Superparamagnetic γ-Fe2O3 nanoparticles as an easily recoverable catalyst for the chemical recycling of PET†
Leian
Bartolome
a,
Muhammad
Imran
b,
Kyoung G.
Lee
c,
Arvin
Sangalang
a,
Jeong Keun
Ahn
d and
Do Hyun
Kim
*a
aDepartment of Chemical & Biomolecular Engineering (BK21 Program), KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 305-701, Republic of Korea. E-mail: DoHyun.Kim@kaist.ac.kr; Fax: +82 42 350 3910; Tel: +82 42 350 3929
bChemical Engineering Department, King Saud University, Saudi Arabia
cDepartment of Chemical Engineering, University of Michigan, Ann Arbor, MI 48109-2136, USA
dDepartment of Microbiology, School of Bioscience & Biotechnology, Chungnam National University, Daejeon, 305-764, Republic of Korea
First published on 7th October 2013
Abstract
There have been numerous studies to develop catalysts for the chemical recycling of poly(ethylene terephthalate) (PET) via glycolysis. However, in the field of PET glycolysis, only a few have attempted to recover and reuse the catalysts. This research utilized easily recoverable superparamagnetic γ-Fe2O3 nanoparticles as a reusable catalyst for PET glycolysis. γ-Fe2O3 nanoparticles were produced by calcining Fe3O4 nanoparticles prepared by the co-precipitation method. The produced γ-Fe2O3 nanoparticles had an average size of 10.5 ± 1.4 nm, and a very high surface area reaching 147 m2 g−1. Its superparamagnetic property was also confirmed. Glycolysis reactions were carried out, and the γ-Fe2O3 catalysts were recovered after the reactions by simple magnetic decantation. The use of magnetic iron oxide allowed the easy recovery of the catalyst from the glycolysis products. At 300 °C and a 0.05 catalyst/PET weight ratio, the maximum bis(2-hydroxyethlyl) terephthalate (BHET) monomer yield reached more than 90% in 60 min. At 255 °C and a 0.10 catalyst/PET weight ratio, the BHET yield reached more than 80% in 80 min. The catalyst was reused 10 times, giving almost the same BHET yield each time.
1. Introduction
Poly(ethylene terephthalate), more commonly known as PET, is an indispensable material due to its excellent physical and chemical properties. It is an important material in the textile industry, and in food packaging where it has noticeably become the choice for beverage containers.1 Due to its extensive use, the increasing consumption of PET has led to the creation of large amounts of waste, inspiring research on recycling. In view of sustainability, chemical recycling, which leads to the formation of the raw materials from which PET was originally made, remains the most viable option.2Various processes have been developed and utilized in PET chemical recycling. Among them, glycolysis is the most established and popular, because of its simplicity and low cost. In addition, glycolysis produces the monomer BHET, which can be used in both dimethyl terephthalate (DMT) and terephthalic acid (TPA) production lines to make virgin PET, or other advanced materials.2
Studies on the kinetics show that without catalysts, PET glycolysis is very slow, and complete conversion of PET to monomer BHET is almost impossible.3,4 Metal acetates were the first reported catalysts for PET glycolysis.5–8 Later, researchers developed more environment-friendly alternatives like mild alkalies,9,10 sulfates,10,11 metal chlorides,12 and zeolites.13 However, even with these aforementioned catalysts, the PET glycolysis still required long reaction times, and gave low BHET monomer yields. These catalysts could only increase the glycolysis rate at temperatures below the melting point of PET, and had no catalytic activity in melt-phase glycolysis due to mass transfer limitations.14 A list of all the studied catalysts for PET glycolysis can be found in previous work.15
Recently, our group has developed thermally stable and highly selective silica nanoparticle-supported metal (Mn, Zn, Ce) oxide16,17 and graphene oxide–manganese oxide18 catalysts for melt-phase PET glycolysis. Catalysts fabricated at nanoscale have increased catalytic efficiency, due to the increased number of active sites. In addition, the change of intrinsic properties of the catalysts at nanoscale usually leads to better catalytic performance.19 As a result, the metal oxide catalysts gave a higher monomer yield, exceeding 90%, at a shorter reaction time compared to the other previously studied catalysts.16
In spite of previous research efforts, there is still a problem as the toxic metals in these catalysts are harmful when released to the environment. The difficulty of separation of nanomaterials after the reaction makes the problem even more challenging. The failure to separate the catalyst from the glycolysis products not only poses environmental risks, but may also change the properties of the products. Thus, there has been a need to find a catalyst that is both efficient and easily recoverable.
There have been efforts recently to utilize recoverable catalysts for PET glycolysis, yet there are still issues that need to be addressed. Wang et al. have reported that various ionic liquid catalysts for PET glycolysis could be recovered, and then used repeatedly.20–22 In a separate study, Yue et al. used basic ionic liquids.23 These ionic liquid catalysts had less than 80% BHET yield, which is evidently lower than the yield from metal oxide catalysts.16–18 More recently, Wang et al. reported urea to be a reusable catalyst with good low-temperature catalytic activity for the glycolysis of PET.24 However, the catalyst was recovered using vacuum distillation, which is an energy intensive process. Thus, to date, the issues of catalyst efficiency and environmental sustainability have not yet been resolved simultaneously. Eco-friendly catalysts, such as mild alkalies and chlorides, show less efficiency compared to the not-so-eco-friendly ones (e.g. metal oxides) usually containing toxic metals.
In this paper, we present superparamagnetic γ-Fe2O3 nanoparticles as an efficient and environment-friendly catalyst for a sustainable PET chemical recycling. γ-Fe2O3 nanoparticles are easily recoverable and reusable. By recovering the catalyst, the danger of releasing harmful catalyst components to the environment is reduced. By reusing the catalyst, the process becomes more cost-effective and sustainable.
As a reusable catalyst, γ-Fe2O3 nanoparticles have shown good performance in a number of reactions.25–27 However, to the best of our knowledge, superparamagnetic γ-Fe2O3 nanoparticles have never been used in any glycolysis reactions, particularly in PET. In fact, to date, no magnetic nanomaterial has been applied as a recoverable and reusable solid catalyst in PET glycolysis. Among the magnetic materials, we selected nano-γ-Fe2O3, because of its stability, high catalytic activity and magnetic susceptibility.25 In addition, it is cheap, nontoxic, environment-friendly and abundant. Most importantly, due to its superparamagnetic property, it can be easily recovered by simple magnetic decantation.
2. Experimental
Materials
For the preparation of γ-Fe2O3 nanoparticles, FeCl2 and FeCl3 precursors were purchased from Sigma-Aldrich. HCl, citric acid granules, and NaOH pellets were from Junsei Chemical. For the PET glycolysis, PET pellets were provided by HUVIS. The pellets were ground to fine powder with a mean diameter of 150 μm. Ethylene glycol (EG) and the standard BHET were purchased from Sigma-Aldrich, and THF used as a solvent was purchased from J. T. Baker. The chemicals were used without any further purification. Deoxygenated deionized (DI) water was used all throughout the experiment.Synthesis of γ-Fe2O3 as a catalyst and its characterization
The γ-Fe2O3 nanoparticles were produced by calcining Fe3O4 nanoparticles prepared through the traditional co-precipitation method28,29 with slight modifications. To make the Fe3O4 nanoparticles, an aqueous solution of Fe2+/Fe3+ with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 was prepared by dissolving pre-determined amounts of FeCl2 and FeCl3 precursors in an HCl solution (0.4 M, 25 mL). To this Fe2+/Fe3+ aqueous solution, NaOH (1.5 M, 250 mL) was added rapidly with vigorous stirring. The addition of NaOH solution instantly produced a black precipitate, a clear indication that Fe3O4 had formed. Citric acid (0.2 M, 50 mL) was added as a surfactant to control the nanoparticle size. This procedure was done under a nitrogen atmosphere.
:2 was prepared by dissolving pre-determined amounts of FeCl2 and FeCl3 precursors in an HCl solution (0.4 M, 25 mL). To this Fe2+/Fe3+ aqueous solution, NaOH (1.5 M, 250 mL) was added rapidly with vigorous stirring. The addition of NaOH solution instantly produced a black precipitate, a clear indication that Fe3O4 had formed. Citric acid (0.2 M, 50 mL) was added as a surfactant to control the nanoparticle size. This procedure was done under a nitrogen atmosphere.
The black Fe3O4 precipitate was then separated via magnetic decantation. It was washed with water, and centrifuged at 4000 rpm for 4 min. This was done four times. Finally, the precipitate was washed with 0.01 M HCl solution to neutralize the anionic charges on the surface of the nanoparticles.30 The obtained Fe3O4 precipitate was dried and then calcined at 210 °C for 3 hours to produce γ-Fe2O3.
The morphology of the produced nanoparticles was observed using a JEOL JEM-2100F 200 kV transmission electron microscope (TEM). Its specific surface area was estimated using the N2 adsorption isotherm obtained by a Micrometrics ASAP 2020 BET surface area and porosimetry analyzer. To evaluate the crystallinity of the material, X-ray diffraction (XRD) spectra with 2θ ranging from 10° to 80° were obtained using a RIGAKU D/MAX-2500(18 kW) Micro-Area X-ray Diffractometer. X-ray photoelectron spectroscopy (XPS) measurements were carried out using a Thermo VG Scientific Sigma Probe Multi Purpose X-Ray Photoelectron Spectrometer. To confirm its superparamagnetic nature, a Lakeshore EM4-HVA Vibrating Sample Magnetometer (VSM) was used to obtain the magnetization curve of the nanoparticles. A thermogravimetric curve was obtained using a NETCH TG209F3 Thermogravimetric Analyzer (TGA). Temperature profile desorption with NH3 as a molecular probe (NH3-TPD) was also done to investigate the acidity of the produced material.
PET glycolysis using γ-Fe2O3
The glycolysis procedure was based on previous studies.17,18 To compare the performance of γ-Fe2O3 with other previously reported metal oxides, glycolysis reactions were carried out in a 10 mL stainless steel batch-type reactor at 300 °C, and approximately 1.1 MPa. A mixture of 0.3 g of virgin PET, 1 mL of 99.9% anhydrous EG, and a designated amount of catalyst (3, 4.5, 6, 15 mg) were loaded into the reactor, which was then placed in a furnace. After the desired reaction time (40–80 min), the reactor was removed from the furnace and quenched with cold water.The performance of γ-Fe2O3 was also compared with urea. The glycolysis reactions were carried out at 255 °C, with a 0.1 catalyst/PET weight ratio.
The BHET monomer yield was measured with Agilent 1000 series Reverse Phase High Performance Liquid Chromatography (HPLC). The use of HPLC for the qualitative and quantitative analysis of the BHET monomer was discussed in detail in previous work.28 After quenching the reaction, the glycolysis product was dissolved in 20 mL THF. From this solution, the catalysts were recovered via magnetic decantation and reused. For the HPLC sample, 0.1 mL was taken out from the solution and diluted to 24 mL 50/50 (v/v) THF–H2O solution. This solution was used in the HPLC analysis. The UV detector was set to 254 nm, and the flow rate of the mobile phase was set to 1.0 mL min−1. The mobile phase used was 50/50 (v/v) THF–H2O solution. The BHET monomer yield was calculated using the equation:
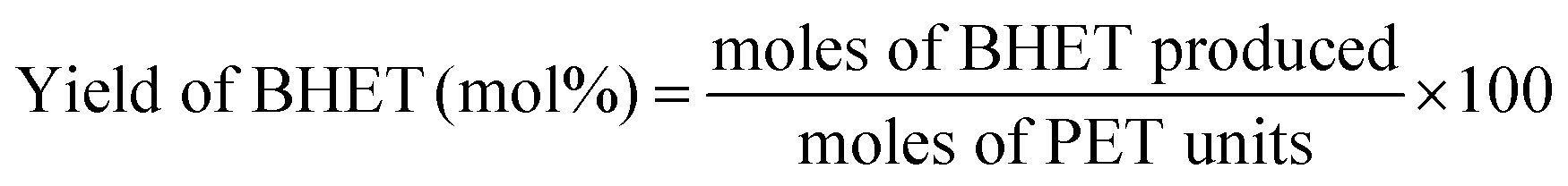
Recovery of the γ-Fe2O3 catalyst and BHET
To evaluate the recoverability of the catalyst, another set of glycolysis reactions were carried out. After the reaction, the glycolysis product was dissolved in water at 90 °C, in which BHET, its dimers, and EG are soluble. From this solution, the catalyst was recovered via simple magnetic decantation. The recovered catalyst was washed with deoxygenated DI water three times, dried, and finally weighed to measure the amount recovered.BHET from the glycolysis product was also recovered for further characterization. After the catalyst was recovered, the solution was kept at 90 °C for 45 min under vigorous stirring. The PET residual was separated from the mixture through filtration. When the resulting filtrate reached 60 °C, it was re-filtered to remove the dimers from the BHET monomers. The resulting filtrate from the second filtration was refrigerated for 24 h, and then filtered to recover the monomer BHET. The recovered BHET crystals were dried, and characterized by Nuclear Magnetic Resonance Spectroscopy (NMR).
Reuse of the γ-Fe2O3 catalyst
The reusability of the γ-Fe2O3 catalyst is another key parameter in PET recycling. The recovered catalyst was reused 10 times under the same reaction conditions.3. Results and discussion
γ-Fe2O3 catalyst characterization
For the application of γ-Fe2O3 as a catalyst, it is important to use superparamagnetic nanoparticles. The superparamagnetic property of the catalyst is essential for the reversible control of dispersion and flocculation. Small crystallites of ferromagnetic and ferrimagnetic materials (ranging from 1 to 30 nm) exhibit superparamagnetism.31,32 In this research, slight modifications of the conventional co-precipitation method were made in order to obtain γ-Fe2O3 nanoparticles small enough to exhibit superparamagnetism. The TEM images and the particle size histogram of the produced γ-Fe2O3 in Fig. 1 show that the size of the nanoparticles ranges from 8 to 14 nm, with an average size of 10.5 nm ± 1.4 nm. The small size and monodispersity of the produced γ-Fe2O3 nanoparticles resulted from (a) the initiation of rapid nucleation by adding the base to the Fe2+/Fe3+ solution instead of adding the latter dropwise to the former as in the traditional co-precipitation method, and (b) the control of crystal growth through the addition of citric acid surfactant.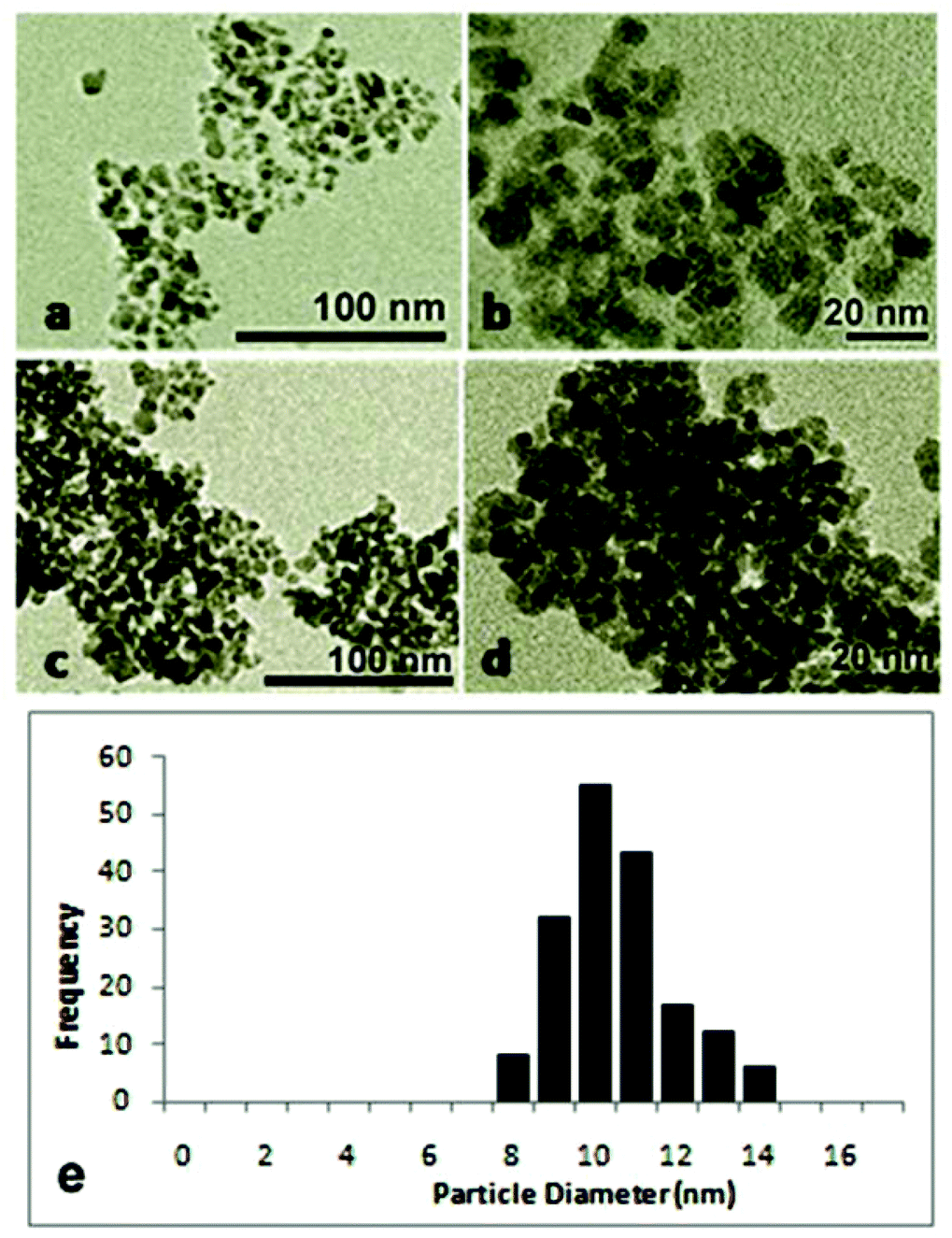 | ||
| Fig. 1 TEM images of (a, b) pristine and (c, d) used γ-Fe2O3, and (e) the TEM histogram for the pristine catalyst. | ||
Due to the small size of the γ-Fe2O3 nanoparticles produced, it exhibited superparamagnetism, as confirmed by the magnetization curve from the VSM. It can be seen in Fig. 2 that when magnetic field was applied, the material showed a strong response, and the saturation magnetization reached 47 emu g−1. More importantly, the curve did not have any hysteresis loop, a unique property of superparamagnetic materials. The absence of hysteresis loop implies that the catalyst will re-disperse when used again in the subsequent reactions.
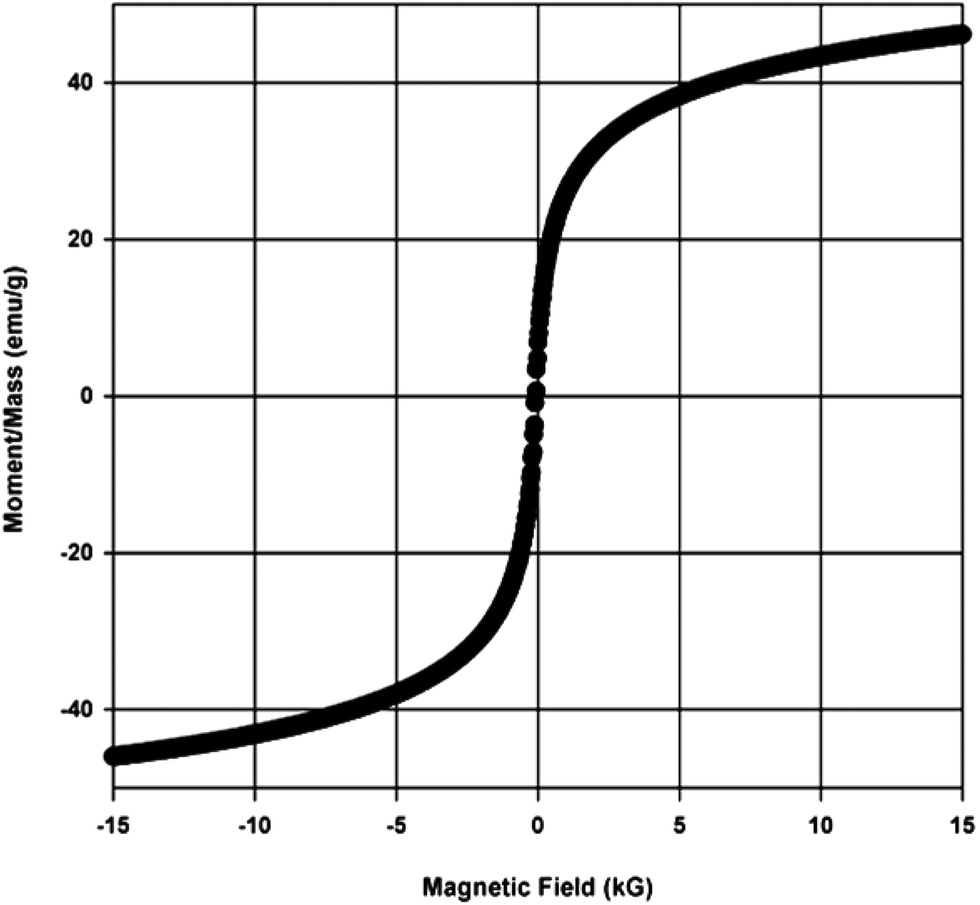 | ||
| Fig. 2 Magnetization curve of the synthesized γ-Fe2O3 confirming its superparamagnetic nature. | ||
The XRD pattern in Fig. 3a shows sharp peaks, with major peaks at (311) and (440), indicating good crystallinity of the obtained product. The peaks in the XRD pattern coincided with those of standard γ-Fe2O3 (JCPDS 39-1346, space group P4132). The XRD pattern suggests that the material is γ-Fe2O3. However, since Fe3O4 and γ-Fe2O3 have the same crystalline structure, they give the same XRD pattern. Hence, further analysis was done to confirm the nature of the material. The confirmation of the transformation of Fe3O4 during the calcinations to γ-Fe2O3 is important, since Fe3O4 is unstable at high temperatures, and may affect the glycolysis reaction; starting at 200 °C, Fe3O4 transforms to γ-Fe2O3, and at 375 °C, γ-Fe2O3 begins to transform to α-Fe2O3.33
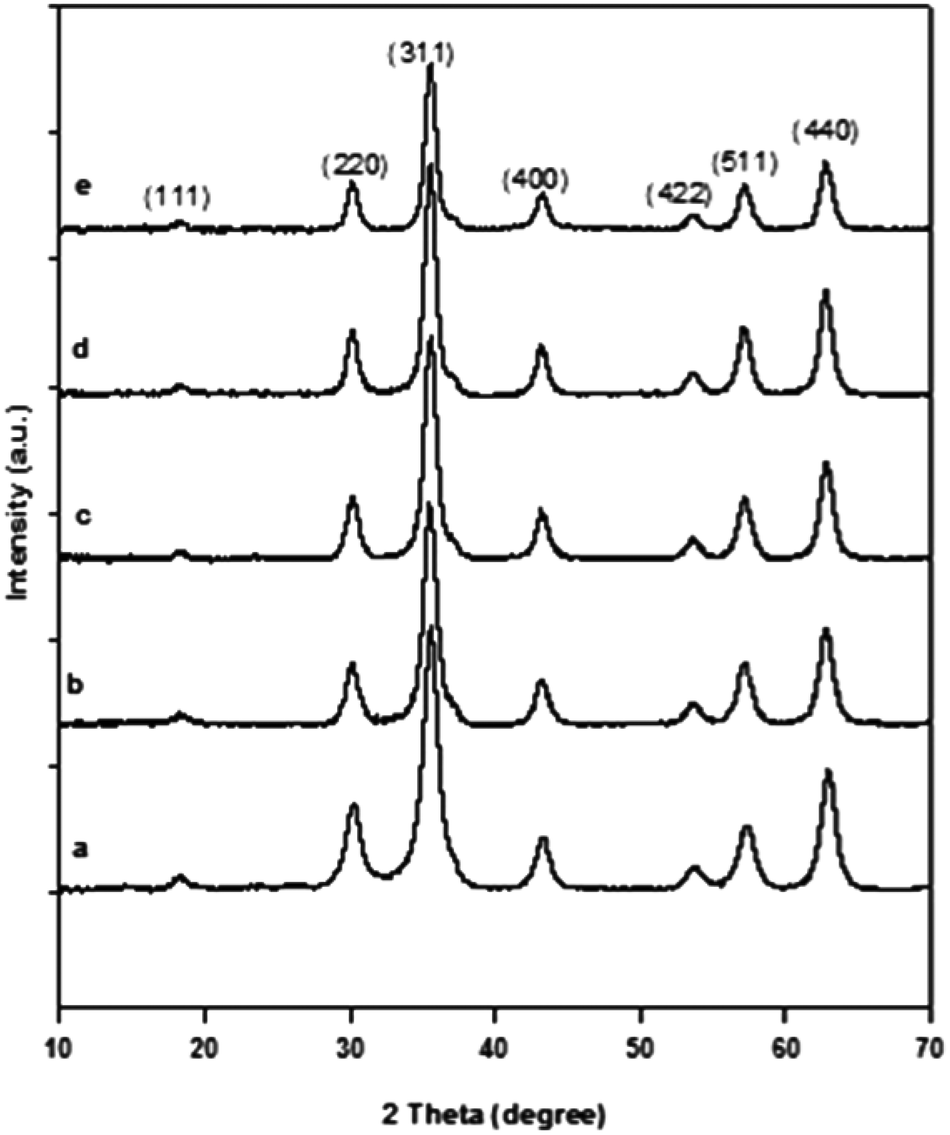 | ||
| Fig. 3 XRD pattern of (a) pristine γ-Fe2O3, and after the (b) first, (c) third, (d) sixth, (e) and ninth time of use with diffraction peaks corresponding to γ-Fe2O3 (JCPDS 39-1346, space group P4132). | ||
The unstable nature of Fe3O4 was also one of the reasons why γ-Fe2O3 was used as a catalyst instead of Fe3O4. To confirm that the produced nanoparticles after calcination were indeed γ-Fe2O3 and not Fe3O4, XPS was done. The Fe2p XPS patterns of the material before and after calcinations are presented in Fig. 4. Before calcination, the Fe2p3/2 and Fe2p1/2 were located at approximately 711.6 eV and 724.8 eV, respectively. After calcination, they were located at approximately 710.9 eV and 724.2 eV, respectively. The set of higher binding energies before calcination (Fig. 4a) is a characteristic of Fe3O4. The shift to relatively lower binding energies after the calcination implies the transformation from Fe3O4 to γ-Fe2O3. Additionally, the satellite peak at approximately 720 eV in Fig. 4b is characteristic of γ-Fe2O3.34 Thus, the Fe3O4 nanoparticles produced from the precipitation method did transform to γ-Fe2O3 during calcination.
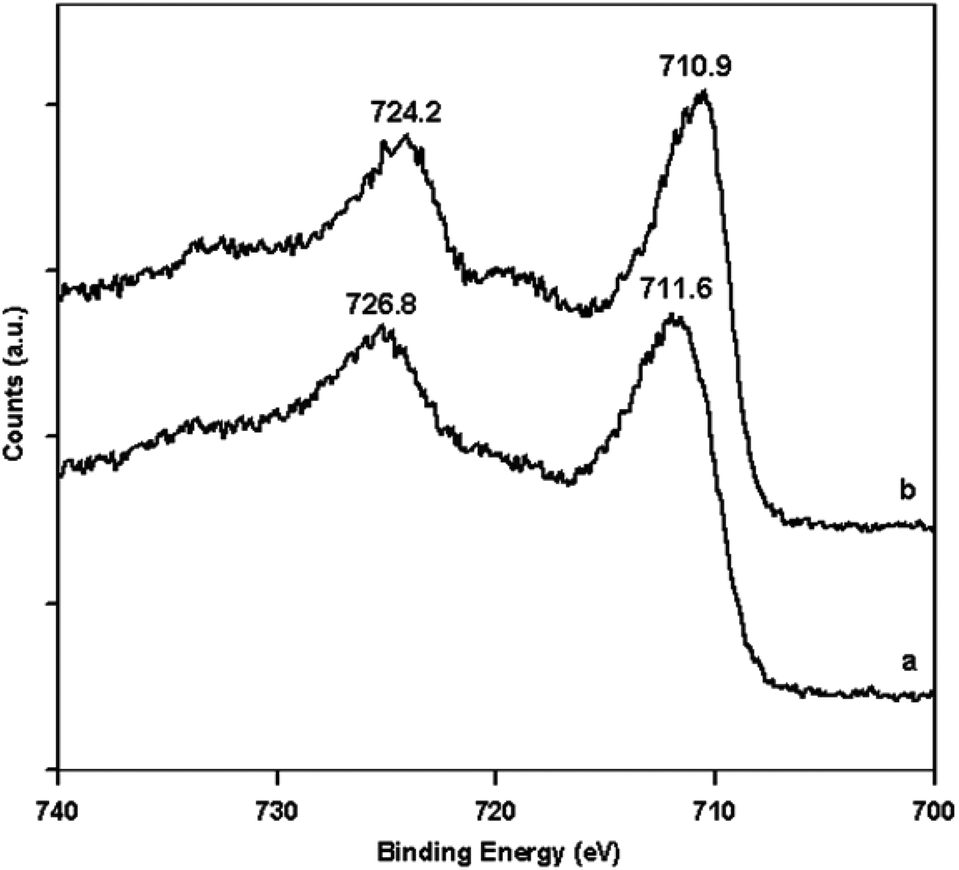 | ||
| Fig. 4 Fe2p XPS patterns of the material (a) before and (b) after calcinations. | ||
The thermal stability of the catalyst is also essential in melt-phase glycolysis, because it affects catalyst performance and reusablity. The thermo-gravimetric curve in Fig. 5 shows only one stage of weight loss. The drop in weight below 200 °C is a result of the evaporation of the adsorbed water from the surface. Aside from this, there were no other transformations observed, indicating that the material did not degrade, and maintained its oxidation state at temperatures up to 800 °C. Thus, the thermal stability of the catalyst was confirmed. Although transformation from γ-Fe2O3 to α-Fe2O3 is also possible, it did not occur during the reactions as confirmed by the XRD spectrum of the used catalyst shown in Fig. 3b–3e.
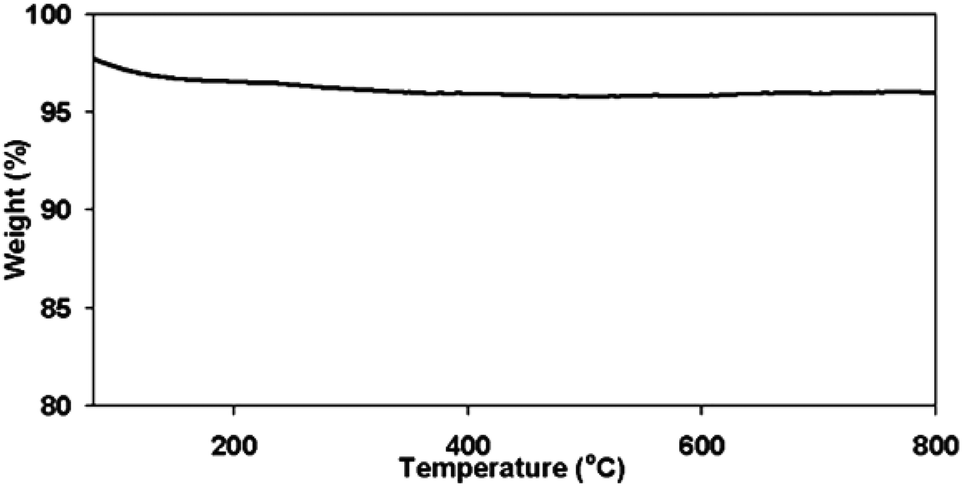 | ||
| Fig. 5 Thermogravimetric analysis for γ-Fe2O3 showing weight loss only at temperature below 200 °C. | ||
Reaction mechanism of γ-Fe2O3-catalyzed glycolysis
Previous studies show that in a transesterification reaction as in PET glycolysis, metal oxides can act as either base catalysts or acid catalysts. The positive metal ions in the structure of metal oxides possess Lewis acidity and the negative oxygen ions behave as Brönsted bases. When acting as a base catalyst, the metal oxide provides sufficient adsorption sites for the alcohol (i.e. ethylene glycol) to transform it into catalytically active (and more nucleophilic) species RO−. The catalytically active species RO− then attacks the carbonyl group in the ester. In the case of PET glycolysis, the RO− attack will lead to the breaking of the PET chain, producing oligomers and dimers, then finally to BHET monomers. On the other hand, when acting as an acidic catalyst, the metal oxide protonates the carbonyl group of the ester, making the carbonyl group more electrophilic. When the carbonyl group is more electrophilic, it is more vulnerable to nucleophilic attack.35 In the case of PET glycolysis, the carbonyl carbon will be more vulnerable to the attack of nucleophile EG, resulting in the breaking of the PET chain.36 The reaction mechanism of γ-Fe2O3 catalyzed PET glycolysis can therefore be postulated by determining if γ-Fe2O3 acts as an acidic or a basic catalyst.The acidity of the γ-Fe2O3 catalyst was evaluated using NH3-TPD analysis, where NH3 is adsorbed on the surface of the material at low temperature, and desorbed at higher temperature. The temperature at which NH3 desorbs depends on the strength of the acid sites in the material.37 The TPD profile in Fig. 6 shows three distinct peaks. By deconvolution, the peaks were determined to be at 177.0, 282.0, and 384.8 °C. The peak at 177.0 °C corresponds to weak acid sites, while the intense peaks at 282.0 °C and 306.0 °C correspond to high concentrations of medium strength acid sites. Thus, the γ-Fe2O3 nanoparticles act predominantly as a mildly acidic catalyst. From this, the glycolysis reaction mechanism for PET glycolysis with γ-Fe2O3 acting as an acidic catalyst may be postulated (Fig. 7). The carbonyl group of the polyester is more vulnerable to attack by any foreign species. The metal (Fe+3) interacts with the oxygen of the carbonyl group and drags more electrons towards itself, creating highly partial positive charge on the carbon. A free lone pair on the EG oxygen then attacks the carbonyl carbon of the ester group of the polyester, resulting in the formation of C–O bond with the oxygen of the EG while breaking the C–O bond of the polyester. In this way, the longer PET chains break down to shorter chain oligomers, dimers, and finally to monomer BHET.36 The transformation of dimers to BHET monomers is a reversible process. Longer reaction after the equilibration of the two is attained will cause the reaction to shift backwards, increasing the amount of dimers and oligomers at the expense of the BHET monomer.30 It is thus important to determine the optimum reaction time for the glycolysis reaction.
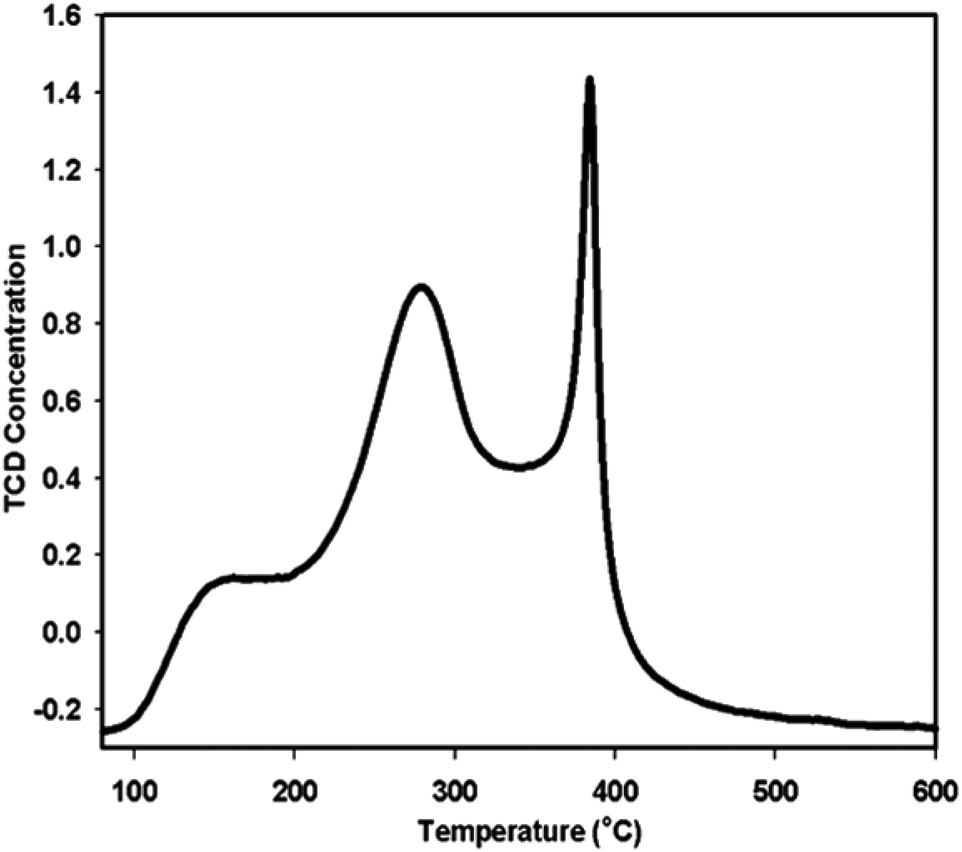 | ||
| Fig. 6 NH3-TPD pattern of γ-Fe2O3. | ||
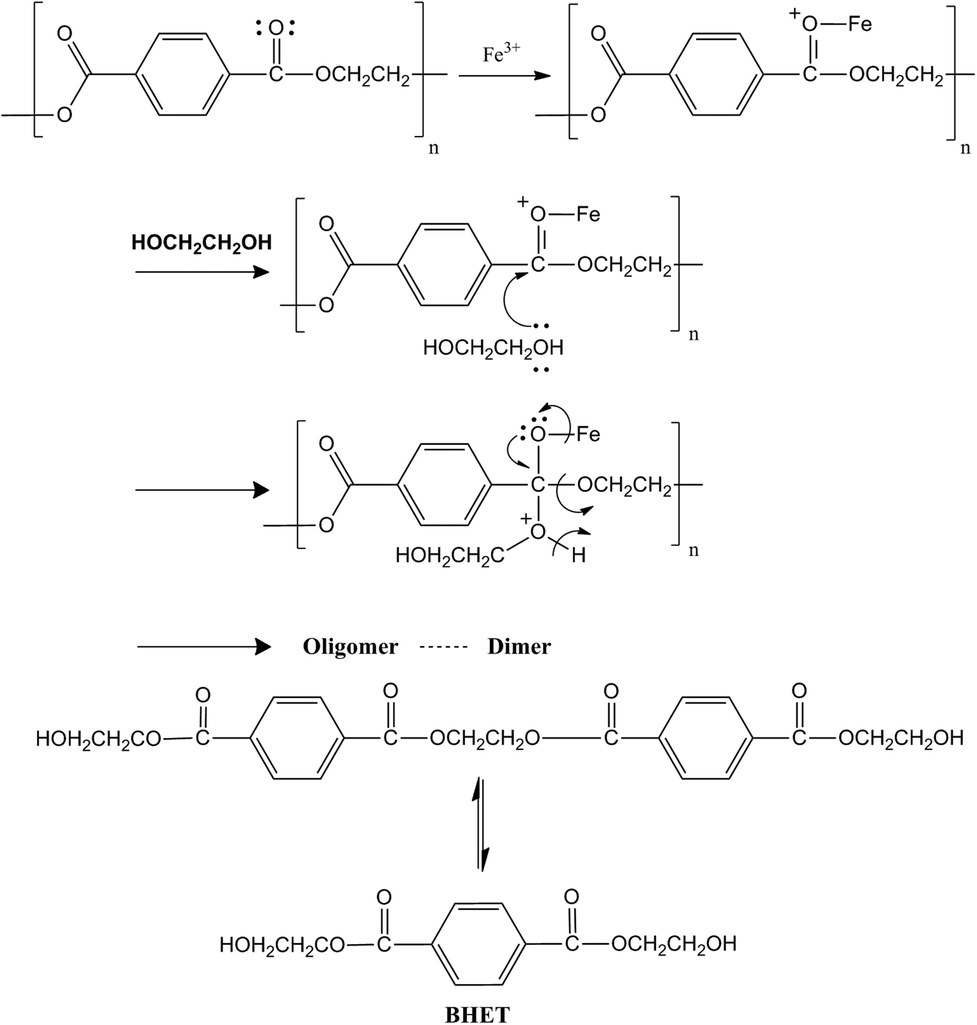 | ||
| Fig. 7 Proposed mechanism for PET glycolysis with γ-Fe2O3 as a catalyst. | ||
γ-Fe2O3 catalyst performance
To evaluate the performance of the catalyst, the BHET monomer yield was measured via HPLC. HPLC, together with the NMR spectra (see Fig. S1†), also confirmed that the main product of the reaction was BHET. The elution time of the main peak (2.8 min) for the monomer product matched with that of the standard BHET (Fig. 8). Fig. 9 shows the BHET yield from the HPLC analysis for different amounts of catalyst. The significant difference between the monomer yield of the reaction with and without γ-Fe2O3 shows that the nanoparticles successfully catalyzed the reaction. Moreover, under the same reaction conditions and catalyst/PET weight ratio with those of the previously reported silica nanoparticles-supported metal oxide catalysts such as ZnO, Mn3O4, and CeO2,16,17 γ-Fe2O3 yielded the same degree of performance (Fig. 10). At a 0.010 catalyst/PET weight ratio, the BHET yield reached higher than 90% in 70 min. The remarkable catalytic performance of the γ-Fe2O3 nanoparticles may be credited to the high surface area, which renders greater accessibility to the active sites. The measured surface area of the nanoparticles in this research is 147 m2 g−1; bulk γ-Fe2O3 has about only 9 m2 g−1.24 In comparison, the high catalytic efficiency (high monomer yield in our case) with metal oxide catalysts may also be attributed to the nature of the metal cation, its oxidation state, reduction potential, its distribution in the crystal structure, and the metal oxide spinel geometry. The γ-Fe2O3 (maghemite) possesses an inverse cubic spinel structure with the formula (Fe+3)[Fe+340/3□1/8] O32, where (Fe+3) and [Fe+3] represent the tetrahedral and octahedral coordination of Fe+3 in the spinel structure. The □ presents the iron vacancy sites on the octahedra.38 Cheng et al.39 reported that the type of spinel structure (cubic, tetragonal) explained the significant effect of the oxygen binding capability on the catalyst surface. The faster attack of Fe+3 cations on the oxygen of carbonyl group of polyester due to their distribution on tetrahedral and octahedral sites combined with the inverse cubic spinel structure is therefore anticipated.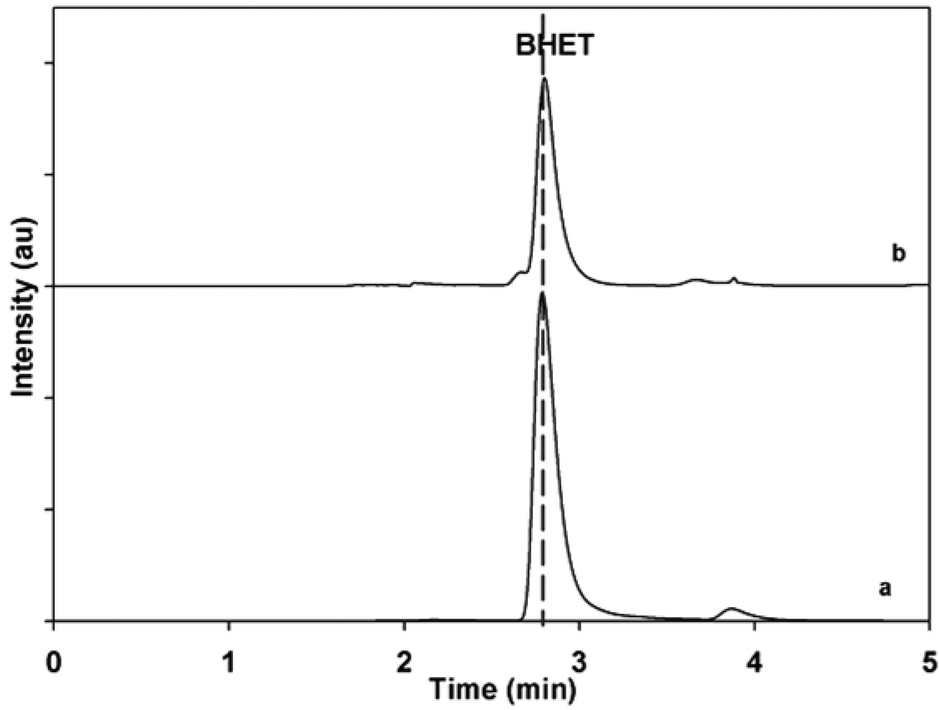 | ||
| Fig. 8 HPLC spectra of (a) standard BHET and (b) the glycolysis product. | ||
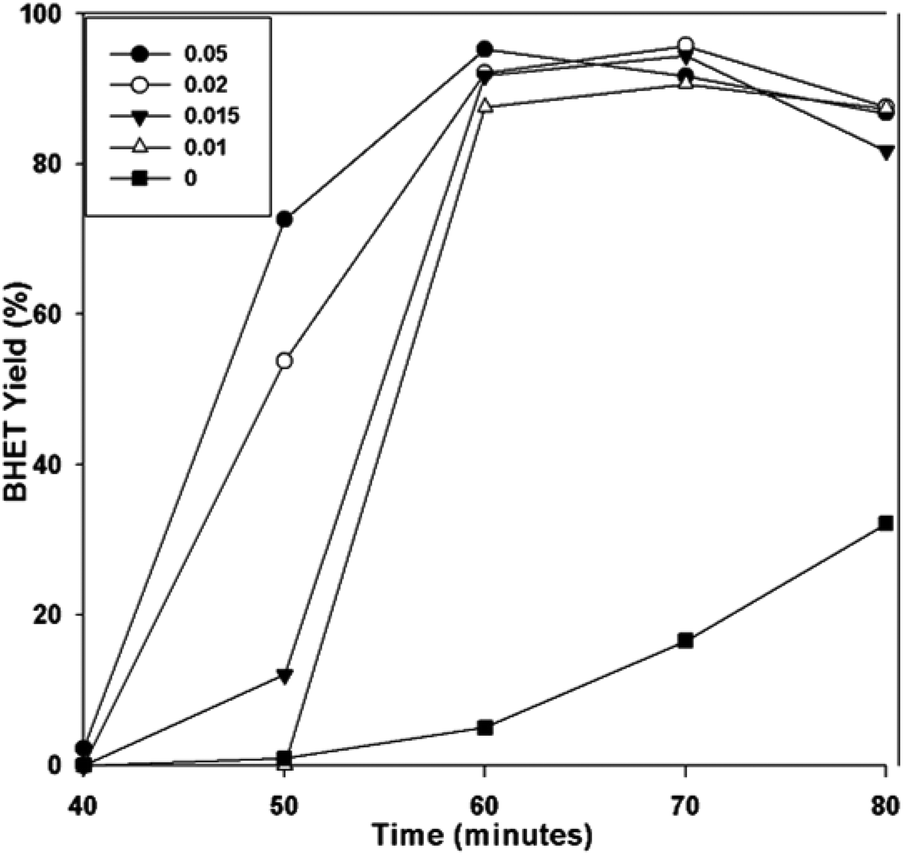 | ||
| Fig. 9 BHET yield with time for different catalyst/PET weight ratios. | ||
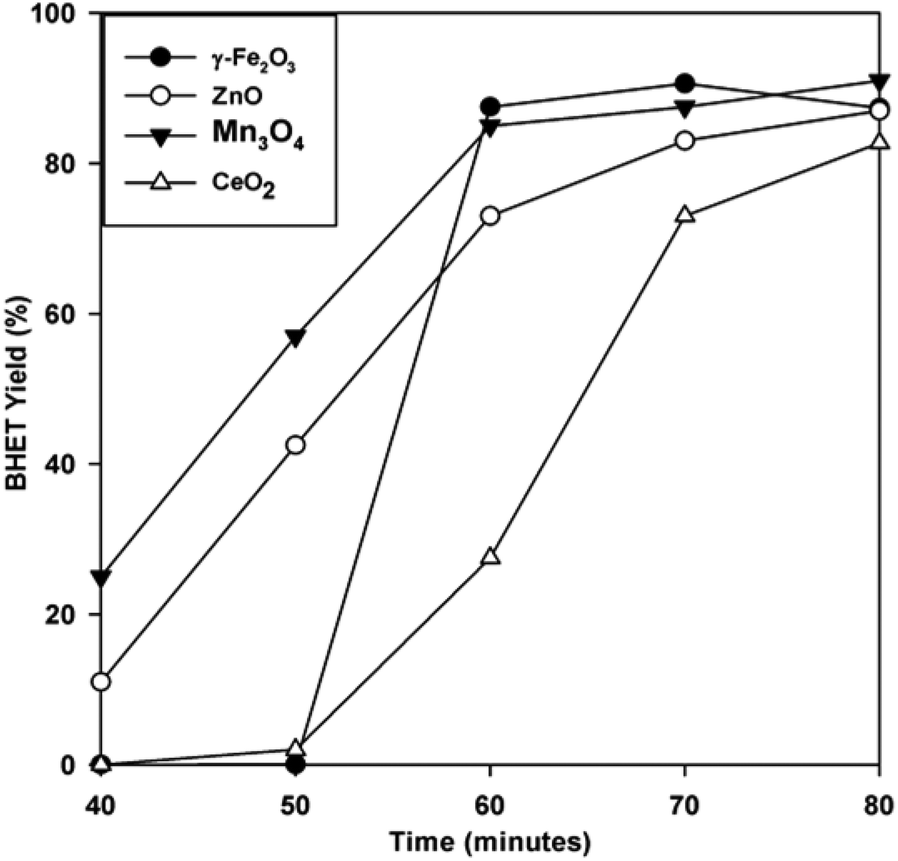 | ||
| Fig. 10 BHET yield with time for glycolysis with γ-Fe2O3 and various silica nanoparticle-supported metal oxides16,17 at a 0.01 catalyst/PET weight ratio and 300 °C. | ||
It is also observed from Fig. 9 that for the reactions with different amounts of catalyst, maximum BHET yield was reached at different times. The possibility of the catalyst's easy recovery and reuse allowed the use of a higher amount of catalyst. As a result, the higher catalyst/PET weight ratio permitted the reaction to reach equilibrium earlier. The glycolysis reaction with a 0.05 catalyst/PET weight ratio only required 60 min to achieve BHET yield greater than 90%. The shorter reaction time translates to a more energy-efficient and cost-effective process compared to the other metal oxide nanocatalysts studied.
We also evaluated the performance of γ-Fe2O3 at milder temperature, and compared its performance with that of another recently reported recyclable catalyst, urea (Fig. 11). It is apparent that γ-Fe2O3 increased the glycolysis reaction rate. At 255 °C, the γ-Fe2O3-catalyzed reaction gave >80% BHET yield in only 80 min. It also showed better performance compared to urea, which attained the maximum BHET yield in 120 min. Thus, Fe2O3 is effective even at mild temperatures.
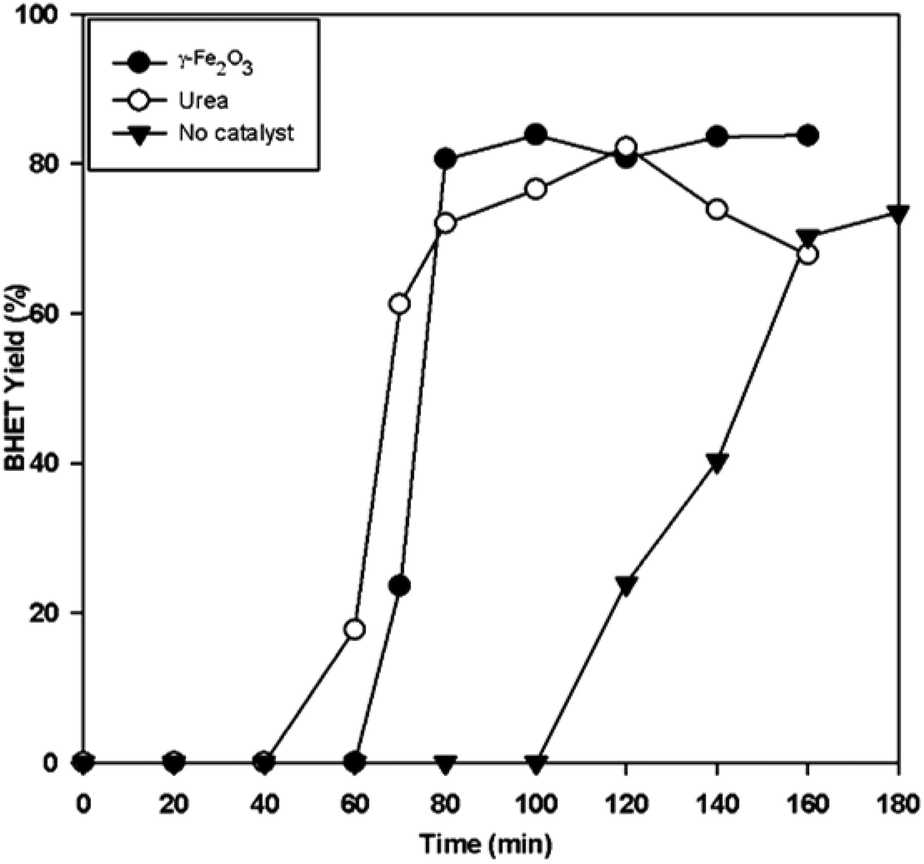 | ||
| Fig. 11 BHET yield with time for non-catalyzed, γ-Fe2O3 catalyzed, and urea-catalyzed glycolysis at 255 °C, and a 0.1 catalyst/PET weight ratio. | ||
For industrial applications, the catalyst should be easily separated from the product mixture. The superparamagnetic property of γ-Fe2O3 nanoparticles provides a unique condition to serve as a catalyst for PET glycolysis as well as easy separation from the product using a magnet. The separation process does not require any energy-intensive or complicated separation method. The average amounts of recovered catalysts are presented in Table 1. There is a minimal amount of lost catalyst with increased amounts of catalyst used. Since the 15 mg catalyst delivered the highest glycolysis and recovery rate, this amount was used for the evaluation of the reusability of the catalyst. Each time the catalyst was reused, the yield was almost the same (Fig. 12), without any significant drop. Statistical analysis using analysis of variance (ANOVA) at 99.9% confidence level showed that the number of times the catalyst was reused did not significantly affect the BHET monomer yield (see Table S1†). This result confirmed the reusability of the catalyst, which may be attributed to its stability, as evidenced by the TGA (Fig. 5), and the XRD spectra of the used catalyst in Fig. 3b. Thus, the repeated use of the γ-Fe2O3 catalyst for PET glycolysis at high temperature does not affect its performance as a catalyst.
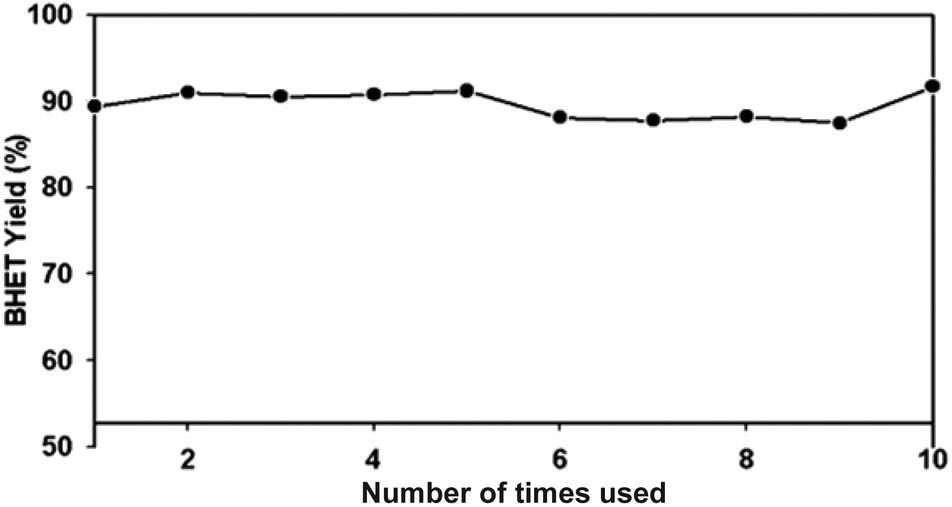 | ||
| Fig. 12 BHET yield for re-used catalyst. | ||
| Catalyst/PET weight ratio | Amount of catalyst (g) | Amount of recovered catalyst (g) | Amount of lost catalyst (g) | % Recovery |
|---|---|---|---|---|
| 0.01 | 3 | 2.6 | 0.4 | 87 |
| 0.015 | 4.5 | 4.1 | 0.4 | 91 |
| 0.02 | 6 | 5.2 | 0.8 | 87 |
| 0.05 | 15 | 14.1 | 0.9 | 94 |
Conclusions
The application of superparamagnetic γ-Fe2O3 nanoparticles as an easily recoverable and reusable catalyst for PET glycolysis was evaluated. The use of a magnetic iron oxide catalyst facilitated easy recovery. Results showed that γ-Fe2O3 delivered excellent performance in catalytically depolymerizing PET back to monomer BHET. This may be attributed to its ability to catalyze the glycolysis through redox reactions, high surface area rendering more active sites, thermal stability, and good crystallinity. Also, the easy recoverability and reusability of the γ-Fe2O3 permitted the use of a larger amount of catalyst for the glycolysis reaction. The higher catalyst/PET weight ratio resulted in a better performance, speeding up the reaction, and thereby reaching maximum monomer yield (>90% BHET) at an earlier time (60 min), which translates to a more energy-efficient and cost-effective process. The catalyst also proved to be reusable, making the process more sustainable and environment-friendly.Acknowledgements
This work was supported by the Basic Science Research Program through a National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (2010-0025671).Notes and references
- F. Welle, Conservation Recycling, 2011, 55, 865 CrossRef PubMed.
- D. Achilias and G. Karayannidis, Water, Air, Soil Pollut., 2004, 4, 385 CrossRef CAS.
- J. Campanelli, M. Kamal and D. Cooper, J. Appl. Polym. Sci., 1994, 54, 1731 CrossRef CAS.
- J. Chen and L. Chen, J. Appl. Polym. Sci., 1999, 73, 35 CrossRef CAS.
- S. Baliga and W. Wong, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2071 CrossRef CAS.
- M. Ghaemy and K. Mossaddegh, Polym. Degrad. Stab., 2005, 90, 570 CrossRef CAS PubMed.
- C. Chen, J. Appl. Polym. Sci., 2003, 87, 2004 CrossRef CAS.
- G. Xi, M. Lu and C. Sun, Polym. Degrad. Stab., 2005, 87, 117 CrossRef CAS PubMed.
- S. Shukla and K. Kulkarni, J. Appl. Polym. Sci., 2002, 85, 1765 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022 CrossRef PubMed.
- S. Shukla and A. Harad, J. Appl. Polym. Sci., 2005, 97, 513 CrossRef CAS.
- N. Pingale, V. Palekar and S. Shukla, J. Appl. Polym. Sci., 2010, 115, 249 CAS.
- S. Shukla, V. Palekar and N. Pingale, J. Appl. Polym. Sci., 2008, 110, 501 CrossRef CAS.
- C. Y. Kao, W. H. Cheng and B. Z. Wan, Thermochim. Acta, 1997, 292, 95 CrossRef CAS.
- L. Bartolome, M. Imran, B. G. Cho, W. Al-Masry and D. H. Kim, Material Recycling – Trends and Perspectives, InTech, 2012 Search PubMed.
- M. Imran, K. G. Lee, Q. Imtiaz, B. Kim, M. Han, B. Cho and D. Kim, J. Nanosci. Nanotechnol., 2011, 11, 824 CrossRef CAS PubMed.
- R. Wi, M. Imran, K. G. Lee, S. Yoon, B. Cho and D. Kim, J. Nanosci. Nanotechnol., 2011, 11, 6544 CrossRef CAS PubMed.
- G. Park, L. Bartolome, K. G. Lee, S. J. Lee, D. H. Kim and T. J. Park, Nanoscale, 2012, 4, 3879 RSC.
- U. Heiz and U. Landman, Nanocatalysis, Springer-Verlag, New York, 2007 Search PubMed.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568 RSC.
- H. Wang, Z. Li, Y. Liu, X. Zhang, S. Zhang and Y. Zhang, Eur. Polym. J., 2009, 45, 1535 CrossRef CAS PubMed.
- H. Wang, R. Yan, Z. Li, X. Zhang and S. Zhang, Catal. Commun., 2010, 11, 763 CrossRef CAS PubMed.
- Q. Yue, Z. Wang, L. Zhang, Y. Ni and Y. Jin, Polym. Degrad. Stab., 2011, 96, 399 CrossRef CAS PubMed.
- Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559 RSC.
- N. Koukabi, E. Kolvari, A. Khazaei, M. Zolfigol, B. Shirmadi-Shaghamesi and H. Khavasi, Chem. Commun., 2011, 47, 9230 RSC.
- A. Garade, M. Bharadwaj, S. Bhagwat, A. Athawale and C. Rode, Catal. Commun., 2009, 10, 485 CrossRef CAS PubMed.
- G. Picasso, A. Quintilla, M. Pina and J. Herguido, Appl. Catal., B, 2003, 46, 133 CrossRef CAS.
- Y. Kang, S. Risbud, J. Rabolt and P. Stroeve, Chem. Mater., 1996, 8, 2209 CrossRef CAS.
- J. Jeong, S. Lee, J. Kim and S. Shin, Phys. Status Solidi, 2004, 241, 1593 CrossRef CAS.
- M. Imran, B. Kim, M. Ha, B. Cho and D. Kim, Polym. Degrad. Stab., 2010, 95, 1686 CrossRef CAS PubMed.
- K. Troev, G. Grancharov, R. Tsevi and I. Gitsov, J. Appl. Polym. Sci., 2003, 90, 1148 CAS.
- N. Burke, H. Stover and F. Dawson, Chem. Mater., 2002, 14, 4752 CrossRef CAS.
- H. Lepp, The American Minerologist, 1957, 42, 679 CAS.
- J. Lu, X. Jiao, D. Chen and W. Li, J. Phys. Chem., 2009, 113, 4012 CAS.
- A. A. Refaat, Int. J. Environ. Sci. Tech., 2011, 8, 203 CrossRef CAS.
- J. Scheirs and T. E. Long, Modern Polyesters: Chemistry and Technology of Polyesters and Copolyesters, John Wiley and Sons, Ltd, West Sussex, 2003 Search PubMed.
- D. Dahlan, I. Marsih, I. Makertihartha, P. Praserthadam, J. Panpranot and I. Ismunander, Chem. Mater. Res., 2012, 2, 31 Search PubMed.
- V. Hiremath and A. Venkataraman, Bull. Mater. Sci., 2003, 26, 391 CrossRef CAS.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ANOVA for BHET yield, 13C and 1H NMR spectra of the BHET obtained from the glycolysis reaction. See DOI: 10.1039/c3gc41834k |
| This journal is © The Royal Society of Chemistry 2014 |