Depolymerization of organosolv lignin to aromatic compounds over Cu-doped porous metal oxides†
Katalin
Barta
,
Genoa R.
Warner
,
Evan S.
Beach
and
Paul T.
Anastas
*
Center for Green Chemistry & Green Engineering, Yale University, 225 Prospect Street, New Haven, CT, USA. E-mail: paul.anastas@yale.edu; Fax: +1-203-436-8574; Tel: +1-203-432-5215
First published on 17th October 2013
Abstract
Isolated, solvent-extracted lignin from candlenut (Aleurites moluccana) biomass was subjected to catalytic depolymerization in methanol with an added pressure of H2, using a porous metal oxide catalyst (PMO) derived from a Cu-doped hydrotalcite-like precursor. The Cu-PMO was effective in converting low-molecular weight lignin into simple mixtures of aromatic products in high yield, without char formation. Gel permeation chromatography was used to track changes in molecular weight as a result of the catalytic treatments and product mixtures were characterized by 1H and 13C NMR spectroscopy. In the temperature range 140–220 °C, unusual C9 catechols were obtained with high selectivity. Lignin conversion of >90% and recovery of methanol-soluble products in yields of was >70% was seen at 180 °C with optimized catalyst and biomass loadings. At 140 °C, 4-(3-hydroxypropyl)-catechol was the major product and could be isolated in high purity.
Introduction
Depolymerization of lignocellulosic materials found in renewable biomass has been put forward as an important strategy for replacing petroleum-derived platform chemicals with more sustainable alternatives,1,2 in accordance with the principles of green chemistry.3,4 Efficient conversion of bio-derived polymers is challenging due to the variability and complexity of starting materials; this requires careful control of reaction conditions and use of catalysts capable of selective bond cleavage reactions.Transformations of lignin have been developed with the goal of preserving aromatic structure that is difficult to otherwise derive from renewable resources and is desirable for functional performance in a variety of fields ranging from medicinal chemistry and drug design to polymers and coatings. Many approaches have been reported for lignin depolymerization, including pyrolysis and liquefaction techniques, acidic or basic hydrolysis and more selective depolymerization via catalytic cleavage of C–C or C–O bonds in lignin interunit linkages.5–9 Recently, Lercher and co-workers described base-catalyzed depolymerization of lignin to aromatic monomers with high yield, with product mixtures depending strongly on reaction conditions.10 Efficient hydrogenolysis of in situ or isolated lignins was reported by Torr and co-workers using palladium on carbon resulting in good yields of bio-oils mainly comprised of aromatic monomers and dimers.11 Depolymerization and hydrodeoxygenation of switchgrass lignin with formic acid was described by Jones.12 Further, cleavage and hydrodeoxygenation of model chemicals relevant to lignin conversion with synergistic Pd/Zn catalysts was reported by Abu-Omar et al.13
Growing understanding of metal scarcity issues and vulnerability of existing supplies14 has led to increasing study of catalysts based on earth-abundant metals. There are several reports of Ni-promoted transformations mainly applied to lignin model compounds,15–19 but also biomass samples, for example Ni/AlSBA15-catalyzed depolymerisation of organosolv lignin to generate phenolic monomers with low char formation.20
Ford and co-workers described quantitative conversion of organosolv lignin21 as well as cellulose and crude lignocellulosic biomass22 over a hydrotalcite-derived Cu-doped porous metal oxide (PMO) catalyst, with no char formation. Those processes take place in supercritical methanol and the hydrogen equivalents needed for depolymerization and further reduction are obtained by solvent reforming. Those studies targeted liquid fuels as products therefore extensive reduction and deoxygenation was desired; although aromatic structures were observed as intermediates in hydrogenolysis of organosolv lignin, these rapidly underwent further reduction to cyclohexanol derivatives.21 A summary of reaction conditions from selected studies of the conversion of lignin and lignocellulosic biomass is given in Table 1. In this work, we have shown that Cu-PMO in conjunction with milder reaction pressures and temperatures promotes more selective chemistry and generation of relatively clean product mixtures of aromatic compounds.
| Authora | Biomass source | Catalyst | Time (h) | Temp. (°C) | Conversion | Products |
|---|---|---|---|---|---|---|
| a Citation in parentheses. b Combined yield of liquid products and estimated volatiles. | ||||||
| Lercher (10) | Organosolv lignin (Sigma-Aldrich) | NaOH | <1 | 240–340 | 85% | Monomers (syringol and guaiacol derivatives), oligomers, water-soluble byproducts |
| Torr (11) | Organosolv lignin, wood, or steam-exploded wood fiber (P. radiata) | Pd/C | 2–24 | 195 | 79–89% | Dihydroconiferyl alcohol, 4-propylguaiacol, predominantly dimers and oligomers |
| Jones (12) | Organosolv lignin (switchgrass) | Pt/C | 1–20 | 350 | >96% | Monomers (phenols and guaiacol derivatives), oligomers, char |
| Abu-Omar (13) | β-O-4 synthetic polymer | Pd/C + ZnCl2 | 2 | 150 | >99% | Monomers (guaiacol derivatives) |
| Labidi (20) | Organosolv lignin (O. eurpoea) | Ni/SBA15 | 0.5 | 150 | 35–45% | Monomers (phenols, guaiacol derivatives), oligomers |
| Ford (21) | Organosolv lignin (poplar) | Cu-PMO | 2–12 | 300 | >99% | Monomers (partially oxygenated cyclohexyl derivatives) |
| Ford (22) | Wood flour (ponderosa pine), sawdust (maple) | Cu-PMO | 8 | 300–320 | >99% | Monomers (partially oxygenated cyclohexyl derivatives, C2–C6 aliphatic alcohols and ethers) |
| This work | Organosolv lignin (A. moluccana) | Cu-PMO | 8–20 | 140–220 | 60–93%b | Predominantly monomers (catechol derivatives), some oligomers |
Experimental
Materials
Candlenut nutshells (Aleurites moluccana; Euphorbiaceae) were provided by African Power Initiative (Kampala, Uganda). The nutshells were rinsed with water to remove soil and debris then air-dried at room temperature. The moisture content of the nutshells was determined to be 9.6% by drying to constant weight at 105 °C. The air-dried material was milled and passed through standard sieves (ASTME-11) to obtain particles between 0.25–1 mm in diameter (60–18 mesh).23 Methanol (99.8%, anhydrous) used as a reaction medium was purchased from ACROS Organics. Tetrahydrofuran (OMNISOLV, >99.9%, uninhibited) used for chromatography was purchased from VWR. Solvents for extraction procedures and catalyst washing were reagent grade. Mg(NO3)2·4H2O (≥99.0%), Cu(NO3)2·2.5H2O (98%), Cr(NO3)3·9H2O (99.0%), and Mn(NO3)2·4H2O (>97.0%) were purchased from Sigma Aldrich, La(NO3)3·6H2O (>99.0%) was obtained from Fluka, Al(NO3)3·9H2O (98.7%) and Na2CO3·H2O (p.a.) were from J. T. Baker and NaOH (98.6%) was purchased from Mallinckrodt Chemicals. The stationary phase for column chromatography was SiliaFlash F60 silica gel (SiliCycle Inc., 40–63 μm, 60 Å). All chemicals were used as received without further purification.Equipment
NMR spectra were recorded at 25 °C using an Avance 500 MHz spectrometer (Bruker Biospin, Billerica, MA). GPC was performed using a Shimadzu Prominence Series HPLC (LC-20AD) with column oven (CTO-20A) and refractive index detector (RID-10A) both maintained at 40 °C. Two Phenogel columns (Phenomex, Torrance, CA, 5 μm, 300 × 7.8 mm) were placed in series, one with 50 Å and one with 100 Å pore size. The system was calibrated with polystyrene standards purchased from Sigma-Aldrich. Chromatography was performed with injections on a 200 μL sample loop and 1 mL min−1 flow rate of THF.Lignin extraction, isolation, and purification
A scheme of the overall procedure is given in the ESI (Fig. S1†). In a 2 L round-bottom flask with condenser, 500 mL methanol and 110 g of pre-ground biomass was combined. The mixture was stirred under reflux conditions for 12 h then cooled to room temperature. Residue was removed by filtration in vacuo and washed with additional small portions of methanol. The filtrate was concentrated to <200 mL then poured over ice in 25 mL aliquots with vigorous stirring, causing light brown solid to precipitate. This lignin (with residual ice) was collected by filtration, washed with a small portion of ice-cold water, then dried under vacuum. The yield of dried, crude lignin was 13.62 g (12% by mass) (ESI, Table S1†).To remove residual fats and resins, 8.39 g of the crude lignin and 150 mL dichloromethane was placed in a 1 L round-bottom flask with condenser and refluxed for 12 h. The mixture of solid and pale yellow liquid was filtered under vacuum and the residue was washed with additional solvent. The recovered solid was then dried yielding 8.077 g purified lignin (96% by mass) which was characterized by GPC (ESI, Table S1 and Fig. S2†).
Lignin fractionation by solvent
Purified lignin (7.29 g) was placed in a 2 L round-bottom flask with condenser and refluxed with 400 mL ethyl acetate for 12 h then cooled to room temperature. The mixture was filtered and the residue was washed with ethyl acetate until the washings were clear. The combined filtrate and washings were evaporated yielding 4.98 g of ethyl acetate-soluble lignin (68% by mass) (ESI, Table S2†). The fractions were characterized by gel permeation chromatography and NMR spectroscopy (ESI, Fig. S2–S4†).Catalyst preparation
The catalyst was prepared based on a previously described procedure for synthesis of hydrotalcite-like materials.24 The precursor contained Cu2+, Mg2+ and Al3+ in the ratio of 0.03![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.12:0.05. The pre-catalyst was calcined for 24 h at 460 °C to yield the PMO, which is nearly amorphous in structure. Representative XRD data for these materials are provided in the ESI (Fig. S5†).
:0.12:0.05. The pre-catalyst was calcined for 24 h at 460 °C to yield the PMO, which is nearly amorphous in structure. Representative XRD data for these materials are provided in the ESI (Fig. S5†).
Catalytic depolymerisation in methanol
In a typical reaction, the desired amount of the dried lignin recovered from ethyl acetate filtrate (as described above) was dissolved in 30 mL methanol and combined with the desired amount of solid PMO catalyst in a 100 mL Parr reactor equipped with a magnetic drive stirrer and controller. The reactor was pressurized with 4 MPa H2 at room temperature then heated to 140–220 °C (as indicated in the Results section) and the desired temperature was established within 30–40 min. The vessel was stirred for the indicated time period at 700 rpm and then cooled in an ice-water bath. Upon reaching room temperature, the pressure was noted and the vessel carefully depressurized. (Caution! Release of flammable vapors!) The reaction mixture was observed as a colorless solution that turned dark upon exposure to air, with purple solids at the bottom of the vessel. The entire contents of the reactor were transferred to centrifuge tubes with additional portions of methanol for rinsing as needed, and the suspensions were centrifuged. The solutions were decanted, the solids were washed with 2 × 10 mL portions of methanol and the washings combined with the decanted liquid. The solvent was removed yielding dark, viscous material that was sampled for NMR spectroscopy and GPC measurements. The solids were further washed with 2 × 5 mL diethyl ether and dried in a desiccator in vacuo prior to determining the mass of the solids.NMR spectral details for a typical crude product mixture consistent with literature data for 4-propylcatechol (1),25 4-(3-hydroxypropyl)catechol (2),26 2,3-dihydro-1H-indene-5,6-diol (3),27 and predicted signals for 4-(3-methoxypropyl)catechol (4).
1H NMR (CD3OD, 500 MHz), δ (ppm): 0.85 (3H, t, J = 14 Hz) (1-CH3); 1.52 (m, 2H, J = 14 Hz) (1-CH2); 1.70–1.75 (m, 2.6H, J = 13 Hz) (2-CH2 and 4-CH2); 1.94 (m, 1.28H, J = 15 Hz) (3-CH2); 2.37 (t, 2H, J = 15 Hz) (1-CH2); 2.44–2.49 (m, 2.88H, J = 16 Hz) (2-CH2 and 4-CH2); 2.68 (t, 2.51H, J = 15 Hz) (3-CH2); 3.26 (b, 0.63H) (4-OCH3); 3.31 (m, 1.79H) (4-CH2); 3.50 (t, 1.52H, J = 13 Hz) (2-CH2); 6.50 (m, 2.51H, J = 15 Hz, 4 Hz); 6.64–6.68 (m, 6.8H).
13C NMR (CD3OD, 125 MHz), δ (ppm): 13.2 (1-CH3); 25.0 (1-CH2); 25.9 (3-CH2); 31.3 (4-CH2); 31.5 (2-CH2Ar and 4-CH2Ar); 32.6 (3-CH2Ar); 34.8 (2-CH2); 37.5 (1-CH2Ar); 57.8 (4-OCH3); 61.5 (2-CH2OH); 72.2 (4-CH2OCH3); 112.1 (Ar-H); 116.1 (Ar-H); 116.2 (Ar-H); 116.5 (Ar-H); 120.6 (Ar-H); 120.7 (Ar-H); 135.0 (Ar-CH2); 135.5 (Ar-CH2); 136.0 (Ar-CH2); 144.0 (Ar-OH); 144.1 (Ar-OH); 144.6 (Ar-OH); 145.9 (Ar-OH); 146.0 (Ar-OH).
Column chromatography on silica gel was performed on crude mixtures of the liquid products, using chloroform–methanol (20:1) as the initial eluent. Fractions were collected in 1–2 mL portions and monitored by TLC. After the first two spots eluted, the solvent ratio was changed to 3:1 and a further set of fractions was collected in 10 mL portions. Methanol was then used to elute any residual organic material into a single fraction.
Results and discussion
Lignin isolation and pre-treatment
This work focuses on a methanol-extracted lignin from the nutshells of candlenut (A. moluccana). Candlenut was selected for several reasons: (1) the nuts are used as a source of oilseed-derived biodiesel; therefore development of the waste material represents progress toward a biorefinery concept; (2) its methanol-extracted lignin has been previously characterized;23 and (3) its high Klason lignin content (∼60%) provides potentially higher extraction yields than would be expected from conventional woody biomass (lignin content ∼22%).21,28 Indeed, we observed that simple extraction with refluxing methanol gave excellent recovery (ESI, Table S1†) compared to previously reported results for sawdust.21 The crude lignin was purified using well-established techniques based on dichloromethane extraction.28 Analysis of the dichloromethane-soluble material by 1H NMR spectroscopy showed that the removed contaminants were not lignin-like but rather aliphatic in nature, with gel permeation chromatography (GPC) giving Mw = 595 g mol−1 and Mn = 242 g mol−1. GPC of the purified lignin showed Mw of 1162 g mol−1 and Mn of 862 (Ð = 1.35). To narrow the dispersity and minimize potential solubility issues, we applied a solvent fractionation technique to remove higher-weight species (scheme is depicted in ESI, Fig. S1†). Thring et al. have previously shown that ethyl acetate, as a relatively polar solvent, dissolved most of an organosolv lignin sample but left a residue that was insoluble due to its high molecular weight.29 Accordingly, in this work we isolated an ethyl acetate-soluble lignin that by GPC showed a Mw of 1133 g mol−1 and Mn of 736 (Ð = 1.18). GPC data and 1H NMR spectra for the lignin fractions are provided in the ESI (Fig. S2–S4, Table S3†). The ethyl acetate-soluble lignin was used as the substrate for all catalytic treatments.Catalyst synthesis and characterization
The PMO catalyst was prepared by calcination of a hydrotalcite-like precursors resulting from modification of the hydrotalcite structure30 with Cu dopant keeping a 1:3 molar ratio of M3+/M2+ by replacing 20 mol% of the Mg2+ ions with Cu2+. Similarly-doped catalysts have been previously used in various biomass transformations.21,22,24 The X-ray power diffraction spectrum of the precursor shows highly ordered crystalline structures with distinct hydrotalcite-like features consistent with literature precedents (ESI, Fig. S5†). The resulting Cu-PMO was found to have a surface area of 142 m2 g−1 by Brunauer–Emmett–Teller (BET) methodology.
Catalytic depolymerisation in subcritical methanol
The solvent-fractionated lignin was completely soluble in the amounts of methanol used for catalytic treatments. Experiments in Table 2 were conducted in the temperature range 140–220 °C with an initial pressure of H2 added. The absolute and relative amounts of lignin and catalyst were varied. Biomass conversion was calculated based on the amount of insoluble organic residue recovered.| Entry | Catalyst (g) | Lignin (g) | Time (h) | T (°C) | Initial H2 pressure (MPa) | Initial volume MeOH (mL) | MeOH-soluble productsb (g) | MeOH-insoluble residuec (g) | Conversiond (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 100 mL vessel, Cu-PMO catalyst unless otherwise noted. b Residual after removal of methanol. c Comprised of catalyst and methanol-insoluble organic material. d Calculated as [mass initial lignin − (mass solid residue − mass catalyst added)/mass initial lignin]. e Cu-free, [Mg/Al]-PMO catalyst used. | |||||||||
| 1 | 0.5 | 1 | 8 | 220 | 4.0 | 30 | 0.468 | 0.844 | 65.6 |
| 2 | 0.5e | 1 | 18 | 220 | 4.0 | 30 | 0.184 | 1.127 | 37.3 |
| 3 | 0.5 | 1 | 14 | 180 | 4.0 | 30 | 0.712 | 0.575 | 92.5 |
| 4 | 0.5 | 1 | 20 | 180 | 4.0 | 30 | 0.702 | 0.623 | 87.7 |
| 5 | 0.25 | 1 | 18 | 180 | 4.0 | 30 | 0.699 | 0.561 | 68.9 |
| 6 | 0.5 | 1 | 18 | 180 | — | 30 | 0.278 | 1.204 | 29.6 |
| 7 | 1 | 1 | 12 | 180 | — | 50 | 0.159 | 1.767 | 23.3 |
| 8 | — | 1 | 18 | 180 | 4.0 | 30 | 0.538 | 0.223 | 77.7 |
| 9 | 0.5 | 1 | 18 | 140 | 4.0 | 30 | 0.633 | 0.787 | 71.3 |
| 10 | 1 | 2 | 20 | 140 | 4.0 | 30 | 0.908 | 1.810 | 59.5 |
| 11 | 3 | 5 | 18 | 180 | 3.0 | 40 | 2.482 | 4.180 | 76.4 |
| 12 | 1 | 4 | 14 | 180 | 6.0 | 37 | 1.656 | 3.696 | 32.6 |
Controls for Cu-free PMO (entry 2), reaction in the absence of H2 (entries 6–7) and reaction in the absence of catalyst (entry 8) and are included in the above results. Comparing entries 1 and 2, it is apparent that the absence of Cu sharply lowers yield of low-molecular weight products; instead a mixture of dark, degraded lignin and char was obtained. Similarly, when the Cu-PMO is used without H2 gas present, yield of methanol-soluble products is poor (entries 6–7). Very low amounts of insoluble organics are seen at 180 °C (entries 3–5) with around 70% recovery of crude soluble products. In the absence of catalyst under similar conditions (entry 8), a higher amount of insoluble organics is obtained. Moderate yields were maintained with a catalyst-to-lignin ratio of 1:4 on a mass basis (entries 5 and 12), lower reaction temperatures (entries 9–10), or lignin loadings up to 5 g (entry 11). It is clear from these results that Cu-PMO in the presence of H2 can minimize formation of high molecular weight products.
Identification of liquid phase products
A comparison of GPC traces for Cu-PMO, Cu-free PMO, and uncatalyzed treatments (Table 2, entries 1–3 and 8) shows that the Cu-promoted reaction leads to sharper GPC peaks, with dramatically lower molecular weights at lower temperature and extended reaction times (ESI, Fig. S6–S8†). Similarly, the NMR spectra of the crude liquid products from the Cu-PMO treatments show clean, distinct signals (ESI, Fig. S9–S11†). This is in contrast to the uncatalyzed reaction (Table 2, entry 8) which gives 1H and 13C NMR spectra appearing relatively unchanged compared to the starting lignin (ESI, Fig. S12–13†). In the Cu-PMO product mixtures, we could assign peaks corresponding to 4 principal components, C9 catechols 1–4 (Fig. 1); the amounts varied depending on reaction conditions and are tabulated in the ESI (Table S4†). Peak assignments were supported by 13C and 1H–13C NMR techniques, and several of the catechols were additionally confirmed by electrospray-mass spectrometry.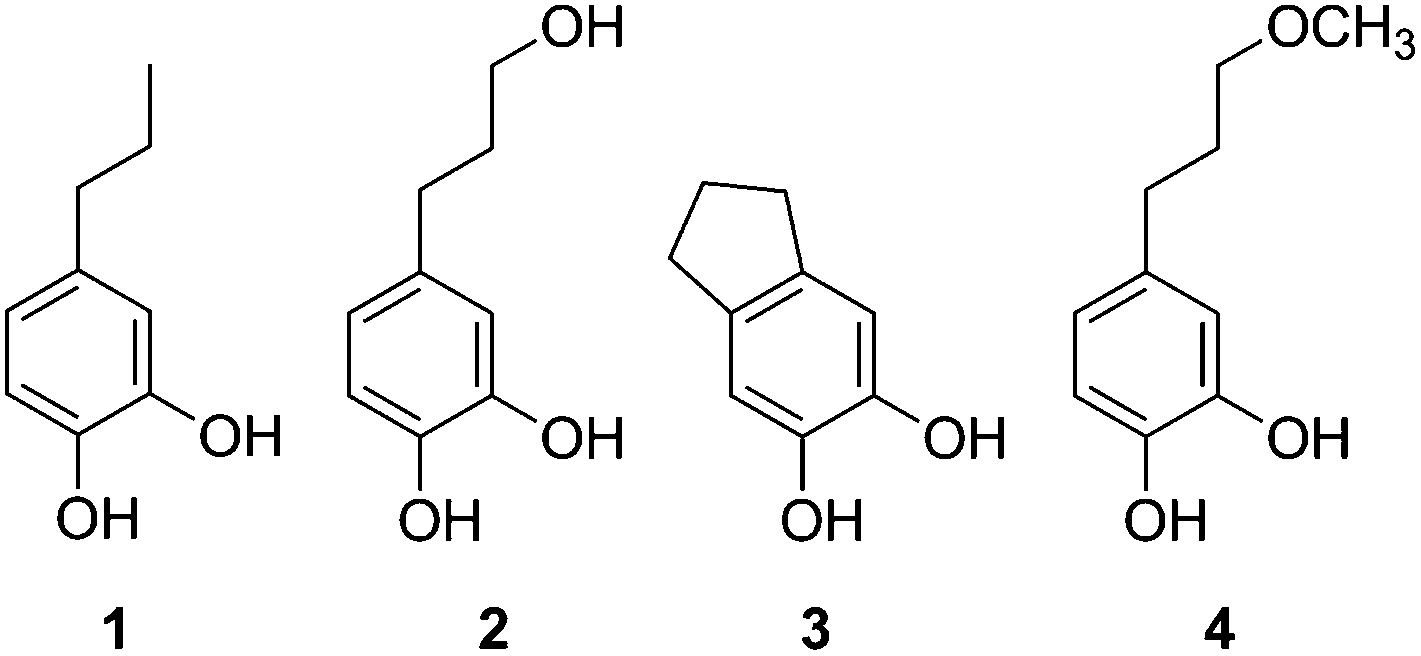 | ||
| Fig. 1 Catechols identified in product mixtures. | ||
These compounds might be formed via a series of steps, for example hydrogenolysis of the common β-O-4 lignin interunit linkage with further hydrogenolysis, dehydration and hydrogenation of the intermediates. Strikingly, aromatic methoxyl groups are cleaved to a very high extent. We observed both higher and lower molecular weight components in the GPC traces, and additional minor peaks in the NMR spectra, which are consistent with the irregular, complex nature of the starting biomass. However, the sharper signals suggest that a limited number of components dominate the Cu-PMO product mixtures. Model compound studies would be necessary to show whether the unique features of candlenut lignin (e.g., relatively low syringyl content)23 are responsible for the observed chemistry. Early indications are that Cu plays an important role in the catalytic mechanism; this is in accordance with the recent findings of Rothenberg et al. who have shown catalytic cleavage of the β-O-4 interunit linkage in lignin models using Cu on Mg and Al oxides.31
Isolation of major products by column chromatography
To obtain purified aromatic compounds, we attempted separation by column chromatography. NMR spectroscopy showed that the first-eluting fractions contained mixtures of catechols 1, 3, and 4 in relatively constant proportion, followed by fractions of 2 alone, and finally higher molecular weight residue (ESI, Fig. S14–S23, Table S4†). Catechol 2 was isolated as a white solid that slowly became beige with prolonged storage. Further attempts to separate 1, 3, and 4via chromatography or Kugelrohr distillation remained unsuccessful. Table 3 shows column conditions and results for separation of liquid products from selected Cu-PMO reactions.| Entry | Catalyst (g) | Lignin (g) | Time (h) | T (°C) | Total yield, catechols 1–4b (%) | Yield, purified catechol 2c (%) |
|---|---|---|---|---|---|---|
| a Column fraction amounts and relative quantities of 1–4 are provided in the ESI. b Includes column fractions containing mixtures of products 1–4 as well as 2 alone; calculated as (mass of isolated catechols/mass crude liquid products applied to column × total mass crude liquid products/mass initial lignin). c Column fractions containing product 2 alone. | ||||||
| 1 | 1 | 1 | 12 | 180 | 49.3 | — |
| 2 | 0.5 | 1 | 14 | 180 | 54.8 | 36.1 |
| 3 | 1 | 2 | 20 | 140 | 63.7 | 43.3 |
There are some literature precedents for generation of catechols from lignins, but they are rarely reported as major components. For example, a variety of catechols including small amounts of C9 compounds have been reported in Kraft pulping liquor.32 Catechols have also been obtained under oxidizing conditions, for example in fungal metabolism of lignins,33 and model compound studies have shown that chemical oxidants can yield catechols via cleavage of methoxyl groups.34 Catechol 1 has been detected as a minor component of product mixtures derived from lignin via pyrolysis35 or supercritical water processing.36 Catechol 2 has been reported as a natural product extractable from leaves and twigs,37 and oxygenated indane derivatives have been found in pyrolysis oils,38 but to the best of our knowledge our results are the first example of catechols 2–4 isolated from lignin.
Conclusions
Cu-PMO in the presence of H2 and methanol under mild conditions achieves near-complete conversion of lignin, high yields of bio-derived liquids, and unusual selectivity for C9 catechols that would typically be derived from petroleum feedstocks via multi-step synthetic pathways. Cu plays an important role, minimizing char formation and apparently favouring the required hydrogenolysis, dehydration, and hydrogenation steps without reducing the aromatic rings, to obtain catechols from lignin biopolymer. These results represent progress in obtaining lignin-derived aromatic compounds that could potentially serve as renewable-carbon alternatives in chemical and material applications.Acknowledgements
This work was supported by the USDA National Institute of Food and Agriculture, Critical Agricultural Materials program, award #2010-38202-21853.Notes and references
- T. Werpy and G. Petersen, ed., Top Value Added Chemicals from Biomass, Volume I – Results of Screening for Potential Candidates from Sugars and Synthetic Gas, Pacific Northwest National Laboratory, National Renewable Energy Laboratory, Office of Biomass Program, 2004 Search PubMed.
- T. Werpy and G. Petersen, ed., Top Value Added Chemicals from Biomass, Volume II – Results of Screening for Potential Candidates from Biorefinery Lignin, Pacific Northwest National Laboratory and the National Renewable Energy Laboratory, 2007 Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- E. S. Beach, Z. Cui and P. T. Anastas, Energy Environ. Sci., 2009, 2, 1038–1049 CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- H. Kobayashi, H. Ohta and A. Fukuoka, Catal. Sci. Technol., 2012, 2, 869–883 CAS.
- V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. Li and J. A. Lercher, Chem.–Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- K. M. Torr, D. J. van de Pas, E. Cazeils and I. D. Suckling, Bioresour. Technol., 2011, 102, 7608–7611 CrossRef CAS PubMed.
- W. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, ChemSusChem, 2012, 5, 667–675 CrossRef CAS PubMed.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttamaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Chem. Sci., 2013, 4, 806–813 RSC.
- T. E. Graedel, R. Barr, C. Chandler, T. Chase, J. Choi, L. Christoffersen, E. Friedlander, C. Henly, C. Jun, N. T. Nassar, D. Schechner, S. Warren, M.-y. Yang and C. Zhu, Environ. Sci. Technol., 2011, 46, 1063–1070 CrossRef PubMed.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994 CAS.
- X. Zhang, T. Wang, L. Ma, Q. Zhang, Y. Yu and Q. Liu, Catal. Commun., 2013, 33, 15–19 CrossRef CAS PubMed.
- A. Toledano, L. Serrano, A. M. Balu, R. Luque, A. Pineda and J. Labidi, ChemSusChem, 2013, 6, 529–536 CrossRef CAS PubMed.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- A. P. Klein, E. S. Beach, J. W. Emerson and J. B. Zimmerman, J. Agric. Food Chem., 2010, 58, 10045–10048 CrossRef CAS PubMed.
- T. S. Hansen, K. Barta, P. T. Anastas, P. C. Ford and A. Riisager, Green Chem., 2012, 14, 2457–2461 RSC.
- A. Ozanne, L. Pouységu, D. Depernet, B. François and S. Quideau, Org. Lett., 2003, 5, 2903–2906 CrossRef CAS PubMed.
- A. Madrid Villegas, L. Espinoza Catalan, I. Montenegro Venegas, J. Villena Garcia and H. Carrasco Altamirano, Molecules, 2011, 16, 4632–4641 CrossRef PubMed.
- C. Huang, N. Ghavtadze, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 17630–17633 CrossRef CAS PubMed.
- E. E. Harris, J. D'Ianni and H. Adkins, J. Am. Chem. Soc., 1938, 60, 1467–1470 CrossRef CAS.
- R. W. Thring, M. N. Vanderlaan and S. L. Griffin, J. Wood Chem. Technol., 1996, 16, 139–154 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- Z. Strassberger, A. H. Alberts, M. J. Louwerse, S. Tanase and G. Rothenberg, Green Chem., 2013, 15, 768–774 RSC.
- K. Niemelä, Holzforschung, 1989, 43, 99 CrossRef.
- T. K. Kirk and E. Adler, Acta Chem. Scand., 1969, 23, 705–707 CrossRef CAS PubMed.
- Y.-P. Sun, A. Wallis and K. L. Nguyen, J. Wood Chem. Technol., 1997, 17, 209–222 CrossRef.
- L. Liguori and T. Barth, J. Anal. Appl. Pyrolysis, 2011, 92, 477–484 CrossRef CAS PubMed.
- H. Pińkowska, P. Wolak and A. Złocińska, Chem. Eng. J., 2012, 187, 410–414 CrossRef PubMed.
- S.-C. Chien, H.-K. Liu and Y.-H. Kuo, Chem. Pharm. Bull., 2004, 52, 762–763 CAS.
- D. W. Page, J. Anal. Appl. Pyrolysis, 2003, 70, 169–183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GPC traces and details of the extraction and purification of lignin from candlenut nutshells; spectroscopic details of low- and high-molecular weight lignins; representative powder XRD data for synthesized hydrotalcite-like catalyst precursor; GPC and NMR spectroscopic data for representative product mixtures after catalytic treatment in the supercritical regime. See DOI: 10.1039/c3gc41184b |
| This journal is © The Royal Society of Chemistry 2014 |