Coordination effect-regulated CO2 capture with an alkali metal onium salts/crown ether system†
Zhen-Zhen
Yang
ab,
De-en
Jiang
b,
Xiang
Zhu
bc,
Chengcheng
Tian
bc,
Suree
Brown
d,
Chi-Linh
Do-Thanh
d,
Liang-Nian
He
*a and
Sheng
Dai
*bd
aState Key Laboratory and Institute of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, P.R. China. E-mail: heln@nankai.edu.cn
bChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
cState Key Laboratory of Chemical Engineering and Department of Chemistry, East China University of Science and Technology, Shanghai 200237, P. R. China
dDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA
First published on 24th September 2013
Abstract
A coordination effect was employed to realize equimolar CO2 absorption, adopting easily synthesized amino group containing absorbents (alkali metal onium salts). The essence of our strategy was to increase the steric hindrance of cations so as to enhance a carbamic acid pathway for CO2 capture. Our easily synthesized alkali metal amino acid salts or phenolates were coordinated with crown ethers, in which highly sterically hindered cations were obtained through a strong coordination effect of crown ethers with alkali metal cations. For example, a CO2 capacity of 0.99 was attained by potassium prolinate/18-crown-6, being characterized by NMR, FT-IR, and quantum chemistry calculations to go through a carbamic acid formation pathway. The captured CO2 can be stripped under very mild conditions (50 °C, N2). Thus, this protocol offers an alternative for the development of technological innovation towards efficient and low energy processes for carbon capture and sequestration.
Introduction
The chemistry of CO2 has attracted much attention from the scientific community because of the increasing atmospheric CO2 concentration associated with global warming.1–9 CO2 capture from fossil fuel combustion represents a critical component of efforts aimed at stabilizing greenhouse gas levels in the atmosphere.10 In addition, removal of CO2 from natural gas is of vital importance to maintain and expand the availability of these clean-burning, efficient fuel sources.5In recent years, worldwide efforts have been devoted to developing various technologies/processes for CO2 capture, including liquid absorbents, solid adsorbents and membranes.4,6,7,9 Among them, chemisorption of CO2 based on C–N bond formation is one of the most efficient strategies,8 utilizing liquid absorbents such as conventional aqueous amine solutions,11–20 chilled ammonia,21–30 amino-functionalized ionic liquids (ILs),31–42 and amino-functionalized solid absorbents.4,43–49 The traditional technology for CO2 capture is chemisorption by aqueous solution of monoethanolamine (high reactivity, low cost and good absorption capacity) to produce ammonium bicarbonate.50,51 However, this process for CO2 capture has some disadvantages, including solvent loss, corrosion and particularly, highly energy intensive nature owing to the thermodynamic properties of water and high enthalpy of absorption.52 Notably, there are inherent drawbacks generally associated with amino group-containing absorbents in the absence of water, that is, the requirement of two amine units to react with one CO2 molecule, leading to the formation of ammonium carbamate (Scheme 1). On the other hand, extensive energy input in desorption processes is needed owing to the relatively higher stability characterization of ammonium carbamate compared with the carbamic acid intermediate. Hence, increasing the undesired 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (CO2:amine) stoichiometry for CO2 capacity to 1:1 and reducing the energy requirement of desorption are essential prerequisites for a breakthrough in absorption techniques.32 Therefore, it will be very desirable to form relatively unstable carbamic acid (1:1) for CO2 capture, that is, to prevent the deprotonation of carbamic acid by another amino group. In this way, equimolar CO2 absorption and simultaneously easy CO2 desorption will be realized due to the relatively unstable nature of carbamic acid.
:2 (CO2:amine) stoichiometry for CO2 capacity to 1:1 and reducing the energy requirement of desorption are essential prerequisites for a breakthrough in absorption techniques.32 Therefore, it will be very desirable to form relatively unstable carbamic acid (1:1) for CO2 capture, that is, to prevent the deprotonation of carbamic acid by another amino group. In this way, equimolar CO2 absorption and simultaneously easy CO2 desorption will be realized due to the relatively unstable nature of carbamic acid.
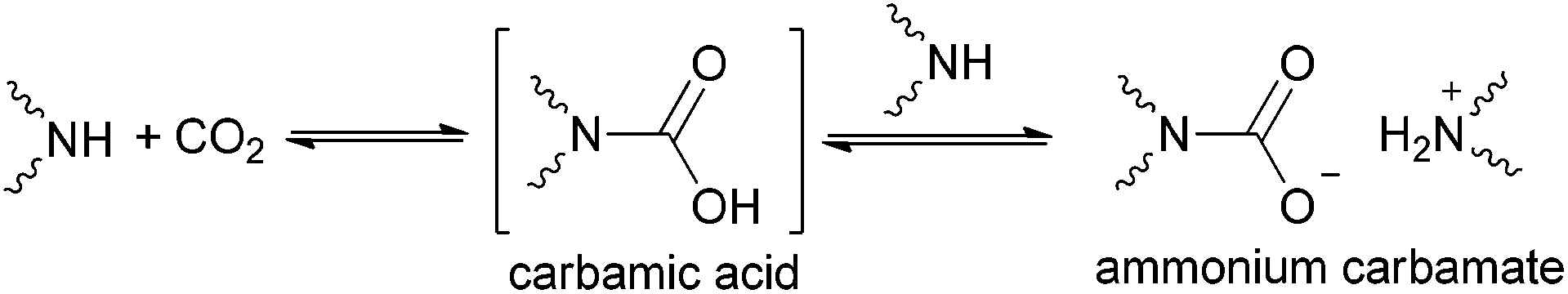 | ||
| Scheme 1 Reactions of CO2 with amino group-containing absorbents. | ||
As readily accessible chiral molecules, natural α-amino acids (AAs) have many merits, such as diverse structures, low cost, high thermal stability, bio-compatibility, easy regeneration and biodegradability.53 Phosphonium and ammonium ionic liquids (ILs) with AAs as anions have been already used for CO2 uptake, but the CO2:AA stoichiometry arising from the formation of ammonium carbamate is still 1:2.34–38 In this respect, specially designed absorbents derived from AAs have been successfully synthesized and applied for 1:1 CO2 capture through a carbamic acid formation pathway. Gurkan et al. have synthesized two AA-based ILs with a long-chain alkyl phosphonium cation, including trihexyl(tetradecyl)phosphonium prolinate([P66614][Pro]) and methioninate ([P66614][Met]), both of which can react with CO2 in a ratio of one CO2 per amine (1:1 stoichiometry) by termination of the reaction sequence at the formation of carbamic acid.33 We also designed AA salts with bulky N-substituents for chemisorption of CO2, approaching almost equimolar absorption in polyethylene glycol (PEG) solution.54 The key idea of these two successful examples leading to the formation of carbamic acid is high steric hindrance, either derived from bulky cations [P66614]+, or bulky N-substituents (iPr), to prevent nucleophilic attack of the amino group on the carboxylic acid group of carbamic acid. Despite such great advances, the development of efficient processes for CO2 capture with simple, easily prepared absorbents is still appealing. Therefore, it is desirable to develop a way to make easily synthesized AA salts with “small” cations and regular anions, such as commercially available alkali metal cations and AA anions, to obtain an equimolar absorption of CO2 through a carbamic acid formation pathway. Moreover, the carboxylate group of the AA salts has the potential to stabilize the formed carbamic acid through intramolecular hydrogen-bond formation.
Crown ethers are cyclic chemical compounds that consist of several ether groups, which can strongly bind certain cations to form complexes, probably possessing some toxicity to creatures,55 thus having the potential to form bulky cations.56–63 Herein, we found that the coordination effect between crown ether and alkali metal cations (increasing steric hindrance) leads to an extremely high CO2 capacity, approaching almost equimolar absorption in PEG solution, adopting easily synthesized onium salts derived from AAs or phenols and alkali metal hydroxide as absorbents. Coordination effect-adjusted CO2 absorption is assumed to proceed via the carbamic acid formation rather than the subsequent ammonium carbamate. On the other hand, the coordination effect leading to the formation of relatively unstable carbamic acid greatly reduces the energy requirement for CO2 desorption in comparison with ILs with the same anions.
Results and discussion
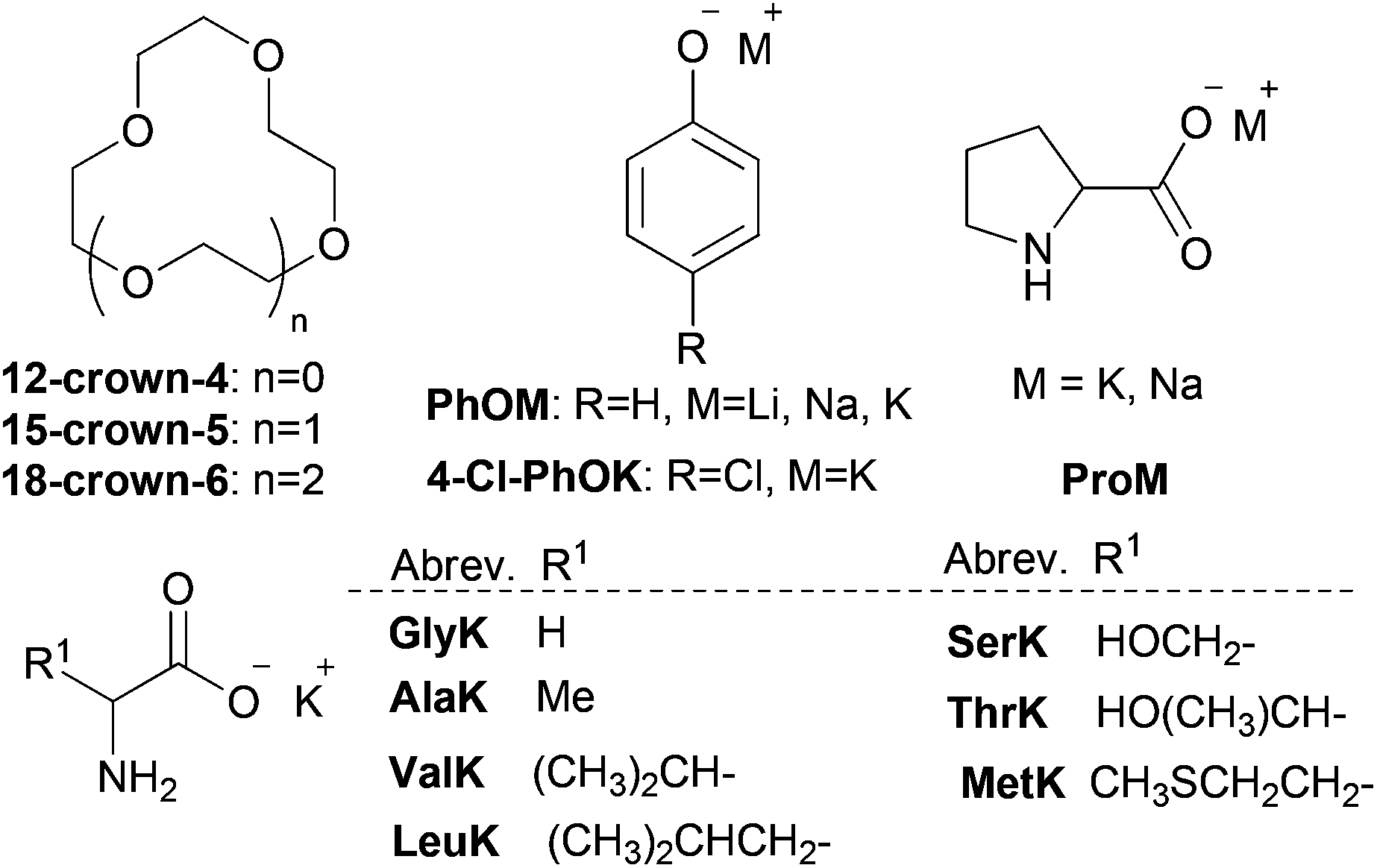
Easily synthesized alkali metal AA salts and phenolate (onium salts) together with different crown ethers were investigated to test our idea about coordination-regulated CO2 absorption (Table 1). PEG300 (M.W. = 300 Da) was selected as a suitable solvent due to its distinctive properties, such as low cost, almost negligible vapor pressure, toxicological innocuity and CO2-philic property.64–66 As shown in Table 1, crown ether/PEG300 gave extremely poor CO2 sorption capacity, implying that only physical interaction was observed (entries 1–3). For various alkali metal cations, the corresponding crown ethers on the basis of size fit within the interior of the macrocycle is needed for effective coordination (18-crown-6 for K+, 15-crown-5 for Na+, 12-crown-4 for Li+),58 and the ionic diameter for cations or diameter of cavity for crown ethers is 1.20 (Li+), 1.90 (Na+), 2.66 (K+), 1.2–1.5 (12-crown-4), 1.7–2.2 (15-crown-5) and 2.6–3.2 (18-crown-6), respectively.62 Alkali metal cations together with corresponding crown ethers, the CO2 capacity of which increases in the order of Na+/15-crown-5 < K+/18-crown-6 for ProM and Li+/12-crown-4 < Na+/15-crown-5 < K+/18-crown-6 for PhOM, could play a key role in CO2 sorption performance (entries 4–5 and 18–20). In particular, coordination of ProK with 18-crown-6 greatly enhanced the CO2 capacity, approaching the 1:1 stoichiometry (CO2 capacity of 0.99), which is consistent with the theoretical formation of carbamic acid, comparable with the IL [P66614][Pro] with the same anion (∼0.90) (entry 5 vs. 6).33 However, disadvantage was existed in our system (e.g. ProK/18-crown-6/PEG300) by valuing the mass of CO2 absorbed per gram of the absorbent, owing to addition of large quantities of PEG300 as a solvent. In fact, the solvent PEG300 itself was able to coordinate with the cation K+ in ProK,54,68 although not as strongly as 18-crown-6, and gave a CO2 capacity of 0.83 (entry 7). However, for other potassium AA onium salts, a CO2 capacity of 0.63–0.79 was attained, probably due to the high viscosity of the system after CO2 absorption, but still much higher than the corresponding ILs with the same AA anions (∼0.50) through an ammonium carbamate formation pathway.37
:1)a
|
|
|||
|---|---|---|---|
| Entry | Absorbent | Timeb (min) | CO2 capacityc |
|
a Absorption conditions: crown ether/alkali metal onium salts (molar ratio 1:1), 1 mmol; PEG300, 2 mmol; CO2, 0.1 MPa; 25 °C.
b Time required to reach absorption equilibrium.
c Moles of CO2 captured per mole of onium salt were estimated from the weight increase of the reaction mixture.
d Data from ref. 33.
e 0.2643 g PEG300 (the same weight as 1 mmol 18-crown-6) was added instead of 18-crown-6.
f Data from ref. 67.
|
|||
| 1 | 18-crown-6 | 5 | 0.04 |
| 2 | 15-crown-5 | 5 | 0.03 |
| 3 | 12-crown-4 | 5 | 0.03 |
| 4 | ProNa/15-crown-5 | 25 | 0.89 |
| 5 | ProK/18-crown-6 | 10 | 0.99 |
| 6d | [P66614][Pro] | — | ∼0.90 |
| 7e | ProK/PEG300 | 30 | 0.83 |
| 8 | ValK/18-crown-6 | 10 | 0.79 |
| 9 | MetK/18-crown-6 | 20 | 0.75 |
| 10 | GlyK/18-crown-6 | 20 | 0.75 |
| 11 | ThrK/18-crown-6 | 15 | 0.73 |
| 12 | AlaK/18-crown-6 | 15 | 0.71 |
| 13 | LeuK/18-crown-6 | 10 | 0.68 |
| 14 | SerK/18-crown-6 | 15 | 0.63 |
| 15 | PhOLi/12-crown-4 | 5 | 0.75 |
| 16 | PhONa/15-crown-5 | 15 | 0.84 |
| 17 | PhOK/18-crown-6 | 10 | 0.90 |
| 18f | [P66614][PhO] | 30 | 0.85 |
| 19 | 4-ClPhOK/18-crown-6 | 10 | 0.95 |
| 20f | [P66614][4-Cl-PhO] | 30 | 0.82 |
Interaction between CO2 (carbon atom) and ether linkages (oxygen atom) in both PEG and crown ethers leads to lowered viscosity and increased gas/liquid diffusion rates, and thus assumedly facilitates CO2 sorption with faster sorption kinetics and increasing capacity.38 Indeed, a CO2 capacity of 0.90 was obtained within 10 min adopting PhOK/18-crown-6, while 30 min was needed for [P66614][PhOK] to get a comparable CO2 capacity (0.85) (entry 17 vs. 18). In addition, 4-Cl-PhOK/18-crown-6 gave a much better performance for CO2 capture (CO2 capacity of 0.95 in 10 min) than the corresponding IL with the same anion [P66614][4-Cl-PhOK] (CO2 capacity of 0.82 in 30 min) (entry 19 vs. 20).
Extensive energy input into the desorption process would be a crucial barrier to realize practical CO2 capture, and also the stability and reusability of the absorbent are two important factors to identify whether it can find potential practical application in industry. The stability of ProK/18-crown-6/PEG300 after CO2 capture was studied by thermogravimetric analysis (TGA) (Fig. 1(a)). It can be seen that the thermal stability of the absorbent system ProK/18-crown-6/PEG300 was increased compared with 18-crown-6, which can also account for the coordination effect between 18-crown-6 and K+. Notably, the captured CO2 was easy to remove just at a temperature as low as 50 °C by bubbling N2 within 25 min. Since higher temperatures are required for CO2 removal from the common conventional amine-based absorbents (e.g. 120 °C, N2, 75 min),69 this can prove that our system proceeds through the carbamic acid pathway (Scheme 2). Notably, no significant drop in CO2 capacity was detected after six successive absorption–desorption cycles (Fig. 1(b)). For the 4-Cl-PhOK/18-crown-6/PEG300 system, the absorbed CO2 can be removed completely at 80 °C by bubbling N2 within 20 min, much more easily than IL [P66614][4-Cl-PhO] (80 °C, N2, 60 min) (Fig. S2(b)†).
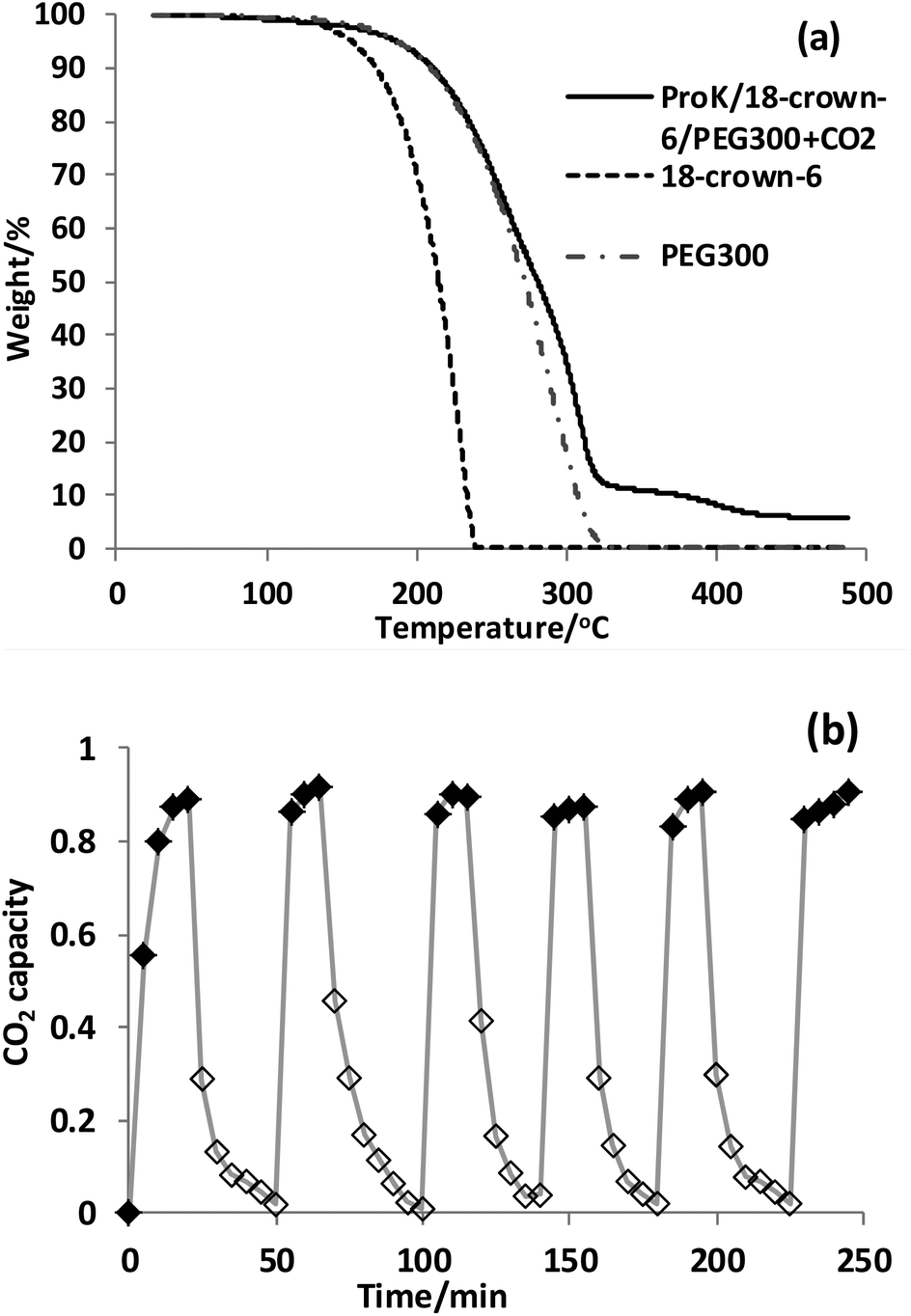 | ||
| Fig. 1 (a) The scanning thermogravimetric analysis (TGA) results for 18-crown-6, PEG300 and ProK/18-crown-6/PEG300+CO2 with a 10 °C min−1 temperature ramping rate to 500 °C. (b) Six consecutive cycles of CO2 absorption (◆ 15 min, 25 °C, CO2 0.1 MPa) and release (◇ 25 min, 50 °C, N2 0.1 MPa) by the ProK/18-crown-6/PEG300 system; data were obtained every 5 min. | ||
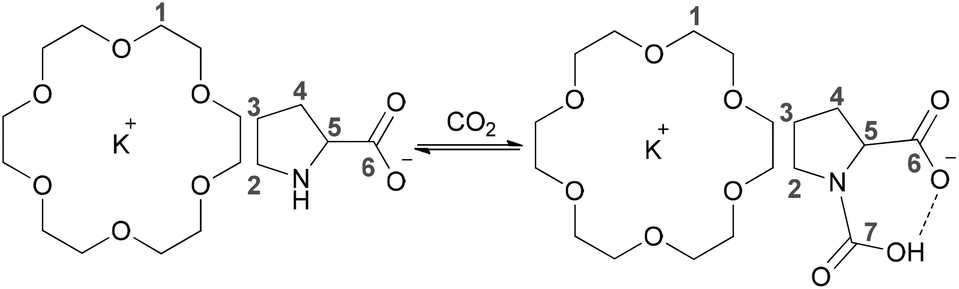 | ||
| Scheme 2 Reaction of CO2 with ProK/18-crown-6 to produce carbamic acid with PEG300 as a solvent. | ||
To further gain insight into the absorption mechanism, coordination-regulated absorption involving the formation of carbamic acid (Scheme 2) was identified by NMR and FT-IR. It was found that in the 1H NMR spectrum all the protons of Pro− and 18-crown-6 (H1–H5) showed an upfield shift after mixing them together, due to the coordination of 18-crown-6 with the K+ cation (Fig. 2(a)). Afterwards, the downfield shift of all the protons in Pro− (H2–H5) after CO2 absorption could indicate that CO2 is chemically bound to the nitrogen center (Fig. 2(a)). Simultaneously, the new peak in the 13C NMR spectrum at 157.7 ppm after CO2 uptake would support the formation of the carbamic acid structure between the secondary amine and CO2 (C7), while C6 of the carbonyl group in Pro− showed an upfield shift from 177.7 ppm to 175.9 ppm after CO2 absorption (Fig. 2(b)). Notably, bicarbonate anions formed from the reaction between CO2 and aqueous (D2O) solution of Prok showed a new peak at 160.2 ppm, while C6 of the carbonyl group in ammonium cation (protonated Pro-) resulted in an upfield shift from 177.7 ppm to 174.7 ppm, which was quite different from the 13C NMR spectrum of carbamic acid (Fig. 2(b)). For the 4-Cl-PhOK/18-crown-6 system, coordination between 18-crown-6 and K+ can also be detected by 1H NMR (Fig. S1(a)†). After CO2 capture, all the protons of 4-Cl-PhO− showed a downfield shift and the new peak in the 13C NMR spectrum at 158.7 ppm could prove the formation of phenylcarbonate [PhOCO2]− (Fig. S1(b)†).
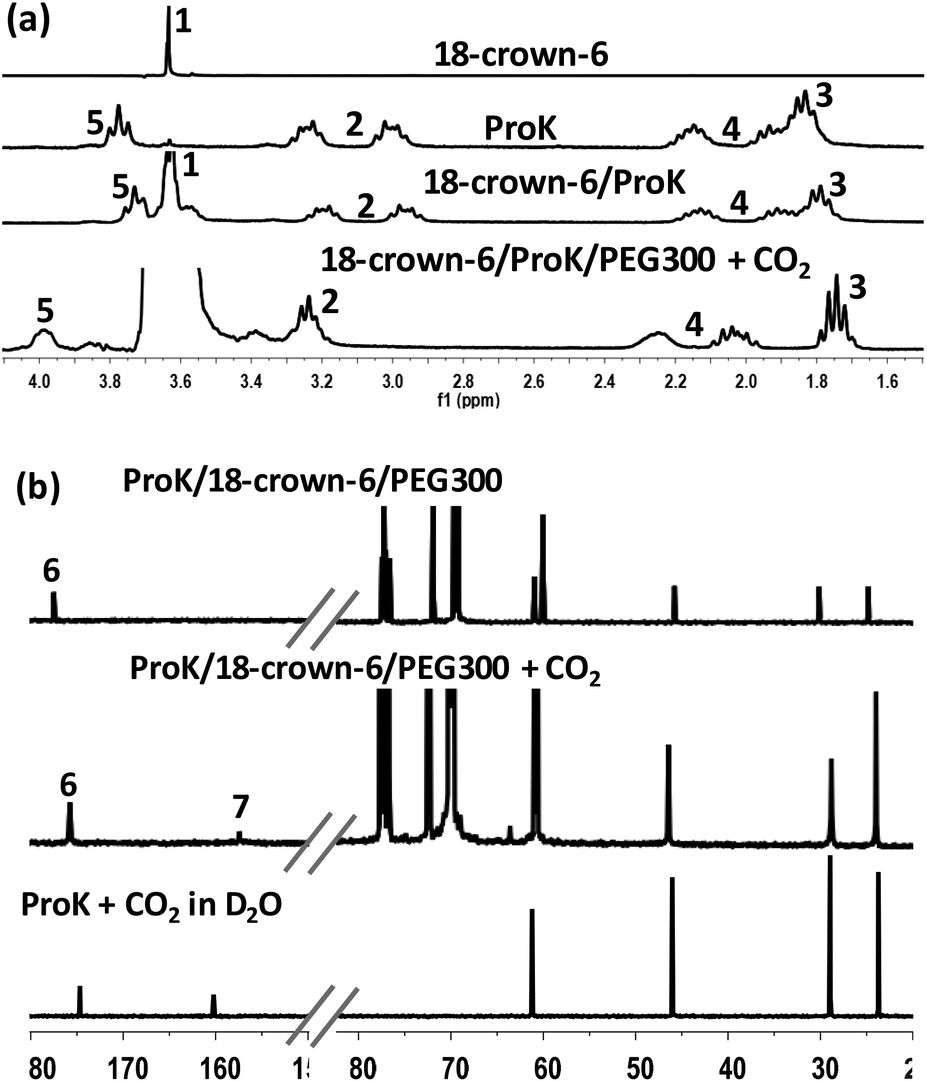 | ||
| Fig. 2 (a) 1H NMR spectra (CDCl3, 300 MHz) of 18-crown-6, ProK, ProK/18-crown-6 and ProK/18-crown-6/PEG300+CO2 (from top to bottom); (b) 13C NMR (CDCl3, 75 MHz) spectra of ProK/18-crown-6/PEG300 before (top) and after (middle) CO2 absorption, and 13C NMR (D2O, 75 MHz) spectra of CO2 capture by D2O solution of ProK (bottom). | ||
In the in situ FT-IR spectrum, there are three important features. Firstly, the strength of O–H stretching from PEG increased after CO2 absorption due to the formation of carbamic acid (O–H containing group), and blueshifted from 3298 cm−1 to 3472 cm−1, also indicating that PEG could be chemically inert during the process (Fig. 3(a)). Secondly, distinct bands corresponding to the ammonium cation in the 2000–3000 cm−1 region are not observed, while the new peak at 2338 cm−1 was derived from physically dissolved CO2. Finally, the original vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the carbonyl group in Pro− was shown at 1598 cm−1, and then after CO2 uptake, two characteristic peaks centered at 1635 cm−1 and 1667 cm−1 can be assigned to asymmetric CO vibrations of the carboxylate anion and the COOH moiety of the carbamic acid, respectively (Fig. 3(b)). For the 4-Cl-PhOK/18-crown-6/PEG300 system, the carbonyl group absorption was observed at 1647 cm−1 after CO2 capture, indicating the formation of phenylcarbonate [PhOCO2]− (Fig. S1(c)†).
O bond of the carbonyl group in Pro− was shown at 1598 cm−1, and then after CO2 uptake, two characteristic peaks centered at 1635 cm−1 and 1667 cm−1 can be assigned to asymmetric CO vibrations of the carboxylate anion and the COOH moiety of the carbamic acid, respectively (Fig. 3(b)). For the 4-Cl-PhOK/18-crown-6/PEG300 system, the carbonyl group absorption was observed at 1647 cm−1 after CO2 capture, indicating the formation of phenylcarbonate [PhOCO2]− (Fig. S1(c)†).
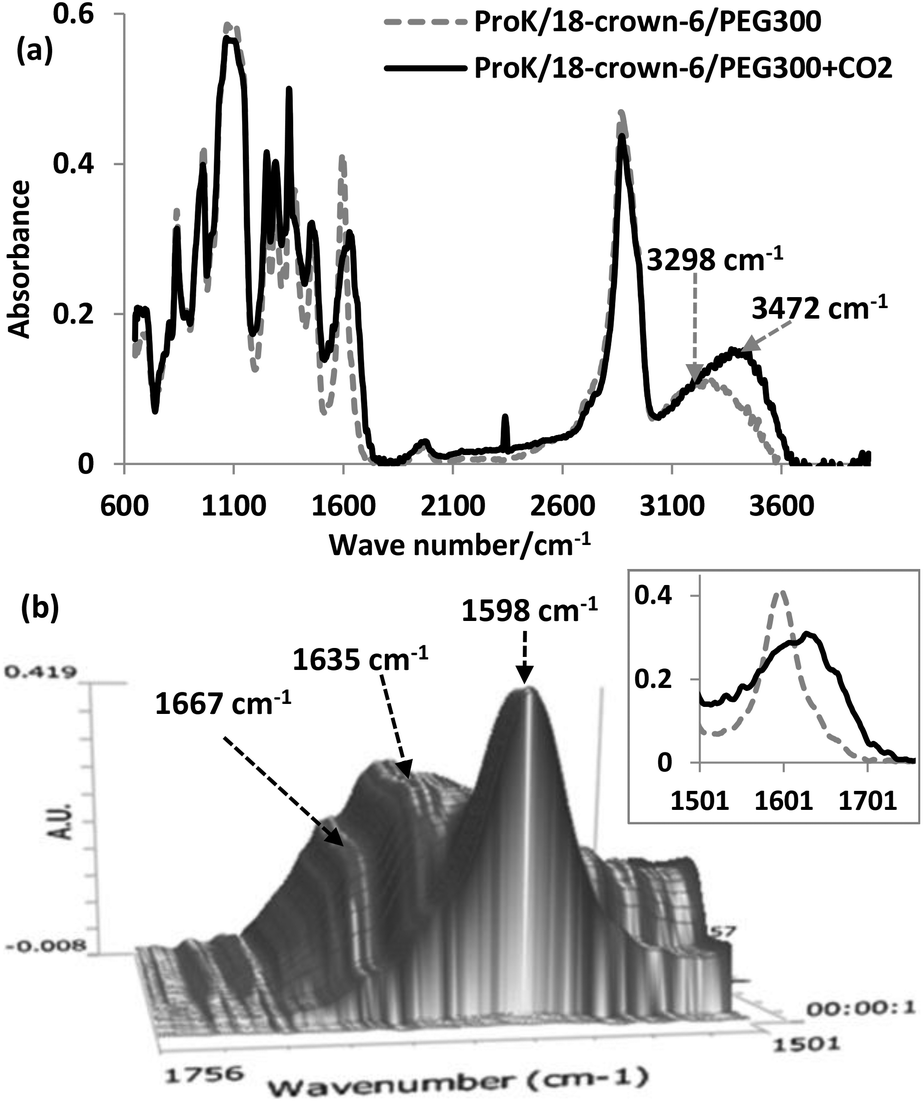 | ||
| Fig. 3 Results of in situ IR spectroscopy under 0.1 MPa CO2 pressure monitoring at various reaction times. (a) ProK/18-crown-6/PEG300 (molar ratio 1:1:2) before and after CO2 uptake at 25 °C; (b) magnified three-dimensional spectra of the absorption mixture with reaction time. | ||
Quantum chemistry calculations, being performed with the program Turbomole V6.0,70 were employed to calculate the enthalpy changes for the present equimolar CO2 capture (Scheme 3). The PEG solvent was simulated as a continuum medium with a dielectric constant of 13.471 within the Conductor-like Screening Model (COSMO)72 implemented in Turbomole. Both structure optimization and the solvation calculation were performed at the B3LYP level with the def2-TZVP basis sets.73 The amino acid salt ProK with 18-crown-6 coordinated at the K+ reacts with CO2 at the secondary amine to form the carbamic acid, which is stabilized through an intramolecular hydrogen bond with the carboxylate anion (bond length: 1.593 Å). In addition, formation of the carbamic acid has a calculated enthalpy change of −39.0 kJ mol−1, indicating that the absorption process is thermodynamically favourable, but not too strong. This is consistent with the easy desorption we have found (Fig. 1b).
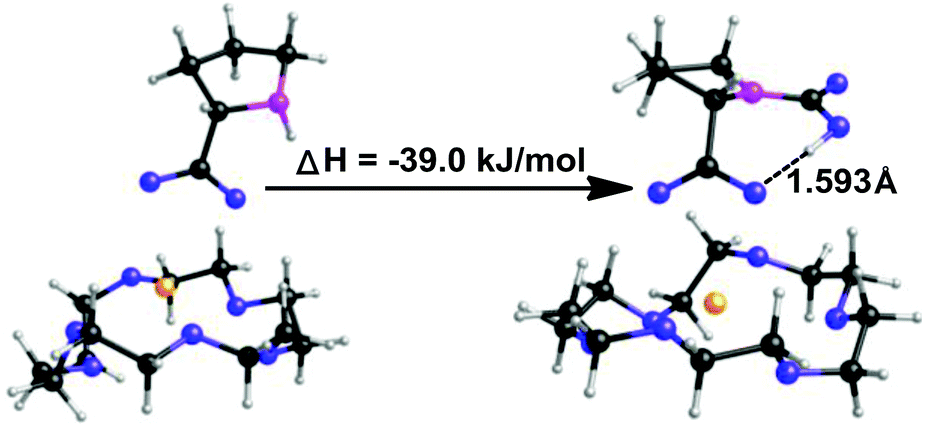 | ||
| Scheme 3 The quantum chemistry calculations of the reaction between CO2 and ProK/18-crown-6 in a PEG300 solution to produce carbamic acid; the PEG300 is modelled as a continuum medium with a dielectric constant of 13.4. H: gray, C: black, N: pink, O: blue, K+: yellow. | ||
Conclusions
In summary, a coordination effect was employed to realize equimolar CO2 absorption, adopting an easily synthesized amino group containing absorbents (alkali metal onium salts), and the essence was to increase the steric hindrance of cations and go through a carbamic acid pathway. Our strategy is to utilize easily synthesized alkali metal AA salts together with crown ethers, in which highly sterically hindered cations were obtained through the strong coordination effect of crown ethers with alkali metal cations. For example, a CO2 capacity of 0.99 was attained by ProK/18-crown-6, detected by NMR, in situ FT-IR and quantum chemistry calculation to go through a carbamic acid formation pathway. The captured CO2 can be stripped out under very mild conditions (50 °C, N2). In addition, crown ethers together with alkali metal phenolates are also easily synthesized absorbents for efficient CO2 absorption. Thus, this protocol would pave the way for the development of technological innovation towards efficient and low energy demanding practical processes for CO2 capture.Acknowledgements
This research was sponsored by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy.References
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenh. Gas Control, 2008, 2, 9–20 CrossRef CAS.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef CAS PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- J. E. Bara, T. K. Carlisle, C. J. Gabriel, D. Camper, A. Finotello, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2009, 48, 2739–2751 CrossRef CAS.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 CAS.
- Z.-Z. Yang, Y.-N. Zhao and L.-N. He, RSC Adv., 2011, 1, 545–567 RSC.
- Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 CAS.
- L. Li, N. Zhao, W. Wei and Y. Sun, Fuel, 2013, 108, 112–130 CrossRef CAS PubMed.
- M. R. Raupach, G. Marland, P. Ciais, C. Le Quere, J. G. Canadell, G. Klepper and C. B. Filed, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10288–10293 CrossRef CAS PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- C. Lastoskie, Science, 2010, 330, 595–596 CrossRef CAS PubMed.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- N. Mac Dowell, F. Llovell, C. S. Adjiman, G. Jackson and A. Galindo, Ind. Eng. Chem. Res., 2010, 49, 1883–1899 CrossRef CAS.
- T. Lewis, M. Faubel, B. Winter and J. C. Hemminger, Angew. Chem., Int. Ed., 2011, 50, 10178–10181 CrossRef CAS PubMed.
- J. E. Rainbolt, P. K. Koech, C. R. Yonker, F. Zheng, D. Main, M. L. Weaver, J. C. Linehan and D. J. Heldebrant, Energy Environ. Sci., 2011, 4, 480–484 CAS.
- M. Caplow, J. Am. Chem. Soc., 1968, 90, 6795–6803 CrossRef CAS.
- J. Wang, X. Zhang and Y. Zhou, Environ. Chem. Lett., 2011, 9, 1–3 CrossRef CAS.
- D. J. Heldebrant, P. K. Koech, M. T. C. Ang, C. Liang, J. E. Rainbolt, C. R. Yonker and P. G. Jessop, Green Chem., 2010, 12, 713–721 RSC.
- J. Im, S. Y. Hong, Y. Cheon, J. Lee, J. S. Lee, H. S. Kim, M. Cheong and H. Park, Energy Environ. Sci., 2011, 4, 4284–4289 CAS.
- J. K. You, H. Park, S. H. Yang, W. H. Hong, W. Shin, J. K. Kang, K. B. Yi and J.-N. Kim, J. Phys. Chem. B, 2008, 112, 4323–4328 CrossRef CAS PubMed.
- H. Bai and A. C. Yeh, Ind. Eng. Chem. Res., 1997, 36, 2490–2493 CrossRef CAS.
- X. Li, E. Hagaman, C. Tsouris and J. W. Lee, Energy Fuels, 2003, 17, 69–74 CrossRef CAS.
- A. C. Yeh and H. Bai, Sci. Total Environ., 1999, 228, 121–133 CrossRef CAS.
- Y.-F. Diao, X.-Y. Zheng, B.-S. He, C.-H. Chen and X.-C. Xu, Energy Convers. Manage., 2004, 45, 2283–2296 CrossRef CAS PubMed.
- L. Meng, S. Burris, H. Bui and W.-P. Pan, Anal. Chem., 2005, 77, 5947–5952 CrossRef CAS PubMed.
- H. Konghak, Korean Chem. Eng. Res., 2007, 45, 258–263 Search PubMed.
- J. T. Yeh, K. P. Resnik, K. Rygle and H. W. Pennline, Fuel Process. Technol., 2005, 86, 1533–1546 CrossRef CAS PubMed.
- N. Wen and M. H. Brooker, J. Phys. Chem., 1995, 99, 359–368 CrossRef CAS.
- A. Bandyopadhyay, Clean Technol. Environ. Policy, 2011, 13, 269–294 CrossRef CAS.
- K. E. Gutowski and E. J. Maginn, J. Am. Chem. Soc., 2008, 130, 14690–14704 CrossRef CAS PubMed.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS PubMed.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS PubMed.
- J. Zhang, S. Zhang, K. Dong, Y. Zhang, Y. Shen and X. Lv, Chem.–Eur. J., 2006, 12, 4021–4026 CrossRef CAS PubMed.
- Y. Zhang, S. Zhang, X. Lu, Q. Zhou, W. Fan and X. Zhang, Chem.–Eur. J., 2009, 15, 3003–3011 CrossRef CAS PubMed.
- Y.-Y. Jiang, G.-N. Wang, Z. Zhou, Y.-T. Wu, J. Geng and Z.-B. Zhang, Chem. Commun., 2008, 505–507 RSC.
- H. Yu, Y.-T. Wu, Y.-Y. Jiang, Z. Zhou and Z.-B. Zhang, New J. Chem., 2009, 33, 2385–2390 RSC.
- X. Li, M. Hou, Z. Zhang, B. Han, G. Yang, X. Wang and L. Zou, Green Chem., 2008, 10, 879–884 RSC.
- M. D. Soutullo, C. I. Odom, B. F. Wicker, C. N. Henderson, A. C. Stenson and J. H. Davis, Chem. Mater., 2007, 19, 3581–3583 CrossRef CAS.
- G. Yu, S. Zhang, X. Yao, J. Zhang, K. Dong, W. Dai and R. Mori, Ind. Eng. Chem. Res., 2006, 45, 2875–2880 CrossRef CAS.
- D. Camper, J. E. Bara, D. L. Gin and R. D. Noble, Ind. Eng. Chem. Res., 2008, 47, 8496–8498 CrossRef CAS.
- H. Xie, S. Zhang and S. Li, Green Chem., 2006, 8, 630–633 RSC.
- S. Kim, J. Ida, V. V. Guliants and Y. S. Lin, J. Phys. Chem. B, 2005, 109, 6287–6293 CrossRef CAS PubMed.
- G. Osei-Prempeh, H.-J. Lehmler, S. E. Rankin and B. L. Knutson, Ind. Eng. Chem. Res., 2011, 50, 5510–5522 CrossRef CAS.
- N. Hiyoshi, K. Yogo and T. Yashima, Chem. Lett., 2004, 510–511 CrossRef CAS.
- M. R. Mello, D. Phanon, G. Q. Silveira, P. L. Llewellyn and C. M. Ronconi, Microporous Mesoporous Mater., 2011, 143, 174–179 CrossRef CAS PubMed.
- W. Chaikittisilp, J. D. Lunn, D. F. Shantz and C. W. Jones, Chem.–Eur. J., 2011, 17, 10556–10561 CrossRef CAS.
- G. Qi, L. Fu, X. Duan, B. H. Choi, M. Abraham and E. P. Giannelis, Greenhouse Gas. Sci. Technol., 2011, 1, 278–284 CAS.
- M. L. Gray, Y. Soong, K. J. Champagne, J. Baltrus, R. W. Stevens Jr., P. Toochinda and S. S. C. Chuang, Sep. Purif. Technol., 2004, 35, 31–36 CrossRef CAS.
- N. McCann, M. Maeder and M. Attalla, Ind. Eng. Chem. Res., 2008, 47, 2002–2009 CrossRef CAS.
- J. Alejandre, J. L. Rivera, M. A. Mora and V. de la Garza, J. Phys. Chem. B, 2000, 104, 1332–1337 CrossRef CAS.
- D. J. Heldebrant, C. R. Yonker, P. G. Jessop and L. Phan, Energy Environ. Sci., 2008, 1, 487–493 CAS.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS PubMed.
- A.-H. Liu, R. Ma, C. Song, Z.-Z. Yang, A. Yu, Y. Cai, L.-N. He, Y.-N. Zhao, B. Yu and Q.-W. Song, Angew. Chem., Int. Ed., 2012, 51, 11306–11310 CrossRef CAS PubMed.
- W.-W. Tso, W.-P. Fung and M.-Y. W. Tso, J. Inorg. Biochem., 1981, 14, 237–244 CrossRef CAS.
- H. Høiland, J. A. Ringseth and T. S. Brun, J. Solution Chem., 1979, 8, 779–792 CrossRef.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- J. A. Danis, M. R. Lin, B. L. Scott, B. W. Eichhorn and W. H. Runde, Inorg. Chem., 2001, 40, 3389–3394 CrossRef CAS PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- R. M. Izatt, Chem. Rev., 1974, 74, 351–384 CrossRef.
- J. S. Bradshaw and R. M. Izatt, Acc. Chem. Res., 1997, 30, 338–345 CrossRef CAS.
- D. J. Heldebrant and P. G. Jessop, J. Am. Chem. Soc., 2003, 125, 5600–5601 CrossRef CAS PubMed.
- J. Zhang, B. Han, Y. Zhao, J. Li, M. Hou and G. Yang, Chem. Commun., 2011, 47, 1033–1035 RSC.
- N. P. Patel, M. A. Hunt, S. Lin-Gibson, S. Bencherif and R. J. Spontak, J. Membr. Sci., 2005, 251, 51–57 CrossRef CAS PubMed.
- C. Wang, H. Luo, H. Li, X. Zhu, B. Yu and S. Dai, Chem.–Eur. J., 2012, 18, 2153–2160 CrossRef CAS PubMed.
- D.-L. Kong, L.-N. He and J.-Q. Wang, Synlett, 2010, 1276–1280 CAS.
- C. Wang, Y. Guo, X. Zhu, G. Cui, H. Li and S. Dai, Chem. Commun., 2012, 48, 6526–6528 RSC.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- M. Salomon, M. Xu, E. M. Eyring and S. Petrucci, J. Phys. Chem., 1994, 98, 8234–8244 CrossRef CAS.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, synthetic procedure of alkali metal onium salts, and characterization of the absorption systems (NMR, TGA and IR). See DOI: 10.1039/c3gc41513a |
| This journal is © The Royal Society of Chemistry 2014 |