DOI:
10.1039/C3GC41752B
(Paper)
Green Chem., 2014,
16, 181-190
Understanding the chemical transformations of lignin during ionic liquid pretreatment†
Received
26th August 2013
, Accepted 17th October 2013
First published on 17th October 2013
Abstract
Unveiling the fundamental chemistry of lignin under ionic liquid (IL) pretreatment will facilitate the understanding of biomass recalcitrance involved in pretreatment processes. To examine in greater detail the chemical transformations of lignin under different IL pretreatment conditions without competing reactions from plant polysaccharides, the IL pretreatment of the isolated poplar alkaline lignin (hardwood lignin) under varying IL pretreatment conditions (i.e., 110–170 °C, 1–16 hours) was performed in an appropriate manner. The structural transformations of the lignin have been investigated by elemental analysis, 2D-HSQC spectra, quantitative 13C-NMR spectra, 31P NMR, and GPC analysis. Results revealed that a decrease of aliphatic OH and an increase in phenolic hydroxyl groups occurred in lignin as the pretreatment proceeded. The increased phenolic OH was mainly as a result of cleavage of β-O-4′ linkages, while the reduced aliphatic OH is probably attributed to the dehydration reaction. The cleavage of β-O-4′ linkages, degradation of β–β′ and β-5′ linkages obviously happened at high temperatures and resulted in the decrease of molecular weights. In addition, IL pretreatment selectively degraded the G-type lignin fractions and the condensation reaction took place more easily at S units than G units. Moreover, the demethoxylation preferentially occurred in G units, especially at higher temperatures. It is believed that investigating the fundamental chemistry of lignin during IL pretreatments would be beneficial to optimize and control the pretreatment process.
1 Introduction
A biorefinery is a facility that integrates biomass conversion processes and equipment to produce fuels, power, and chemicals from biomass. Lignocellulosic materials contain high amounts of carbohydrates (cellulose and hemicelluloses), which can be converted into fermentable sugars.1 However, the effective conversion of biomass into fuels (i.e. ethanol) is always impeded by many natural factors, such as crystalline domains of cellulose, degree of lignification, the structural heterogeneity and complexity of cell-wall constituents as well its complex cross-linking, which are defined as biomass recalcitrance.2 The presence of lignin is one of the most significant contributors to biomass recalcitrance.3 The composition of the functional groups of lignin and physical distribution, such as the ratio of syringyl (S) unit to guaiacyl (G) unit, has also been demonstrated to affect the enzymatic hydrolysis of cellulose.4,5 In general, the yield of enzymatic hydrolysis is believed to be enhanced by a low lignin content and a high S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G ratio of the substrate. Lignin appears to limit cellulose hydrolysis by two distinct mechanisms: first by forming a physical barrier that prevents enzyme access and second by non-productive binding cellulolytic enzymes.6
:G ratio of the substrate. Lignin appears to limit cellulose hydrolysis by two distinct mechanisms: first by forming a physical barrier that prevents enzyme access and second by non-productive binding cellulolytic enzymes.6
To break down the biomass recalcitrance, a pretreatment step is extremely needed. Generally, the pretreatment technologies can be classified into biological, physical, chemical, and physicochemical methods. The advantages and disadvantages of different pretreatment methods have been profoundly reviewed recently.7 The purpose of pretreatment generally is as follows: (1) breaking down the compact plant cell wall; (2) increasing the pore size of substrates; (3) removing lignin during pretreatment. Among these pretreatments, ionic liquids (ILs) have attracted significant attention as promising “green” solvents with environmental friendliness.8–10 ILs have been widely studied for the deconstruction and fractionation of lignocellulosic biomass.9 However, the practical applications of ILs are obstructed by various difficulties, such as the responding mechanism of lignin to IL pretreatments. Among numerous ILs, 1-ethyl-3-methylimidazolium acetate, [C2mim][OAc], was demonstrated to effectively extract lignin from wood.11 It has been demonstrated that the IL pretreatment facilitates the “delignification process” and significantly increases the enzymatic hydrolysis efficiency of pretreated biomass. In addition, a part of the dissociated lignin could be collected after the IL pretreatment.12,13 Actually, the structural changes of the lignin in the IL pretreated substrates have also been diagnosed by in situ NMR technique.14,15 More recently, the changes of lignin during the [C2mim][OAc] pretreatment processes were preliminarily achieved by investigating the pretreated biomass via pyrolysis-GC/MS, and two different mechanisms of [C2mim][OAc] pretreatment at different temperatures were tentatively proposed by comparing the changes of the S/G ratios of the pretreated substrates.16 However, the information obtained from in situ NMR and Py-GC/MS of the pretreated substrates is inadequate and imprecise when considering the heterogeneity of the lignin in the plant cell wall.
It is well known that the [C2mim][OAc] pretreatments under different conditions could result in diverse structural changes of cellulose and lignin, which will affect the enzymatic hydrolysis of the pretreated substrates as well as further applications of the obtained lignin fractions in a biorefinery process.14,15,17–19 However, some limitations still existed when investigating the lignin transformations during [C2mim][OAc] pretreatment.15,16 For instance, the accompanying plant polysaccharides will somewhat impede the tracking of changes of lignin during the IL pretreatment. In addition, milled wood lignin (MWL) and cellulolytic enzyme lignin (CEL) are often chosen as references to investigate the structural transformation of the IL pretreatment.12,13 However, this methodology is inappropriate. The main reasons for this are as follows: (1) the isolated native lignin samples (MWL and CEL) are mainly extracted from different locations of the plant cell wall, such as middle lamella (more guaiacyl-type units, MWL) and secondary wall S2 (mainly syringyl-type unit, CEL),20 while the lignin rich in H units has been observed to be laid down in the cell wall very early in the lignification process, which is difficult to extract with traditional methods.21,22 (2) The origin of the degraded IL lignin is still unknown.
Based on the limitations mentioned, it is desired that an improved understanding of the chemical transformations involved in the [C2mim][OAc] pretreatment should be more deeply investigated. Therefore, to examine in greater detail the changes of lignin under [C2mim][OAc] pretreatment without competing reactions from plant polysaccharides, the pretreatments of the isolated alkaline lignins (Triploid of Populus tomentosa Carr.) were carried out under varying IL pretreatment conditions (i.e. 110–170 °C, 1–16 hours).11–13,16,23,24 The structural changes of the pretreated lignin samples were thoroughly investigated by elemental analysis, quantitative 13C-, two-dimensional heteronuclear single quantum coherence (2D-HSQC), 31P-NMR, and gel permeation chromatography (GPC) analysis. To the best of our knowledge, this is the first report on the investigation of the structural transformations of lignin under [C2mim][OAc] pretreatments apart from the biomass matrix. It was anticipated to provide fundamental information on the chemistry of lignin under [C2mim][OAc] pretreatments in the absence of the plant polysaccharide cell wall polymers.
2 Experimental
2.1 Material
Ionic liquid, 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc] (≥98%)), was purchased from Lanzhou Institute of Chemical Physics, Lanzhou, China. All the chemical reagents used were of analytical grade or the best available. Triploid of Populus tomentosa Carr., a fast-growing poplar tree, 6 years old, was harvested from the Shandong province, China. The wood sample was ground to pass through a 0.8 mm size screen and was then extracted with toluene–ethanol (2:1, v/v) in a Soxhlet instrument for 6 h. The extracted poplar sawdust (25 g) was then milled using a planetary ball mill (Fritsch, Germany) in a 500 mL ZrO2 bowl with mixed balls (10 balls of 2 cm diameter and 25 balls of 1 cm for 5 hours). Alkaline lignin fraction (AL) was isolated from the ball-milled wood with 1% sodium hydroxide with a solid-to-liquid ratio of 1:15 (g mL−1) at 75 °C for 3 h according to a previous publication.25 The purification procedure was performed according to the method of Sun et al.26 The chemical composition of the raw poplar wood and AL was determined according to the standard laboratory analytical procedures developed by the National Renewable Energy Laboratory.27 The composition of the poplar wood is cellulose (44.3%), hemicelluloses (19.7%), acid-insoluble lignin (23.8%), acid-soluble lignin (4.1%), and others (8.1%). The composition of the AL is lignin (97.3%), total carbohydrates (1.1%), and others (1.6%).
2.2 Ionic liquid pretreatment
10 g IL was transferred into a 50 mL three-necked round-bottom flask. Then, AL (0.3 g) was slowly added into the IL under quick stirring. The mixture was then placed in an oil bath and heated on a hot plate (IKA RCT basic, Germany) with vigorous magnetic stirring (600 rpm) at given temperatures (110–170 °C) for desired periods (1–16 hours) under a N2 atmosphere (Table 1). After the pretreatment, the regeneration of the dissolved lignin was carried out by precipitation of the IL-lignin solution into 200 mL deionized water (pH = 2.0, 80 °C) under vigorous magnetic stirring. The regenerated lignin was obtained by filtering through a cellulose nitrate membrane filter (pore size 0.45 μm, Whatman®), thoroughly washing with deionized water (pH = 2.0, 60 °C), and freeze-drying.
Table 1 The pretreated condition and quantificational results of the lignins by 2D-HSQC spectra (results expressed per 100Ar)
| Samples |
T (°C) |
T (h) |
Yielda |
β-O-4′ |
β–β′ |
β-5′ |
S/G |
|
The yield was calculated in terms of mass of lignin before and after the IL pretreatment.
|
| AL |
0 |
0 |
100 |
55.8 |
15.6 |
3.4 |
2.25 |
| IL-1 |
130 |
3 |
90 |
52.9 |
14 |
3.1 |
2.43 |
| IL-2 |
110 |
16 |
93 |
53.1 |
11.5 |
2.3 |
2.49 |
| IL-3 |
150 |
1 |
83 |
47.4 |
11 |
2.8 |
2.76 |
| IL-4 |
150 |
3 |
75 |
44.4 |
9.8 |
2.5 |
3.11 |
| IL-5 |
170 |
1 |
60 |
40.3 |
8 |
2.1 |
3.42 |
2.3. Structure elucidation of the lignins
Elemental analysis of the lignins was carried out using an elemental analyzer Vario EL III (Elementar, Hanau, Germany). The oxygen content was deduced from the difference with respect to the total sample. The C900 formula and degree of unsaturation was calculated. The weight-average (Mw) and number-average (Mn) molecular weights of the lignins were determined by GPC according to a previous publication.25 The NMR spectra of the lignins were recorded at 25 °C on a Bruker AVIII 400 MHz (Bruker, Germany) spectrometer according to the previous literature.13,25,28
3 Results and discussion
3.1 2D-HSQC NMR spectra
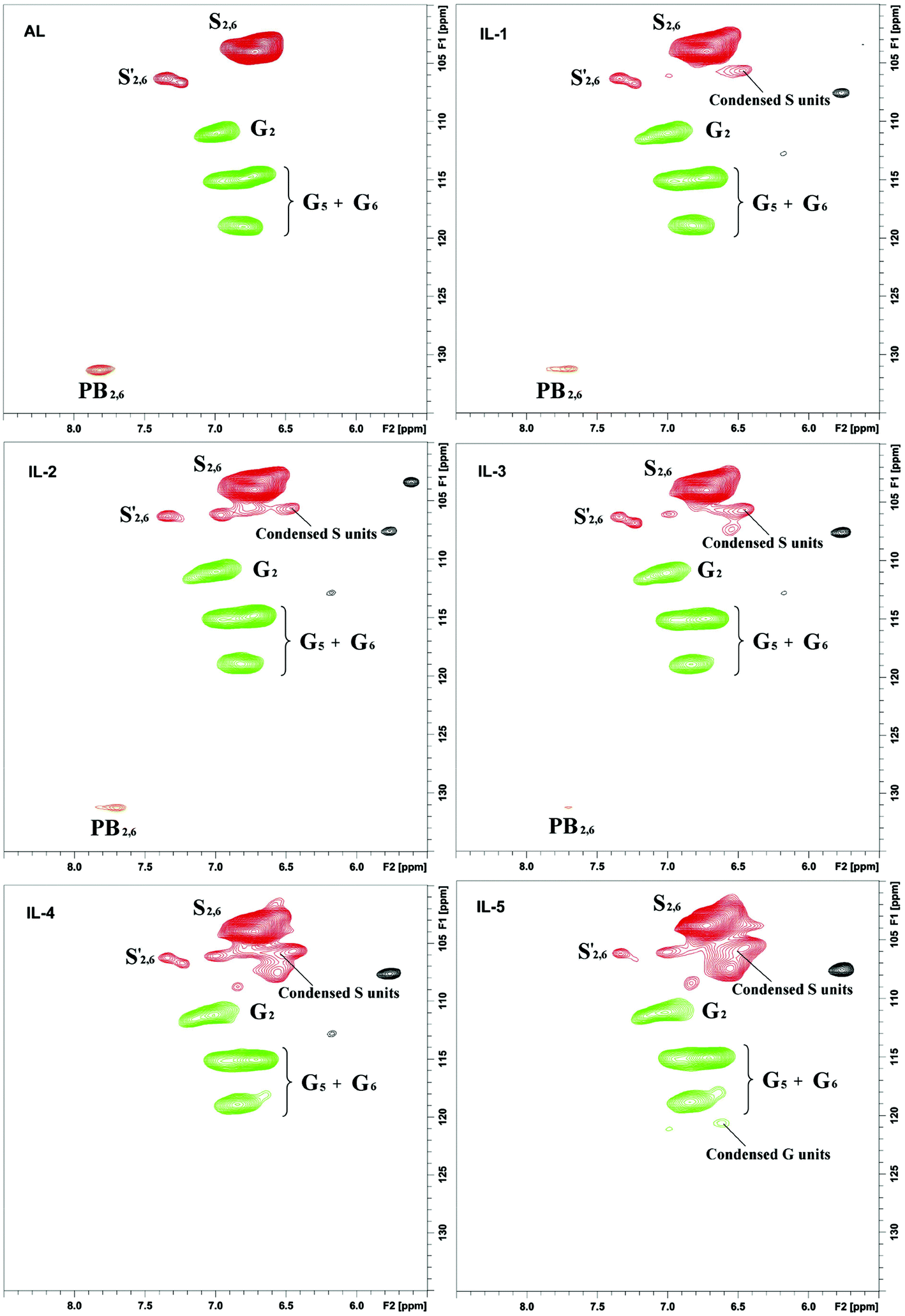
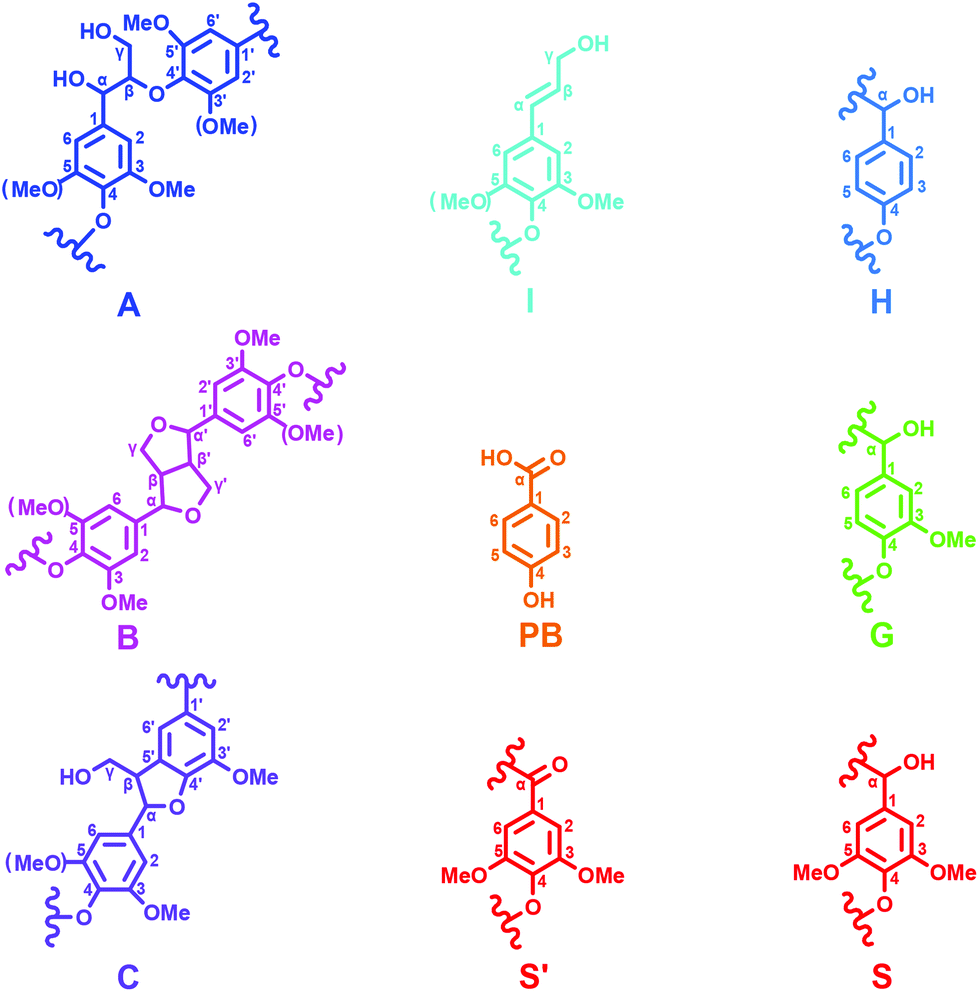
2D-HSQC NMR analysis of untreated (AL) and IL pretreated lignin (samples IL-1–5) provided evidence of structural changes of lignin after IL pretreated. The HSQC spectra of AL and IL-1–5 samples are given in Fig. 1 and 2 and the spectra were annotated with peak assignments based on previous publications (Table S1†).25,29,30Fig. 3 depicts the structures of the identified lignin sub-units in the AL. The inter-unit linkages in AL, β-O-4′ aryl ether (A), resinol (β–β′, B) and phenylcoumaran (β-5′, C) were identified by the cross peaks at δC/δH 71.8/4.88 (Aα), 83.6/4.32 (Aβ(G/H)), 85.8/4.12 (Aβ(s)), 84.8/4.69 (Bα), 86.7/5.49 (Cα), and 53.5/3.10 (Bβ), respectively (Fig. 1). The aromatic lignin units syringyl (S), guaiacyl (G), and p-hydroxybenzoate (PB) units showed prominent correlations at δC/δH 104.0/6.72 (S2/6), 110.9/6.98 (G2), 114.7/6.74 (G5), 119.0/6.80 (G6), 131.3/7.82 (PB2/6), respectively (Fig. 2). Minor amounts of oxidized syringyl units were detected due to the presence of a correlation at δC/δH 106.3/7.35 (S′2/6). Furthermore, a minor C2,6–H2,6 aromatic correlation from H units (δC/δH 127.9/7.19) could only be discovered at lower contour levels (not shown).
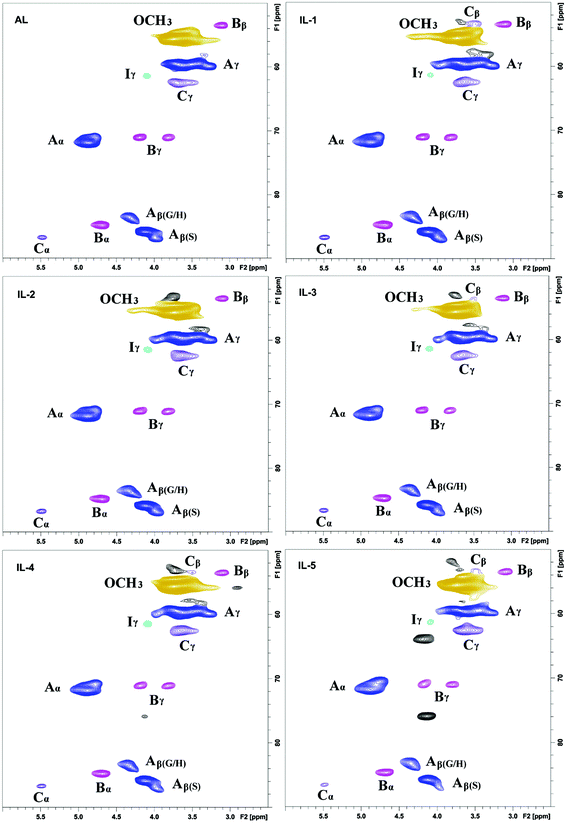 |
| | Fig. 1 Side-chain in the 2D HSQC NMR spectra of the lignins. | |
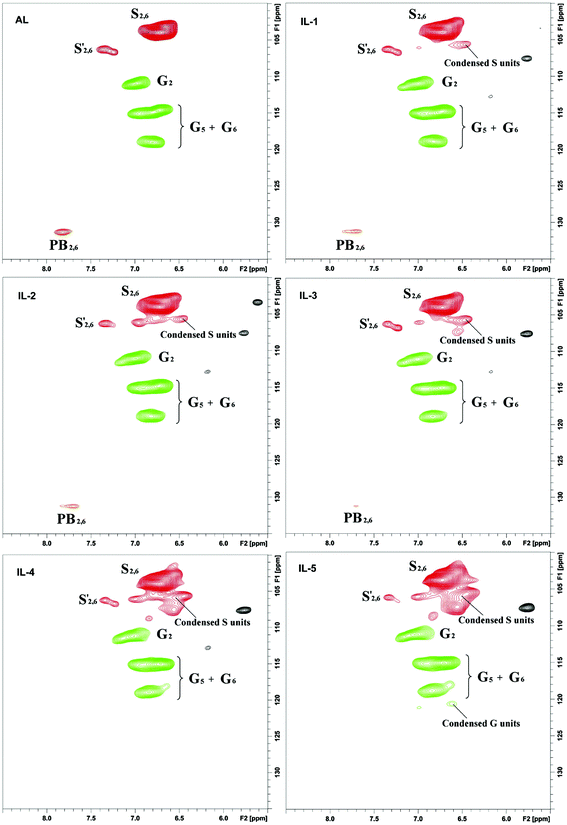 |
| | Fig. 2 Aromatic ring in the 2D HSQC NMR spectra of the lignins. | |
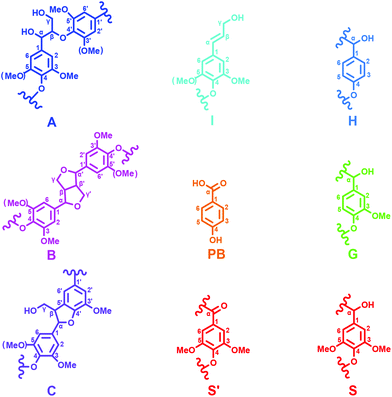 |
| | Fig. 3 Main classical substructures, involving different side-chain linkages, and aromatic units identified by 2D NMR of poplar alkaline lignin: (A) β-O-4′ aryl ether linkages with a free –OH at the γ-carbon; (B) resinol substructures formed by β–β′, α-O-γ′, and γ-O-α′ linkages; (C) phenylcoumaran substructures formed by β-5′ and α-O-4′ linkages; (PB) p-hydroxybenzoic acid; (I) p-hydroxycinnamyl alcohol end groups; (H) p-hydroxyphenyl units; (G) guaiacyl units; (S) syringyl units; (S′) oxidized syringyl units with a Cα ketone. | |
The HSQC spectra of IL pretreated lignin samples illustrate the relative degradation of the lignin side chains and the aromatic units with increasing the pretreated temperature and time as compared to that of AL. According to the computing method of the literature,28,31 the different linkages could be expressed by a comparative mode. For example, the content of β-O-4′ aryl ether in AL is 55.8/100Ar, while it has decreased to 53.1/100Ar in IL-2. As the temperature increased from 130 to 150 °C, the β-O-4′ content decreased to 47.4 and 44.4/100Ar for IL-3 and IL-4, respectively. The content of β-O-4′ content dramatically decreased to 40.3/100Ar under severe conditions (170 °C for 1 h). All of the data herein suggested that the cleavage of β-O-4′ aryl ether (depolymerization) is the predominant reaction during the IL pretreatment at higher temperatures. Besides the cleavage of β-O-4′, other carbon–carbon linkages (such as β–β′ and β-5′), had also undergone degradation. As shown in Table 1, the content of β–β′ and β-5′ diminished as the temperature rose and time prolonged. Moreover, the correlations from the condensed S and G units increased with the elevated pretreated temperatures. It means that a recondensation reaction of the lignin took place during the IL pretreatment.
Besides β-O-4′ aryl ether linkages cleavage and carbon–carbon linkages degradation, change of the S/G ratio in lignin was another prominent structural alteration observed after the IL pretreatment. Most publications about the IL pretreatment indicated that the G-type lignin was more easily degraded and collected than the S-type lignin during treatment of the biomass with ionic liquid at 110–120 °C for some time.12,16 Interestingly, our data also suggested that the G-type lignin was more easily degraded, as revealed by a slightly elevated S/G ratio in IL-1 and IL-2. With the temperature increase, the S/G ratio gradually increased and reached a plateau (3.42) in IL-5. The increased S/G ratio at least suggested that the G-type lignin was more easily degraded during the IL pretreatment, but the degraded G-type lignin could not be collected after the IL pretreatment. Moreover, the issue of whether the IL pretreatment leads to condensation of lignin aromatic units needs to be confirmed. After examining the 2D-HSQC spectra of the lignin fractions before and after the IL pretreatment, it was found that the S units were more easily condensed as compared to the G units in the recovered lignin (IL-1–5, Fig. 2). Furthermore, some newly appeared signals at δC/δH 103.2/5.61 and 107.4/5.76 (not shown in current contour level) suggested that the conjugated double bonds increased in IL pretreated lignin fractions (samples IL-1–5), which were also evidence of dehydration reaction during the pretreatment.
3.2 Quantitative 13C-NMR spectra
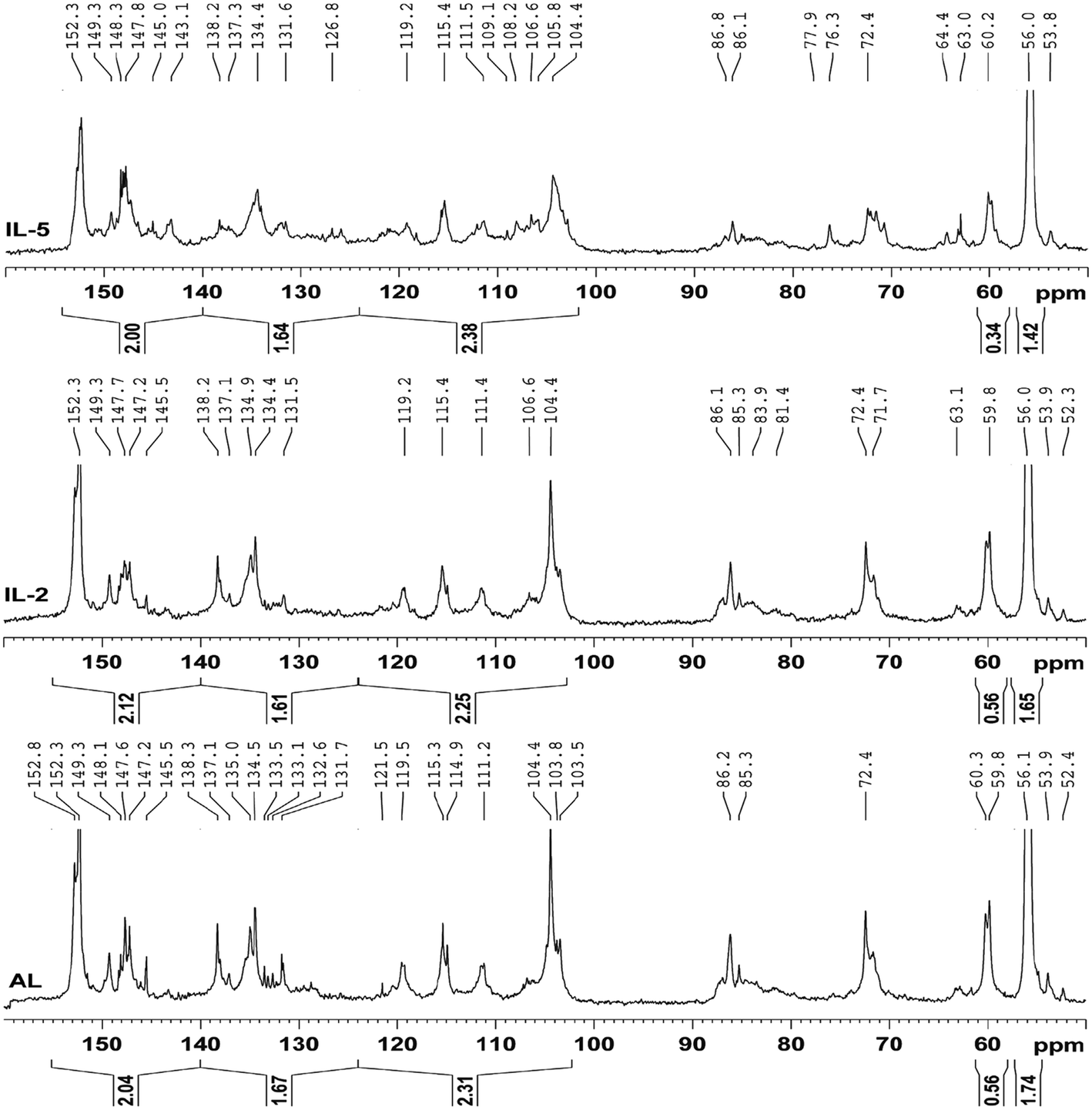
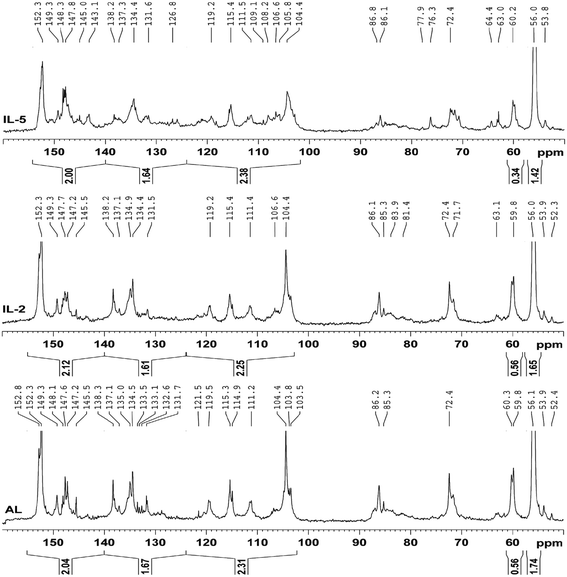
Quantitative 13C-NMR is frequently used to determine the changes of the linkages in lignin during the pretreatments.13,31,32 The 13C-NMR spectra (Fig. 4) of the IL pretreated lignin samples were analyzed and compared with that of the control lignin (AL). It was observed that the three spectra were similar except for different intensities of these signals, for example, the reduced signals for etherified S3,5 in IL-5 suggested that β-O-4′ was cleaved to some extent during the IL pretreatment at the higher temperature. The reduced signal at 138.2 ppm (C-4 in etherified S) in IL-5 was also attributed to the cleavage of etherified S units. In addition, the newly appeared signals at 145.0 and 126.8 ppm in the pretreated lignin samples were probably assigned to the C-4 in etherified 5-5′ and C-5/C-5′ in non-etherified 5-5′ linkages, suggesting that the 5-5′ linkages were formed during the IL pretreatment. Moreover, the broadened signals (121, 105–110 ppm) for G and S units in IL-5 are probably attributed to the newly generated G and S units, implying that some homologues of normal S and G units were formed. By contrast, there was no obvious difference between AL and IL-2, implying that treatment under mild temperature (110 °C, 16 h) had no obvious effect on the structural features of the lignin. This seems to be inconsistent with some previous publications,12,13 which stated that the lignin degraded and recovered after the IL pretreatment is somewhat different from native MWL, for example, a decrease in the molecular weight, S/G ratio, and β-O-4′ linkages occurred in IL lignin (ILL). However, it should be noted that ILL was extracted with 50% acetone from the IL-biomass mixture, which was different from 96% dioxane-extracted MWL. Thus, the samples obtained are still inadequate to investigate the effect of the IL pretreatment on the structural feature of lignin.
 |
| | Fig. 4 Quantitative 13C NMR spectra of the lignins. | |
A further comparison of the integration values (Table 2) of the protonated aromatic (δ 124.0–102.0 ppm), condensed aromatic (δ 140.0–124.0 ppm) and oxygenated aromatic (δ 155.0–140.0 ppm) regions can provide detailed information on the structural features of the lignin fractions.33 However, it was found that there were no obvious differences between the contents of aromatic C–O, aromatic C–C, and aromatic C–H in these lignin samples, suggesting that the IL pretreatment had a slight effect on the content of aromatic rings. By contrast, the content of β-O-4′ linkages was unaltered in IL-2 but decreased sharply from 0.56/Ar in AL to 0.34/Ar in IL-5, suggesting that the pretreatment under mild temperature (110 °C) has a mild effect on the content of β-O-4′ linkages, while higher temperature (170 °C) has a significant effect on the content of β-O-4′ linkages. In addition, the content of OCH3 was reduced from 1.74/Ar in AL to 1.65/Ar in IL-2 and further decreased to 1.42/Ar in IL-5, implying that demethoxylation occurred in the IL pretreatment process. Considering the higher S/G ratios (from 2D-HSQC) of IL pretreated lignin, it was concluded that demethoxylation preferentially took place in G units, especially at higher temperatures.
Table 2 The assignment and quantification of the signals of the 13C-NMR spectra (results expressed per Ar)
|
δ
|
Assignment |
AL |
IL-2 |
IL-5 |
| 155.0–140.0 |
Aromatic C–O |
2.04 |
2.12 |
2.00 |
| 140.0–124.0 |
Aromatic C–C |
1.67 |
1.61 |
1.64 |
| 124.0–102.0 |
Aromatic C–H |
2.31 |
2.25 |
2.38 |
| 61.3–58.0 |
β-O-4′ linkages |
0.56 |
0.56 |
0.34 |
| 58.0–54.0 |
CH3O |
1.74 |
1.65 |
1.42 |
3.3
31P-NMR spectra
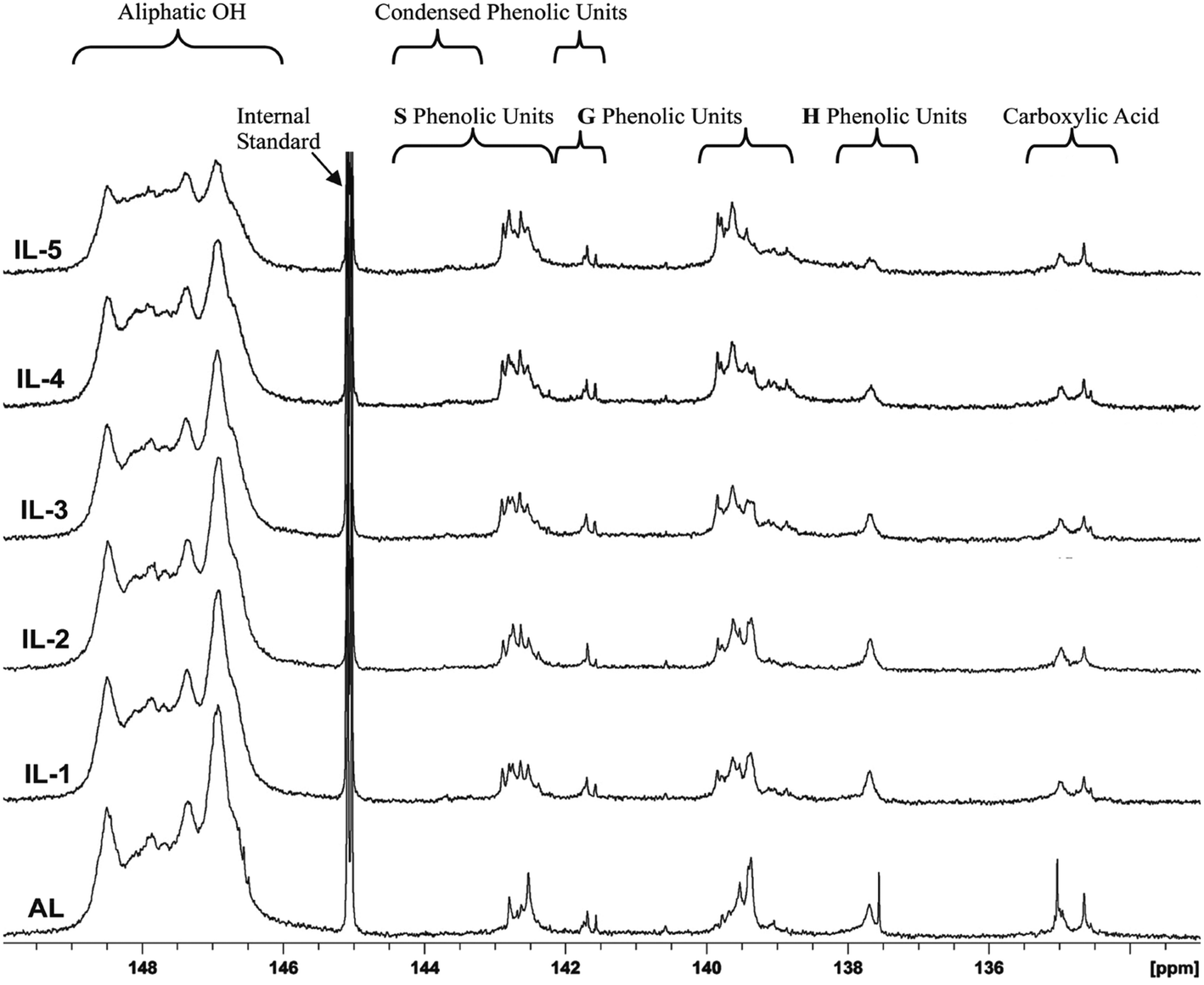
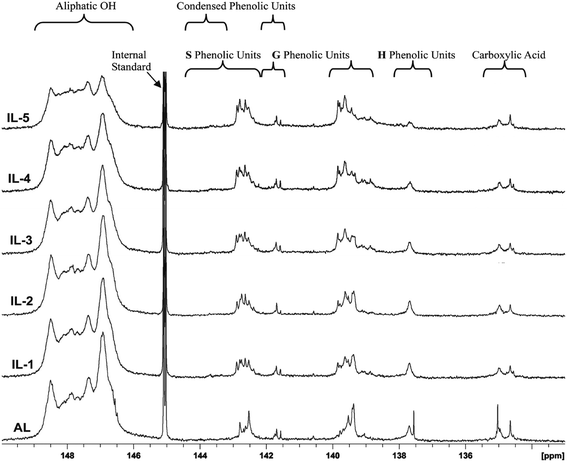
31P NMR spectroscopy is a facile and direct analysis tool for quantifying the major hydroxyl groups in lignin.25,34,35 To follow the changing of different hydroxyl groups in these lignin fractions, typical 31P-NMR spectra of phosphitylated poplar lignin and the different hydroxyl group contents in these lignins are shown in Fig. 5 and Table 3, respectively. It was observed that the aliphatic hydroxyl groups in AL were mainly from the side-chain of lignin since AL is free of carbohydrates. After the IL pretreatment, the content of aliphatic OH was found to be decreased under the severe pretreatment conditions. By contrast, the phenolic hydroxyl greatly increased after pretreatment, reached 1.7 mmol g−1 in IL-5, which is nearly equivalent to two times of AL. The increased phenolic OH was mainly as a result of cleavage of β-O-4′ linkages, while the reduced aliphatic OH is attributed to the dehydration reaction. However, the COOH content was relatively stable or slightly decreased under pretreatment probably because the oxidation reaction was not evident under the conditions given, which further confirmed that the dehydration reaction rather than oxidation reaction occurred during the IL pretreatment. In addition, it was also observed that the increase of non-condensed G-OH and non-condensed S-OH was slow under mild conditions (samples IL-1–2), but the increment was obvious in the samples treated under harsh conditions (samples IL-3–5), especially in the content of non-condensed S-OH. This suggested that the cleavage of β-O-4′ linkage was obvious under harsh conditions, which also explains that the fact that preferential breakdown of S-lignin in both eucalyptus and switchgrass occurred at 160 °C in a recent publication because β-O-4′ linkage is mainly composed of S-type lignin units.16 Moreover, the content of non-condensed H-OH did not increase even if the demethoxylation reaction took place, suggesting that the product of demethoxylation of G-type lignin units was not similar to normal H units, thus it was not detected by 31P-NMR spectra.
 |
| | Fig. 5 Quantitative 31P NMR spectra of the lignins. | |
Table 3 Quantification of the functional groups in the lignins by quantitative 31P-NMR method
| Samples |
AL |
IL-1 |
IL-2 |
IL-3 |
IL-4 |
IL-5 |
| Aliphatic |
5.36 |
4.84 |
5.10 |
4.81 |
4.49 |
3.66 |
| Condensed S |
0 |
0.06 |
0.04 |
0.02 |
0.05 |
0.10 |
| Non-condensed S |
0.29 |
0.34 |
0.34 |
0.39 |
0.45 |
0.54 |
| Condensed G |
0.07 |
0.10 |
0.09 |
0.09 |
0.12 |
0.15 |
| Non-condensed G |
0.46 |
0.49 |
0.49 |
0.58 |
0.71 |
0.78 |
| Non-condensed H |
0.14 |
0.13 |
0.12 |
0.10 |
0.11 |
0.13 |
| Total phenolic |
0.96 |
1.12 |
1.08 |
1.18 |
1.44 |
1.70 |
| COOH |
0.18 |
0.18 |
0.16 |
0.13 |
0.16 |
0.19 |
3.4 Elemental analysis
Table 4 shows the elemental composition and C900 formula as well as the degree of unsaturation in the control and IL pretreated lignin samples. The degree of unsaturation in the AL is 365, while it decreased to 364 and 361 in IL-1 and IL-2, respectively. This result suggested that the lignin samples pretreated under mild conditions have less unsaturated bonds, such as unsaturated double bonds in the side-chain of the lignin. However, the lignin pretreated under harsh conditions had a higher degree of unsaturation, implying that more double bonds were formed during the IL pretreatments, which were also revealed by recent publications.13,15 This was also in line with the results of 2D-HSQC spectra analysis in this study.
Table 4 Elemental analysis and C900 empirical formula of the lignins
| Samples |
C (%) |
H (%) |
O (%) |
C900 formula |
Degree of unsaturation |
| AL |
60.17 |
5.97 |
33.86 |
C900H1071O379 |
365 |
| IL-1 |
60.02 |
5.97 |
34.01 |
C900H1074O382 |
364 |
| IL-2 |
60.39 |
6.04 |
33.57 |
C900H1080O375 |
361 |
| IL-3 |
60.37 |
5.75 |
33.88 |
C900H1028O378 |
387 |
| IL-4 |
60.86 |
5.92 |
33.22 |
C900H1050O368 |
376 |
| IL-5 |
61.30 |
5.85 |
32.85 |
C900H1030O361 |
386 |
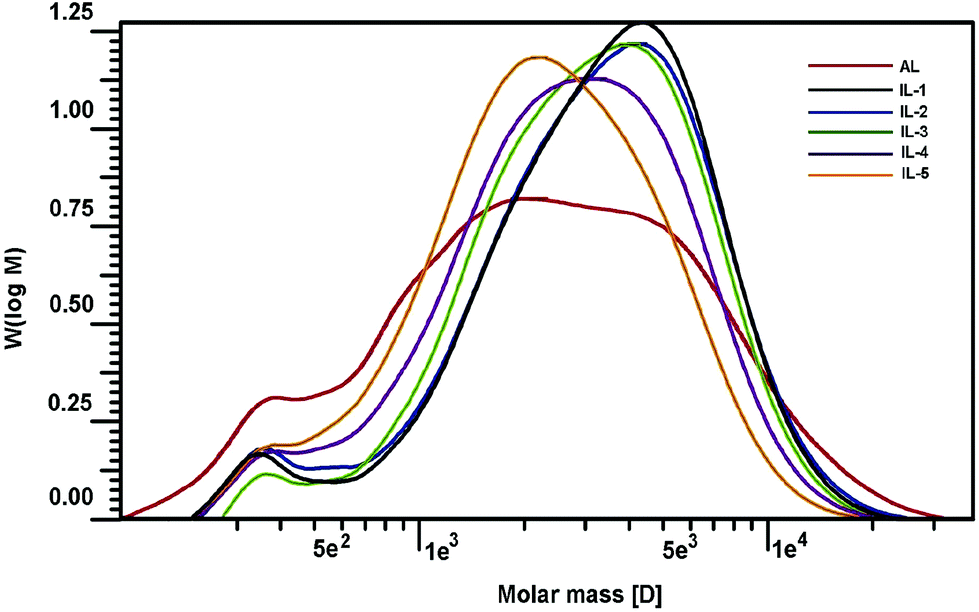
3.5 Molecular weights
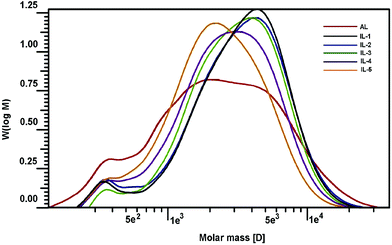
Changes in molecular weights of lignin can provide important insights into lignin's fragmentation and recondensation reactions during the IL pretreatment. While cleavage of β-O-4′ linkages can result in a decrease in the molecular weight of lignin, condensation reactions usually lead to a condensed and heterogeneous lignin structure with an increase in the molecular size.36 Two recent publications also demonstrated that lignin subunits after the pretreatment of [C2mim][OAc] were released via dissociation or depolymerization, thus resulting in the reduced size and shape of the lignin.37,38 To determine the effect of the IL pretreatment on the molecular weight of lignin, the AL and pretreated lignin samples (IL-1–5) were acetylated and subsequently analyzed by GPC. Table 5 and Fig. 6 illustrate the changes in the molecular weight of the lignins with respect to temperature and extent of the IL pretreatment. As shown in Table 5, the molecular weight (Mw) of the starting lignin (AL) was 3550 g mol−1, and its polydispersity (Mw/Mn) was 2.71. After pretreatment under mild conditions (IL-1, IL-2, and IL-3), it was observed that the Mw increased to 4080, 4090, and 3830 g mol−1, respectively. With further pretreatment under harsh conditions (IL-4 and IL-5), the Mw was decreased (3380 and 2890 g mol−1) as compared to AL. The depolymerization and recondensation of lignin were two competing reactions during [C2mim][OAc] pretreatment, which was observed by 2D-HSQC analysis aforementioned. When the pretreatment was done under mild conditions, the recondensation reaction occurred (confirmed by corresponding 2D-HSQC spectra). Meanwhile, some “low-molecular” fractions may be lost during the dissolution and regeneration process, which was supported by the reduced polydispersities of the pretreated samples. All these led to the higher molecular weights of the recovered samples (IL-1–3). In addition, the reduced yield (83–93%) of the recovered samples might be an indication of fractionation upon regeneration. However, the depolymerization reaction became the leading reaction under harsh conditions, which leads to the reduced Mw of samples IL-4 and IL-5 even if some lignin fragments were lost during the IL pretreatment (yield, 60–75%). Besides, all the polydispersities of the pretreated lignins were reduced, suggesting that the regenerated lignin had homogeneous molecular structures, which was also observed by the SEM technique in a recent literature.39
 |
| | Fig. 6 GPC curves of the lignins. | |
Table 5 Weight-average (Mw) and number-average (Mn) molecular weights (g mol−1) and polydispersity (Mw/Mn) of the lignins
| Samples |
AL |
IL-1 |
IL-2 |
IL-3 |
IL-4 |
IL-5 |
|
M
w
|
3550 |
4080 |
4090 |
3830 |
3380 |
2890 |
|
M
n
|
1310 |
2120 |
2070 |
2200 |
1790 |
1580 |
|
M
w/Mn |
2.71 |
1.92 |
1.98 |
1.74 |
1.89 |
1.83 |
3.6 Implications
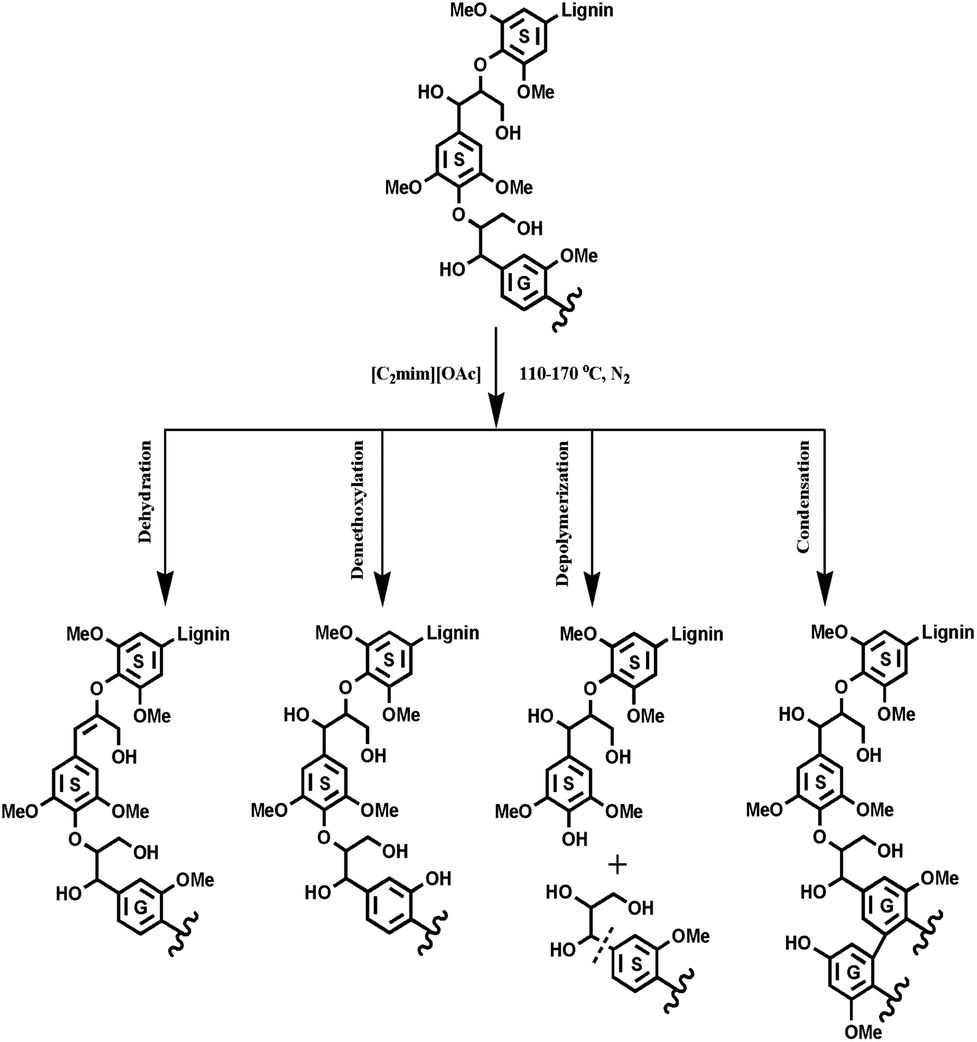
With respect to the applications of IL ([C2mim][OAc]) in biomass pretreatment, exploration of the inherent mechanism of lignin transformations is an important topic. The previous publications reported that the IL pretreatment selectively degraded the G-type lignin fractions under mild conditions (110 and 120 °C), however, at higher temperatures, an opposite tendency was found, that is, it is easier to remove the S-type lignin than the G-type lignin at 160 °C. The potential reason for this still waits to be proved although two different mechanisms were proposed to explain the experimental results of IL pretreated biomass.16 However, in this study, IL pretreatment was found to selectively degrade the G-type lignin fractions in all pretreatments, no matter under mild or higher temperatures, as revealed by increasing the S/G ratio of the pretreated lignin samples. By combining all the data herein, we seemingly obtain a slightly different mechanism as compared to the previous publication,16 in which the authors found that the preferential breakdown of the S-lignin in both eucalyptus and switchgrass at higher pretreatment temperatures and prolonged times, whereas, breakdown of the G-lignin for eucalyptus and no preferential break down of either the S- or the G-lignin in switchgrass was observed at lower pretreatment temperatures (120 °C). In fact, the conclusion is appropriate and convincing for eucalyptus wood, in which lignin is a typical G–S type and similar to poplar lignin. However, the switchgrass lignin is a G-rich lignin (S/G < 0.2), which presents an abnormal tendency during the IL pretreatments as compared to the eucalyptus lignin. The abnormal tendency was also attributed to the partial effect of polysaccharides in the compact biomass matrix, which may facilitate autohydrolysis of the acetyl group at high temperature, thus accelerating the hydrolysis of β-O-4′ linkages, which are mainly composed of the G- and S-type units in switchgrass. In summary, the following results could be obtained by this study: (1) the increased phenolic OH was mainly as a result of cleavage of β-O-4′ linkages, while the reduced aliphatic OH is probably attributed to the dehydration reaction. (2) The cleavage of β-O-4′ linkages, degradation of β–β′ and β-5′ linkages obviously happened at high temperatures and resulted in the decrease of the molecular weights. (3) [C2mim][OAc] pretreatment selectively degraded the G-type lignin fragments and the condensation reaction took place more easily at the S units than the G units. (4) The demethoxylation preferentially occurred in G units, especially at higher temperatures. Based on these results, the proposed [C2mim][OAc] pretreatment mechanism of lignin is shown in Fig. 7.
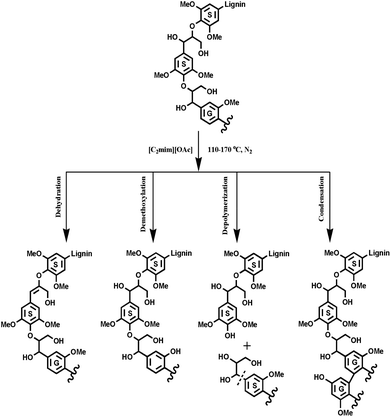 |
| | Fig. 7 The proposed [C2mim][OAc] pretreatment mechanism of lignin. | |
4 Conclusions
The present study demonstrates the impact of [C2mim][OAc] pretreatment at different temperatures and times on the structural features of lignin apart from the plant cell wall matrix. The results suggest that there is a decrease of aliphatic OH, which is accompanied by a significant increase in the phenolic hydroxyl groups in lignin with increasing temperature and time of the IL pretreatment. The cleavage of the β-O-4′ linkage, degradation of β–β′ and β-5′ linkages as well as demethoxylation reactions occurred at higher temperatures. Meanwhile, cleavage of the β-O-4′ linkages accompanied with reduced molecular weight was distinct at higher temperatures. In addition, [C2mim][OAc] pretreatment selectively degraded the G-type lignin fragments and the condensation reaction took place more easily at the S units than the G units. This study provides an enhanced understanding of the fundamental [C2mim][OAc] pretreatment lignin chemistry and will facilitate improved methodology to optimize the pretreatment of lignocellulosic materials for bio-ethanol production.
Acknowledgements
We are grateful for the financial support of this research from National Science Foundation of China (31110103902) and Major State Basic Research Projects of China (973-2010CB732204, 863-2012AA023204).
References
- A. K. Sangha, L. Petridis, J. C. Smith, A. Ziebell and J. M. Parks, Environ. Prog. Sustain., 2012, 31, 47–54 CrossRef CAS.
- M. E. Himmel, S. Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS PubMed.
- T. Q. Yuan, F. Xu and R. C. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 346–352 CrossRef CAS.
- S. Nakagame, R. P. Chandra, J. F. Kadla and J. N. Saddler, Biotechnol. Bioeng., 2011, 108, 538–548 CrossRef CAS PubMed.
- M. H. Studer, J. D. DeMartini, M. F. Davis, R. W. Sykes, B. Davison, M. Keller, G. A. Tuskan and C. E. Wyman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6300–6305 CrossRef CAS PubMed.
- X. J. Pan, D. Xie, N. Gilkes, D. J. Gregg and J. N. Saddler, Appl. Biochem. Biotechnol., 2005, 121, 1069–1079 CrossRef.
-
(a) X. B. Zhao, L. H. Zhang and D. H. Liu, Biofuel. Bioprod. Bior., 2012, 6, 465–482 CrossRef CAS;
(b) X. B. Zhao, L. H. Zhang and D. H. Liu, Biofuel. Bioprod. Bior., 2012, 6, 561–579 CrossRef CAS.
- T. Welton, Green Chem., 2011, 13, 225–225 RSC.
- A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550–583 RSC.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen, R. Sjöholm and J. P. Mikkola, Ind. Crop. Prod., 2010, 32, 175–201 CrossRef PubMed.
- N. Sun, M. Rahman, Y. Qin, M. L. Maxim, H. Rodríguez and R. D. Rogers, Green Chem., 2009, 11, 646–655 RSC.
- J. Y. Kim, E. J. Shin, I. Y. Eom, K. Won, Y. H. Kim, D. Choi, I. G. Choi and J. W. Choi, Bioresour. Technol., 2011, 102, 9020–9025 CrossRef CAS PubMed.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, J. Agric. Food Chem., 2013, 61, 635–645 CrossRef CAS PubMed.
- Ö. P. Çetinkol, D. C. Dibble, G. Cheng, M. S. Kent, B. Knierim, M. Auer, D. E. Wemmer, J. G. Pelton, Y. B. Melnichenko and J. Ralph, Biofuels, 2010, 1, 33–46 CrossRef.
- K. M. Torr, K. T. Love, Ö. P. Çetinkol, L. A. Donaldson, A. George, B. M. Holmes and B. A. Simmons, Green Chem., 2012, 14, 778–787 RSC.
- P. Varanasi, P. Singh, R. Arora, P. D. Adams, M. Auer, B. A. Simmons and S. Singh, Bioresour. Technol., 2012, 126, 156–161 CrossRef CAS PubMed.
- G. Cheng, P. Varanasi, C. Li, H. Liu, Y. B. Melnichenko, B. A. Simmons, M. S. Kent and S. Singh, Biomacromolecules, 2011, 12, 933–941 CrossRef CAS PubMed.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368–1376 CrossRef CAS PubMed.
- C. Li, L. Sun, B. A. Simmons and S. Singh, BioEnerg. Res., 2013, 6, 14–23 CrossRef CAS.
- P. Whiting and D. Goring, Wood Sci. Technol., 1982, 16, 261–267 CrossRef CAS.
- A. Ziebell, K. Gracom, R. Katahira, F. Chen, Y. Q. Pu, A. J. Ragauskas, R. A. Dixon and M. Davis, J. Biol. Chem., 2010, 285, 38961–38968 CrossRef CAS PubMed.
- J. Ralph, T. Akiyama, H. D. Coleman and S. D. Mansfield, BioEnergy Res., 2012, 5, 1009–1019 CrossRef CAS.
- W. Y. Li, N. Sun, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 2038–2047 RSC.
- J. A. Perez-Pimienta, M. G. Lopez-Ortega, P. Varanasi, V. Stavila, G. Cheng, S. Singh and B. A. Simmons, Bioresour. Technol., 2013, 127, 18–24 CrossRef CAS PubMed.
- T. Q. Yuan, S. N. Sun, F. Xu and R. C. Sun, J. Agric. Food Chem., 2011, 59, 6605–6615 CrossRef CAS PubMed.
- R. C. Sun, J. M. Fang and J. Tomkinson, J. Wood Chem. Technol., 1999, 19, 335–356 CrossRef CAS.
-
A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter,D. Templeton and D. Crocker, Laboratory analytical procedure (LAP): Determination of structural carbohydrates and lignin in biomass, Technical Report: NREL/TP-510-42618, National Renewable Energy Laboratory, Golden, CO, USA, 2008.
- M. Sette, R. Wechselberger and C. Crestini, Chem.–Eur. J., 2011, 17, 9529–9535 CrossRef CAS PubMed.
- T. Q. Yuan, S. N. Sun, F. Xu and R. C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- J. C. Del Rio, J. Rencoret, G. Marques, J. B. Li, G. Gellerstedt, J. Jiménez-Barbero, Á. T. Martínez and A. Gutiérrez, J. Agric. Food Chem., 2009, 57, 10271–10281 CrossRef CAS PubMed.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, Materials, 2013, 6, 359–391 CrossRef CAS.
-
J. Ralph and L. L. Landucci, NMR of lignins, CRC Press, 2010, pp. 137–244 Search PubMed.
- B. B. Hallac, Y. Pu and A. J. Ragauskas, Energy Fuels, 2010, 24, 2723–2732 CrossRef CAS.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154–3166 CAS.
- C. Crestini and D. S. Argyropoulos, J. Agric. Food Chem., 1997, 45, 1212–1219 CrossRef CAS.
- Y. Pu, F. Hu, F. Huang, B. H. Davison and A. J. Ragauskas, Biotechnol. Biofuels, 2013, 6, 1–13 CrossRef PubMed.
- G. Cheng, M. S. Kent, L. He, P. Varanasi, D. Dibble, R. Arora, K. Deng, K. Hong, Y. B. Melnichenko, B. A. Simmons and S. Singh, Langmuir, 2012, 28, 11850–11857 CrossRef CAS PubMed.
- A. George, K. Tran, T. J. Morgan, P. I. Benke, C. Berrueco, E. Lorente, B. C. Wu, J. D. Keasling, B. A. Simmons and B. M. Holmes, Green Chem., 2011, 13, 3375–3385 RSC.
- W. Ji, Z. Ding, J. Liu, Q. Song, X. Xia, H. Gao, H. Wang and W. Gu, Energy Fuels, 2012, 26, 6393–6640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Assignment of HSQC spectra and experimental section. See DOI: 10.1039/c3gc41752b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.