key-intermediate of the production of hexamethylene diamine or caprolactam for the polyamide industry. Two main pathways have been described in the literature.62–64 The first goes through the formation of the diacids that are further esterified with an alcohol (methanol in the example). The second takes place in the gas phase and first consists in the formation of the imide derivative that is then reacted with methanol to form the desired diesters. The two routes lead to similar global yields. However, the first path generates stoichiometric amounts of salts even if ammonia (by-product) is recycled and it requires extraction procedures to recover products, which leads to a theoretical E-factor higher than 0.8 kg salt per kg of the product. The second is preferred since the only by-product is ammonia that can be recovered and valorized. The theoretical E-factor for path 2 is almost zero if ammonia is fully recycled. Atom economy is also more favorable for pathway 2 since it can reach 100% if ammonia is fully recycled while it is only of 79% for pathway 1. Thus, the D criterion of pathway 1 is lower than the D criterion of pathway 2.
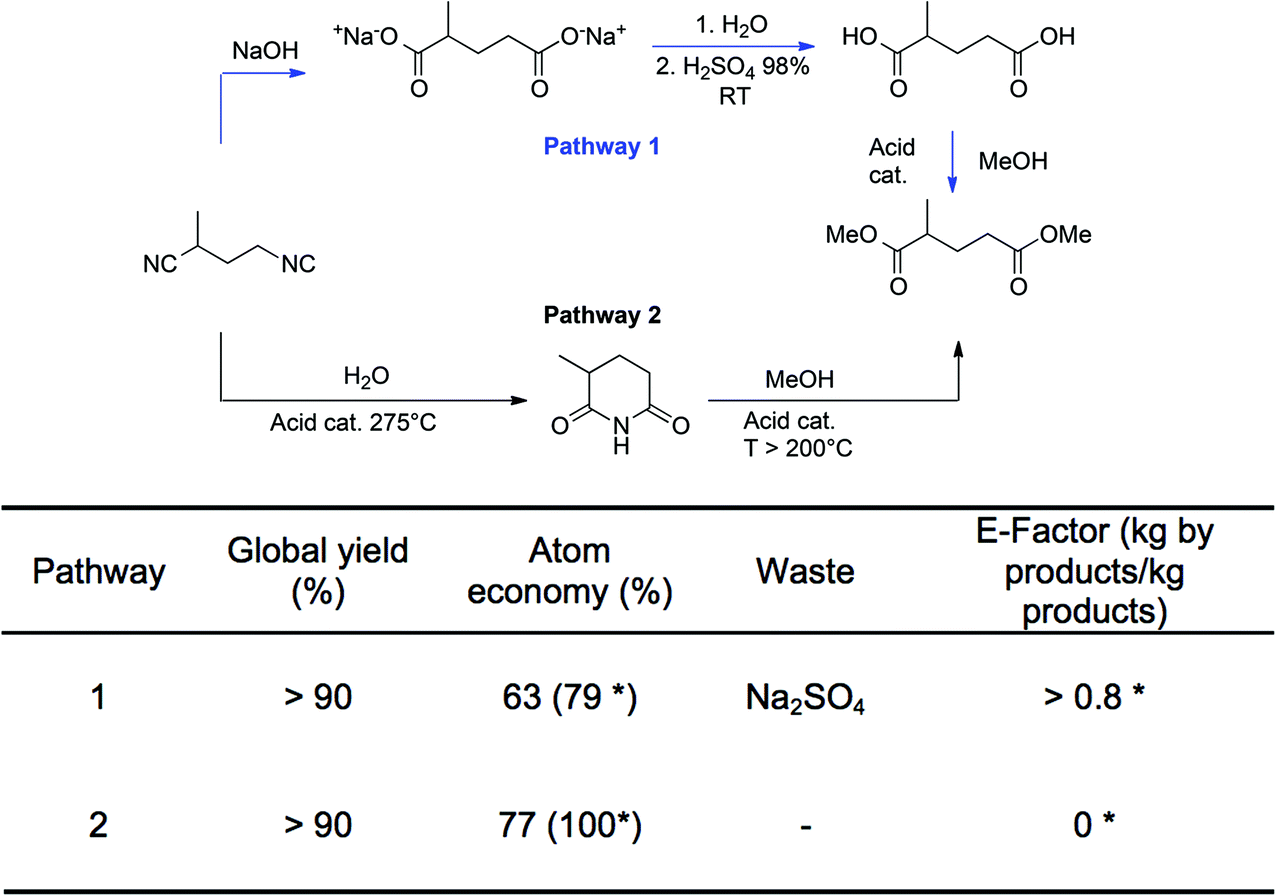 | ||
| Fig. 3 The two possible pathways to the dimethylester of methyl glutaric acid starting from the methyl-2 glutaronitrile, and their yields, their theoretical atom economies and their E-factors. (*Values of atom economy in brackets and E-factors have been calculated when NH3 is recycled (to HCN process for example).) | ||
The same considerations could be applied to caprolactam65 or methylmethacrylate66 productions, for which new routes allowed a strong reduction of salt generation.
With the same considerations, we gave a 0 to 4 ranking (0: non-acceptable; 4: very good) to each criterion for the 104 transformations. Then, a DEF mark – deriving from the notion of desirability function67 – was given to each transformation as presented below:
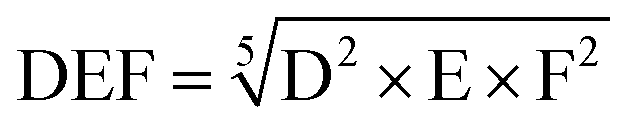
A higher weight was given to the D and F criteria that were considered as more consistently evaluated and of greater importance for the preparation of commodity chemicals. If a transformation is not acceptable, a score penalty (0) is applied, which makes the DEF plummets and excludes the reaction (see Table 1 for details). The transformations having a DEF higher than 2 have been held up to form the “TOP transformation list”.
7 transformations involving halogenated compounds (indicated in grey in Table 1) have been deliberately excluded from the “TOP transformation list” even though their DEFs are higher than 2. Chlorinated derivatives are unlikely to meet the requirements of green solvents since most of them are listed among HAP (e.g. dichloromethane) or CMR (e.g. chloroform) substances according to US and/or European legislations. As regards the use of halogenated compounds as starting materials for synthesis, they are often undesirable despite their low cost and excellent reactivity, because of handling problems or side-products generation (salts) and, at the industrial scale, alternative processes are usually preferred. That is why ether formation by a reaction between an alcohol and a halogenated derivative in Williamson's conditions (transformation 29) has not been retained within the “TOP transformation list” even if this is the conventional pathway used at the laboratory scale. At the industrial scale, simple ethers as diethyl ether are formed, starting from the corresponding alcohols under heterogeneous catalysis (transformation 43). Recently, Lemaire and Co. described eco-friendly accesses to ethers via the Pd-catalyzed reductive alkylation of alcohols starting from an aldehyde, a ketone and even an acid or an ester.68–71 However, as these reactions have not been up-scaled at the industrial level yet, they have not been included in the list. It is worth mentioning at this point that this “TOP transformation list” is proposed as a snapshot of the transformations that currently meet the specifications in terms of DEF criteria. It is quite possible, indeed desirable that this list undergoes improvements based on scientific advances.
The “TOP transformation list” is thus made up of the 53 first transformations that are presented in Table 1, above the bold line. The transformations listed are mainly functional conversions.
The strength of this approach lies in the fact that the choice of transformations is based on the industrial reality for the chosen application, i.e. commodity chemicals, which implies low cost and simple syntheses. It also takes into account reactions applicable to polyfunctional substrates as the bio-based building blocks. There are, however, some intrinsic limitations, the main one being that relative reactivities are not taken into account. Also, the transformations selected do not include reactions specific to aromatic cores and specific to alkynes. They could be implemented to the set if needed.
Even if quasi-all the chemical processes today are based on the transformation of starting materials from oil (plant-derived materials represented altogether only 5% of industry feedstock inputs in 2004),72 it is expected that by 2050, a considerable quantity of base chemicals, at least 30%, will be produced from biomass.72 The “biorefinery” concept, i.e. the transposition of the petrorefinery scheme for the processing of biomass, is gaining importance and a spectrum of products is expected to be obtained from the biomass feedstock in the forthcoming years.
Fig. 4 shows a schematic route from biomass to defined chemicals. The main biomass sources providing incomes for biorefineries are forestry, dedicated crops and vegetable residues (cobs, straw, sugar cane bagasse, etc.). The exploitation of aquatic biomass (algae) is also a promising source of renewable carbon.
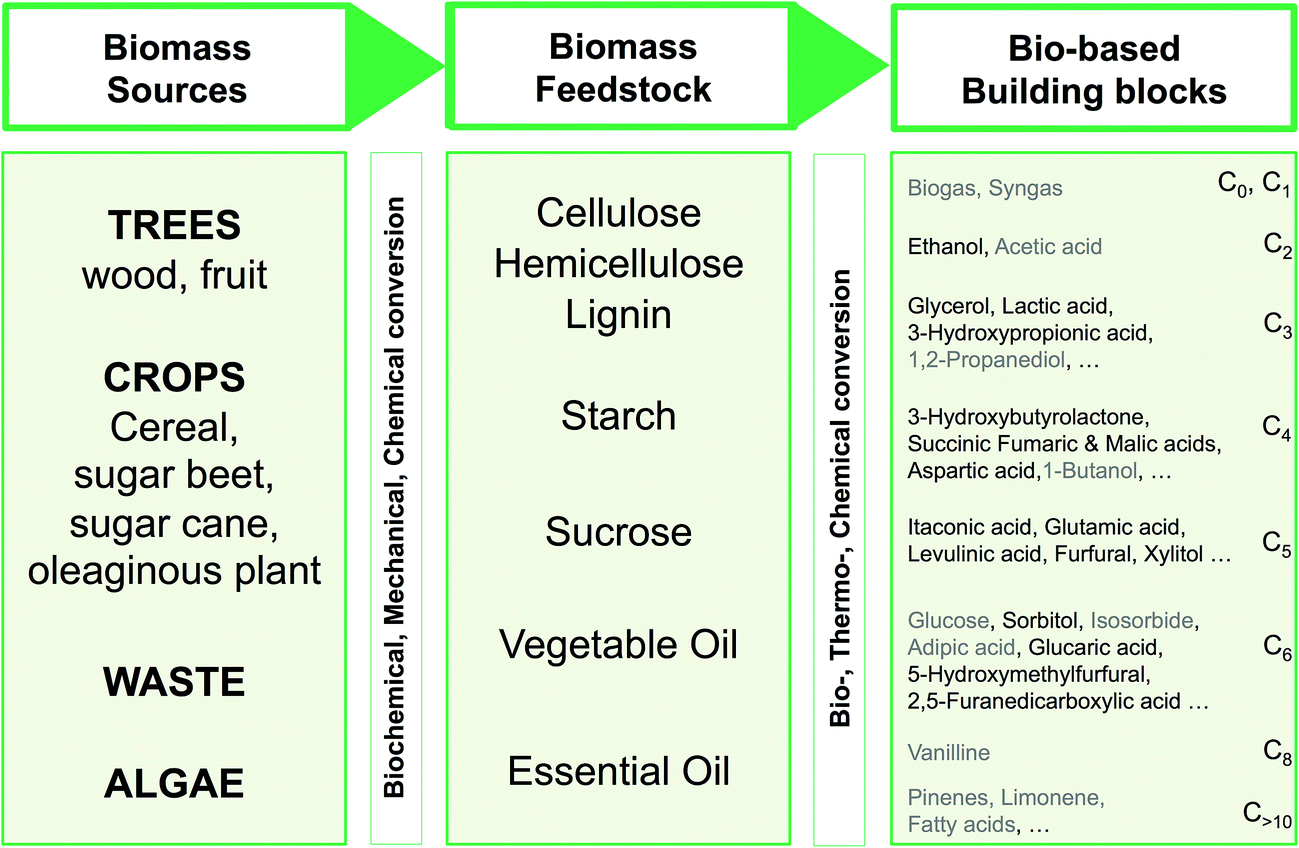 | ||
| Fig. 4 Bio-based building blocks (ordered by ascending number of carbon atoms) obtainable from the biomass feedstock, through biochemical, mechanical or chemical conversions of various sources. Recommended building blocks (US Dept. of Energy and Bozell and Petersen)18,74 have been indicated in black. | ||
From a chemical point of view, the main biomass feedstocks are polysaccharides. Cellulose (50% of total biomass) and hemicellulose (24%) come from the ligno-cellulosic biomass (wood, residues, some crops such as switchgrass) and can be processed more or less easily, chemically or biochemically, to simple sugars and derivatives; lignin, the third polymeric component of ligno-cellulosic biomass, is a phenolic polymer that accounts for 20% of the total biomass but is not at the moment chemically exploited. Starch is a polysaccharide produced by many plants for energy storage. It is easily hydrolyzed – chemically or enzymatically – to glucose that is then processed to a range of well-defined chemicals. Starch, however, represents only 1% of the total biomass on the earth. Sucrose and vegetable oils are extracted from dedicated crops and can be used as starting materials for chemistry even if they each represent only 0.1% of the total biomass worldwide. Other components (proteins and essential oils) can also be viewed as potential starting materials for chemistry. Three biomass conversion strategies are usually described: (i) conversion of biomass to a mixture of molecules used without separation, (ii) one-step conversion of biopolymers to introduce new functionalities, and (iii) conversion of biomass into platform molecules (building blocks) that can be subsequently transformed.73 In this work, only the latter strategy (iii) will be considered.
The chemical or biochemical transformation of this biomass feedstock may lead to a spectrum of bio-based building blocks available for chemistry. As the main sources are polysaccharides, the available building blocks are currently sugars and sugar derivatives (polyols, organic acids obtained by fermentation). In 2004, the US Department of Energy released a list of “Top 12 chemicals” obtained from the ligno-cellulosic and starch feedstocks that could be employed as platform molecules for the synthesis of bio-based chemicals.18 These top 12 were considered as the most promising in a list of 30 that were considered as building-blocks among 300 molecules initially screened. In 2010, Bozell and Petersen74 established a revisited list of top chemical opportunities, obtained from the carbohydrate platform. The building blocks enlightened by these publications are shown in black in Fig. 4 and are classified according to the number of carbon atoms.
To illustrate the methodology proposed to generate commodity chemicals from biomass, itaconic acid, which is an unsaturated carboxylic diacid obtained by the fermentation of sugars, has been chosen. Any available bio-based building block, however, could be used as a starting synthon for GRASS.
| Co-reactant | Selected compound | Obtainable from biomass |
|---|---|---|
| Alcohol |
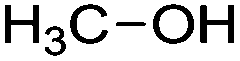
|
Sugar fermentation |
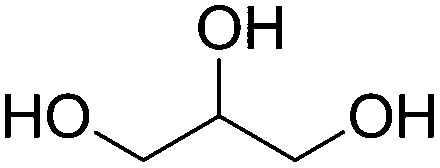
|
Triglyceride conversion | |
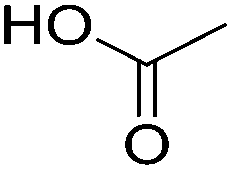
| Carboxylic acid |
|
Ethanol oxidation |
| Amine |
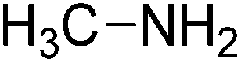
|
No process currently described |
| Epoxide |

|
Ethylene oxidation |
| Ketone |
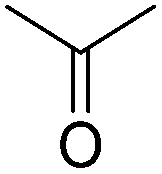
|
Starch fermentation |
| Alkene |
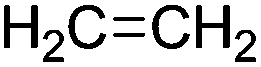
|
Ethanol dehydration |
Transformation of itaconic acid into virtual solvents
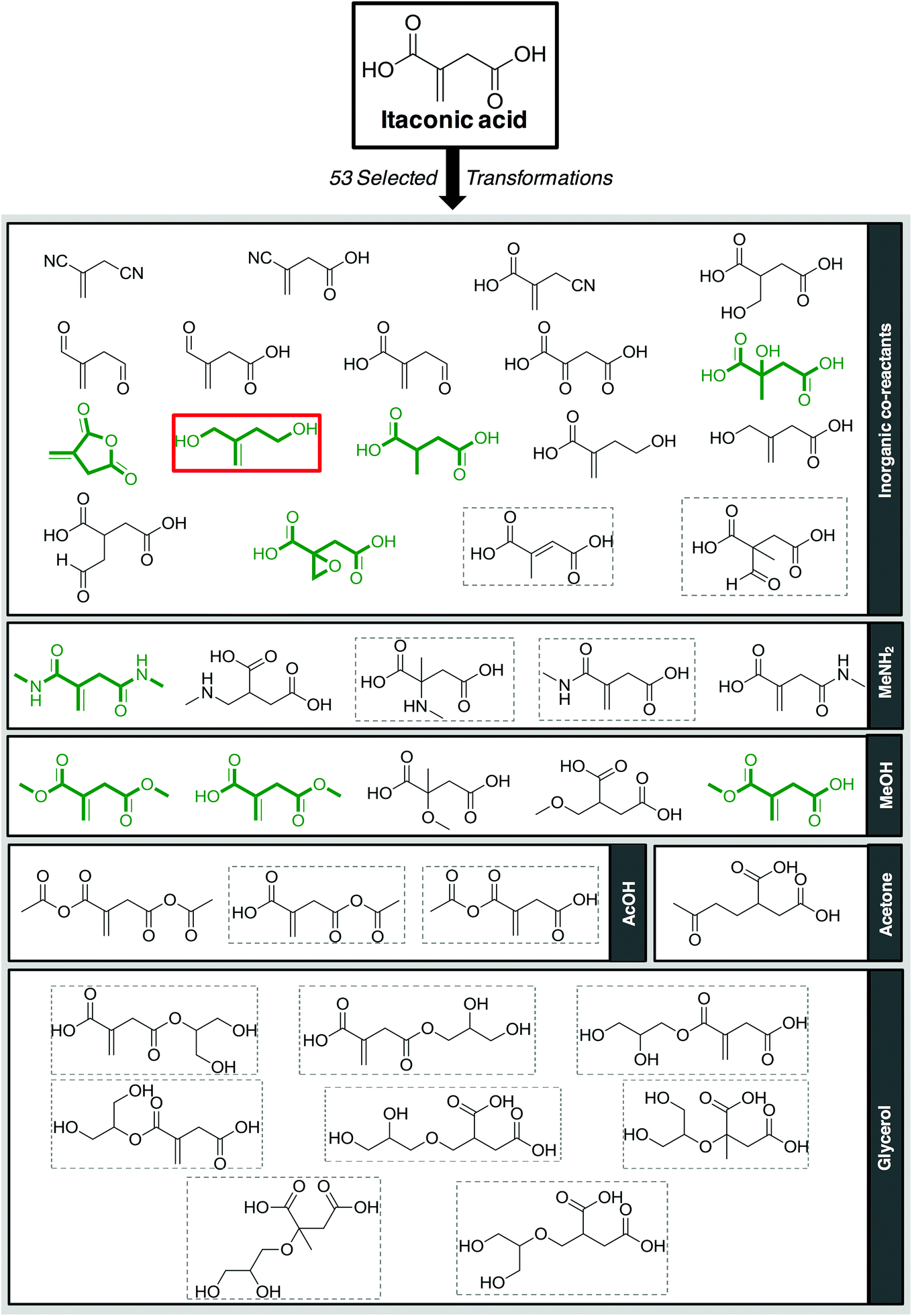 | ||
| Fig. 5 40 First generation compounds resulting from the 53 selected transformations applied to itaconic acid and co-reactants. Compounds without CAS registry number are surrounded by grey dotted line (14); compounds described in the literature as itaconic acid derivatives are drawn in bold green (9) and the only compound that is liquid at room temperature is surrounded in red (1). | ||
A bibliographic search has been carried out for each compound in order to find information about CAS registry number, experimental melting and boiling points, and synthetic pathways. 14 compounds out of the 40 derivatives do not have a CAS registry number and are surrounded by a dotted line. They could be of potential interest depending on their physico-chemical properties. Most of these 14 compounds are in fact mono-substituted derivatives and we can guess that they have not been synthesized yet because of selectivity issues. Among the compounds that do have a CAS registry number, only 9 (drawn in green) are indeed described as itaconic acid derivatives, the others being obtained from other starting materials. Here again, it does not mean that they could not be obtained from itaconic acid, but the synthetic pathways have not been developed so far, perhaps because itaconic acid has been available at a non-negligible scale only recently. The sole compound that is known to be liquid at room temperature is framed in red.75 It is 2-methylenebutan-1,4-diol, for which an experimental melting point of 8 °C is indicated by SciFinder.
Within this first generation, only 2-methylenebutan-1,4-diol (2-MBD) thus stands out for the chosen application because it is liquid at room temperature and has already been experimentally obtained from itaconic acid. To the best of our knowledge, no applications as solvent are claimed for 2-MBD and it could be of interest to get some information on its solvent properties. The COSMO-RS approach16 has already been used to describe and to classify classical organic and green solvents17,49in silico, without any prerequisite experimental data, only from the calculation of the so-called “σ-potentials” that can be considered as solvent footprints. The σ-potential of 2-MBD is represented in black in Fig. 6 and the limit σ-potentials of the closest classical organic solvents are superimposed in grey. 2-MBD exhibits an electron pair donor ability (red area on the COSMOsurface) as well as a hydrogen bond donor ability (blue area on the COSMOsurface) due to its two alcohol functions. The previously described classification procedure17 locates 2-MBD within the polar protic solvents family, among classical organic solvents such as 2-aminoethanol, ethylene glycol, 1,3-propanediol, and furfuryl alcohol. Thus, 2-MBD could a priori be an interesting alternative to replace these solvents.
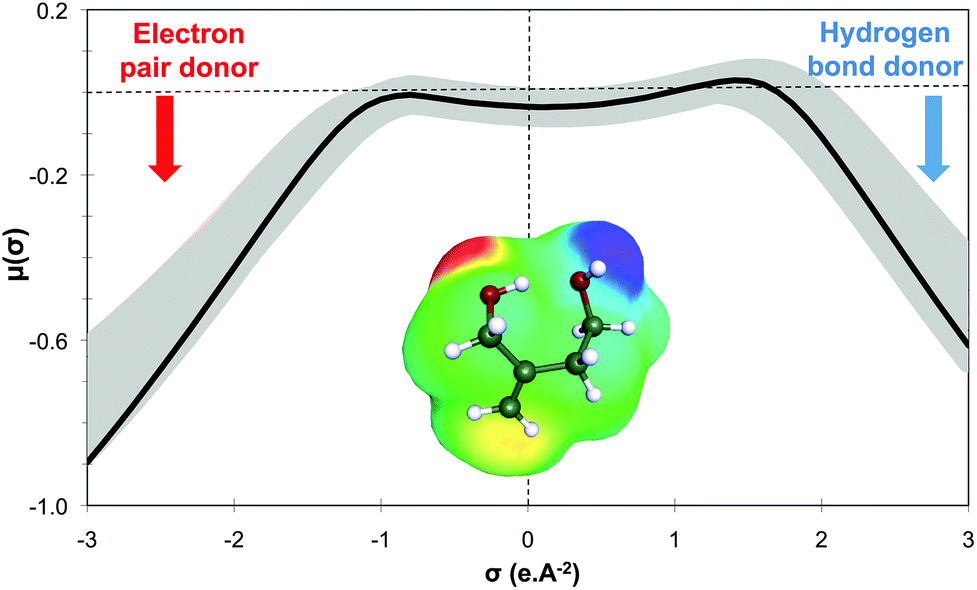 | ||
| Fig. 6 Sigma-potential μ(σ) (kcal mol−1 A−2) of 2-methylenebutan-1,4-diol (black line) obtained thanks to the COSMO-RS approach and compared to those of classical polar protic solvents (in grey). The COSMOsurface that has been used is also represented. | ||
For this compound, it is also interesting to compare the virtual access from itaconic acid proposed by GRASS and the experimental preparation described in the literature (Fig. 7). Clearly, it would be experimentally difficult to reduce selectively the acid functions without reducing the methylene group of itaconic acid, as suggested by GRASS. This example highlights the differences existing between GRASS and experimental76 pathways: here, one step within GRASS corresponds to two steps experimentally. GRASS allows generating compounds and gives directions concerning the synthetic pathway, but knowledge of chemistry is needed to carry out experimental synthesis properly. However, even though the synthesis has already been described in the literature, this pathway is still not acceptable for the preparation of commodity chemicals considering experimental conditions of the second step (AlH3, −30 °C).
 | ||
| Fig. 7 Two synthetic pathways to obtain 2-methylenebutan-1,4-diol (2-MBD): in dotted line, the “GRASS” pathway that involves transformation no. 27 (hydrogenation of a carboxylic acid into an alcohol) and the two-step experimental procedure described in the literature ((a) 2-propanol, H2SO4, reflux, 30 h, yield 98% and (b) AlH3, Et2O, −30 °C, 5 min, yield 98%).76 | ||
Because the first generation is only made up of 40 compounds, further generations have been considered in order to obtain a larger compounds set.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds respectively), which impedes a human structure exploration. Within these lists however, it was possible to retrieve the five molecular structures that have already been reported in the literature as solvents derived from itaconic acid. These compounds are indicated in bold in Fig. 8 and their GRASS and experimental synthetic pathways are described.
000 compounds respectively), which impedes a human structure exploration. Within these lists however, it was possible to retrieve the five molecular structures that have already been reported in the literature as solvents derived from itaconic acid. These compounds are indicated in bold in Fig. 8 and their GRASS and experimental synthetic pathways are described.
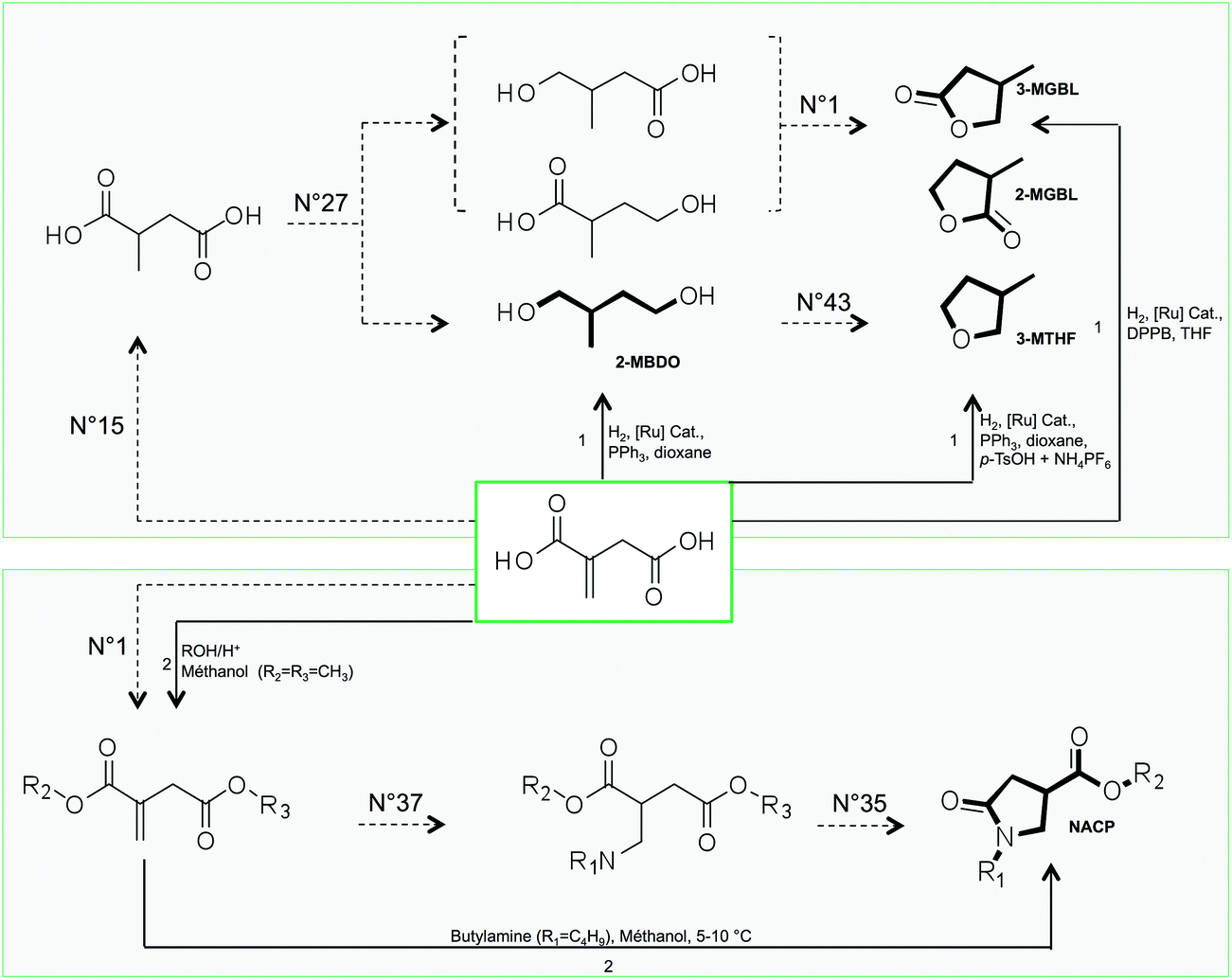 | ||
| Fig. 8 GRASS (dotted arrow) and experimental (black arrow) pathways to obtain compounds that are claimed as solvents derived from itaconic acid (bold compounds are called 2-MBDO, 2-MGBL, 3-MGBL, 3-MTHF and NACP). The numbers above dotted arrows refer to the number of TOP list transformations within Table 1 while numbers above black arrows refer to experimental conditions described in the literature (1: Geilen et al.77; 2: Wu et al.78). | ||
In 2010, Geilen et al.77 described selective and flexible access to levulinic and itaconic acid derivatives by using a multifunctional catalytic system based on ruthenium. They showed that itaconic acid could be selectively transformed into lactones (2-MGBL and 3-MGBL), 2-methylbutanediol (2-MBDO) or 3-methyltetrahydrofuran (3-MTHF), by tuning the nature of the catalyst. The lactones can be obtained with 93% yield by using the ruthenium catalyst based on the bidentate ligand DPPB (1,4-diphenylphosphinobutane).
The use of tridentate ligand triphos(1,1,1-tris(diphenylphosphinomethyl)-ethane) leads to a more active catalyst, which provides diol 2-MBDO in 93% yield. The cyclisation of diol 2-MBDO to 3-methyltetrahydrofuran (3-MTHF) is achieved by the addition of acidic species as NH4PF6 and p-toluenesulfonic acid (p-TsOH) with a global yield of 97% from itaconic acid. Of course, these direct catalytic accesses to itaconic acid derivatives cannot be provided by GRASS. However, the compounds are indeed virtually obtained after two or three virtual transformation steps, as described in Fig. 8. It is also worth pointing out that the virtual route that GRASS takes to access lactones 3-MGBL and 2-MGBL (no. 15–no. 27–no. 1) corresponds to the mechanism proposed by the authors, even if the intermediates were not isolated. On the other hand, GRASS proposes the direct reduction of 2-methyl succinic acid into 2-methylbutanediol (2-MBDO), which does not seem to be experimentally feasible as such but requires esterification before reduction.
Another class of itaconic acid-based solvents are the N-alkyl-4-carboxypyrrolidinone esters (NACP), for which synthesis from itaconic acid was described by Wu et al. as early as in 1961.78 The experimental synthesis is a two-step procedure (esterification followed by a one-pot amine addition and cyclic amide formation), while the GRASS pathway decomposes it into three virtual transformations. It should be noted that both experimentally and virtually, the first two steps can be switched and still lead to the obtention of NACP (no. 1–no. 37–no. 35). Tools have been developed to detect the most promising reaction pathways considering constraints (such as mass balance criteria, energy criteria, and cost criteria) and could be very powerful since it has already been described for the production of 3-MTHF from itaconic acid.79
The GRASS software is thus able to generate these molecular structures of existing solvents, or at least the corresponding head of series, which is a strong argument to validate the proposed methodology. Using N-alkyl-4-carboxypyrrolidinone ester (NACP) derivatives as solvents is particularly interesting since this solvent family has already been described as cosmetically acceptable,80i.e. harmless to human health, which is a very convincing argument for claiming these derivatives as “green”. Moreover, NACP structures are particularly interesting since they greatly differ from those of green solvents that are currently on the market.17 The σ-potential of methyl 1-butyl-5-oxopyrrolidine-3-carboxylate (R1: C4H9 and R2: CH3) is shown in Fig. 9. This solvent belongs to the weak electron pair donor base family since it exhibits an electron pair donor ability (red area on the COSMOsurface) while it does not exhibit any hydrogen bond donor ability. Current green solvents are scarcely represented within this family, which is essentially made up of classical organic solvents such as DMF, N,N-dimethylacetamide or NMP (for which σ-potentials are represented in grey in Fig. 9). This structure of green solvent is thus of the utmost interest for the substitution of banned solvents with specific properties such as NMP or DMF.
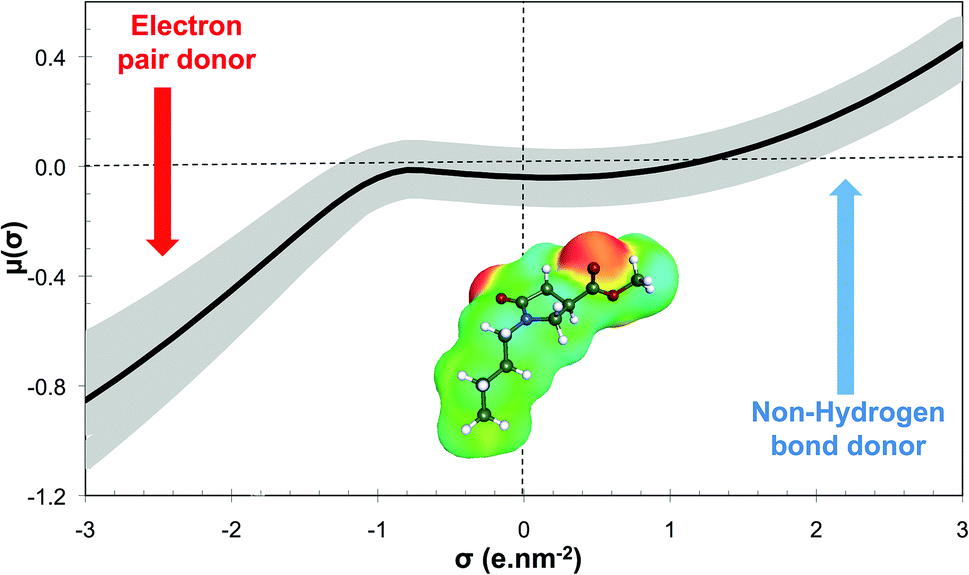 | ||
| Fig. 9 Sigma-potential μ(σ) (kcal mol−1 A−2) obtained thanks to COSMO-RS approach of NACP (in black) and compared to those of classical weak electron pair donor bases (in grey). COSMOsurface that has been used within COSMOtherm calculations is also represented. | ||
Finally, among the huge number of virtual compounds generated, it should be possible to point out potential new structures with interesting properties by combining this computer-assisted organic synthesis program to properties prediction models (e.g., rough sorting using group contribution, QSAR or UNIFAC models, then more accurate predictions using COSMO-RS approach). This approach will be described in forthcoming papers.
Conclusion
In this work, a computer-assisted organic synthesis program has been developed to generate commodity chemicals from platform molecules of the biomass feedstock. With this end in view, a list of bio-based building blocks and selected co-reactants is proposed, based on literature and economical surveys. The core of the methodology lies in the careful selection of 53 chemical transformations that can be applied to these substrates, relying on the review of commodity and fine chemistry literature evaluated through criteria of durability and workability.The program has been used in this work to generate virtual derivatives of itaconic acid that can be used as solvents. All the structures described as such in the literature have been encountered within the list of compounds generated, which validates the methodology. The program also proposes an extended list of structures that are not described yet as solvents, but should be screened by looking at their predicted physico-chemical properties. This subsequent step will be described in forthcoming papers.
Acknowledgements
This work was performed within the framework of the InBioSynSolv project supported by the French ANR CP2D program (no. ANR-CP2D-2009-08).References
- N. A. Owen, O. R. Inderwildi and D. A. King, Energy Policy, 2010, 38, 4743–4749 CrossRef.
- K. Salmenkivi and H. Jaaskelainen, ICIS, 2013 Search PubMed.
- T. L. Brewer, Bridges Trade BioRes Rev., 2013, 7 Search PubMed.
- D. Forster and J. Perks, Climate impact of potential shale gas production in the EU, Oxford, 2012 Search PubMed.
- M. Eissen, J. O. Metzger, E. Schmidt and U. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414–436 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- Responsible Care Global Charter (http://www.icca-chem.org/Global/Initiatives/RC_GlobalCharter2006%5b1%5d.pdf), Brussel, 2006.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- P. T. Anastas and J. B. Zimmerman, Environ. Sci. Technol., 2003, 37, 94A–101A CrossRef PubMed.
- R. Höfer and M. Selig, in Polymer Science: A Comprehensive Reference, ed. M. Krzysztof and M. Martin, Elsevier, Amsterdam, 2012, pp. 5–14 Search PubMed.
- D. J. Adams, P. J. Dyson and S. J. Tavener, Chemistry In Alternative Reaction Media, John Wiley & Sons, Chichester, UK, 2004 Search PubMed.
- K. Mikami, Green Reaction Media in Organic Synthesis, Blackwell, Oxford, UK, 2005 Search PubMed.
- C. Estévez, in Sustainable Solutions for Modern Economies, ed. R. Höfer, RSC, Cambridge, UK, 2009, ch. 10, pp. 407–423 Search PubMed.
- F. M. Kerton, Alternative Solvents for Green Chemistry, RSC, Cambridge, UK, 2009 Search PubMed.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- A. Klamt, COSMO-RS: From Quantum Chemistry to Fluid Phase Thermodynamics and Drug Design, Elsevier Science, Amsterdam, Netherlands, 2005 Search PubMed.
- L. Moity, M. Durand, A. Benazzouz, C. Pierlot, V. Molinier and J.-M. Aubry, Green Chem., 2012, 14, 1132–1145 RSC.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Report DOE/GO-102004-1992, 2004 Search PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- M. H. Todd, Chem. Soc. Rev., 2005, 34, 247–266 RSC.
- R. Barone and M. Chanon, in Encyclopedia of Computational Chemistry, ed. P. Ragué Schleyer, John Wiley & Sons, Chichester, UK, 1998, vol. 5, pp. 2931–2948 Search PubMed.
- R. Barone and M. Chanon, in Handbook of Chemoinformatics, ed. J. Gasteiger, Wiley-VCH Verlag, Weinheim, Germany, 2008, pp. 1428–1456 Search PubMed.
- A. Cook, A. P. Johnson, J. Law, M. Mirzazadeh, O. Ravitz and A. Simon, WIREs Comput. Mol. Sci., 2012, 2, 79–107 CrossRef CAS.
- E. J. Corey and T. Wipke, Science, 1969, 166, 178–192 CAS.
- D. A. Pensak and E. J. Corey, in Computer-Assisted Organic Synthesis, ACS, Washington, US, 1977, vol. 61, ch. 1, pp. 1–32 Search PubMed.
- W. T. Wipke, G. I. Ouchi and S. Krishnan, Artif. Intell., 1978, 11, 173–193 CrossRef.
- H. L. Gelernter, A. F. Sanders, D. L. Larsen, K. K. Agarwal, R. H. Boivie, G. A. Spritzer and J. E. Searleman, Science, 1977, 197, 1041–1049 CAS.
- J. Gasteiger, M. Hutchings, H. Saller and P. Löw, in Chemical Structures, ed. W. Warr, Springer Berlin, Heidelberg, Berlin, Germany, 1988, ch. 36, pp. 343–359 Search PubMed.
- J. B. Hendrickson, Angew. Chem., Int. Ed., 1990, 29, 1286–1295 CrossRef.
- R. Barone, M. Chanon and J. Metzger, Tetrahedron Lett., 1974, 15, 2761–2764 CrossRef.
- F. Choplin, C. Laurenco, R. Marc, G. Kaufmann and W. T. Wipke, Nouv. J. Chim., 1978, 2, 285–293 CAS.
- F. Barberis, R. Barone and M. Chanon, Tetrahedron, 1996, 52, 14625–14630 CrossRef CAS.
- I. Dogane, T. Takabatake and M. Bersohn, Recl. Trav. Chim. Pays-Bas, 1992, 111, 291–296 CrossRef CAS.
- T. D. Salatin and W. L. Jorgensen, J. Org. Chem., 1980, 45, 2043–2051 CrossRef CAS.
- P. Jauffret, C. Laurenço and G. Kaufmann, Tech. Sci. Inform., 1986, 5, 375–390 Search PubMed.
- P. Jauffret, C. Laurenço and G. Kaufmann, Tech. Sci. Inform., 1987, 6, 129–143 Search PubMed.
- J. B. Hendrickson and C. A. Parks, J. Chem. Inf. Comput. Sci., 1992, 32, 209–215 CrossRef CAS.
- I. M. Socorro, K. Taylor and J. M. Goodman, Org. Lett., 2005, 7, 3541–3544 CrossRef CAS PubMed.
- R. Barone, M. Chanon and J. Metzger, Riv. Ital., EPPOS, 1980, 3, 136–145 Search PubMed.
- G. Vernin, J. Metzger, P. Azario, R. Barone, M. Arbelot and M. Chanon, in Food Science and Human Nutrition, ed. G. Charalambous, Elsevier Science Limited, Amsterdam, Netherlands, 1992, pp. 75–97 Search PubMed.
- J. H. Chen and P. Baldi, J. Chem. Inf. Model., 2009, 49, 2034–2043 CrossRef CAS PubMed.
- R. Barone, C. Chanon, G. Vernin and C. Parkanyi, in Food Flavor and Chemistry; Explorations into the 21st Century, ed. A. M. Spanier, F. Shahidi, T. H. Parliment, C. Mussinan and E. Tratras Contis, RSC, Cambridge, UK, 2005, pp. 175–212 Search PubMed.
- R. Barone, M. Chanon, G. Vernin and C. Parkanyi, in Recent Advances in Food and Flavor Chemistry: Food Flavors and Encapsulation, Health Benefits, Analytical Methods, and Molecular Biology of Functional Foods, ed. C. T. Ho, E. T. Contis, C. J. Mussinan and F. Shahidi, RSC, Cambridge, UK, 2010, pp. 81–127 Search PubMed.
- S. Ghosh, A. Nie, J. An and Z. Huang, Curr. Opin. Chem. Biol., 2006, 10, 194–202 CrossRef CAS PubMed.
- A. Klamt, F. Eckert and M. Diedenhofen, J. Phys. Chem. B, 2009, 113, 4508–4510 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- O. Treutler and R. Ahrlichs, J. Chem. Phys., 1995, 102, 346 CrossRef CAS.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369–385 CrossRef CAS.
- M. Durand, V. Molinier, W. Kunz and J.-M. Aubry, Chem.–Eur. J., 2011, 17, 5155–5164 CrossRef CAS PubMed.
- K. G. Joback and R. C. Reid, Chem. Eng. Commun., 1987, 57, 233–243 CrossRef CAS.
- R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253–258 CrossRef CAS.
- T. W. J. Cooper, I. B. Campbell and S. J. F. Macdonald, Angew. Chem., Int. Ed., 2010, 49, 8082–8091 CrossRef CAS PubMed.
- M. Butters, D. Catterick, A. Craig, A. Curzons, D. Dale, A. Gillmore, S. P. Green, I. Marziano, J.-P. Sherlock and W. White, Chem. Rev., 2006, 106, 3002–3027 CrossRef CAS PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 CAS.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- N. I. Vasilevich, R. V. Kombarov, D. V. Genis and M. A. Kirpichenok, J. Med. Chem., 2012, 55, 7003–7009 CrossRef CAS PubMed.
- H.-J. Arpe and K. Weissermel, Industrial organic chemistry, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed.
- S. Luxon, Chemical safety data sheets, Solvents, RCS, Cambridge, UK, 1989, vol. 1 Search PubMed.
- B. M. Trost, Science, 1991, 254, 1471–1477 CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- P. Leconte and C. Denis, Rhodia Recherches et Technologies, Process for converting nitrile compounds to carboxylic acids and corresponding esters, France Pat., WO 2007/141404 A1, 2007 Search PubMed.
- P. Leconte, P. Marion and R. Jacquot, Rhodia Operations, Process for the preparation of diesters, France Pat., WO 2008/009792 A1, 2008 Search PubMed.
- R. Jacquot and P. Marion, Rhodia Operations, Process for the preparation of imides, of derivatives thereof and uses, France Pat., WO 2012/059680 A2, 2012 Search PubMed.
- M. Morgan and C. Ordiscoll, Eur. Chem. News, 2004, 81, 18 Search PubMed.
- K. Nagai, Appl. Catal., A, 2001, 221, 367–377 CrossRef CAS.
- G. Derringer and R. Suich, J. Qual. Technol., 1980, 12, 214–219 Search PubMed.
- Y. Shi, W. Dayoub, G.-R. Chen and M. Lemaire, Green Chem., 2010, 12, 2189–2195 RSC.
- M. Sutter, W. Dayoub, E. Métay, Y. Raoul and M. Lemaire, ChemSusChem, 2012, 5, 2397–2409 CrossRef CAS PubMed.
- V. Bethmont, F. Fache and M. Lemaire, Tetrahedron Lett., 1995, 36, 4235–4236 CrossRef CAS.
- G. Mignani, R. Jacquot, P. Marion, Y. Raoul, M. Lemaire, W. Dayoub and E. Da Silva, CNRS, FIDOP, Rhodia Operations, Université Claude Bernard de Lyon 1, Preparation of (Poly)glycerol ethers, France Pat., WO 2012/120017 A1, 2012 Search PubMed.
- J. O. Metzger and M. Eissen, C. R. Chim., 2004, 7, 569–581 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- J. Fuchs and G. Szeimies, Chem. Ber., 1992, 125, 2517–2522 CrossRef CAS.
- T. Mendgen, T. Scholz and C. D. Klein, Bioorg. Med. Chem. Lett., 2010, 20, 5757–5762 CrossRef CAS PubMed.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed.
- Y.-H. Wu and R. F. Feldkamp, J. Org. Chem., 1961, 26, 1519–1524 CrossRef CAS.
- A. Voll and W. Marquardt, AIChE J., 2012, 58, 1788–1801 CrossRef CAS.
- H. Richard and B. Muller, L'Oreal, Ester solvents derived from 4-Carboxy-2-pyrrolidinone formulated into UV-screening cosmetic compositions, France Pat., US 2010/0143276 A1, 2010 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |